
EMEND®
APREPITANT
Cápsulas
1 Caja, 1 Cápsulas, 125 Miligramos
1 Caja, 2 Cápsulas, 80 Miligramos
1 Caja, 1+2 Cápsulas, 125+80 mg/mg
III. FORMA FARMACÉUTICA Y FORMULACIÓN
Cápsula.
El aprepitant es un antagonista de los receptores neurocinina 1 (NK1) de la sustancia P estructuralmente nuevo, cuyo nombre químico es 5-[[(2R,3S)-2-[(1R)-1-[3,5-bis(trifluorometil)fenil]etoxi]-3-(4-fluorofenil)-4-morfolinil]metil]-1,2-dihidro-3H-1,2,4-triazol-3-ona.
Su fórmula empírica es C23H21F7N4O3, y su fórmula estructural es:
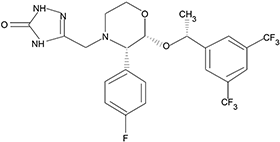
El aprepitant es un sólido cristalino blanco o blanquecino, y tiene un peso molecular de 534.43. Es prácticamente insoluble en agua, escasamente soluble en etanol y en acetato de isopropilo, y ligeramente soluble en acetonitrilo.
Ingredientes activos
Cada cápsula de EMEND para administración por vía oral contiene 80 mg o 125 mg de aprepitant.
Ingredientes inactivos
Cada cápsula de EMEND contiene los siguientes ingredientes inactivos: sacarosa, celulosa microcristalina, hidroxipropilcelulosa y lauril sulfato de sodio. La cápsula está compuesta de gelatina y dióxido de titanio, puede contener lauril sulfato de sodio y dióxido de silicio. La cápsula de 125 mg también contiene óxido férrico rojo y óxido férrico amarillo.
XIX. NÚMERO DE REGISTRO
Reg. No. 130M2003 SSA IV
Tracer Number: MK0869-MEX-2016-012919
IV. INDICACIONES TERAPÉUTICAS
EMEND está indicado, en combinación con otros fármacos antieméticos, para la prevención de la náusea y el vómito agudos y tardíos asociados con los ciclos inicial y siguientes de:
• quimioterapia anticancerosa altamente emetogénica (véase XIII. DOSIS Y VÍA DE ADMINISTRACIÓN).
• quimioterapia anticancerosa moderadamente emetogénica (véase XIII. DOSIS Y VÍA DE ADMINISTRACIÓN).
V. FARMACOCINÉTICA Y FARMACODINAMIA
Mecanismo de acción
El aprepitant tiene un mecanismo de acción único; es un antagonista selectivo de gran afinidad en los receptores neurocinina 1 (NK1) de la sustancia P. Las pruebas de contraselección mostraron que es por lo menos 3,000 veces más selectivo por los receptores NK1 que por otras enzimas, transportadores, canales de iones y sitios receptores, incluyendo los receptores de dopamina y de serotonina, en los cuales actúan los tratamientos existentes contra la náusea y el vómito inducidos por quimioterapia (NVIQ) y contra la náusea y el vómito postoperatorios (NVPO).
En las pruebas preclínicas se ha mostrado que los antagonistas de los receptores NK1 inhiben por acciones centrales el vómito inducido por fármacos quimioterapéuticos citotóxicos (como el cisplatino). Estudios preclínicos y en seres humanos mediante tomografía por emisión de positrones han mostrado que aprepitant penetra en el tejido cerebral y ocupa los receptores NK1. Los estudios preclínicos muestran que aprepitant tiene una acción central prolongada, inhibe tanto la fase aguda como la tardía del vómito inducido por el cisplatino, y aumenta la actividad antiemética del antagonista de los receptores de serotonina ondansetrón y del corticosteroide dexametasona contra el vómito inducido por el cisplatino.
PROPIEDADES FARMACOCINÉTICAS
Absorción
El promedio de biodisponibilidad absoluta de aprepitant (cápsulas de 80 o 125 mg) es de aproximadamente 60 a 65%, y el de concentración plasmática máxima (Cmáx) ocurrió a las cuatro horas aproximadamente (Tmáx). La administración oral de la cápsula con un desayuno estándar no tuvo ningún efecto de importancia clínica sobre la biodisponibilidad de aprepitant.
La farmacocinética de aprepitant no es lineal en el rango de las dosis clínicas. En adultos jóvenes sanos, el aumento del ABC0-∞ fue 26% mayor que el proporcional a la dosis entre las dosis únicas de 80 mg y de 125 mg administradas después de ingerir alimentos.
Tras la administración oral de una sola dosis de 125 mg de EMEND el Día 1 y de 80 mg una vez al día los Días 2 y 3, el ABC0-24 h fue aproximadamente de 19.5 μg• h/mL el Día 1 y de 20.1 μg• h/mL el Día 3. Concentraciones máximas de 1.5 μg/mL el Día 1 y de 1.4 μg/mL el Día 3 se alcanzaron en cuatro horas (Tmáx) aproximadamente.
Distribución
Más del 95% de aprepitant se une a las proteínas plasmáticas. El promedio geométrico del volumen aparente de distribución en el estado de equilibrio es de 66 litros aproximadamente en los seres humanos.
El aprepitant pasa a través de la placenta en las ratas, y a través de la barrera hematoencefálica en las ratas y los hurones. Los estudios de tomografía por emisión de positrones indican que también atraviesa la barrera hematoencefálica en los seres humanos (véase V. FARMACOCINÉTICA Y FARMACODINAMIA, Mecanismo de acción).
Metabolismo
El aprepitant experimenta una extensa transformación metabólica. En adultos jóvenes sanos constituye aproximadamente el 24% de la radiactividad en el plasma en las 72 horas siguientes a la administración de una sola dosis oral de 300 mg de [14C]-aprepitant, lo cual indica una considerable presencia de metabolitos en el plasma. En el plasma humano se han identificado siete metabolitos de aprepitant, los cuales son sólo débilmente activos. El aprepitant se metaboliza en gran parte por oxidación en el anillo morfolínico y en sus cadenas laterales. Estudios in vitro con microsomas hepáticos humanos indican que aprepitant es metabolizado principalmente por CYP3A4, poco por CYP1A2 y CYP2C19, y nada por CYP2D6, CYP2C9 o CYP2E1.
Eliminación
El aprepitant es eliminado principalmente por transformación metabólica; no es excretado por los riñones. Tras la administración de una sola dosis oral de 300 mg de [14C]-aprepitant a sujetos sanos, se recuperó el 5% de la radiactividad en la orina y el 86% en las heces.
La depuración plasmática aparente de aprepitant varió entre 60 y 84 mL/min, y su vida media terminal aparente varió entre nueve y trece horas.
Características en los pacientes
Sexo
Tras la administración oral de una sola dosis de EMEND, el ABC 0-24 h y la Cmáx de aprepitant es 9% y 17% mayor, respectivamente, en las mujeres que en los hombres. Su vida media es aproximadamente 25% menor en ellas, y el Tmáx es aproximadamente el mismo en los dos sexos. Esas diferencias no se consideran clínicamente importantes. No es necesario ajustar la dosificación de EMEND según el sexo del paciente.
Pacientes de edad avanzada
Después de administrar por vía oral una sola dosis de 125 mg de EMEND el Día 1 y 80 mg una vez al día los Días 2 a 5, el ABC0-24 h de aprepitant fue 21% mayor el Día 1 y 36% mayor el Día 5 en los pacientes de edad avanzada (≥ 65 años) que en los menores de esa edad. La Cmáx fue 10% mayor el Día 1 y 24% mayor el Día 5 en personas mayores en comparación con adultos más jóvenes. Esas diferencias no se consideran clínicamente importantes. No es necesario ajustar la dosificación de EMEND en los pacientes de edad avanzada.
Niños
No se ha determinado la farmacocinética de EMEND en pacientes menores de 18 años.
Raza
Tras la administración oral de una sola dosis de EMEND en pacientes Hispanos, el ABC0-24 h de aprepitant es aproximadamente 27% mayor que en los de raza blanca y 31% mayor que en los de raza negra. En los pacientes de origen hispanoamericano la Cmax de aprepitant es 19% mayor que en los de raza blanca y 29% mayor que en los de raza negra. La administración de una dosis única de aprepitant oral en pacientes Asiáticos resultó en un aumento del 74% y 47% del ABC0-24 y Cmax, respectivamente, en comparación con los pacientes Caucásicos. Esas diferencias no se consideran clínicamente importantes. No es necesario ajustar la dosificación de EMEND según la raza del paciente.
Índice de Masa Corporal (IMC)
El Índice de Masa Corporal (IMC) no tuvo ningún efecto clínicamente significativo en la farmacocinética del aprepitant.
Insuficiencia hepática
EMEND fue bien tolerado en pacientes con insuficiencia hepática de leve a moderada. Tras la administración oral de una sola dosis de 125 mg de EMEND el Día 1 y de 80 mg una vez al día los Días 2 y 3 a pacientes con insuficiencia hepática leve (puntuación de Child-Pugh de 5 a 6), el ABC0-24 h de aprepitant fue 11% menor el Día 1 y 36% menor el Día 3, que en los sujetos sanos que recibieron esas mismas dosis. En los pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh de 7 a 9), el ABC0-24 h de aprepitant fue 10% mayor el Día 1 y 18% mayor el Día 3 que en los sujetos sanos que recibieron esas mismas dosis. Esas diferencias en el ABC0-24 h no se consideran clínicamente importantes, por lo que no es necesario ajustar la dosificación de EMEND en los pacientes con insuficiencia hepática de leve a moderada.
No hay datos clínicos o farmacocinéticos en pacientes con insuficiencia hepática ver (puntuación de Child-Pugh mayor de 9).
Insuficiencia renal
Se administró una sola dosis de 240 mg de EMEND a pacientes con insuficiencia renal grave (depuración de creatinina <30 mL/min) y a pacientes con nefropatía terminal que requerían hemodiálisis.
En comparación con los sujetos sanos, en los pacientes con insuficiencia renal grave el ABC0-∞ de aprepitant total (el libre y el unido a las proteínas) disminuyó 21% y la Cmáx disminuyó 32%, y en los pacientes con nefropatía terminal sometidos a hemodiálisis el ABC0-∞ de aprepitant total disminuyó 42% y la Cmáx disminuyó 32%. Debido a pequeñas disminuciones de la unión de aprepitant a las proteínas en los pacientes con nefropatía, en los pacientes con insuficiencia renal el ABC de aprepitant libre farmacológicamente activo no cambió significativamente en comparación con los sujetos sanos. La hemodiálisis efectuada 4 o 48 horas después de la administración no tuvo ningún efecto significativo sobre la farmacocinética de aprepitant; se recuperó del dializado menos del 0.2% de la dosis.
No es necesario hacer ningún ajuste de la dosificación de EMEND para los pacientes con insuficiencia renal grave o con nefropatía terminal tratados con hemodiálisis.
FARMACODINAMIA
Fosaprepitant es el profármaco de aprepitant, cuando se administra intravenoso es rápidamente convertido a aprepitant.
Electrofisiología cardiaca
En un estudio de distribución al azar, doble ciego, con control positivo, de QTc completo, una dosis única de 200 mg de fosaprepitant no tuvo efecto en el intervalo QTc.
Ocupación del receptor NK1 cerebral Evaluado por tomografía por Emisión de Positrones
En un estudio de tomografía por emisión de positrones, en hombres jóvenes saludables, se administró vía oral una dosis única de 165 mg de aprepitant o una dosis de 150 mg de fosaprepitant demostrando una ocupación del receptor NK1 cerebral a un Tmax, (≥ 99%), 24 horas (≥ 99%), 48 horas (≥ 97%), y 120 horas (37 a 76%) posterior a la dosis. La ocupación de los receptores NK1 cerebrales por aprepitant se correlaciona bien con las concentraciones plasmáticas del mismo.
ESTUDIOS CLÍNICOS
PREVENCIÓN DE LA NÁUSEA Y EL VÓMITO INDUCIDOS POR QUIMIOTERAPIA (CINV por sus siglas en inglés)
En estudios clínicos bien controlados, se ha mostrado que la administración oral de EMEND en combinación con ondansetrón y dexametasona previene la náusea y el vómito agudos y tardíos asociados con la quimioterapia altamente emetogénica (HEC por sus siglas en inglés) y moderadamente emetogénica (MEC por sus siglas en inglés).
Quimioterapia altamente emetogénica (HEC por sus siglas en inglés)
En dos estudios clínicos multicéntricos comparativos, doble ciego, de grupos paralelos y con distribución al azar, se comparó el régimen con aprepitant con el tratamiento control en 1,094 pacientes tratados con quimioterapia que incluía cisplatino a dosis de 70 mg/m2 o más. Algunos de esos pacientes recibían también otros fármacos quimioterapéuticos, como gemcitabina, etopósido, fluorouracilo, tartrato de vinorelbina, doxorrubicina, ciclofosfamida, paclitaxel, o docetaxel. El régimen con aprepitant consistió en 125 mg de EMEND el Día 1 y 80 mg diarios los Días 2 y 3 en combinación con 32 mg de ondansetrón por vía intravenosa el Día 1 y 12 mg de dexametasona el Día 1 y 8 mg una vez al día los Días 2 a 4. El tratamiento control consistió en un placebo en combinación con 32 mg de ondansetrón por vía intravenosa el Día 1 y 20 mg de dexametasona el Día 1 y 8 mg dos veces al día los Días 2 a 4. A pesar que en los estudios clínicos se usó una dosis de ondansetrón de 32 mg IV, ésta puede ya no ser la dosis actualmente recomendada. Consulte la información para prescribir del antagonista de 5-HT3 seleccionado para una dosificación apropiada.
Se evaluó la actividad antiemética de EMEND durante la fase aguda (0 a 24 horas después de la administración de cisplatino), durante la fase tardía (25 a 120 horas después de la administración de cisplatino) y total (0 a 120 horas después de la administración de cisplatino) en el Ciclo 1. La evaluación de la eficacia se basó en los siguientes parámetros compuestos:
• Respuesta completa (definida como ningún episodio de vómito y ningún tratamiento de rescate)
• Protección completa (definida como ningún episodio de vómito, ningún tratamiento de rescate, y una puntuación máxima de náusea en la escala análoga visual <25 mm)
• Efecto de la náusea y el vómito sobre la vida diaria (puntuación total en el Índice Funcional de Vida-Emesis >108).
La evaluación de la eficacia se basó también en los siguientes parámetros individuales:
• Ningún episodio de vómito (definido como ningún episodio de vómito con o sin tratamiento de rescate)
• Ningún episodio de náusea significativa (puntuación máxima en la escala análoga visual <25 mm).
Los resultados se evaluaron en cada estudio por separado y en los dos estudios combinados.
La tabla 1 muestra un resumen de los resultados principales del análisis combinado de los dos estudios.
Tabla 1 Porcentaje de pacientes con quimioterapia altamente emetogénica que respondieron, por grupo de tratamiento y por fase – Ciclo 1
|
PARÁMETROS COMPUESTOS |
Régimen con aprepitant* (n=521)† % |
Tratamiento control* (n=524)† % |
Valor de p |
|
Respuesta completa (sin vómito y sin tratamiento de rescate) |
|||
|
Total‡ |
67.7 |
47.8 |
<0.001 |
|
Fase aguda§ |
86.0 |
73.2 |
<0.001 |
|
Fase tardía" |
71.5 |
51.2 |
<0.001 |
|
Protección completa (sin vómito, sin tratamiento de rescate, y puntuación máxima de la náusea en la escala análoga visual¶ <25 mm) |
|||
|
Total |
59.5 |
44.9 |
<0.001 |
|
Fase aguda |
82.4 |
69.6 |
<0.001 |
|
Fase tardía |
63.7 |
47.8 |
<0.001 |
|
Sin efecto sobre la vida diaria (puntuación total en el Índice Funcional de Vida-Vómito >108) |
|||
|
Total |
74.4 |
63.9 |
<0.001 |
|
PARÁMETROS INDIVIDUALES |
|||
|
Sin vómito (con o sin tratamiento de rescate) |
|||
|
Total |
71.9 |
49.7 |
<0.001 |
|
Fase aguda |
86.8 |
74.0 |
<0.001 |
|
Fase tardía |
76.2 |
53.5 |
<0.001 |
|
Sin náusea significativa (puntuación máxima en la escala análoga visual <25 mm) |
|||
|
Total |
72.1 |
64.9 |
0.014 |
|
Fase tardía |
74.0 |
66.9 |
0.013 |
* Régimen con aprepitant: EMEND, 125 mg por vía oral el Día 1 y 80 mg por vía oral una vez al día los Días 2 y 3, más ondansetrón, 32 mg por vía intravenosa el Día 1, y dexametasona, 12 mg por vía oral el Día 1 y 8 mg por vía oral una vez al día los Días 2 a 4.
** Tratamiento control: Placebo más ondansetrón, 32 mg IV el Día 1, más dexametasona, 20 mg por vía oral el Día 1 y 8 mg por vía oral dos veces al día los Días 2 a 4.
† N: Número de pacientes que recibieron cisplatino, el medicamento en estudio y tuvieron por lo menos una evaluación de la eficacia después del tratamiento.
‡ Total: De 0 a 120 horas después de la administración de cisplatino.
§ Fase aguda: De 0 a 24 después de la administración de cisplatino.
" Fase tardía: De 25 a 120 horas después de la administración de cisplatino.
¶ Puntuación en la escala análoga visual: 0= sin náusea; 100= náusea máxima.
En el análisis combinado, la proporción de pacientes que tuvieron una respuesta completa y protección completa fue significativamente mayor con el régimen con aprepitant que con el tratamiento control en el Ciclo 1. Se observó una diferencia estadísticamente significativa en los porcentajes de pacientes que tuvieron una respuesta completa y protección completa a favor de los que recibieron el régimen con aprepitant durante la fase aguda y durante la fase tardía en el ciclo 1, comparado con los pacientes que recibieron el tratamiento control. También se obtuvieron esos resultados en cada uno de los dos estudios individuales.
En el análisis combinado, la proporción de pacientes que no tuvieron vómito alguno fue significativamente mayor en los que recibieron el régimen con aprepitant en el Ciclo 1, tanto en la fase temprana como en la fase tardía, que en los que recibieron el tratamiento control. Se observó una diferencia estadísticamente significativa en sin vómito alguno, en los pacientes que recibieron el régimen de aprepitant durante las fases aguda y tardía en el Ciclo 1, comparados con los pacientes que recibieron el tratamiento control. Se obtuvieron esos mismos resultados en cada uno de los dos estudios individuales.
Además, en el análisis combinado la proporción de pacientes que no tuvieron náusea significativa en total ni durante la fase tardía, independientemente del uso de tratamiento de rescate, fue significativamente mayor en los que recibieron el régimen con aprepitant en el Ciclo 1 que en los que recibieron el tratamiento control.
El impacto de la náusea y el vómito sobre la vida cotidiana de los pacientes se evaluó por medio del Índice Funcional de Vida-Emesis, que es una medición validada del resultado reportada por los pacientes. En el análisis combinado, la proporción de pacientes que reportaron que la náusea y el vómito no tuvieron ningún efecto en su vida cotidiana (Índice Funcional de Vida-Emesis >108) fue significativamente mayor en los que recibieron el régimen con aprepitant en el Ciclo 1 que en los que recibieron el tratamiento control. Se obtuvieron esos mismos resultados en cada uno de los dos estudios individuales.
En el análisis combinado, el tiempo estimado transcurrido entre el inicio de la administración de cisplatino y el primer vómito fue significativamente mayor (p<0.001) y la incidencia de vómito fue menor con el régimen con aprepitant que con el tratamiento control, como se muestra en la figura 2.
Figura 2: Porcentajes de pacientes con quimioterapia altamente emetogénica que permanecieron sin vómito al paso del tiempo – Ciclo 1
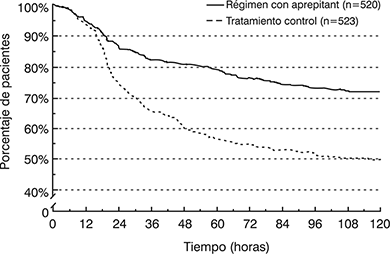
Régimen con aprepitant: EMEND, 125 mg por vía oral el Día 1 y 80 mg por vía oral una vez al día los Días 2 y 3, más ondansetrón, 32 mg lV el Día 1, y dexametasona, 12 mg por vía oral el Día 1 y 8 mg por vía oral una vez al día los Días 2 a 4.
Tratamiento control: Placebo más ondansetrón, 32 mg por vía intravenosa el Día 1, más dexametasona, 20 mg por vía oral el Día 1 y 8 mg por vía oral dos veces al día los Días 2 a 4.
Extensión a ciclos múltiples: En esos mismos dos estudios clínicos, 851 pacientes continuaron en la extensión a ciclos múltiples por hasta seis ciclos de quimioterapia. La eficacia del régimen con aprepitant se mantuvo durante todos los ciclos. En la figura 3 se muestran las tasas de respuesta de pacientes que en el análisis combinado no presentaron vómito ni náusea significativa en los seis ciclos de quimioterapia después de iniciar el tratamiento con cisplatino. Durante los ciclos 2 a 6, el punto final de ninguna náusea significativa se determinó por la respuesta de los pacientes a una pregunta directa, en vez de usar la escala análoga visual como en el Ciclo 1.
Figura 3: Porcentajes de pacientes con quimioterapia altamente emetogénica sin vómito ni náusea significativa, por grupo de tratamiento y por ciclo
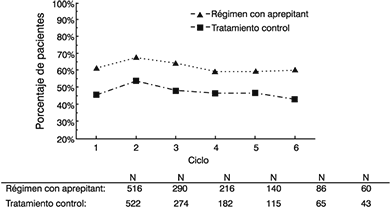
Régimen con aprepitant: EMEND, 125 mg por vía oral el Día 1 y 80 mg por vía oral una vez al día los Días 2 y 3, más ondansetrón, 32 mg IV el Día 1, y dexametasona, 12 mg por vía oral el Día 1 y 8 mg por vía oral una vez al día los Días 2 a 4.
Tratamiento control: Placebo más ondansetrón, 32 mg por vía intravenosa el Día 1, más dexametasona, 20 mg por vía oral el Día 1 y 8 mg por vía oral dos veces al día los Días 2 a 4.
Quimioterapia moderadamente emetogénica (MEC por sus siglas en inglés)
En un estudio clínico, multicéntrico, con distribución al azar, doble ciego, de grupos paralelos, se comparó el régimen con aprepitant con el tratamiento control en 866 pacientes con cáncer de mama, que estaban bajo quimioterapia que incluía ciclofosfamida 750-1500 mg/m2 o ciclofosfamida 500-1500 mg/m2 y dexorobicina (≤60 mg/m2) o epirubicina (≤100 mg/m2). Algunos pacientes también recibieron otros fármacos quimioterapéuticos como fluorouracilo, metotrexato, docetaxel o paclitaxel. El régimen con aprepitant consistió en EMEND 125 mg el Día 1 y 80 mg/día los Días 2 y 3 en combinación con ondansetrón 8 mg por vía oral el Día 1 más dexametasona 12 mg por vía oral el Día 1. El tratamiento control consistió en placebo en combinación con ondansetrón 8 mg orales (dos veces al día el Día 1 y cada doce horas en los Días 2 y 3), más dexametasona 20 mg orales en el Día 1.
La actividad antiemética de EMEND se evaluó durante la fase aguda (de 0 a 24 horas después de la quimioterapia), la fase tardía (de 25 a 120 horas después de la quimioterapia) y total (de 0 a 120 horas después de la quimioterapia) en el Ciclo 1. La eficacia se basó en la evaluación de los siguientes parámetros compuestos:
• Respuesta completa (definida como sin episodio de vómito y sin tratamiento de rescate)
• Efecto de la náusea y del vómito sobre la vida diaria (puntuación total en el Índice Funcional de Vida-Emesis >108).
La evaluación de la eficacia se basó también en los siguientes parámetros individuales:
• Ningún episodio de vómito (definido como ningún episodio de vómito con o sin tratamiento de rescate)
• Sin tratamiento de rescate
Un resumen de los resultados del estudio clave se muestra en la Tabla 2.
Tabla 2 Porcentaje de pacientes con quimioterapia moderadamente emetogénica que respondieron, por grupo de tratamiento y por fase-Ciclo 1
|
PARÁMETROS COMPUESTOS |
Régimen con aprepitant* (n=433)† % |
Tratamiento control** (n=424)† % |
Valor de p |
|
Respuesta completa (sin vómito y sin tratamiento de rescate) |
|||
|
Total‡ Fase aguda§ Fase tardía" |
51 76 55 |
42 69 49 |
0.015 0.034 0.064 |
|
Sin efecto sobre la vida diaria (puntuación total en el Índice Funcional de Vida-Emesis >108) |
|||
|
Total |
64 |
56 |
0.019 |
|
PARÁMETROS INDIVIDUALES |
|||
|
Sin vómito |
|||
|
Total Fase aguda Fase tardía |
76 88 81 |
59 77 69 |
<0.001 <0.001 <0.001 |
|
Sin tratamiento de rescate |
|||
|
Total Fase aguda Fase tardía |
59 83 63 |
56 80 60 |
0.480 0.366 0.407 |
* Régimen con aprepitant: EMEND, 125 mg por vía oral el Día 1 y 80 mg por vía oral los Días 2 y 3, más ondansetrón, 8 mg por vía oral dos veces al día el Día 1, y dexametasona, 12 mg por vía oral el Día 1.
** Tratamiento control: Placebo más ondansetrón, 8 mg por vía oral (dos veces al día el Día 1 y cada 12 horas los Días 2 y 3), más dexametasona, 20 mg por vía oral el Día 1.
† N: Número de pacientes incluidos en el análisis primario de respuesta completa.
‡ Total: De 0 a 120 horas después de la quimioterapia.
§ Fase aguda: De 0 a 24 después de la quimioterapia.
" Fase tardía: De 25 a 120 horas después de la quimioterapia.
En este estudio, una proporción estadística y significativamente (p=0.015) mayor de pacientes con régimen con aprepitant (51%) en el Ciclo 1 tuvo respuesta completa (punto final primario) durante la fase total en comparación con los pacientes con tratamiento control (42%). La diferencia absoluta no ajustada en la respuesta completa (8.3%) representa un 20% de mejoría relativa (tasa de riesgo relativo = 1.2, régimen con aprepitant comparado con el tratamiento control). Una proporción mayor de pacientes con régimen con aprepitant en el Ciclo 1 tuvo respuesta completa durante las fases aguda y tardía en comparación con los pacientes con tratamiento control.
En este estudio, el tiempo estimado para el primer vómito después del inicio de la quimioterapia fue significativamente (p<0.001) más largo con el régimen con aprepitant, y la incidencia del primer vómito se redujo en el grupo con el régimen con aprepitant en comparación con el grupo con tratamiento control como se muestra en la Figura 4.
Figura 4: Porcentaje de pacientes con quimioterapia moderadamente emetogénica que permanecen sin vómito al paso del tiempo – Ciclo 1

Régimen con aprepitant: EMEND, 125 mg por vía oral el Día 1 y 80 mg por vía oral los Días 2 y 3, más ondansetrón, 8 mg por vía oral dos veces al día el Día 1, y dexametasona, 12 mg por vía oral el Día 1.
Tratamiento control: Placebo más ondansetrón, 8 mg por vía oral (dos veces al día el Día 1 y cada 12 horas los Días 2 y 3), más dexametasona, 20 mg por vía oral el Día 1.
En este estudio, una proporción estadística y significativamente mayor de pacientes con régimen con aprepitant en el Ciclo 1 no tuvo efecto alguno de náusea ni vómito en la vida diaria, como se midió mediante la puntuación total en el Índice Funcional de Vida-Emesis >108, comparado con los pacientes con tratamiento control.
Extensión a ciclos múltiples: Un total de 744 pacientes con quimioterapia anticancerosa moderadamente emetogénica continuó en la extensión de ciclos múltiples hasta cuatro ciclos de quimioterapia. La eficacia del régimen con aprepitant se mantuvo durante todos los ciclos. En la figura 5 se muestran las tasas de respuesta.
Figura 5: Porcentaje de pacientes con quimioterapia moderadamente emetogénica sin vómito ni tratamiento de rescate, por grupo de tratamiento y por ciclo
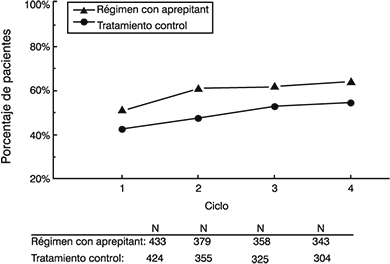
Régimen con aprepitant: EMEND, 125 mg por vía oral el Día 1 y 80 mg por vía oral los Días 2 y 3, más ondansetrón, 8 mg dos veces al día el Día 1, y dexametasona, 12 mg por vía oral el Día 1.
Tratamiento control: Placebo más ondansetrón, 8 mg por vía oral (dos veces al día el Día 1 y cada 12 horas los Días 2 y 3), más dexametasona, 20 mg por vía oral el Día 1.
En un segundo estudio clínico multicéntrico, aleatorizado, doble ciego, de grupos paralelos, el régimen con aprepitant se comparó con tratamiento control en 848 pacientes que recibían quimioterapia, la cual incluyó cualquier dosis IV de oxaliplatino, carboplatino, epirubicina, idarubicina, ifosfamida, irinotecan, daunorubicina, doxorubicina; ciclofosfamida IV (<1500 mg/m2) o citarabina IV (>1 g/m2). Los pacientes aleatorizados para recibir el régimen de aprepitant eran 76% mujeres y 24% hombres. Los pacientes que recibieron el régimen de aprepitant estaban recibiendo quimioterapia por una variedad de tipos de tumores que incluían 52% con cáncer de mama, 21% con cánceres gastrointestinales que incluían cáncer colorrectal, 13% con cáncer de pulmón y 6% con cánceres ginecológicos. El régimen con aprepitant consistió en EMEND 125 mg el Día 1 y 80 mg/día los Días 2 y 3 en combinación con ondansetrón 8 mg por vía oral, dos veces el Día 1 más dexametasona 12 mg por vía oral el Día 1. El tratamiento control consistió en placebo en combinación con ondansetrón 8 mg por vía oral (dos veces el Día 1 y cada 12 horas los Días 2 y 3) más dexametasona 20 mg por vía oral el Día 1.
La actividad antiemética de EMEND se evaluó durante la fase general (de 0 a 120 horas después del tratamiento de quimioterapia) en el Ciclo 1. La eficacia se basó en la evaluación de los siguientes puntos finales:
Punto final primario:
• ningún vómito en el periodo general (de 0 a 120 horas después de la quimioterapia)
Otros puntos finales pre-especificados:
• respuesta completa (definida como sin vómito y sin uso de terapia de rescate) en el periodo general (de 0 a 120 horas después de la quimioterapia)
• tiempo hasta el primer episodio de vómito general (de 0 a 120 horas después de la quimioterapia)
• sin vómito–agudo (de 0 a 24 horas después del inicio de la infusión de quimioterapia) y tardío (de 25 a 120 horas después del inicio de la infusión de quimioterapia)
• respuesta completa – agudo y tardío, como se define arriba
• sin uso de terapia de rescate–General, agudo y tardío, como se define arriba
• sin impacto en la Vida Diaria (Índice Funcional de Vida-Emesis puntuación total >180 – General, como se define arriba)
• sin vómito y sin náusea significativa (VAS <25 mm) – General, como se define arriba
Un resumen de los resultados clave del estudio se muestra en la Tabla 3
Tabla 3 Porcentaje de Pacientes que Recibían Quimioterapia Moderadamente Emetogénica que Respondieron por Tratamiento, Grupo y Fase para el Estudio 2 – Ciclo 1
|
OBJETIVOS FINALES |
Régimen Aprepitant (N = 430) % |
Tratamiento Control (N = 418) % |
Valor-p‡ |
|
OBJETIVO FINAL PRIMARIO |
|||
|
Sin Vómito |
|||
|
General§ |
76 |
62 |
<0.0001 |
|
OBJETIVO FINAL SECUNDARIO CLAVE |
|||
|
Respuesta Completa |
|||
|
General§ |
69 |
56 |
0.0003 |
|
OTROS OBJETIVOS FINALES SECUNDARIOS |
|||
|
Sin Vómito |
|||
|
Fase aguda |
92 |
84 |
0.0002 |
|
Fase tardía |
78 |
67 |
0.0005 |
|
Sin Impacto en la Vida Diaria (calificación total FLIE >108) |
|||
|
General |
73 |
66 |
0.035 |
|
Respuesta Completa |
|||
|
Fase aguda# |
89 |
80 |
0.0005 |
|
Fase tardíaÞ |
71 |
61 |
0.0042 |
|
Sin Uso de Terapia de Rescate |
|||
|
General |
81 |
75 |
0.0427ß |
|
Fase aguda |
0.0179ß |
||
|
Masculinoà |
97 |
100 |
|
|
Femeninoà |
95 |
88 |
|
|
Fase tardía |
84 |
79 |
0.0922ß |
|
Sin vómito y sin Náusea Significativa (VAS <25 mm) |
|||
|
General |
65 |
53 |
0.0011 |
* Régimen con Aprepitant: EMEND 125 mg por vía oral en el Día 1 y 80 mg por vía oral en el Día 2 y 3 más ondansetrón 8 mg por vía oral dos veces en el Día 1 más dexametasona 12 mg por vía oral en el Día 1.
** Tratamiento Control: Placebo más ondansetrón 8 mg por vía oral (dos veces en el Día 1 y cada 12 horas en los Días 2 y 3) más dexametasona 20 mg por vía oral en el Día 1.
+ N = Número de Pacientes que recibieron tratamiento de quimioterapia, fármaco en estudio y tuvieron cuando menos una evaluación de eficacia después del tratamiento.
‡ El procedimiento de Hochberg se utilizó como ajuste de multiplicidad cuando se probaron los puntos finales secundarios por importancia.
§ General: de 0 a 120 horas de tratamiento después de la quimioterapia.
¶ Respuesta Completa = Sin vómito y sin terapia de rescate
# Fase aguda: de 0 a 24 horas después del inicio de la infusión de quimioterapia
Þ Fase tardía: de 25 a 120 después del inicio de la infusión de quimioterapia
ß Estadísticamente no significativo
à Los datos se muestran por separado para hombres y mujeres por el plan analítico pre-especificado
El rango de puntuación de la escala visual análoga (VAS): 0 mm = sin náusea; 100 mm = náusea tan mala como se pueda.
En este estudio, una proporción estadísticamente significativa (p<0.0001) de pacientes que recibieron el régimen con aprepitant (76%) en el Ciclo 1, no tuvieron vómito (objetivo final primario) durante la fase general, comparados con pacientes que recibieron el tratamiento estándar (62%). Además, una proporción más alta de pacientes que recibieron el régimen con aprepitant en el Ciclo 1 tuvieron una respuesta completa en la fase general (0-120 horas) comparados con pacientes que recibieron el tratamiento control. Aprepitant fue numéricamente superior contra el tratamiento control a pesar de la edad, género o tipo de tumor (mama, gastrointestinal, pulmón u otro) como se evaluó en los objetivos finales Sin Vómito y Respuesta Completa.
En este estudio, el tiempo estimado hasta el primer vómito, después del inicio del tratamiento de quimioterapia, fue más largo con el régimen de aprepitant y la incidencia se redujo en el grupo del régimen con aprepitant comparado con el grupo de tratamiento control como se describe en las curvas de Kaplan-Meier en la Figura 6.
Figura 6: Curvas Kaplan-Meier para el Tiempo hasta el Primer Episodio de Vómito Desde el Inicio de la Administración de Quimioterapia en la Fase General-Ciclo 1 (Conjunto de Análisis Completo de la Población de Pacientes)
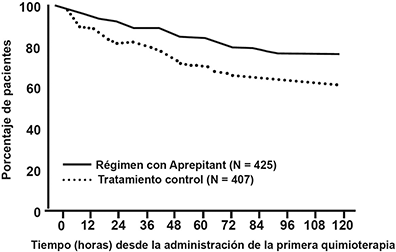
En este estudio, una proporción de pacientes significativamente más alta que recibieron el régimen con aprepitant en el Ciclo 1 no reportaron impacto de náusea y vómito en la vida diaria, como se mide por una puntuación total FLIE >108, comparados con los pacientes que recibieron el tratamiento control.
VI. CONTRAINDICACIONES
EMEND está contraindicado en pacientes con hipersensibilidad a cualquiera de los componentes del producto.
EMEND no se debe emplear al mismo tiempo que pimocida, terfenadina, astemizol o cisaprida. La inhibición dependiente de la dosis de la isoenzima 3A4 del citocromo P450 (CYP3A4) por aprepitant puede aumentar las concentraciones plasmáticas de esos medicamentos y causar reacciones graves o mortales (véase X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO).
VII. PRECAUCIONES GENERALES
EMEND, un inhibidor dependiente de la dosis de CYP3A4, se debe usar con precaución en pacientes que están recibiendo concomitantemente medicamentos administrados por vía oral, que son metabolizados principalmente por la enzima CYP3A4; algunos fármacos quimioterapéuticos son metabolizados por la enzima CYP3A4 (véase X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO). La inhibición moderada de CYP3A4 por aprepitant, régimen de 125 mg/80 mg en un esquema de tratamiento de 3 días, puede aumentar las concentraciones plasmáticas de esos medicamentos administrados oralmente (véase X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO). El efecto de EMEND en la farmacocinética de los sustratos administrados por vía oral de CYP3A4 es mayor que el efecto de EMEND en la farmacocinética de los sustratos administrados vía intravenosa de CYP3A4 (véase X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO).
La co-administración de EMEND y warfarina puede provocar una disminución clínicamente significativa en el tiempo de protrombina, reportado como Razón Normalizada Internacional (INR, por sus siglas en inglés). En los pacientes bajo tratamiento crónico con warfarina, el INR debe ser monitorizado de cerca en un periodo de 2 semanas, particularmente de 7 a 10 días ser después de iniciar el régimen de tres días de administración de EMEND (125 mg/80 mg) en cada ciclo de quimioterapia (véase X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO).
La eficacia de los anticonceptivos hormonales puede disminuir durante y 28 días después de la administración de EMEND. Durante el tratamiento con EMEND y un mes después de su última dosis se deben utilizar métodos anticonceptivos opcionales o de respaldo (véase X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO).
EMPLEO EN NIÑOS
No se han determinado la seguridad y la eficacia de EMEND en niños.
EMPLEO EN PACIENTES DE EDAD AVANZADA
En los estudios clínicos la eficacia y la seguridad de EMEND fueron similares en los pacientes de edad avanzada (≥ 65 años) y en los de menos edad (<65 años). No es necesario ajustar la dosificación en los pacientes de edad avanzada.
VIII. RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA
EMBARAZO
No hay estudios adecuados y bien controlados en mujeres embarazadas. Sólo se debe usar EMEND durante el embarazo si el posible beneficio justifica el riesgo potencial para la madre y el feto.
LACTANCIA
El aprepitant es excretado en la leche de ratas lactantes. No se sabe si este medicamento es excretado en la leche humana. Debido a que muchos medicamentos son excretados en la leche humana y a los posibles efectos adversos de EMEND en los lactantes, se debe decidir si se suspende la lactancia o la administración de EMEND, teniendo en cuenta la importancia de éste para la madre.
IX. REACCIONES SECUNDARIAS Y ADVERSAS
Se evaluó la seguridad general de aprepitant en 6,500 personas aproximadamente.
PREVENCIÓN DE LA NÁUSEA Y EL VÓMITO INDUCIDOS POR QUIMIOTERAPIA (CNIV)
Quimioterapia altamente emetogénica (HEC)
En dos estudios clínicos bien controlados en pacientes que recibían quimioterapia anticancerosa altamente emetogénica, se trató con el régimen de tres días de tratamiento con aprepitant a 544 pacientes durante el Ciclo 1 y 413 de ellos continuaron en la extensión de Ciclos múltiples hasta por seis ciclos de quimioterapia. El régimen de 3 días de EMEND por vía oral se administró en combinación con ondansetrón y dexametasona y fue generalmente bien tolerado. La mayoría de las reacciones adversas reportadas en esos estudios clínicos fueron descritas como de intensidad leve a moderada.
En el Ciclo 1 se reportaron reacciones adversas clínicas relacionadas con el medicamento en aproximadamente 19% de los pacientes tratados con el régimen oral de aprepitant de 3 días y en aproximadamente 14% de los que recibieron el tratamiento control. Se suspendió la administración del tratamiento a causa de reacciones adversas clínicas relacionadas con el medicamento en 0.6% de los pacientes tratados con el régimen de 3 días con aprepitant y en 0.4% de los que recibieron el tratamiento control.
Las reacciones adversas relacionadas con el medicamento que se reportaron con más frecuencia en los pacientes tratados con el régimen de 3 días con aprepitant que con el tratamiento control fueron: hipo (4.6%), aumento de la alanina-aminotransferasa (ALT) (2.8%), dispepsia (2.6%), estreñimiento (2.4%), cefalea (2.0%), y disminución del apetito (2.0%).
En un estudio clínico adicional con control activo en 1,169 pacientes que recibieron el régimen de aprepitant por tres días y HEC, el perfil de reacciones adversas fue generalmente similar al observado en los otros estudios de HEC con aprepitant oral por tres días.
Quimioterapia moderadamente emetogénica (MEC)
En 2 estudios clínicos bien controlados en pacientes que recibían quimioterapia anticancerosa moderadamente emetogénica (MEC), 868 pacientes fueron tratados con un régimen de aprepitant por tres días durante el Ciclo 1 de quimioterapia y 686 de ellos continuaron en las extensiones hasta por cuatro ciclos de quimioterapia. En ambos estudios se administró EMEND en régimen de tres días de tratamiento, en combinación con ondansetrón y dexametasona y fue generalmente bien tolerado. La mayoría de las reacciones adversas reportadas en esos estudios clínicos fueron descritas de leves a moderadas en intensidad.
En el análisis combinado en el Ciclo 1 de los datos de esos 2 estudios se reportaron reacciones adversas clínicas relacionadas con el medicamento en aproximadamente 14% de los pacientes tratados con el régimen de 3 días con aprepitant y en aproximadamente 15% de los que recibieron el tratamiento control. Se suspendió la administración del tratamiento a causa de reacciones adversas relacionadas con el medicamento en 0.7% de los pacientes tratados con el régimen de 3 días con aprepitant y en 0.2% de los que recibieron el tratamiento control. La reacción adversa más común relacionada con el medicamento y que se reportó con una mayor incidencia en los pacientes tratados con el régimen de 3 días con aprepitant que con el tratamiento control fue fatiga (1.4%).
Quimioterapia alta y moderadamente emetogénica
En un análisis conjunto de estudios de HEC y MEC se reportaron las siguientes reacciones adversas asociadas al medicamento en pacientes tratados con el régimen de tres días con aprepitant , con una incidencia mayor que en los que recibieron el tratamiento control:
[Comunes (≥1/100, <1/10), Poco comunes (≥ 1/1,000, <1/100), Raros (≥1/10,000, <1/1,000)]
Infecciones e infestaciones:
Raros: candidiasis, infección estafilocócica.
Trastornos sanguíneos y linfáticos:
Poco comunes: anemia, neutropenia febril.
Trastornos metabólicos y nutricionales:
Comunes: disminución del apetito.
Raros: polidipsia.
Trastornos psiquiátricos:
Poco comunes: ansiedad.
Raros: desorientación, euforia.
Trastornos del sistema nervioso:
Poco comunes: mareo, somnolencia.
Raros: trastornos cognitivos, letargo, disgeusia.
Trastornos oculares:
Raros: conjuntivitis.
Trastornos del oído y el laberinto:
Raros: tinnitus.
Trastornos cardiacos:
Poco comunes: palpitaciones.
Raros: bradicardia, trastorno cardiovascular.
Trastornos vasculares:
Poco comunes: bochorno.
Trastornos respiratorios, torácicos y mediastinales:
Comunes: hipo.
Raros: dolor orofaríngeo, estornudos, tos, goteo retronasal, irritación de la garganta.
Trastornos gastrointestinales:
Comunes: dispepsia.
Poco comunes: eructos, náusea, enfermedad por reflujo gastroesofágico, vómito, dolor abdominal, boca seca, flatulencia.
Raros: heces duras, úlcera duodenal perforante, colitis neutropénica, estomatitis, distensión abdominal.
Trastornos cutáneos y subcutáneos:
Poco comunes: exantema, acné.
Raros: fotosensibilidad, hiperhidrosis, seborrea, lesión cutánea, urticaria pruriginosa.
Trastornos musculoesqueléticos y del tejido conectivo:
Raros: espasmos musculares, debilidad muscular.
Trastornos renales y urinarios:
Poco comunes: disuria
Raros: polaquiuria.
Trastornos generales y condiciones en el sitio de administración:
Comunes: fatiga.
Poco comunes: astenia, malestar general
Raros: edema, malestar en el pecho, trastornos de la marcha.
Trastornos en las pruebas de laboratorio:
Comunes: aumento de la ALT.
Poco comunes: incremento en la AST, aumento en la fosfatasa alcalina.
Raros: aumento en la producción de orina, eritrocitos en orina, disminución de sodio sérico, disminución de peso, presencia de glucosa en orina, disminución de la cuenta de neutrófilos.
Las reacciones adversas observadas en las extensiones de Múltiples ciclos de los estudios de HEC y MEC hasta por seis ciclos de quimioterapia fueron generalmente similares a las observadas en el Ciclo 1.
En otro estudio en CINV se reportó un caso de Síndrome de Stevens-Johnson como reacción secundaria grave en un paciente que recibió aprepitant con quimioterapia anticancerosa.
Otros estudios
Las dosis únicas de 40 mg de EMEND también se han estudiado para la prevención de náusea y vómito post-operatorio (PONV) en pacientes que no están bajo tratamiento con quimioterapia y que reciben anestesia general balanceada. En estos estudios, las reacciones adversas que se observaron con una incidencia mayor en comparación con el activo (ondansetrón) incluyen: aumento de ALT, dolor abdominal superior, sonidos intestinales anormales, disartria, disnea, hipoestesia, insomnio, miosis, náusea, trastornos sensoriales, molestias en el estómago, disminución de la agudeza visual, sibilancias.
Adicionalmente, en estudios clínicos para la prevención de PONV se registraron dos experiencias adversas graves en pacientes que tomaban una dosis mayor de aprepitant: un caso de estreñimiento y un caso de sub-íleon.
En un estudio con pacientes sin CINV o PONV se reportaron un caso de angioedema y de urticaria como experiencias adversas graves.
Experiencia Post-comercialización:
Las siguientes reacciones adversas han sido identificadas durante la post-comercialización con aprepitant. Debido a que estas reacciones son reportadas voluntariamente por una muestra de población no determinada, no es posible generalmente una estimación confiable de la frecuencia o establecer una relación causal con el fármaco.
Trastornos cutáneos y subcutáneos: prurito, erupción, urticaria y, rara vez: Síndrome de Stevens-Johnson / necrólisis epidérmica tóxica
Trastornos del sistema inmune: reacciones de hipersensibilidad, incluyendo reacciones anafilácticas.
X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO
El aprepitant es un sustrato, un inhibidor débil a moderado (dependiente de la dosis) y un inductor de la enzima CYP3A4. También es un inductor de CYP2C9.
EFECTO DE APREPITANT SOBRE LA FARMACOCINÉTICA DE OTROS MEDICAMENTOS
Como inhibidor moderado (125 mg/80 mg) de la enzima CYP3A4, aprepitant puede aumentar las concentraciones plasmáticas de los medicamentos metabolizados por esa enzima que se administren por vía oral al mismo tiempo. Aprepitant (125 mg/80 mg) puede aumentar en menor grado las concentraciones plasmáticas de los medicamentos metabolizados por la enzima CYP3A4 que se administren por vía intravenosa de manera concomitante.
EMEND no se debe emplear al mismo tiempo que pimocida, terfenadina, astemizol o cisaprida. La inhibición dependiente de la dosis de CYP3A4 por aprepitant puede aumentar las concentraciones plasmáticas de esos medicamentos y causar reacciones potencialmente graves o mortales (véase VI. CONTRAINDICACIONES).
Se ha mostrado que aprepitant induce el metabolismo de la S(-)warfarina y la tolbutamida, que son metabolizadas por la enzima CYP2C9. La co-administración de EMEND con estos u otros medicamentos que son metabolizados por CYP2C9, como la fenitoína, puede disminuir las concentraciones plasmáticas de esos medicamentos.
Es improbable que EMEND interactúe con medicamentos que son sustratos para el transportador de glucoproteína-P, demostrado por la ausencia de interacción de EMEND con la digoxina en un estudio clínico de interacciones farmacológicas.
Antagonistas de la serotonina: En estudios clínicos de interacciones farmacológicas, cuando aprepitant se administró como un régimen de 125 mg el Día 1 y 80 mg los Días 2 y 3, no tuvo efectos de importancia clínica sobre la farmacocinética de ondansetrón, granisetrón o -hidrodolasetrón (el metabolito activo de dolasetrón).
Corticosteroides:
Dexametasona: Cuando se co-administraron por vía oral 125 mg de EMEND y 20 mg de dexametasona el Día 1 y 80 mg diarios de EMEND y 8 mg/día de dexametasona los Días 2 a 5, aumentó 2.2 veces el área bajo la curva (ABC) de la dexametasona, un substrato de CYP3A4, los Días 1 y 5. Las dosis orales usuales de dexametasona se deben disminuir 50% aproximadamente cuando se co-administra con EMEND (régimen de 125 mg/80 mg), para alcanzar concentraciones de dexametasona similares a las obtenidas cuando se administra sin EMEND. La dosis diaria de dexametasona que se administró en los estudios clínicos con EMEND sobre náusea y vómito inducidos por quimioterapia refleja una reducción de aproximadamente el 50% de la dosis de dexametasona (véase XIII. DOSIS Y VÍA DE ADMINISTRACIÓN).
Metilprednisolona: Cuando EMEND fue administrado como un régimen de 125 mg el Día 1 y 80 mg/día los Días 2 y 3, el ABC de metilprednisolona (que es un sustrato de la CYP3A4) aumentó a 1.3 veces el Día 1 y a 2.5 veces el Día 3, cuando la etil-prednisolona fue co-administrada por vía intravenosa como 125 mg el Día 1 y 40 mg por vía oral los Días 2 y 3. Cuando se co-administra con EMEND (régimen de 125 mg/80 mg), la dosis intravenosa usual de metilprednisolona se debe disminuir 25% aproximadamente y su dosis oral usual se debe disminuir 50% aproximadamente, para producir concentraciones de metilprednisolona similares a las obtenidas cuando se administra sin EMEND.
Fármacos quimioterapéuticos: En los estudios clínicos se co-administró EMEND (régimen 125 mg/80 mg) con los siguientes fármacos quimioterapéuticos que son metabolizados principalmente o en parte por CYP3A4: etopósido, vinorelbina, docetaxel, ifosfamida, ciclofosfamida, irinotecan y paclitaxel. Las dosis de estos medicamentos no se ajustaron para suprimir las interacciones farmacológicas potenciales. Se recomienda precaución y un monitoreo cuidadoso de los pacientes que reciben estos fármacos u otros agentes quimioterapéuticos metabolizados principalmente por CYP3A4. En eventos post-comercialización de neurotoxicidad fue reportada una reacción adversa potencial de ifosfamida, después de la administración conjunta de aprepitant e ifosfamida (ver VII. PRECAUCIONES GENERALES).
Docetaxel: En un estudio farmacocinético separado EMEND (régimen 125 mg/80 mg) no influyó en la farmacocinética de docetaxel.
Vinorelbina: En un estudio farmacocinético separado EMEND (régimen 125 mg/80 mg) no influyó en la farmacocinética de vinorelbina.
Warfarina: Se administró una sola dosis de 125 mg de EMEND el Día 1 y 80 mg diarios los Días 2 y 3 a sujetos sanos estabilizados bajo tratamiento crónico con warfarina. Aunque EMEND no tuvo ningún efecto sobre el ABC de la warfarina R(+) o S(-) calculada el Día 3, cinco días después de terminar la administración de EMEND había una disminución de 34% de la concentración mínima de warfarina S(-) (un sustrato de CYP2C9) y de 14% del tiempo de protrombina (reportado como Razón Normalizada Internacional, INR). En los pacientes con tratamiento crónico con warfarina, al terminar el régimen de tres días de administración de EMEND en cada ciclo de quimioterapia se debe vigilar estrechamente el tiempo de protrombina (INR) en un periodo de dos semanas, en particular de los siete a los diez días, después de iniciar el régimen de 3 días de EMEND (125 mg/80 mg) de EMEND en cada ciclo de quimioterapia.
Tolbutamida: cuando se administró a una dosis de 125 mg de EMEND el Día 1 y 80 mg/día los Días 2 y 3, disminuyó el ABC de tolbutamida (un sustrato de CYP2C9) en 23% el Día 4, 28% el Día 8 y 15% el Día 15, al ser administrada una sola dosis oral de tolbutamida 500 mg antes de administrar el régimen de 3 días de EMEND y en los Días 4, 8 y 15.
Anticonceptivos orales: La administración de una cápsula de 100 mg de aprepitant una vez al día durante 14 días junto con un anticonceptivo oral con 35 μg de etinilestradiol y 1 mg de noretindrona disminuyó 43% el ABC de etinilestradiol y 8% la de noretindrona.
En otro estudio, una dosis única de un anticonceptivo oral que contenía etinilestradiol y noretindrona se administró los Días 1 al 21 concomitantemente con EMEND, como un régimen de 125 mg el Día 8 y 80 mg/día los Días 9 y 10 con ondansetrón, a 32 mg IV el Día 8 y dexametasona oral, 12 mg el Día 8 y 80 mg/día los Días 9, 10 y 11. En ese estudio, el ABC de etinilestradiol disminuyó 19% el Día 10 y hasta 64% las concentraciones valle de etinil estradiol los Días 9 a 21. Aunque EMEND no afectó el ABC de noretindrona el Día 10, sí disminuyeron hasta 60% las concentraciones valle de noretindrona los Días 9 a 21.
La eficacia de los anticonceptivos hormonales puede disminuir durante y 28 días después de la administración de EMEND. Durante el tratamiento con EMEND y un mes después de su última dosis se deben utilizar métodos anticonceptivos opcionales o de respaldo.
Midazolam: EMEND aumentó el ABC de midazolam, un sustrato sensible de CYP3A4, 2.3 veces más el Día 1 y 3.3 veces más el Día 5 cuando se co-administró una dosis única de 2 mg de midazolam el Día 1 y el Día 5 de un régimen de 125 mg de EMEND el Día 1 y 80 mg/día los Días 2 a 5. Se deben considerar los efectos potenciales del aumento de las concentraciones plasmáticas de midazolam o de otras benzodiacepinas que son metabolizadas por CYP3A4 (alprazolam, triazolam) cuando se co-administren estos medicamentos con EMEND (125 mg/80 mg).
En otro estudio en el que se administró midazolam por vía intravenosa, EMEND se administró en una dosis de 125 mg el Día 1 y de 80 mg/día los Días 2 y 3 y midazolam 2mg IV fue administrado antes de la administración del régimen de 3 días de EMEND y en los Días 4, 8 y 15. EMEND aumentó el ABC de midazolam en 25% el Día 4 y disminuyó el ABC de midazolam en 19% el Día 8 en relación con la dosificación de EMEND los Días 1 al 3. Estos efectos no se consideraron de importancia clínica. El ABC de midazolam el Día 15 fue similar a aquella observada a nivel basal.
Se completó un estudio adicional con la administración de midazolam y EMEND por vía intravenosa. Se administró midazolam 2 mg por vía intravenosa una hora después de la administración por vía oral de una dosis única de EMEND 125 mg. El ABC plasmática de midazolam incrementó 1.5 veces. Este efecto no se consideró clínicamente importante.
EFECTO DE OTROS MEDICAMENTOS SOBRE LA FARMACOCINÉTICA DE APREPITANT
El aprepitant es un sustrato de CYP3A4, por lo que la co-administración de EMEND con medicamentos que inhiben la actividad de CYP3A4 puede aumentar las concentraciones plasmáticas de aprepitant. Por lo tanto, se debe tener precaución al co-administrar EMEND con inhibidores potentes de CYP3A4 (por ejemplo, ketoconazol), pero la co-administración de EMEND con inhibidores moderados de CYP3A4 (como diltiazem) no causa cambios de importancia clínica en las concentraciones plasmáticas de aprepitant.
Por ser aprepitant un sustrato de CYP3A4, la co-administración de EMEND con medicamentos que son inductores potentes de la actividad de CYP3A4 (como la rifampicina) puede disminuir las concentraciones plasmáticas de aprepitant y, como consecuencia, la eficacia de EMEND.
Ketoconazol: Cuando se administró una sola dosis de 125 mg de EMEND el Día 5 de un régimen de diez días de tratamiento con 400 mg diarios de ketoconazol (un inhibidor potente de CYP3A4), el ABC de aprepitant aumentó cinco veces aproximadamente y su vida media terminal promedio aumentó tres veces aproximadamente. Se debe tener precaución al co-administrar EMEND con inhibidores potentes de CYP3A4.
Rifampicina: Cuando se administró una dosis única de 375 mg de EMEND el Día 9 de un régimen de 14 días de tratamiento con 600 mg diarios de rifampicina (un potente inductor de CYP3A4), el ABC de aprepitant disminuyó once veces aproximadamente y su vida media terminal promedio disminuyó tres veces aproximadamente. La co-administración de EMEND con medicamentos que inducen la actividad de CYP3A4 puede disminuir la concentración plasmática y la eficacia de EMEND.
OTRAS INTERACCIONES
Diltiazem: En pacientes con hipertensión de leve a moderada, la co-administración de aprepitant una vez al día en una tableta equivalente a una cápsula de 230 mg de aprepitant y 120 mg de diltiazem tres veces al día durante cinco días aumentó al doble el ABC de aprepitant y 1.7 veces el ABC de diltiazem. Estos efectos farmacocinéticos no causaron cambios clínicamente significativos en el electrocardiograma, la frecuencia cardiaca ni la presión arterial, que los inducidos por el diltiazem solo.
Paroxetina: La co-administración una vez al día de aprepitant en una tableta equivalente a una cápsula de aprepitant con 85 mg o 170 mg y 20 mg de parotexina una vez al día disminuyó aproximadamente 25% las ABCs y aproximadamente 20% las Cmáx de ambos medicamentos.
XI. ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO
Para mayor información respecto a los pacientes tratados con aprepitant de donde se desprenden estos resultados véase la Sección IX. REACCIONES SECUNDARIAS Y ADVERSAS.
[Comunes (≥1/100, <1/10); raras (>1/1000, <1/100)]
Comunes: Aumento de la ALT, aumento de la AST.
Raras: Aumentos de la fosfatasa alcalina, hiperglucemia, hematuria microscópica, hiponatremia, pérdida de peso.
XIII. DOSIS Y VÍA DE ADMINISTRACIÓN
PREVENCIÓN DE NÁUSEA Y VÓMITO INDUCIDOS POR QUIMIOTERAPIA (CINV)
EMEND (aprepitant) está disponible en cápsulas de 80 y 125 mg para administración por vía oral.
EMEND se administra por 3 días como parte de un régimen que incluye un corticosteroide y un antagonista de 5-HT3. Consulte la información para prescribir del antagonista 5-HT3 para la co-administración previa al inicio del tratamiento con EMEND. La dosis recomendada de EMEND para el régimen oral de 3 días es de 125 mg 1 hora antes del tratamiento con quimioterapia (Día 1) y 80 mg una vez al día en la mañana los Días 2 y 3.
Debe usarse el régimen siguiente para la prevención de náusea y vómito relacionados con quimioterapia anticancerosa altamente emetogénica:
|
Día 1 |
Día 2 |
Día 3 |
Día 4 |
|
|
EMEND* |
125 mg oral |
80 mg oral |
80 mg oral |
nada |
|
Dexametasona** |
12 mg por vía oral |
8 mg por vía oral |
8 mg por vía oral |
8 mg por vía oral |
|
Antagonista de 5-HT3 |
Consulte la información para prescribir del antagonista de 5-HT3 seleccionado para una dosificación apropiada. |
nada |
nada |
nada |
* Administrar EMEND por vía oral una hora antes de la quimioterapia el Día 1 y en la mañana los Días 2 y 3.
** La dexametasona debe administrarse 30 minutos antes de la quimioterapia el Día 1 y en la mañana los Días 2 a 4. Las interacciones farmacológicas dependen de la dosis de dexametasona.
Debe usarse el siguiente régimen para la prevención de náusea y vómito relacionados con quimioterapia anticancerosa moderadamente emetogénica:
|
Día 1 |
Día 2 |
Día 3 |
|
|
EMEND* |
125 mg oral |
80 mg oral |
80 mg oral |
|
Dexametasona** |
12 mg por vía oral |
nada |
nada |
|
Antagonista del 5-HT3 |
Consulte la información para prescribir del antagonista del 5-HT3 seleccionado para una dosificación apropiada. |
nada |
nada |
* Administrar EMEND por vía oral una hora antes de la quimioterapia el Día 1 y en la mañana los Días 2 y 3.
** La dexametasona debe administrarse 30 minutos antes de la quimioterapia el Día 1. Las interacciones farmacológicas dependen de la dosis de dexametasona.
También está disponible EMEND 150 mg (fosaprepitant), una prodroga liofilizada de aprepitant para administración intravenosa. EMEND 150 mg está disponible como dosis única y puede ser usado como una alternativa al régimen oral de 3 días de EMEND.
INFORMACIÓN GENERAL
Vea en X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO información adicional sobre la co-administración de EMEND y corticosteroides.
Consulte la información completa para prescribir de los fármacos antieméticos que se administren de manera concomitante.
EMEND se puede tomar con o sin alimentos.
No es necesario ajustar la dosificación según edad, sexo, raza o Índice de Masa Corporal (IMC) del paciente.
No es necesario ajustar la dosificación en los pacientes con insuficiencia renal grave (depuración de creatinina <30 mL/min) ni en los pacientes con nefropatía terminal tratados con hemodiálisis.
No es necesario ajustar la dosificación en los pacientes con insuficiencia hepática de leve a moderada (puntuación de Child-Pugh de 5 a 9). No hay información clínica en pacientes con insuficiencia hepática grave (puntuación de Child-Pugh mayor de 9).
XIV. MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL
No hay información específica sobre el tratamiento de la sobredosificación con EMEND. Dosis únicas de hasta 600 mg de aprepitant fueron generalmente bien toleradas por sujetos sanos. El aprepitant fue generalmente bien tolerado cuando se administró a dosis de 375 mg una vez al día hasta por 42 días a pacientes en estudios que no eran de CINV. En 33 pacientes con cáncer, la administración de una sola dosis de 375 mg de aprepitant el Día 1 y 250 mg una vez al día los Días 2 a 5 fue generalmente bien tolerada.
Se reportaron somnolencia y cefalea en un paciente que ingirió 1,440 mg de aprepitant.
En el caso de una sobredosificación con EMEND, se debe suspender su administración, aplicar tratamiento general de apoyo y vigilar al paciente. Debido a la actividad antiemética de aprepitant, puede resultar ineficaz la administración de eméticos.
La hemodiálisis no extrae el aprepitant.
XII. PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD
TOXICOLOGÍA ANIMAL
Toxicidad aguda
La DL50 aproximada de aprepitant por vía oral fue mayor de 2,000 mg/kg en ratones y ratas hembra. La DL50 aproximada de aprepitant por vía peritoneal fue mayor de 800 mg/kg, pero menor de 2,000 mg/kg en ratas hembra y mayor de 2,000 mg/kg en ratonas.
Toxicidad crónica
Se evaluó la toxicidad potencial de aprepitant en una serie de estudios de hasta un año de duración con dosis repetidas por vía oral en ratas y en perros.
En las ratas, la administración de aprepitant por vía oral durante seis meses a dosis de hasta la máxima posible de 1,000 mg/kg dos veces al día (aproximadamente equivalente en las hembras a la dosis en personas adultas basándose en la exposición sistémica después de 125 mg de aprepitant por vía oral, y menores que esa dosis en los machos) produjo aumentos del peso del hígado relacionados con hipertrofia hepatocelular, aumentos del peso de la tiroides relacionados con hipertrofia y/o hiperplasia celular folicular, y vacuolización celular hipofisiaria. Esos cambios son una consecuencia, específica de esa especie, de la inducción de las enzimas hepáticas del citocromo P-450 en la rata, y concuerdan con los cambios observados en ratas con otros compuestos estructural y farmacológicamente distintos que inducen dichas enzimas.
En los perros que recibieron aprepitant por vía oral durante nueve meses a dosis de 5 mg/kg o más dos veces al día (13 o más veces mayores que la dosis en personas adultas basándose en la exposición sistémica al medicamento, después de 125 mg de aprepitant por vía oral), la toxicidad se caracterizó por ligeros aumentos de la actividad de la fosfatasa alcalina en suero y disminuciones de la relación albúmina/globulina. Se observaron disminución significativa del aumento de peso corporal, degeneración testicular y atrofia prostática a dosis de 25 mg/kg o más dos veces al día (31 o más veces mayores que la dosis en personas adultas basándose en la exposición sistémica al medicamento después de 125 mg de aprepitant por vía oral). Con 500 mg/kg dos veces al día (70 veces más que la dosis en personas adultas basándose en la exposición sistémica al medicamento después de 125 mg de aprepitant por vía oral) hubo un ligero aumento del peso del hígado, sin cambios histológicos correlacionados. No se observó ninguna toxicidad en los perros que recibieron 32 mg/kg/día (seis veces más que la dosis en personas adultas basándose en la exposición sistémica al medicamento después de 125 mg de aprepitant por vía oral) durante un año.
Carcinogenicidad
Se hicieron estudios de carcinogenicidad en ratones y ratas durante dos años con aprepitant por vía oral. Los ratones desarrollaron adenomas hepatocelulares y/o carcinomas a dosis de 500 a 2,000 mg/kg/día (en hembras) y carcinomas hepatocelulares a dosis de 1,000 y 2,000 mg/kg/día (en machos). La exposición sistémica a estas dosis en ratones fue aproximadamente de 2.5 a 3.6 veces la exposición en humanos a la dosis recomendada. En las ratas se desarrollaron adenomas hepatocelulares a dosis de 5 a 1,000 mg/kg dos veces al día (en las hembras), y de 125 mg/kg dos veces al día (en los machos), carcinomas hepatocelulares a dosis de 125 a 1,000 mg/kg dos veces al día (en hembras), adenomas foliculares tiroideos a dosis de 125 a 1,000 mg/kg dos veces al día (en hembras y en los machos), y carcinomas foliculares tiroideos a dosis de 125 a 1,000 mg/kg dos veces al día (en los machos). A esas dosis, las exposiciones sistémicas en las ratas fueron menores que o arriba de aproximadamente 2 veces la exposición en humanos a la dosis recomendada. Los tumores de esos tipos en hígado y tiroides son una consecuencia específica de esa especie, de la inducción de las enzimas del citocromo P-450 hepáticas en roedores, y concuerdan con los cambios observados en roedores con otros compuestos estructural y farmacológicamente distintos que inducen dichas enzimas.
Mutagenicidad
El aprepitant no fue mutagénico ni genotóxico en las pruebas realizadas para detectar mutagenicidad, roturas del filamento de ADN y aberraciones cromosómicas, y resultó negativo en las pruebas in vitro de mutagénesis microbiana y de células linfoblastoides humanas TK6, de rotura del filamento de ADN en hepatocitos de rata en elución alcalina y de aberraciones cromosómicas en células de ovario de hámster chino, y en la prueba in vivo de micronúcleos en médula ósea de ratón.
Reproducción
El aprepitant administrado a ratas hembras a dosis de hasta la máxima posible de 1,000 mg/kg dos veces al día (aproximadamente equivalente a la dosis en personas adultas basándose en la exposición sistémica al medicamento después de 125 mg de aprepitant por vía oral) no tuvo ningún efecto sobre la conducta reproductiva, la fertilidad ni la supervivencia de los embriones y los fetos.
La administración de aprepitant a ratas macho a dosis de hasta la máxima posible de 1,000 mg/kg dos veces al día (menor que la dosis en personas adultas basándose en la exposición sistémica al medicamento después de 125 mg de aprepitant por vía oral) no tuvo ningún efecto sobre la conducta reproductiva, la fertilidad, la supervivencia de los embriones y los fetos, el número y la movilidad de los espermatozoides, el peso de los testículos, ni el aspecto microscópico de testículos y epidídimos.
Desarrollo
En ratas que recibieron dosis orales de aprepitant de hasta 1,000 mg/kg dos veces al día y en las conejas que recibieron 25 mg/kg/día (hasta 1.5 veces más que la exposición sistémica en los seres humanos adultos, después de 125 de aprepitant por vía oral) no hubo ningún indicio de toxicidad sobre el desarrollo fetal, según indicó la supervivencia de los embriones y los fetos y el peso y la morfología externa, visceral y esquelética de los fetos. A esas dosis, aprepitant pasó a través de la placenta en ratas y conejas.
Se encontraron concentraciones significativas de aprepitant en la leche de las ratas que recibieron 1,000 mg/kg dos veces al día. A esa dosis, el promedio de concentración del medicamento en la leche fue de 90% del promedio de concentración en el plasma materno.
XVIII. NOMBRE Y DOMICILIO DEL LABORATORIO:
SCHERING - PLOUGH, S.A. de C.V.
Av. 16 de Septiembre No. 301, Col. Xaltocan.
C.P. 16090, Ciudad de México, México.
XV. PRESENTACIÓN
Caja con 1 cápsula de 125 mg y 2 cápsulas de 80 mg
Caja con 1 cápsula de 125 mg
Caja con 2 cápsulas de 80 mg
Estuche con 1 cápsula de 125 mg y 2 cápsulas de 80 mg
Estuche con 1 cápsula de 125 mg
Estuche con 2 cápsulas de 80 mg
XVI. RECOMENDACIONES SOBRE ALMACENAMIENTO
Consérvese a no más de 30°C y en lugar seco.
XVII. LEYENDAS DE PROTECCIÓN
Literatura exclusiva para médicos. No se deje al alcance de los niños. Su venta requiere receta médica. El uso en embarazo y lactancia queda bajo la responsabilidad del médico
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
dpoc.mexico@msd.com


