ACTILYSE - Solución inyectable
Sustancia(s):
- Alteplasa
Presentaciones:
- 1 Caja, 2 Frasco ámpula con liofilizado y ampolleta de diluyente, 50 mL,
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula con polvo liofilizado contiene:
Alteplasa 50 mg
Excipiente cbp
Activador de plasminógeno tisular de origen ADN recombinante expresado en células de ovario de hámster chino (CHO)
El frasco ámpula con diluyente contiene:
Agua estéril para uso inyectable 50 mL
La solución reconstituida contiene 1 mg de alteplasa por mL
INDICACIONES TERAPÉUTICAS:
Tratamiento trombolítico del infarto agudo de miocardio (IAM):
• Régimen de tratamiento acelerado de 90 minutos, en los pacientes para los cuales el tratamiento puede comenzar dentro de las primeras 6 horas del inicio de la sintomatología.
• Régimen de tratamiento de 3 horas, para los pacientes en los que el tratamiento puede comenzar entre 6-12 horas después del inicio de los síntomas.
Tratamiento trombolítico de la tromboembolia pulmonar aguda masiva, en pacientes con inestabilidad hemodinámica:
El diagnóstico debe ser confirmado en la medida de lo posible con elementos diagnósticos certeros como la angiografía pulmonar o procedimientos no invasivos como la gammagrafía pulmonar de ventilación/perfusión.
Tratamiento trombolítico del evento vascular cerebral (EVC) isquémico agudo:
El tratamiento debe comenzar tan temprano como sea posible dentro de las 4.5 horas del inicio de los síntomas de EVC y después de excluir la hemorragia intracraneal mediante técnicas de imagen adecuadas (p. ej., tomografía axial computarizada [TAC] de cráneo u otros métodos de imagen diagnósticos sensibles para detectar la presencia de hemorragia). El efecto del tratamiento es tiempo-dependiente, por lo tanto, tratamientos más tempranos incrementan la probabilidad de un resultado favorable.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacológicas:
Mecanismo de acción:
El principio activo de ACTILYSE® es la alteplasa, un activador tisular del plasminógeno humano, una glucoproteína que activa el plasminógeno directamente a plasmina. Cuando se administra por vía intravenosa, la alteplasa permanece relativamente inactiva en el sistema circulatorio. Una vez que se une a la fibrina, se activa induciendo la conversión de plasminógeno a plasmina, conduciendo a la disolución del coágulo de fibrina.
Farmacodinamia:
Debido a su relativa especificidad por la fibrina, la alteplasa, a una dosis de 100 mg, lleva a una escasa reducción de los niveles de fibrinógeno circulante, hasta cerca del 60% en 4 horas, lo cual, generalmente es revertido hasta más del 80% después de 24 horas. El plasminógeno y la alfa-2-antiplasmina disminuyen a cerca de 20 y 35%, respectivamente, después de 4 horas, y aumentan, de nuevo, a más del 80% a las 24 horas. Una reducción marcada y prolongada del nivel de fibrinógeno circulante sólo se observa en unos pocos pacientes.
Estudios clínicos:
Pacientes con infarto agudo del miocardio (IAM):
ACTILYSE® ha probado reducir la mortalidad al día 30 en pacientes con IAM.
Se han estudiado dos regímenes de dosis con ACTILYSE®, en pacientes con infarto agudo al miocardio. No se ha evaluado la eficacia comparativa de estos dos regímenes.
Infusión acelerada en pacientes con IAM:
La infusión acelerada de ACTILYSE®, se evaluó en un estudio multicéntrico internacional (GUSTO) en el cual 41,021 pacientes con infarto agudo de miocardio fueron aleatorizados a cuatro regímenes trombolíticos. La administración de 100 mg de ACTILYSE®, durante 90 minutos con una infusión intravenosa de heparina concomitante, condujo a una menor mortalidad al cabo de 30 días (6.3%) en comparación con la administración de estreptoquinasa, 1.5 millones de UI en 60 minutos, con heparina subcutánea o intravenosa (7.3%). El descenso absoluto del 1% de la mortalidad a los 30 días observado para ACTILYSE®, en comparación con la estreptoquinasa, fue estadísticamente significativo (p = 0.001). Los pacientes tratados con ACTILYSE®, mostraron mayores índices de permeabilidad de los vasos sanguíneos relacionados con un infarto a los 60 y 90 minutos después de la trombólisis comparados con los pacientes tratados con estreptoquinasa. No se observaron diferencias en las tasas de permeabilidad a los 180 minutos o después.
Un estudio de mortalidad a gran escala (ASSENT 2) que incluyó cerca de 17,000 pacientes mostró que alteplasa y tenecteplasa son terapéuticamente equivalentes para disminuir la mortalidad (6.2% para ambos tratamientos, a los 30 días). El uso de tenecteplasa estuvo asociado con una incidencia significativamente menor de hemorragias no intracraneales en comparación con la alteplasa (26.4% frente a 28.9%, p = 0.0003). La reducción del riesgo de sangrado probablemente esté relacionada con la mayor especificidad por la fibrina que tiene la tenecteplasa y el hecho de que su régimen sea adaptado en función del peso del paciente.
Infusión de 3 horas en pacientes con IAM:
En un estudio doble ciego aleatorizado (5 013 pacientes) que comparó ACTILYSE® con placebo (estudio ASSENT), los pacientes que recibieron infusión de ACTILYSE® en las primeras 5 horas del inicio de los síntomas del infarto agudo de miocardio tuvieron una mejor tasa de sobrevida a los 30 días en comparación con aquellos tratados con placebo. Al mes 1, las tasas de mortalidad generales fueron del 7.2% para el grupo tratado con ACTILYSE® y del 9.8% para el grupo tratado con placebo (p = 0.001). Este beneficio se mantuvo a los 6 meses en el caso de los pacientes tratados con ACTILYSE® (10.4%) en comparación con los pacientes tratados con placebo (13.1%, p = 0.008).
En un estudio aleatorizado doble ciego (721 pacientes) que comparó ACTILYSE®con placebo, los pacientes que recibieron la infusión con ACTILYSE® en las primeras 5 horas del inicio de los síntomas tuvieron mejor función ventricular 10 a 22 días después del tratamiento comparados con el grupo placebo, momento en el cual la fracción de eyección global se midió mediante ventriculografía con contraste (50.7% frente a 48.5%, p = 0.01). Los pacientes tratados con ACTILYSE® tuvieron una reducción del 19% en las dimensiones del infarto, según lo medido por la actividad de liberación acumulativa de HBD (α-hidroxibutirato deshidrogenasa) en comparación con los pacientes tratados con placebo (p = 0.001). Los pacientes tratados con ACTILYSE® tuvieron un número significativamente menor de episodios de choque cardiogénico (p = 0.02), fibrilación ventricular (p < 0.04) y pericarditis (p = 0.01) en comparación con los pacientes tratados con placebo. La mortalidad a los 21 días en los pacientes tratados con ACTILYSE® se redujo al 3.7% en comparación con un 6.3% en los pacientes tratados con placebo (p unilateral = 0.05). Si bien estos datos no demuestran de manera inequívoca una reducción significativa de la mortalidad para este estudio, sí indican una tendencia que es apoyada con los resultados del estudio ASSET.
En un estudio controlado con placebo (LATE) realizado en 5 711 pacientes con IAM con inicio de los síntomas entre las 6 y las 24 horas, una infusión de 100 mg ACTILYSE® administrada en un lapso de 3 horas con placebo. Se observó una reducción no significativa del 14.1% (IC 95% 0-28.1%, p > 0.05) en la tasa de mortalidad a los 30 días con ACTILYSE®. En el análisis de sobrevida predeterminado en pacientes tratados en las 12 horas del inicio de los síntomas, se observó una reducción significativa del 25.6% en la mortalidad a favor de ACTILYSE® (IC 95% 6.3 - 45%; p = 0.023).
Pacientes con tromboembolia pulmonar masiva aguda:
En un estudio comparativo aleatorizado de alteplasa frente a urocinasa en 63 pacientes con tromboembolia pulmonar masiva aguda documentada por angiografía, ambos grupos experimentaron una reducción significativa de la hipertensión pulmonar inducida por embolia pulmonar. La hemodinamia pulmonar mejoró significativamente más rápido con ACTILYSE® que con la urocinasa.
No existen estudios clínicos sobre la mortalidad y morbilidad posterior relacionadas con el embolismo pulmonar.
Pacientes con Enfermedad Vascular Cerebral (EVC) isquémico agudo:
Se han realizado varios estudios en el campo del EVC isquémico agudo. El estudio NINDS es el único sin un límite superior de edad; que también incluyó pacientes mayores de 80 años. Todos los demás estudios aleatorios excluyeron a los pacientes mayores de 80 años. Por lo tanto, las decisiones terapéuticas en este grupo de pacientes requieren atención particular individualizada.
En dos estudios doble ciego comparados con placebo (Estudio NINDS t-PA Stroke, Parte 1 y Parte 2) se incluyeron pacientes con déficit neurológico medible que pudieron completar la selección e iniciar el tratamiento en las 3 horas siguientes al inicio de los síntomas. Se realizó una tomografía computarizada (CT) craneal antes del tratamiento para descartar la presencia de una hemorragia intracraneal sintomática (HIS). Se excluyeron pacientes con condiciones relacionadas a riesgo de sangrado, déficit neurológico menor, mejoría rápida de los síntomas antes de iniciar el tratamiento de estudio o glicemia < 50 mg/dL o > 400 mg/dL. Los pacientes se aleatorizaron para recibir 0.9 mg/kg de ACTILYSE® (máximo 90 mg) o placebo. Se administró ACTILYSE® como bolo inicial del 10% de la dosis, durante 1 minuto de duración, seguido de una infusión intravenosa continua del resto de la dosis durante 60 minutos.
El estudio inicial (NINDS-Parte 1, n = 291) evaluó la mejoría neurológica a las 24 horas después del inicio del EVC. El criterio de valoración principal, la proporción de pacientes con una mejoría de 4 puntos o más en la calificación de escala NIHSS Institutos Nacionales de Salud (National Institutes of Health Stroke Scale, NIHSS) o una recuperación completa (calificación NIHSS = 0), no tuvo diferencia significativa entre ambos grupos terapéuticos. Un análisis secundario sugirió mejoría en el resultado a 3 meses asociado al tratamiento con ACTILYSE® con las siguientes escalas de valoración de enfermedad vascular cerebral (EVC): Índice de Barthel (Barthel Index), Escala de Rankin Modificada (Modified Rankin Scale, mRS), Escala de Resultado de Glasgow (Glasgow Outcome Scale) y la escala del NIHSS. Un segundo estudio (NINDS-Parte 2, n = 333) valoró el resultado clínico a los 3 meses como resultado primario. Un resultado favorable se definió como una discapacidad mínima o nula usando las cuatro escalas de valoración de EVC: Índice de Barthel (calificación ≥ 95), Escala de Rankin Modificada (calificación ≤ 1), Escala de Resultado de Glasgow (calificación = 1) y escala del NIHSS (calificación ≤ 1). El índice de probabilidad para resultado favorable en grupo con tratamiento con ACTILYSE® fue 1.7 (odds ratio, OR) (IC 95% 1.2-2.6). En comparación con el placebo, hubo un aumento absoluto del 13% en el número de pacientes con discapacidad mínima o nula (mRS 0-1) 1.7 (odds ratio, OR) (IC 95% 1.1-2.6). También hubo un beneficio consistente con ACTILYSE® en otras escalas neurológicas y de discapacidad. Los análisis secundarios demostraron mejoría funcional y neurológica consistente en las 4 escalas de EVC como lo indicaron las medianas de las calificaciones. Estos resultados fueron altamente consistentes con el resultado a los 3 meses de los efectos terapéuticos en el estudio Parte 1. La incidencia de mortalidad a los 90 días por todas las causas, SICH y un nuevo EVC isquémico después del tratamiento con ACTILYSE® en comparación con placebo indicó un aumento significativo en la SICH sintomática (según la definición de NINDS) en las 36 horas siguientes al tratamiento con ACTILYSE® (ACTILYSE® 6.4%, placebo, 0.65%). En los pacientes tratados con ACTILYSE®, no hubo aumento en comparación con placebo en la incidencia de mortalidad a los 90 días, ni discapacidad severa (ACTILYSE® 20.5%; placebo, 17.3%).
En un análisis acumulado de 2 775 pacientes de 6 estudios clínicos aleatorizados grandes (NINDS parte 1 y 2, dos estudios ECASS y ATLANTIS parte A y B) evaluó el estado de la discapacidad en pacientes tratados con ACTILYSE® o placebo. En este análisis, la probabilidad de un resultado favorable a los 3 meses aumentó conforme disminuía el tiempo de inicio de tratamiento con ACTILYSE®. Se observó un índice de SICH de 5.9% en pacientes tratados con ACTILYSE® frente a un 1.1% de los controles (p < 0.0001), lo cual se relacionó con la edad, pero no con el tiempo para iniciar el tratamiento. Este análisis confirma fuertemente que el tratamiento rápido con ACTILYSE® se relaciona con mejores resultados a los 3 meses. También proporciona evidencia de que la ventana terapéutica puede extenderse hasta un lapso de 4.5 horas.
En un estudio observacional extenso por (SITS-MOST: The Safe Implementation of Thrombolysis in Stroke-Monitoring Study) se valoraron la seguridad y eficacia de ACTILYSE® para el tratamiento de EVC agudo en las primeras 3 horas en un ambiente clínico de rutina y se compararon con los resultados de estudios clínicos aleatorizados. Todos los pacientes tuvieron que apegarse con la información para prescribir (europea) de ACTILYSE®. Se recopilaron los datos del tratamiento y el resultado de 6,483 pacientes de 285 centros en 14 países europeos. Los desenlaces principales fueron la hemorragia intracraneal sintomática en las 24 horas y la mortalidad a los 3 meses. El índice de SICH observada en el estudio SITS-MOST fue comparable a la tasa de SICH reportados en estudios aleatorizados; 7.3% (IC 95% 6.7-8.0) en SITS-MOST frente a 8,6% (IC 95% 6.1-11.1) en los estudios clínicos aleatorizados. La mortalidad fue del 11.3% (IC 95% 10.5-12.1) en el estudio SITS-MOST frente a un 17% (IC 95% 13.9-20.7) en los estudios clínicos aleatorizados. Los resultados del SITS-MOST indican que el uso clínico de rutina de ACTILYSE® dentro de las 3 horas del inicio del EVC es tan seguro como se reporta en los estudios clínicos aleatorizados.
El estudio ECASS III es un estudio doble ciego controlado con placebo que se realizó en pacientes con EVC agudo en un tiempo de ventana de 3 a 4.5 horas. El estudio incluyó pacientes con déficit neurológico medible apegados a la información para prescribir (europea) del producto, excepto por el tiempo de ventana. Después de excluir la hemorragia cerebral o infarto mayor mediante una tomografía computarizada, los pacientes con EVC isquémico agudo, se distribuyeron al azar 1:1 y en forma doble ciego para recibir alteplasa por vía intravenosa (0.9 mg/kg de peso corporal) o placebo. El criterio de valoración primario fue la discapacidad a los 90 días, separada según resultado favorable (calificación de 0 a 1 en la escala de Rankin modificada [mRS]) o desfavorable (calificación de 2 a 6 en la mRS). El criterio de valoración secundario fue un análisis del resultado global de cuatro calificaciones neurológicas y de discapacidad combinado. Los criterios de valoración de seguridad fueron mortalidad, SICH y eventos adversos serios. Se distribuyó al azar un total de 821 pacientes (418 a alteplasa y 403 a placebo). Más pacientes alcanzaron un resultado favorable con la alteplasa (52.4%) en comparación con el placebo (45.2%; o OR, 1.34; IC 95% 1.02-1.76; p = 0.038). En el análisis global, el desenlace también mejoró (OR, 1.28; IC 95% 1.00-1.65; p = 0.048). La incidencia de cualquier ICH/SICH fue más alta con alteplasa que con placebo (cualquier SICH: 27.0% frente a 17.6%, p = 0.0012; SICH por definición de NINDS: 7.9% frente a 3.5%, p = 0.006; SICH por definición de ECASS III: 2.4% frente a 0.2%, p = 0.008).
La mortalidad fue baja y no hubo diferencia significativa entre la alteplasa (7.7%) y el placebo (8.4%; p = 0.681). Los resultados del estudio ECASS III indican que ACTILYSE® administrado entre 3 y 4.5 horas después del inicio de los síntomas mejora significativamente los resultados clínicos en los pacientes con EVC isquémico agudo.
La seguridad y la eficacia de ACTILYSE® para el tratamiento del EVC isquémico agudo (EVCIA), hasta 4.5 horas después del inicio de los síntomas se ha evaluado mediante de un registro constantemente actualizado de EVC agudo (SITS-ISTR: The Safe Implementation of Thrombolysis in Stroke Registry). Los datos de desenlace principal y de mortalidad de 21,566 pacientes en la ventana de tiempo de 0 a 3 horas fueron comparados con datos de 2,376 pacientes tratados entre 3 y 4.5 horas después del inicio de EVCIA (datos de 2010). La incidencia de hemorragia intracerebral sintomática (de acuerdo con la definición de NINDS) fue ligeramente mayor en la ventana de tiempo de 3 a 4.5 horas (7.4%) en comparación con la ventana de tiempo de 0-3 horas (7.1%; cociente de probabilidad ajustada IC 95%: 1.18 (0.99-1.41) p = 0.06). Las tasas de mortalidad a los 3 meses fueron similares al comparar la ventana de tiempo de 3 a 4.5 horas (12.4%) con la de 0 a 3 horas (12.3%).
Farmacocinética:
ACTILYSE® se elimina rápidamente del torrente sanguíneo y se metaboliza principalmente por vía hepática (depuración plasmática 550-680 mL/min). En condiciones fisiológicas, la mayor parte de alteplasa en la circulación está unida a inhibidores. La depuración hepática de la alteplasa no se ve alterada por la presencia de otras proteínas, incluidos los inhibidores de la alteplasa. Los complejos de alteplasa y su inhibidor se eliminan como alteplasa libre. La vida media plasmática relevante, el t½ alfa, es de 4 a 5 minutos. Esto significa que, al cabo de 20 minutos, menos del 10% del valor inicial está presente en el plasma. Para la cantidad residual, que permanece en un compartimiento profundo, se midió una semivida beta de aproximadamente 40 minutos.
CONTRAINDICACIONES:
ACTILYSE® está contraindicado en:
- Pacientes con hipersensibilidad conocida al principio activo alteplasa, o a cualquiera de los excipientes.
- Embarazo, lactancia y menores de 18 años.
Como sucede con todos los agentes trombolíticos, y generalmente en todas las indicaciones, ACTILYSE® no debe ser usado en casos donde exista un alto riesgo de hemorragia, tales como:
- Trastorno hemorrágico significativo actual o durante los últimos 6 meses, diátesis hemorrágica conocida.
- Pacientes en tratamiento efectivo con anticoagulantes orales, p. ej., warfarina sódica (INR > 1.3) (ver Precauciones generales, subsección “Sangrado”).
- Antecedentes de daño al sistema nervioso central (p. ej., neoplasia, aneurisma, cirugía intracraneal o de la médula espinal).
- Antecedente, evidencia o sospecha de hemorragia intracraneal incluyendo, la hemorragia subaracnoidea.
- Hemorragia severa o grave, manifiesta o reciente.
- Resucitación cardiopulmonar prolongada o traumática (> 2 minutos), en el transcurso de los últimos 10 días.
- Parto dentro de los últimos 10 días.
- Punción reciente de un vaso sanguíneo no comprimible (p. ej., vena subclavia o yugular).
- Hipertensión arterial severa no controlada.
- Endocarditis bacteriana, pericarditis.
- Pancreatitis aguda.
- Enfermedad gastrointestinal ulcerosa documentada durante los últimos 3 meses.
- Enfermedad hepática severa, incluidas insuficiencia hepática, cirrosis, hipertensión portal (várices esofágicas) y hepatitis activa.
- Cirugía mayor o traumatismo importante en los últimos 10 días (incluido cualquier traumatismo asociado con el infarto agudo de miocardio actual).
- Traumatismo reciente de cabeza o de cráneo.
- Aneurismas arteriales, malformaciones venosas/arteriales.
- Neoplasia con mayor riesgo de sangrado.
En las indicaciones de infarto agudo de miocardio también aplican las siguientes contraindicaciones:
- Antecedentes de EVC hemorrágico o EVC de origen desconocido.
- EVC isquémico o ataques isquémicos transitorios (AIT) dentro de los 6 meses previos, excepto EVC tipo isquémico agudo actual producido dentro de las últimas 4.5 horas.
En la indicación de tromboembolia pulmonar masiva aguda también aplican las siguientes contraindicaciones:
- Antecedentes de EVC hemorrágico o EVC de origen desconocido.
- EVC isquémico o ataques isquémicos transitorios (AIT) dentro de los 6 meses previos, excepto EVC tipo isquémico agudo actual producido dentro de las últimas 4.5 horas.
En la indicación de EVC isquémico agudo también aplican las siguientes contraindicaciones:
- Síntomas de EVC isquémico que comenzaron más de 4.5 horas antes del inicio de la infusión o desconocimiento del momento de inicio de los síntomas.
- Síntomas de EVC isquémico agudo que fueron leves o mejoraron rápidamente antes del inicio de la infusión.
- EVC severo según la evaluación clínica (p. ej., NIHSS > 25) y/o por estudios de diagnóstico por imágenes adecuados.
- Convulsiones al inicio del EVC.
- Antecedentes de EVC previo o traumatismo de cabeza serio en los últimos 3 meses.
- Combinación de EVC previo y diabetes mellitus.
- Administración de heparina dentro de las 48 horas previas al inicio del EVC con tiempo de tromboplastina parcial activada (TTPa) elevado al momento de la presentación.
- Recuento de plaquetas inferior a 100,000/mm3.
- Presión arterial sistólica > 185 mmHg o presión arterial diastólica >110 mmHg, o necesidad de manejo agresivo (medicación IV) para reducir la presión arterial a estos límites.
- Glucemia < 50 mg/dL o > 400 mg/dL. (<2.8 mmol/L o >22.2 mmol/L)
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Existe una limitada cantidad de datos relativos al uso de ACTILYSE® en mujeres embarazadas. En los estudios preclínicos realizados con alteplasa en dosis mayores que las utilizadas en seres humanos se observó inmadurez fetal y/o embriotoxicidad, secundaria a la actividad farmacológica conocida del medicamento. La alteplasa no se considera teratogénica (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
En casos de una enfermedad aguda que ponga en peligro la vida, se debe evaluar el beneficio contra el riesgo potencial.
Lactancia:
Se desconoce si la alteplasa es excretada en la leche materna.
Se debe tener precaución cuando se administra ACTILYSE® a una madre lactante y decidir si se debe interrumpir la lactancia durante las primeras 24 horas después de la administración de ACTILYSE®.
REACCIONES SECUNDARIAS Y ADVERSAS:
La reacción adversa más frecuente asociada con ACTILYSE® es el sangrado (≥ 1:100, ≤ 1:10 sangrado grave; ≥ 1:10 cualquier hemorragia) dando como resultado una caída de los valores del hematocrito y/o hemoglobina. Puede ocurrir hemorragia en cualquier sitio o cavidad corporal, que puede dar como resultado situaciones que pongan en peligro la vida, provoquen incapacidad o muerte.
Los tipos de sangrados asociados con la terapia trombolítica se pueden dividir en dos grandes categorías:
• Sangrado superficial, normalmente de los sitios de venopunción o en vasos sanguíneos lesionados.
• Hemorragia interna, en cualquier sitio o cavidad corporal.
Con la hemorragia intracraneal pueden presentarse síntomas neurológicos asociados tales como somnolencia, afasia, hemiparesia y convulsiones.
El número de pacientes tratados en los estudios clínicos para las indicaciones de embolia pulmonar masiva aguda y EVC isquémico agudo (dentro de la ventana de tiempo de 0-4.5 horas) fue muy reducida en comparación con la cantidad de pacientes del estudio de infarto agudo de miocardio. Por lo tanto, las pequeñas diferencias numéricas observadas en comparación con el número de infartos agudos de miocardio fueron presumiblemente atribuibles a lo reducido del tamaño de la muestra. Excepto por la hemorragia intracraneal como efecto secundario en la indicación de enfermedad cerebral vascular isquémica aguda y de las arritmias por reperfusión en la indicación de infarto agudo de miocardio, no existe ninguna razón médica para suponer que el perfil cuali- cuantitativo de efectos secundarios de ACTILYSE® en las indicaciones de embolia pulmonar masiva aguda y EVC isquémico agudo difieren del perfil de este producto en la indicación de infarto agudo de miocardio.
Los efectos secundarios de ACTILYSE® (alteplasa) enumerados se especificaron por frecuencia y se asignaron a las categorías conforme a la Guía de SmPC de la UE de septiembre de 2009.
|
Muy frecuente |
≥ 1/10 |
≥ 10% |
|
Frecuente |
≥ 1/100 - < 1/10 |
≥ 1%, < 10% |
|
Poco frecuente |
≥ 1/1000 - < 1/100 |
≥ 0,1%, < 1% |
|
Rara |
≥ 1/10.000 - < 1/1000 |
≥ 0,01%, < 0,1% |
|
Muy rara |
< 1/10.000 |
< 0,01% |
|
Frecuencia desconocida |
No se puede estimar a partir de los datos disponibles |
|
Tabla 1. Reacciones adversas enumeradas en la CCDS y frecuencias correspondientes según la Guía de SmPC de la UE (basado en ASSENT II)
|
Reacciones adversas – Indicaciones de infarto agudo de miocardio, embolia pulmonar masiva aguda y accidente cerebrovascular isquémico agudo |
||
|---|---|---|
|
Clasificación por sistema y órgano7 Terminología del MedDRA |
Reacciones adversas (a la alteplasa) según término textual de la CCDS, TP del MedDRA (versión 24.1) |
Frecuencias según la Guía de SPC de la UE |
|
Trastornos del sistema inmunitario |
Reacciones anafilactoides |
Rara |
|
Pueden presentarse como: |
||
|
Exantema |
||
|
Urticaria |
||
|
Broncoespasmo |
||
|
Angioedema |
||
|
Hipotensión |
||
|
Shock o cualquier otro síntoma asociado con hipersensibilidad |
||
|
Trastornos oculares |
Hemorragia ocular |
Rara |
|
Trastornos cardiacos |
Hemorragia pericárdica |
Rara |
|
Trastornos vasculares |
Hemorragia, como hematoma |
Muy frecuente |
|
Emboliaa |
Rara |
|
|
Sangrado de órganos parenquimatosos, como hemorragia hepáticab |
Frecuencia desconocidab |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Hemorragia del aparato respiratorio, como: |
|
|
Hemorragia faríngea |
Frecuente |
|
|
Hemoptisis |
Poco frecuente |
|
|
Epistaxis |
Poco frecuente |
|
|
Hemorragia pulmonar |
Rara |
|
|
Trastornos gastrointestinales |
Hemorragia gastrointestinal, como hemorragia gástrica, hemorragia de úlcera gástrica, hemorragia rectal, hematemesis, melena, hemorragia bucal, sangrado gingival |
Frecuente |
|
Hemorragia retroperitoneal, como hematoma retroperitoneal |
Rara |
|
|
Náuseas |
Rara |
|
|
Vómitosb |
Frecuencia desconocidab |
|
|
Trastornos de la piel y del tejido subcutáneo |
Equimosis |
Frecuente |
|
Trastornos renales y urinarios |
Hemorragia urogenital, como hematuria, hemorragia del aparato urinario |
Frecuente |
|
Trastornos generales y afecciones del lugar de la administración |
Hemorragia en el lugar de la inyección, hemorragia en el lugar de la punción, como hematoma en el sitio de inserción del catéter, hemorragia en el sitio de inserción del catéter |
Frecuente |
|
Exploraciones complementarias |
Disminución de la presión arterial |
Poco frecuente |
|
Aumento en la temperatura corporalb |
Frecuencia desconocidab |
|
|
Lesiones e intoxicaciones |
Embolia grasab |
Frecuencia desconocidab |
|
Procedimientos médicos y quirúrgicos |
Transfusiónb |
Frecuencia desconocidab |
a = BI determinó la frecuencia de estas reacciones adversas sobre la base de los datos que surgen del estudio clínico (ASSENT II).
b = Esta reacción adversa se ha observado en la experiencia posterior a la comercialización. Con una certeza del 95 %, la categoría de frecuencia no es mayor que “rara”, pero podría ser menor. No es posible estimar la frecuencia con precisión, ya que la reacción adversa al medicamento no se presentó en la base de datos de 8299 pacientes de un estudio clínico.
Tabla 2. Reacciones adversas enumeradas en la CCDS y frecuencias correspondientes según la Guía de SmPC de la UE (basado en ASSENT II)
|
Reacciones adversas – Indicaciones de infarto agudo de miocardio |
||
|
Clasificación por sistema y órgano Terminología del MedDRA |
Reacciones adversas (a la alteplasa) según término textual de la CCDS, TP del MedDRA (versión 24.1) |
Frecuencias según la Guía de SPC de la UE |
|
Trastornos del sistema nervioso |
Hemorragia intracraneal (LLT), como hemorragia cerebral, hematoma cerebral, ACV hemorrágico, transformación hemorrágica de ACV, hematoma intracraneal, hemorragia subaracnoidea |
Frecuente |
|
Trastornos cardiacos |
Arritmias por reperfusióna, como arritmia, extrasístoles, fibrilación auricular, bloqueo auriculoventricular de grado I hasta bloqueo auriculoventricular completo, bradicardia, taquicardia, arritmia ventricular, fibrilación ventricular, taquicardia ventricular |
Poco frecuente |
a = BI determinó la frecuencia de estas reacciones adversas sobre la base de los datos que surgen del estudio clínico (ASSENT II).
Tabla 3. Reacciones adversas de HIC enumeradas en la CCDS y frecuencias correspondientes según la Guía de SmPC de la UE (basado en ASSENT II)
|
Reacciones adversas – Indicación embolia pulmonar masiva aguda |
||
|
Clasificación por sistema y órgano Terminología del MedDRA |
Reacciones adversas (a la alteplasa) según término textual de la CCDS, TP del MedDRA (versión 24.1) |
Frecuencias según la Guía de SPC de la UE |
|
Trastornos del sistema nervioso |
Hemorragia intracraneal (LLT), como hemorragia cerebral, hematoma cerebral, ACV hemorrágico, transformación hemorrágica de ACV, hematoma intracraneal, hemorragia subaracnoidea |
Frecuente |
Tabla 4. Reacciones adversas de HIC enumeradas en la CCDS y frecuencias correspondientes según la Guía de SmPC de la UE (basado en NINDS A+B y ECASS III)
|
Reacciones adversas – Indicación evento cerebrovascular isquémico agudo |
||
|
Clasificación por sistema y órgano Terminología del MedDRA |
Reacciones adversas (a la alteplasa) según término textual de la CCDS, TP del MedDRA (versión 24.1) |
Frecuencias según la Guía de SPC de la UE |
|
Trastornos del sistema nervioso |
Hemorragia intracraneal (LLT), como hemorragia cerebral, hematoma cerebral, ACV hemorrágico, transformación hemorrágica de ACV, hematoma intracraneal, hemorragia subaracnoidea |
Frecuente |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Los estudios de carcinogenicidad, mutagenicidad y teratogenicidad no demostraron evidencia de actividad.
No se observaron efectos teratogénicos en animales embarazados después de la infusión intravenosa de dosis efectivas farmacológicamente. Se indujo embriotoxicidad (embrioletalidad, retraso en el crecimiento) en conejos con más de 3 mg/kg/día. No se observaron efectos en el desarrollo peri-postnatal o en los parámetros de fertilidad en ratas con dosis superiores a 10 mg/kg/día.
Fertilidad:
No hay datos clínicos disponibles en la fertilidad para ACTILYSE®. Estudios no clínicos realizados con alteplasa mostraron que no hay reacciones secundarias en la fertilidad.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
No han sido desarrollados estudios de interacción formales con ACTILYSE® y los medicamentos comúnmente administrados en pacientes con infarto agudo de miocardio.
Medicamentos que afectan la coagulación/función plaquetaria:
Los medicamentos que alteran la coagulación o aquellos que alteran la función plaquetaria, pueden incrementar el riesgo de sangrado antes, durante o después del tratamiento con ACTILYSE® y deben evitarse en las primeras 24 horas posteriores al tratamiento del accidente cerebrovascular isquémico agudo (ver sección Contraindicaciones).
Inhibidores de la ECA:
El tratamiento concomitante con inhibidores de la ECA puede incrementar el riesgo de sufrir una reacción de hipersensibilidad (Ver sección precauciones generales).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Se modifican las pruebas de laboratorio relacionadas con los tiempos de sangrado y coagulación. Dicha modificación se manifiesta únicamente durante el periodo de aplicación del medicamento.
PRECAUCIONES GENERALES:
Las siguientes advertencias especiales y precauciones aplican para el tratamiento de Infarto de miocardio agudo, la tromboembolia pulmonar masiva aguda y EVC isquémico agudo.
ACTILYSE® debe ser administrado por médicos experimentados en el uso de tratamientos trombolíticos y medios adecuados para monitorear su uso. Como sucede con otros agentes trombolíticos, se recomienda contar con la infraestructura de resucitación estándar y de medicación disponibles en todo momento.
Trazabilidad: A fin de mejorar la trazabilidad de los medicamentos biológicos, el nombre comercial y el número de lote del producto administrado deben registrarse claramente en la historia clínica del paciente.
Hipersensibilidad:
Las reacciones de hipersensibilidad mediada por el sistema inmunitario asociadas a la administración de ACTILYSE® pueden ser causadas por el principio activo alteplasa, o cualquiera de los excipientes (ver sección Contraindicaciones).
Tras el tratamiento no se ha observado formación sostenida de anticuerpos contra la molécula recombinante activador del plasminógeno tisular humano. No hay experiencia sistemática con la re-administración de ACTILYSE®.
También existe el riesgo de reacciones de hipersensibilidad mediada por un mecanismo no inmunitario.
El angioedema representa la reacción de hipersensibilidad más frecuente informada con ACTILYSE®. Este riesgo puede aumentar en la indicación de enfermedad cerebro vascular isquémico agudo y/o por el tratamiento concomitante con inhibidores de la ECA (ver sección Interacciones medicamentosas y de otro género).
Se debe monitorear a los pacientes tratados por cualquiera de las indicaciones autorizadas a fin de detectar casos de angioedema durante la infusión y hasta las 24 h posteriores.
En el caso de producirse una reacción de hipersensibilidad severa (ej., angioedema), debe suspenderse la infusión e iniciarse de inmediato el tratamiento adecuado, que puede incluir la intubación.
Sangrado:
La complicación más comúnmente encontrada durante la terapia con ACTILYSE® es el sangrado. El uso concomitante de otros principios activos que afectan la coagulación o la función plaquetaria puede contribuir al sangrado. Como la fibrina es lisada durante la terapia con ACTILYSE®, puede ocurrir sangrado en sitios de punción reciente. Por lo tanto, la terapia trombolítica requiere de atención cuidadosa de todos los sitios probables de sangrado (incluyendo aquellos en donde se realice la colocación de catéteres, en venodisecciones o punciones arteriales o venosas, y punciones con agujas). El uso de catéteres rígidos, las inyecciones intramusculares y la manipulación innecesaria del paciente deberán ser evitados durante la administración de ACTILYSE®.
En caso de ocurrir sangrado severo, en particular hemorragia cerebral, la terapia fibrinolítica debe ser suspendida inmediatamente, la administración concomitante de heparina debe ser retirada al mismo tiempo. Se debe de considerar la administración de protamina si la heparina ha sido administrada dentro de las 4 horas anteriores al inicio del sangrado. Puede indicarse el uso racional de productos de transfusión en los pocos pacientes que no respondan a estas medidas conservadoras.
La transfusión de crioprecipitados, plasma fresco congelado y de plaquetas debe de considerarse con base al análisis de laboratorio realizado después de cada administración. Un nivel de fibrinógeno de 1 g/L es el objetivo deseado con la infusión de crioprecipitado. Se debe considerar la utilización probable de agentes antifibrinolíticos.
No se debe exceder la dosis de 100 mg de ACTILYSE® en situaciones de infarto agudo al miocardio, así como en el embolismo pulmonar y de 90 mg en el caso del EVC de origen isquémico agudo, dado que se ha asociado con incremento de hemorragia intracraneal.
Como sucede con todos los agentes trombolíticos, el uso de ACTILYSE® debe ser evaluado cuidadosamente con la intención de ponderar los riesgos potenciales de sangrado con los beneficios esperados bajo las siguientes condiciones:
- Inyección intramuscular reciente o traumas recientes menores, tales como biopsias, punción de vasos mayores, masaje cardiaco para resucitación.
- Condiciones no mencionadas en las contraindicaciones, que aumentan el riesgo de hemorragias.
- Pacientes con tratamiento de anticoagulantes orales.
- El uso de ACTILYSE® puede ser considerado cuando las prueba(s) apropiadas de actividad anticoagulante para el (los) producto(s) en cuestión no muestran actividad clínicamente relevante.
Para el tratamiento del infarto agudo de miocardio aplican además las siguientes advertencias y precauciones especiales:
- Presión sistólica > 160 mmHg (ver sección Contraindicaciones).
- Edad avanzada, lo cual puede incrementar el riesgo de hemorragia intracraneal. Como el beneficio terapéutico así mismo se incrementa en pacientes de edad avanzada, se debe evaluar la relación riesgo beneficio en forma cuidadosa.
Arritmias:
La trombólisis coronaria puede dar como resultado una arritmia asociada con la reperfusión.
Las arritmias de repercusión pueden conducir a un paro cardiaco, que puede amenazar la vida y puede requerir el uso de terapias antiarrítmicas convencionales.
Antagonistas del receptor de la glicoproteína IIb/IIIa:
El uso concomitante de antagonistas del receptor de la GPIIb/IIa incrementa el riesgo de sangrado.
Tromboembolismo:
El uso de trombolíticos puede incrementar el riesgo de eventos tromboembólicos en pacientes con trombosis de ventrículo izquierdo, p. ej. estenosis mitral o fibrilación auricular.
Para el tratamiento de la embolia pulmonar masiva aguda aplican además las siguientes advertencias y precauciones especiales:
- Presión sistólica > 160 mmHg (ver sección Contraindicaciones).
- Edad avanzada, lo cual puede incrementar el riesgo de hemorragia intracraneal. Como el beneficio terapéutico así mismo se incrementa en pacientes de edad avanzada, se debe evaluar la relación riesgo beneficio en forma cuidadosa.
Para el tratamiento del EVC isquémico agudo aplican además las siguientes advertencias y precauciones especiales:
El tratamiento debe ser llevado a cabo bajo la responsabilidad de un médico entrenado y experimentado con el cuidado neurológico. Para la verificación del tratamiento de la indicación, medidas de diagnóstico remotas pueden ser consideradas como apropiadas (ver sección de Indicaciones; Tratamiento trombolítico de la enfermedad vascular cerebral (EVC) isquémica aguda.)
Sangrado:
Las hemorragias intracerebrales representan el principal evento adverso (hasta aproximadamente 15% de los pacientes). Sin embargo, esto no ha evidenciado un aumento en la morbilidad y mortalidad general.
Comparado con otras indicaciones, los pacientes con EVC de origen isquémico agudo tratados con ACTILYSE® presentan un riesgo considerablemente mayor de hemorragia intracraneal ya que el sangrado se produce principalmente en la región infartada.
Esto aplica particularmente en los siguientes casos:
• Todas las situaciones enlistadas en la sección de contraindicaciones y en general todas las situaciones que involucren un alto riesgo de hemorragia.
• Tiempo tardío para iniciar el tratamiento.
• Pacientes pre-tratados con ácido acetilsalicílico (ASA) presentan un mayor riesgo de hemorragia intracerebral, particularmente si el tratamiento con ACTILYSE® se retrasa.
• Pacientes mayores de 80 años de edad pueden tener un riesgo incrementado de hemorragia intracerebral y un beneficio neto reducido del tratamiento comparado con pacientes más jóvenes. Por lo tanto, el uso de ACTILYSE® debe ser medido cuidadosamente contra los riesgos anticipados sobre la base de cada paciente individual.
El tratamiento no debe ser iniciado después de 4.5 horas posteriores a la aparición de los síntomas, debido a la proporción riesgo/beneficio desfavorable, principalmente basada en lo siguiente:
• Los efectos positivos del tratamiento disminuyen con el tiempo.
• Particularmente en pacientes con tratamiento previo con ASA, la tasa de mortalidad aumenta.
• Aumento del riesgo de hemorragia sintomática.
Control de la presión arterial:
Es necesario controlar la presión arterial durante la administración del tratamiento y durante las 24 horas posteriores; se recomienda tratamiento antihipertensivo intravenoso si la presión arterial sistólica es superior a 180 mmHg o la presión arterial diastólica es superior a 105 mmHg.
Grupos especiales de pacientes con relación riesgo-beneficio reducida:
El beneficio terapéutico se reduce en los pacientes que hubieran presentado EVC (ver sección Contraindicaciones) o con diabetes no controlada. La relación riesgo-beneficio en estos pacientes se considera menos favorable pero todavía positiva.
Los pacientes con infartos extensos se encuentran en mayor riesgo de obtener una pobre respuesta, incluyendo la hemorragia y la muerte. En tales pacientes, la relación riesgo/beneficio deberá ser cuidadosamente valorada.
En los pacientes con EVC de origen isquémico, la probabilidad de un resultado favorable disminuye cuanto mayor sea el tiempo transcurrido desde el inicio de los síntomas hasta el tratamiento, con el aumento de la edad, el aumento de la severidad del EVC y el aumento de los niveles de concentración de glucosa sanguínea al momento de su admisión; mientras que la probabilidad de discapacidad severa y muerte o sangrado intracraneal sintomático aumenta, independientemente del tratamiento.
Edema cerebral:
La reperfusión del área afectada por la isquemia puede inducir edema cerebral en la zona infartada.
Población pediátrica:
Por el momento, sólo existe experiencia limitada con el uso de ACTILYSE® en niños.
Efectos en la habilidad para conducir y usar máquinas: No aplica.
DOSIS Y VÍA DE ADMINISTRACIÓN:
ACTILYSE® deberá administrarse a la brevedad posible una vez iniciada la sintomatología.
Vía de administración: Intravenosa.
La administración de este medicamento por vía intravenosa deberá realizarse en bolo seguido de infusión.
La solución reconstituida debe administrarse por vía intravenosa y utilizarse de inmediato.
Este producto es de muy alto riesgo, sólo deberá ser administrado por médicos especialistas. Uso exclusivamente intrahospitalario.
Infarto agudo de miocardio:
a) Régimen de infusión acelerada de 90 minutos.
Para los pacientes con infarto agudo del miocardio, en los cuales el tratamiento puede iniciarse dentro de 6 horas subsiguientes al inicio de los síntomas:
En pacientes con un peso corporal ≥ 65 kg:
- 15 mg como bolo intravenoso, seguidos inmediatamente de 50 mg como infusión intravenosa durante los primeros 30 minutos, seguida inmediatamente de una infusión intravenosa de 35 mg durante 60 minutos, hasta la dosis total máxima de 100 mg.
En pacientes con un peso corporal ≤ 65 kg, la dosis total debe ajustarse en función del peso a:
- 15 mg como bolo intravenoso, seguido inmediatamente de 0.75 mg/kg de peso corporal como infusión intravenosa durante los primeros 30 minutos (máximo 50 mg), seguida inmediatamente de una infusión intravenosa de 0.5 mg/kg durante 60 minutos (hasta un máximo de 35 mg).
b) Régimen de infusión de 3 horas para los pacientes con infarto agudo de miocardio en los cuales el tratamiento puede iniciarse dentro de las 6 a 12 horas subsiguientes al inicio de los síntomas.
En pacientes con un peso corporal ≥ 65 kg:
- 10 mg como bolo intravenoso, seguidos inmediatamente de 50 mg como infusión intravenosa durante la primera hora, seguida inmediatamente de una infusión intravenosa de 40 mg durante las dos horas siguientes, hasta alcanzar la dosis total máxima de 100 mg.
En pacientes con un peso corporal < 65 kg:
- 10 mg como bolo intravenoso, seguidos inmediatamente de una infusión intravenosa a velocidad constante durante 3 horas hasta alcanzar una dosis total máxima de 1.5 mg/kg de peso corporal.
La dosis máxima aceptada para ACTILYSE® es de 100 mg.
Tratamiento coadyuvante:
El tratamiento trombolítico coadyuvante se recomienda de acuerdo a las guías internacionales actuales para el manejo de pacientes con infarto agudo del miocardio con elevación del segmento ST.
Tromboembolia pulmonar masiva aguda:
En pacientes con un peso corporal ≥ 65 kg:
Debe de administrarse una dosis total de 100 mg en 2 horas. La mayor experiencia disponible corresponde al siguiente régimen posológico:
- 10 mg como bolo intravenoso durante 1 - 2 minutos, seguido inmediatamente de 90 mg como infusión intravenosa durante 2 horas hasta alcanzar la dosis total máxima de 100 mg.
En pacientes con un peso corporal < 65 kg:
- 10 mg como bolo intravenoso durante 1-2 minutos, seguido inmediatamente de una infusión intravenosa a velocidad constante durante 2 horas hasta alcanzar una dosis total máxima de 1.5 mg/kg de peso corporal.
Tratamiento coadyuvante:
Después de iniciado el tratamiento con ACTILYSE® se debe iniciar la administración de heparina, una vez que los valores del TPTa sean menores a dos veces el límite máximo normal. La infusión debe ajustarse de manera de mantener los valores del TPTa entre 50-70 segundos (1.5 a 2.5 veces el valor de referencia).
Evento cerebro vascular isquémico agudo:
La dosis total recomendada de ACTILYSE® es de 0.9 mg/kg de peso corporal (dosis máxima 90 mg). Se debe comenzar con el 10% del total de la dosis en forma de bolo intravenoso inicial, seguida inmediatamente del resto de la dosis total administrada por infusión intravenosa durante 60 minutos.
El tratamiento debe iniciarse a la mayor brevedad posible dentro de las 4.5 horas de iniciados los síntomas (ver sección Precauciones generales). El efecto del tratamiento depende del tiempo, por lo tanto, cuanto antes se inicie el tratamiento más temprano incrementa la probabilidad de un resultado favorable.
|
Tabla de dosificación para el evento cerebrovascular isquémico agudo |
|||
|
Peso (kg) |
Dosis total (mg) |
Dosis en bolo (mg) |
Dosis de infusión (mg) |
|
40 |
36.0 |
3.6 |
32.4 |
|
42 |
37.8 |
3.8 |
34.0 |
|
44 |
39.6 |
4.0 |
35.6 |
|
46 |
41.4 |
4.1 |
37.3 |
|
48 |
43.2 |
4.3 |
38.9 |
|
50 |
45.0 |
4.5 |
40.5 |
|
52 |
46.8 |
4.7 |
42.1 |
|
54 |
48.6 |
4.9 |
43.7 |
|
56 |
50.4 |
5.0 |
45.4 |
|
58 |
52.2 |
5.2 |
47.0 |
|
60 |
54.0 |
5.4 |
48.6 |
|
62 |
55.8 |
5.6 |
50.2 |
|
64 |
57.6 |
5.8 |
51.8 |
|
66 |
59.4 |
5.9 |
53.5 |
|
68 |
61.2 |
6.1 |
55.1 |
|
70 |
63.0 |
6.3 |
56.7 |
|
72 |
64.8 |
6.5 |
58.3 |
|
74 |
66.6 |
6.7 |
59.9 |
|
76 |
68.4 |
6.8 |
61.6 |
|
78 |
70.2 |
7.0 |
63.2 |
|
80 |
72.0 |
7.2 |
64.8 |
|
82 |
73.8 |
7.4 |
66.4 |
|
84 |
75.6 |
7.6 |
68.0 |
|
86 |
77.4 |
7.7 |
69.7 |
|
88 |
79.2 |
7.9 |
71.3 |
|
90 |
81.0 |
8.1 |
72.9 |
|
92 |
82.8 |
8.3 |
74.5 |
|
94 |
84.6 |
8.5 |
76.1 |
|
96 |
86.4 |
8.6 |
77.8 |
|
98 |
88.2 |
8.8 |
79.4 |
|
100+ |
90.0 |
9.0 |
81.0 |
* Administrada en una concentración de 1 mg/ml durante 60 minutos.
Tratamiento coadyuvante:
No se ha estudiado suficientemente la administración concomitante de heparina o inhibidores de la agregación plaquetaria tales como ácido acetilsalicílico durante las primeras 24 horas del inicio de los síntomas. Por tanto, la administración de heparina intravenosa o inhibidores de la agregación plaquetaria tales como ácido acetilsalicílico se debe de evitar durante las primeras 24 horas de tratamiento con ACTILYSE® debido al mayor riesgo de hemorragia.
Si fuese necesaria la administración de heparina por alguna otra patología (ej. prevención de trombosis venosa profunda) no se debe exceder la dosis de 10,000 UI al día, administrados por vía subcutánea.
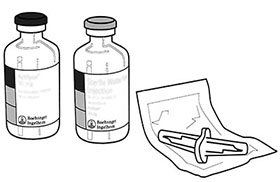
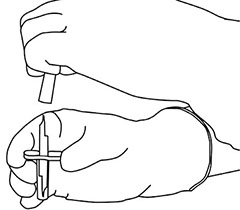
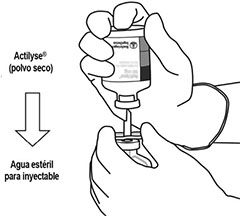
Instrucciones de uso:
Bajo condiciones asépticas, el contenido de un frasco ámpula de ACTILYSE® (50 mg) se disuelve con agua inyectable estéril de acuerdo con la siguiente tabla con la intención de obtener una concentración final de 1 mg de alteplasa por cada 1 mL.
|
Polvo liofilizado de ACTILYSE® |
50 mg |
|
Volumen de agua estéril para uso inyectable para agregar al polvo seco |
50 mL |
|
Concentración final: |
1 mg alteplasa/mL |
Por tanto, para la reconstitución de la concentración final de 1 mg alteplasa/mL, el volumen total del solvente debe ser transferido al frasco ámpula que contiene la sustancia seca ACTILYSE®. Para dicho propósito, se incluye una cánula de transferencia en el empaque del producto.
Instrucciones para la reconstitución de ACTILYSE®
|
1 |
Reconstituir inmediatamente antes de la administración. |
|
|
2 |
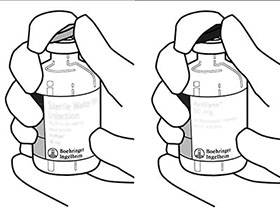
Retire las tapas protectoras de los dos frascos ámpula que contienen el agua estéril y el polvo liofilizado de ACTILYSE® deslizándolas hacia arriba con el pulgar. |
|
|
3 |
Limpie la parte superior de caucho de cada frasco ámpula con un paño impregnado en alcohol. |
|
|
4 |
Retire la cánula de transferencia* de su cubierta. No desinfecte ni esterilice la cánula de transferencia; es estéril. Quite una de las tapas. |
|
|
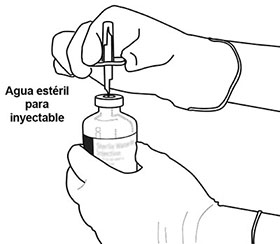
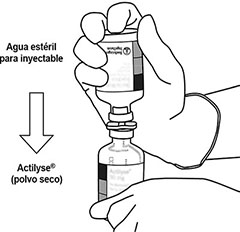
5 |
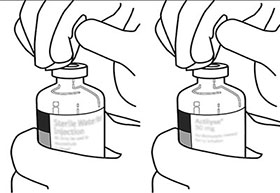
Coloque el frasco ámpula de agua estéril hacia arriba en una superficie estable. Directamente, desde arriba, pinche el centro del tapón de caucho verticalmente con la cánula de transferencia, presionando suave, pero firmemente, sin girar. |
|
|
6 |
Sostenga constantemente el frasco ámpula de agua estéril y la cánula de transferencia con una mano, usando las dos aletas laterales. Retire la tapa que queda en la parte superior de la cánula de transferencia. |
|
|
7 |
Sostenga constantemente el frasco ámpula de agua estéril y la cánula de transferencia con una mano, usando las dos aletas laterales. Mantenga el frasco ámpula con polvo liofilizado de ACTILYSE® en posición vertical encima de la cánula de arriba transferencia y coloque la punta de la cánula de transferencia justo en el centro del tapón. Presione el frasco ámpula con el polvo liofilizado contra la cánula de transferencia directamente desde arriba, pinchando el tapón de caucho en forma vertical y suave, pero firmemente sin girar. |
|
|
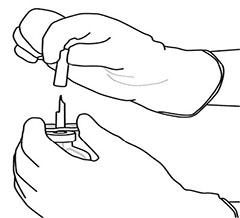
8 |
Invertir los dos frascos ámpula y dejar que el agua drene completamente en el polvo liofilizado. |
|
|
9 |
Retire el frasco ámpula de agua vacío junto con la cánula de transferencia. Ya puede desecharlos. |
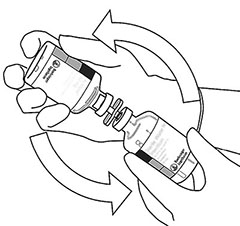
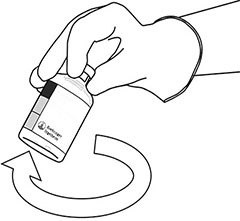
|
|
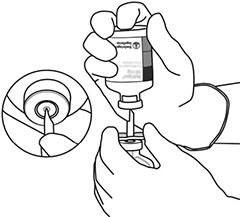
10 |
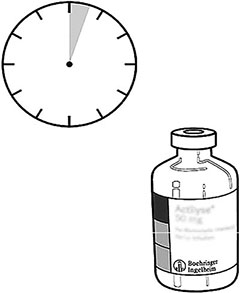
Tome el frasco ámpula con ACTILYSE® reconstituido y agítelo suavemente para disolver el polvo que pudiera quedar, pero no lo agite, ya que eso produce espuma. Si hay burbujas, deje reposar la solución durante unos minutos para que desaparezcan. |
|
|
11 |
La solución reconstituida consiste en 1 mg/mL de ACTILYSE®. Debe ser límpida, entre incolora y ligeramente amarillenta y no debe contener ninguna partícula. |
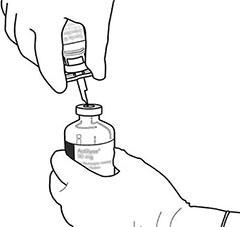
|
|
12 |
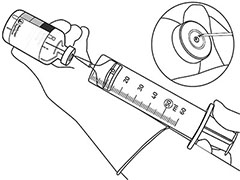
Retire sólo la cantidad requerida usando una aguja y una jeringa. No use el lugar donde pinchó con la cánula de transferencia a fin de evitar filtraciones. |
|
|
13 |
Use inmediatamente. Deseche la solución que no utilice. |
|
* Si el kit incluye una cánula de transferencia. La reconstitución también puede realizarse con una jeringa y una aguja.
La solución reconstituida de 1 mg/mL puede diluirse adicionalmente con solución estéril para inyectables de cloruro de sodio de 9 mg/mL (al 0.9%) hasta una concentración mínima de 0.2 mg/mL, dado que no puede descartarse que la solución reconstituida presente turbidez.
No se recomienda una dilución adicional de la solución reconstituida de 1 mg/mL con agua estéril para inyectables, ni el uso de soluciones de carbohidratos para infusión en general, p. ej., dextrosa, debido a la creciente formación de turbidez de la solución reconstituida.
ACTILYSE® no debe mezclarse con otros fármacos, ni en el mismo frasco ámpula para infusión, ni en la misma vía intravenosa (ni siquiera con heparina).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Síntomas:
Si se excede la dosis máxima recomendada, aumenta el riesgo de sangrado intracraneal.
A pesar de la relativa especificidad por la fibrina, en caso de sobredosis puede ocurrir una significativa reducción clínica del fibrinógeno y de otros componentes de la coagulación sanguínea.
Tratamiento:
En la mayoría de los casos es suficiente con esperar la regeneración fisiológica de estos factores de coagulación después de haber concluido el tratamiento con ACTILYSE®. Sin embargo, si ocurren sangrados graves, se recomienda administrar plasma fresco congelado o sangre fresca, y si es necesario, se pueden administrar antifibrinolíticos sintéticos.
PRESENTACIÓN:
Caja de cartón con:
2 frascos ámpula con 50 mg de polvo liofilizado cada uno.
2 frascos ámpula con 50 mL de diluyente cada uno.
2 cánula de conducción de plástico.
1 instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Estabilidad química y física durante el uso:
Se ha demostrado que la solución reconstituida es estable durante 24 horas a 2-8 °C y durante 8 horas a 25 °C.
Estabilidad microbiológica durante el uso:
Desde el punto de vista microbiológico, el producto debe ser utilizado inmediatamente después de su reconstitución. Si no se utiliza inmediatamente, el tiempo y las condiciones de almacenamiento antes de su uso son responsabilidad del usuario y no deberán ser mayores a 24 horas a una temperatura de 2 °C a 8 °C.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo y en la lactancia, ni en menores de 18 años. Sólo deberá ser administrado por médicos especialistas. Uso exclusivamente intrahospitalario. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se administre si el cierre ha sido violado. Si no se administra todo el producto, deséchese el sobrante.
Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y
farmacovigilancia.mex@boehringer-ingelheim.com
BOEHRINGER INGELHEIM PROMECO, S.A. de C.V.
Calle del Maíz No. 49
Col. Barrio Xaltocan, C.P. 16090
Xochimilco, Ciudad de México, México
Reg. Núm. 166M88 SSA IV
®Marca Registrada