
AFINITOR - Comprimidos
Sustancia(s):
- Everolimus
Presentaciones:
- 1 Caja,1 Envase(s) de burbuja,30 Comprimidos,2.5 mg
- 1 Caja,1 Envase(s) de burbuja,30 Comprimidos,5 mg
- 1 Caja,1 Envase(s) de burbuja,30 Comprimidos,10 mg
- 1 Caja,1 Envase(s) de burbuja,30 Comprimidos,10 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada COMPRIMIDO contiene:
Everolimus 2.5, 5 y 10 mg
Excipiente, c.b.p. 1 comprimido.
INDICACIONES TERAPÉUTICAS:
AFINITOR® está indicado para el tratamiento de:
• Mujeres posmenopáusicas con cáncer de mama avanzado con receptores hormonales positivos en combinación con un inhibidor de aromatasa, posterior a otra terapia endocrina.
• Pacientes con carcinoma de células renales avanzado.
• Pacientes con tumores neuroendocrinos en estado avanzado.
• Pacientes con astrocitomas subependimarios de células gigantes (SEGA por sus siglas en inglés) asociados con esclerosis tuberosa (TS).
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacocinéticas:
Absorción: En los pacientes con tumores sólidos avanzados, las concentraciones máximas de everolimus se alcanzan 1 o 2 horas después de la administración de dosis orales de entre 5 y 70 mg de fármaco en ayunas o tras un refrigerio exento de grasas. La Cmáx es proporcional a la dosis en el intervalo de 5 a 10 mg, tanto en las administraciones diarias como en las semanales. A partir de los 20 mg/semana, el aumento de Cmáx es menos proporcional a la dosis, a pesar de que el área bajo la curva (ABC) es proporcional a la dosis en el intervalo de 5 a 70 mg.
Efecto de los alimentos: En sujetos sanos, los alimentos ricos en grasas redujeron la exposición sistémica a AFINITOR® de 10 mg (medida por el ABC) en 22% y la concentración plasmática máxima Cmáx en 54%. Las comidas ligeras de grasa redujeron en 32% el ABC y la Cmáx en 42%. Los alimentos, sin embargo, no tuvieron un efecto aparente en el perfil de tiempo-concentración de la fase posabsorción, ver Dosis y vía de administración.
Distribución: El cociente sangre/plasma de everolimus, que depende de la concentración en el intervalo de 5 a 5,000 ng/ml, varia entre 17 y 73%. La cantidad de everolimus confinada en el plasma es de 20% en los pacientes con cáncer que toman AFINITOR® en dosis de 10 mg/día.
La unión a proteínas plasmáticas es de 74% tanto en los sujetos sanos como en los pacientes con insuficiencia hepática moderada.
Después de la administración de everolimus por vía intravenosa en un modelo murino, se demostró que everolimus atravesó la barrera hematoencefálica de una manera dosis-dependiente no-lineal, sugiriendo la saturación de una bomba de eflujo en dicha barrera. La penetración encefálica de everolimus se pudo comprobar asimismo en ratas que habían recibido dosis orales de everolimus.
Metabolismo: Everolimus es un sustrato del CYP3A4 y de la glicoproteína P. Luego de la administración oral, es el componente principal en la circulación sanguínea. Se han detectado seis importantes metabolitos de everolimus en la sangre humana, a saber: tres metabolitos monohidroxilados, dos productos de hidrólisis cíclica y un conjugado fosfatodilcolínico de everolimus. Estos metabolitos se habían identificado en las especies animales de los estudios de toxicidad, y su actividad era casi cien veces menor que la del propio everolimus. Por consiguiente, se considera que la mayor parte de la actividad farmacológica corresponde al compuesto inalterado.
Excreción: No se han efectuado estudios de excreción específicos en pacientes con cáncer, pero se tienen datos procedentes de los pacientes con trasplantes. Tras la administración de una dosis única de everolimus radiactivo combinado con ciclosporina, 80% de la radiactividad se recuperó en las heces y 5%, en la orina. No se detectó compuesto inalterado en la orina ni en las heces.
Farmacocinética en el estado estable: Tras la administración diaria o semanal de everolimus a pacientes con tumores sólidos avanzados, el ABC0-8 en el estado estable fue proporcional a la dosis tanto en la gama de concentraciones de 5 a 10 mg con la administración diaria como en la de 5 a 70 mg semanal.
Con la administración diaria, el estado estable se alcanzó en dos semanas. La Cmáx es proporcional a la dosis en el intervalo de 5 a 10 mg en régimen diario o semanal, pero cuando las dosis son iguales o superiores a 20 mg/semana, la Cmáx aumenta de forma menos proporcional a la dosis y se detecta 1 o 2 horas. El tiempo al que se alcanza la concentración máxima (Tmáx) ocurre 1-2 horas después de la administración de la dosis. Hubo una correlación significativa entre ABC0-8 y la concentración mínima anterior a la dosis en el estado estable en el régimen diario. La vida-media de eliminación promedio de everolimus es de aproximadamente 30 horas.
Pacientes con insuficiencia hepática: La seguridad, tolerabilidad y la farmacocinética de everolimus se evaluaron en un estudio de dosis única de everolimus en 34 pacientes con alteración de la función renal en comparación con pacientes con función hepática normal. En comparación con los pacientes con función hepática normal, hubo un aumento de 1.6, 3.3 y 3.6 veces en la exposición (por ejemplo, ABC(0-inf)) para los pacientes con insuficiencia hepática leve (Child-Pugh A), moderada (Child-Pugh B) y severa (Child-Pugh C) respectivamente. Simulaciones de la farmacocinética de dosis múltiples sustenta las recomendaciones de dosificación en pacientes con insuficiencia hepática utilizando como base el estatus de Child Pugh. Se recomienda el ajuste de la dosis en pacientes con insuficiencia hepática (ver Precauciones generales y Dosis y vía de administración).
Pacientes con insuficiencia renal: En el análisis farmacocinético de una población de 170 pacientes afectados de cáncer avanzado, no se observó que la depuración de creatinina (25-178 ml/min) afectara significativamente la de everolimus (CL/F). En los receptores de un trasplante, la insuficiencia renal (depuración de creatinina: 11-107 ml/min) posterior al trasplante no alteró la farmacocinética de everolimus.
Pacientes pediátricos: AFINITOR® no está indicado en la población pediátrica con neoplasias malignas (ver Dosis y vía de administración).
En pacientes con SEGA, las concentraciones sanguíneas en el estado estacionario entre cada paciente fueron proporcionales a la dosis en tratamientos diarios con 1.5-14.6 mg/m2 (ver Dosis y vía de administración).
Pacientes de edad avanzada: En un análisis de farmacocinética de una población de pacientes con cáncer no se apreció un efecto significativo de la edad (27-85 años) en la depuración de everolimus oral (CL/F: entre 4.8 y 54.5 L/h).
Origen étnico: La depuración oral (CL/F) es semejante en pacientes japoneses con cáncer y en pacientes caucásicos con cáncer con funciones hepáticas similares.
Con base en el análisis farmacocinético poblacional, la depuración oral (CL/F) es en promedio 20% mayor, en los pacientes receptores de trasplante de raza negra.
Relación entre la exposición y la respuesta: Tras la administración diaria de 5 o 10 mg de everolimus se observó una moderada correlación entre la disminución de la fosforilación de 4E-BP1 (P4E-BP1) en el tejido tumoral y la Cmín sanguínea media de dicho fármaco en el estado estable. Datos adicionales indican que la inhibición de la fosforilación de la cinasa S6 es muy sensible a la inhibición de mTOR por parte de everolimus. La inhibición de la fosforilación del F-4 G fue completada en todos los valores de concentraciones mínimas (Cmín) que siguieron a las dosis de 10 mg diarios.
En pacientes con SEGA, se han asociado las altas concentraciones de everolimus en sangre con reducciones de importancia en el volumen del SEGA. Sin embargo, al observarse respuesta con concentraciones bajas en sangre como 2 ng/ml, se ha conseguido una eficacia aceptable, por lo cual, puede no ser necesario un aumento adicional en la dosis, (ver Dosis y vía de administración).
Una tendencia que sugiere una mayor supervivencia libre de progresión con una Cmín de everolimus altamente normalizada de tiempo fue evidente en pacientes con tumores neuroendocrinos en estadio avanzado (pNET, taza de riesgo 0.73; 95% Cl: 0.50 a 1.08) y en pacientes con carcinomas en estadio avanzado (taza de riesgo 0.66; 95% Cl: 0.40 a 1.08). La Cmín de everolimus tuvo un impacto en la probabilidad de la reducción del tumor (p < 0.001) con tazas inusuales de 1.62 y 1.46, respectivamente, para un cambio en la exposición desde 5 a 10 ng/ml en pacientes con pNET en estadio avanzado y en pacientes con carcinomas en estadio avanzado.
Farmacodinamia:
Grupo farmacoterapéutico, Código ATC: Inhibidores de la protein-cinasa, ATC L01XE10.
Mecanismo de acción: El everolimus es un inhibidor selectivo de señales de transducción de mTOR (blanco de rapamicina en los mamíferos) y más específicamente del complejo de transducción de señales de mTORC1, (Complejo 1 "diana de rapamicina" en mamíferos). mTOR es una cinasa clave de serina-treonina, la cual participa de manera importante en la regulación del crecimiento, proliferación y supervivencia celular. La regulación de la señalización de mTORC1 es compleja, al ser modulada por mitógenos, factores de crecimiento, requerimientos energéticos y la disponibilidad de nutrientes. mTORC1 es un regulador esencial en la cascada de transducción de señales de P13K/AKT una vía que, como se sabe, está desregulada en la mayoría de las neoplasias malignas humanas.
La activación de la vía mTOR es una adaptación clave que impulsa el cambio de resistencia endocrina en el cáncer de mama. Diferentes vías de transducción de señales se activan para escapar de los efectos de la terapia endocrina. Una vía es la ruta P13K/AKT/mTOR, que es constitutivamente activada en las células de cáncer de mama con resistencia a los inhibidores de la aromatasa (IA) y en condiciones de privación de estrógenos a largo plazo de estas células. En las células de cáncer de mama, la resistencia a inhibidores de la aromatasa, debido a la activación de AKT, puede ser revertida por la administración concomitante de everolimus.
Los complejos -esclerosis-tuberosa 1 & 2 (TSC1, TSC2), supresores de oncogenes, son dos reguladores primarios de la señalización de mTORC1. La pérdida o inactivación de cualquiera de TSC1 o TSC2 conlleva a niveles elevados de GTP Rheb, una GTPasa de la familia ras, que interactúa con el complejo mTORC1 para provocar su activación. La activación de mTORC1 da lugar a una cascada de señalización de quinasas, incluyendo la activación de la S6K1. En el síndrome de esclerosis tuberosa, una enfermedad genética, las inactivaciones generadas por mutaciones tanto en el TSC1 o en el gen TSC2 conllevan a la formación de hamartomas en todo el cuerpo.
Propiedades farmacodinámicas: El everolimus es un inhibidor selectivo de mTOR (la diana de rapamicina en los mamíferos) y más específicamente del complejo de transducción de señales de mTOR-Raptor (mTORC1).
La mTOR es una cinasa clave de serina-treonina en la cascada de transducción de señales de P13K/AKT una vía que, como se sabe, está desregulada en la mayoría de las neoplasias malignas humanas. El everolimus ejerce su actividad mediante una interacción de gran afinidad con el receptor intracelular FKBP12. El complejo FKBP12-everolimus se fija a mTORC1 e inhibe su capacidad transductora de señales. La transducción de señales a través de mTORC1 se efectúa por medio de la modulación de la fosforilación de efectores consecutivos en la serie, entre cuyos componentes mejor caracterizados figuran los reguladores de la traducción de proteínas S6K1 (cinasa 1 de la proteína ribosómica S6) y 4E-BP (proteína de fijación al factor de elongación 4E en los eucariontes). El desmantelamiento de la función de S6K1 y de 4E-BP1, como resultado de la inhibición de mTORC1, interfiere la traducción de los ARNm codificadores de proteínas que son esenciales para la regulación del ciclo celular, la glucólisis y la adaptación a condiciones de baja concentración de oxígeno (hipoxia). Ello inhibe el crecimiento del tumor y la expresión de factores que se inducen en condiciones de hipoxia (como el factor de transcripción HIF-1) y esto último a su vez redunda en una menor expresión de factores implicados en la potenciación de procesos angiogénicos tumorales (como el factor de crecimiento del endotelio vascular, VEGF). El everolimus es un inhibidor potente del crecimiento y la proliferación de células tumorales, células endoteliales, fibroblastos y de las células del músculo liso de la pared de los vasos sanguíneos. En consonancia con la función reguladora central que ejerce mTORC1, se ha visto que el everolimus reduce la proliferación de células tumorales, la glucólisis y la angiogénesis en los tumores sólidos in vivo y de esa forma ofrece dos mecanismos independientes de inhibición del crecimiento tumoral: una actividad antineoplásica directa en las células y una inhibición del compartimiento estromal tumoral.
En un modelo neuronal murino del TSC, en el cual el TSC es extirpado en la mayoría de las neuronas durante el desarrollo cortical, el everolimus mejoró la supervivencia promedio de 33 a más de 100 días, además de mejorar de forma significativa el comportamiento, fenotipo y elevación del peso corporal. Existió penetración cerebral, y se acumuló al paso del tiempo con el tratamiento respectivo, así como una reducción efectiva de los niveles de la fosfo-S6, un marcador involucrado en la cascada de señalización de mTORC1. Hubo mejoras mediadas por el tratamiento en las anormalidades de los filamentos neuronales, en la mielinización y ampliación de las células, aunque las características de displasia neuronal persistieron y hubo cambios moderados en la densidad y longitud de la espina dendrítica. Los ratones tratados con everolimus por 23 días únicamente, (entre 7-30 días de vida) mostraron una mejora continua en el fenotipo, con una supervivencia promedio de 78 días. En resumen, el everolimus fue altamente activo en este modelo neuronal del TSC, en el cual el beneficio es atribuible aparentemente a los efectos en la señalización de mTORC1 y AKT, y consecuentemente, en el tamaño de las células y en la mielinización.
Estudios clínicos:
Cáncer de mama con receptores hormonales positivos: El estudio BOLERO-2 (Estudio CRAD001Y2301) es un estudio aleatorizado, a doble-ciego, multicéntrico de fase III de AFINITOR® + exemestano vs placebo + exemestano, el cual se llevó a cabo en mujeres posmenopáusicas con cáncer de mama avanzado HER2 neu/no amplificado, con receptores positivos de estrógenos, con recurrencia o progresión después de la terapia previa con letrozol o anastrozol. Los pacientes fueron aleatorizados en una proporción de 2:1 al recibir tanto everolimus (10 mg al día) o placebo, además de exemestano no enmascarado (25 mg al día). La aleatorización se estratificó por la sensibilidad documentada a la terapia hormonal previa (sí vs no) y por la presencia de metástasis visceral (sí vs no). La sensibilidad a la terapia hormonal fue definida como (1) beneficio clínico documentado (respuesta completa [RC], respuesta parcial [RP], enfermedad estable > 24 semanas) a por lo menos un tratamiento hormonal previo en fases avanzadas o (2) por lo menos 24 meses de tratamiento hormonal adyuvante previo a la recurrencia.
El objetivo primario del estudio fue la supervivencia sin progresión (SSP) evaluada por los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST, por sus siglas en inglés), basados en la evaluación del investigador (radiología local). Los análisis de soporte de SSP se basaron en una revisión radiológica central independiente.
Los objetivos secundarios incluyeron la supervivencia global (SG), tasa de respuesta general (TRG), tasa de beneficio clínico (TBC), de seguridad, el cambio en la calidad de vida (QoL) y el tiempo de deterioro en ECOG o estado funcional. Los objetivos adicionales incluyen cambios en los marcadores de recambio óseo a las 6 y 12 semanas.
Un total de 724 pacientes fueron aleatorizados en proporción 2:1 a la combinación de everolimus (10 mg al día) + exemestano (25 mg al día) (n = 485) o al grupo de placebo + exemestano (25 mg al día) (n = 239). Los dos grupos de tratamiento fueron equilibrados en general con respecto a los datos demográficos iniciales de las características de la enfermedad y el historial de tratamiento antineoplásico usado previamente. La edad media de las pacientes fue de 61 años (rango de 28 a 93) y el 75% eran caucásicas.
Los resultados de eficacia se obtuvieron de un análisis interno después de 359 eventos de SSP y se observaron 217 eventos centrales SSP. Los pacientes en el grupo placebo + exemestano no se trasladaron al tratamiento con everolimus en el momento de la progresión.
El estudio demostró un beneficio clínico estadísticamente significativo de everolimus+ exemestano frente a placebo + exemestano por una prolongación de 2.4 veces en la mediana de SSP (promedio: 6.93 meses frente a 2.83 meses), lo que resulta en una reducción del riesgo de 57% de progresión o muerte (HR 0.43 SLP IC 95%: 0.35, 0.54, de un solo lado, prueba logarítmica de p < 0.0001 por la evaluación del investigador local (ver la tabla 1 y figura 1).
El análisis de la SSP basado en la evaluación radiológica central independiente fue favorable y mostró una prolongación de 2.6 veces en el promedio de supervivencia libre (10.58 meses frente a 4.14 meses), lo que resulta en una reducción del riesgo de 64% de progresión o muerte (SSP HR 0.36; IC 95%: 0.27 a 0.47; un solo lado, prueba logarítmica de p < 0.0001 (ver la tabla 1 y figura 2).
Se observó en 9.5% de los pacientes una respuesta objetiva basada en la evaluación del investigador y en base de RECIST (95% IC: 7.0, 12.4) en el grupo de everolimus + exemestano vs 0.4% (IC 95%: 0.0 a 2.3) en el grupo de placebo + exemestano (p < 0.0001 para la comparación entre los grupos). La tasa de beneficio clínico de everolimus + exemestano fue de 33.4% frente a 18.0% en el grupo control, p < 0.0001 (ver tabla 1).
Tabla 1. Estudio BOLERO-2 - resultados de eficacia
|
Análisis |
AFINITOR®a n = 485 |
Placebo n = 239 |
Tasa de riesgo |
Valor de p |
|
Promedio de supervivencia libre de progresión (meses, 95% IC) |
||||
|
Revisión radiológica del investigador |
6.93 (6.44 a 8.05) |
2.83 (2.76 a 4.14) |
0.43 (0.35 a 0.54) |
<0.0001 |
|
Revisión radiológica independiente |
10.58 |
4.14 |
0.36 |
<0.0001 |
|
(9.53 a NA) |
(2.83 a 5.75) |
(0.27 a 0.47) |
||
|
Mejor respuesta global (%, 95% IC) |
||||
|
Tasa de respuesta objetiva (TRO)b |
9.5% (7.0 a 12.4) |
0.4% (0.0 a 2.3) |
n/ad |
< 0.0001e |
|
Tasa de beneficio clínico (TBC)c |
33.4% (29.2 a 37.8) |
18.0% (13.3 a 23.5) |
n/ad |
< 0.0001e |
a Más exemestano.
b Tasa de respuesta objetiva = Proporción de pacientes con CR o PR.
c Tasa de beneficio clínico - Proporción de pacientes con CR o PR o SD ³ 24 semanas.
d no aplica.
e el valor de p se obtiene -el valor se obtiene de la prueba exacta de CMH, utilizando una versión estratificada de la prueba de permutación de Cochran-Armitage.
Los datos de supervivencia global (SG) no estaban completos en el momento del análisis interno de SSP. Ochenta y tres muertes fueron reportadas en el análisis interno, lo que representa 10.6 y 13.0% de los pacientes muertos reportados en el grupo de everolimus + exemestano y en el grupo placebo + exemestano, respectivamente.
Figura 1. BOLERO-2 - curva de supervivencia libre de progresión de Kaplan-Meier (revisión radiológica del investigador).
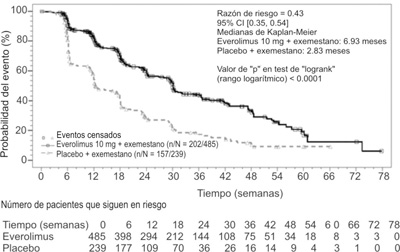
Las tasas de SSP de nueve meses fueron de 40% de los pacientes que recibieron everolimus + exemestano frente a 15% en el grupo de placebo + exemestano en un seguimiento promedio de 7.6 meses.
Figura 2. BOLERO-2 - curva de supervivencia libre de progresión de Kaplan-Meier (revisión radiológica independiente).
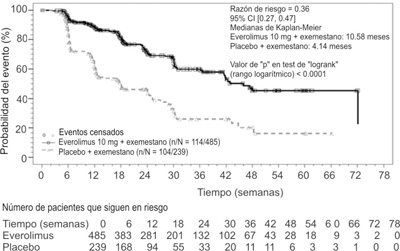
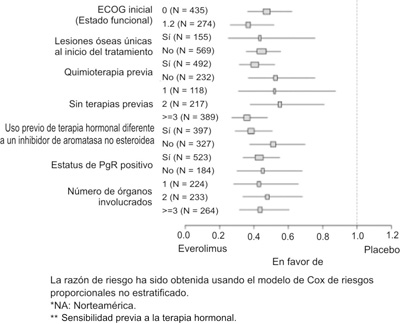
El efecto estimado del tratamiento de SSP fue respaldado por el análisis de subgrupos planificados de la SSP por la evaluación del investigador. Para todos los subgrupos analizados, se observó un efecto positivo del tratamiento con everolimus + exemestano con una proporción de riesgo estimado vs placebo + exemestano oscilando entre 0.25 y 0.60 (ver tabla 2, figuras 3 y 4). Los análisis del subgrupo demostraron un efecto del tratamiento homogéneo y consistente, independientemente de la sensibilidad a la terapia hormonal antes y presencia de metástasis visceral, y en los principales subgrupos demográficos y pronósticos.
|
n |
Everolimus + exemestano SSP promedio (meses) |
Placebo + exemestano |
TR1 |
95% IC |
|
|
Sensibilidad a la terapia hormonal previa |
|||||
|
No |
114 |
6.70 |
2.83 |
0.50 |
0.31, 0.83 |
|
SI |
610 |
6.93 |
2.92 |
0.43 |
0.34, 0.54 |
|
Presencia de metástasis visceral |
|||||
|
No |
318 |
8.48 |
4.24 |
0.43 |
0.30, 0.60 |
|
Si |
406 |
6.18 |
2.69 |
0.44 |
0.34, 0.58 |
|
Grupos de edad |
|||||
|
< 65 años |
449 |
6.97 |
2.83 |
0.40 |
0.31, 0.52 |
|
³ 65 años |
275 |
6.44 |
4.01 |
0.53 |
0.37, 0.76 |
|
Región |
|||||
|
Asia |
137 |
6.97 |
4.14 |
0.60 |
0.38, 0.96 |
|
Europa |
275 |
6.70 |
2.76 |
0.42 |
0.30, 0.60 |
|
América del Norte |
274 |
8.84 |
3.94 |
0.37 |
0.26, 0.52 |
|
Otros |
38 |
4.21 |
1.45 |
0.34 |
0.12, 0.96 |
|
Japoneses |
|||||
|
Japoneses |
106 |
8.41 |
4.14 |
0.59 |
0.35, 1.01 |
|
No-japoneses |
618 |
6.83 |
2.79 |
0.41 |
0.32, 0.51 |
|
Quimioterapia previa |
|||||
|
No |
232 |
6.83 |
3.45 |
0.53 |
0.37, 0.76 |
|
Si |
492 |
6.93 |
2.79 |
0.40 |
0.31, 0.52 |
|
Lesiones óseas únicas al inicio del tratamiento |
|||||
|
No |
569 |
6.70 |
2.76 |
0.44 |
0.35, 0.56 |
|
Si |
155 |
9.89 |
4.37 |
0.43 |
0.25, 0.76 |
|
ECOG inicial (estado funcional) |
|||||
|
0 |
435 |
6.97 |
4.01 |
0.47 |
0.36, 0.62 |
|
1 o 2 |
274 |
6.77 |
2.76 |
0.37 |
0.26, 0.52 |
|
Estatus de PgR |
|||||
|
Negativo |
184 |
6.93 |
2.76 |
0.45 |
0.30, 0.68 |
|
Positivo |
523 |
6.93 |
2.96 |
0.43 |
0.34, 0.55 |
|
Raza |
|||||
|
Asiáticos |
143 |
6.93 |
4.14 |
0.59 |
0.37, 0.93 |
|
Caucásicos |
547 |
6.93 |
2.83 |
0.41 |
0.32, 0.53 |
|
Otros |
34 |
7.03 |
1.41 |
0.25 |
0.09, 0.74 |
|
Uso previo de terapia hormonal diferente a un inhibidor de aromatasa no esteroideo |
|||||
|
No |
327 |
6.83 |
4.01 |
0.51 |
0.37, 0.70 |
|
Sí |
397 |
6.97 |
2.76 |
0.38 |
0.29, 0.51 |
|
Número de órganos involucrados |
|||||
|
1 |
224 |
8.48 |
4.21 |
0.43 |
0.28, 0.66 |
|
2 |
233 |
5.59 |
2.79 |
0.48 |
0.33, 0.68 |
|
³ 3 |
264 |
6.70 |
2.60 |
0.44 |
0.31, 0.61 |
|
Número de terapias previas |
|||||
|
1 |
118 |
6.97 |
4.17 |
0.52 |
0.31, 0.88 |
|
2 |
217 |
6.70 |
2.92 |
0.55 |
0.38, 0.81 |
|
³ 3 |
389 |
7.16 |
2.79 |
0.36 |
0.27, 0.48 |
1 Tasa de riesgo obtenida mediante el método no estratificado de Cox.
Figura 3. Trama del SSP por el investigador por subgrupo (1).
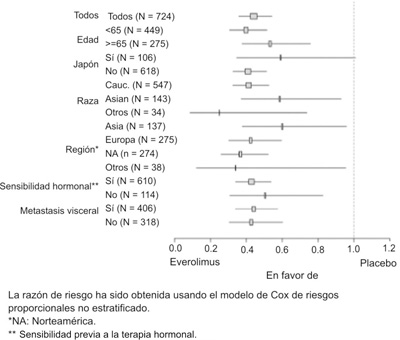
Figura 4. Trama del SSP por el investigador por subgrupo (2).
La reducción del tumor fue también evidente en la trama de cascada correspondiente. Los resultados indican que 68.1% de los pacientes en el grupo de exemestano + everolimus experimentaron una reducción del tumor en comparación con 28.0% para el grupo de placebo + exemestano (figura 5).
Figura 5. Reducción tumoral: mejor cambio porcentual desde el inicio en la suma de los diámetros más largos por cada investigador.
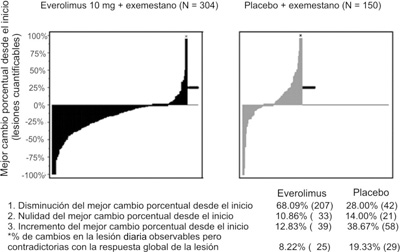
No se observaron diferencias clínicas ni estadísticamente significativas entre los dos grupos de tratamiento en términos de tiempo hasta el deterioro del ECOG PS (³ 1 punto) y el promedio del tiempo hasta el deterioro (a 5%) del QLQ-C30 puntuaciones de dominio.
Efectos óseos: No hay datos a largo plazo sobre el efecto óseo de everolimus. Datos comparativos de BOLERO-2 mostraron una mejoría marcada en los marcadores de recambio óseo séricos durante las primeras 12 semanas de tratamiento, lo que sugiere un efecto favorable sobre el recambio óseo.
Tumores neuroendocrinos de origen gastrointestinal, pulmonar o pancreático en estadio avanzado: El estudio RADIANT-3 (Estudio CRAD001C2324), un estudio de diseño aleatorizado, a doble-ciego, multicéntrico de fase III para AFINITOR® más el tratamiento óptimo de apoyo (TOA) en comparación con placebo más el TOA en pacientes con tumores neuroendocrinos pancreáticos (pNET), ha demostrado un beneficio clínico estadísticamente significativo de AFINITOR® sobre el placebo al registrar una prolongación de 2.4 veces en la supervivencia libre de progresión (SLP) promedio, (11.04 meses frente a 4.6 meses), resultando en una reducción del riesgo del 65% de la SLP (HR 0.35; 95% IC: 0.27, 0.45; p < 0.0001) (ver tabla 3 y figura 6).
El estudio RADIANT-3 reclutó a pacientes con pNET en estadio avanzado cuya enfermedad había progresado en los últimos 12 meses. Los pacientes fueron clasificados por la quimioterapia citotóxica previa (si/no) y por el estatus de rendimiento de la OMS (0 vs 1 y 2). Se le permitió el tratamiento con análogos de la somatostatina como parte de TOA.
La variable primaria del estudio fue la SLP evaluada por RECIST (Criterios de Evaluación de Respuesta en Tumores Sólidos). Después de documentar la progresión radiológica, los pacientes podrían ser informados por el investigador: los asignados a placebo fueron habilitados para recibir AFINITOR® no enmascarado.
Las variables secundarias incluyen la seguridad, la tasa de respuesta objetiva TRO [respuesta completa (RC) o respuesta parcial (RP)], duración de la respuesta y de supervivencia global (SG).
En total, 410 pacientes fueron aleatorizados con una relación 1:1 para recibir ya sea 10 mg/día AFINITOR® (n = 207) o placebo (n = 203). La demografía estaba equilibrada adecuadamente (edad promedio de 58 años, 55% varones, 78.5% caucásicos).
Tabla 3. Estudio RADIANT-3 - Resultados para la supervivencia libre de progresión.
|
Análisis |
n |
AFINITOR® |
Placebo |
Taza de riesgos, (Hazard Ratio) (95% IC) |
Valor de p |
|
410 |
Promedio de la supervivencia libre de progresión (meses) (IC de 95%) |
||||
|
Revisión radiológica del investigador |
11.04 (8.41 a 13.86) |
4.60 (3.06 a 5.39) |
0.35 (0.27 a 0.45) |
< 0.0001 |
|
|
Revisión radiológica independiente |
11.40 (10.84 a 14.75) |
5.39 (4.34 a 5.55) |
0.34 (0.26 a 0.44) |
< 0.0001 |
|
* Incluye la adjudicación para las discrepancias entre las evaluaciones radiológicas obtenidas por la revisión del investigador y la revisión central.
Figura 6. Estudio RADIANT-3 - Curvas de supervivencia libre de progresión de Kaplan-Meier.
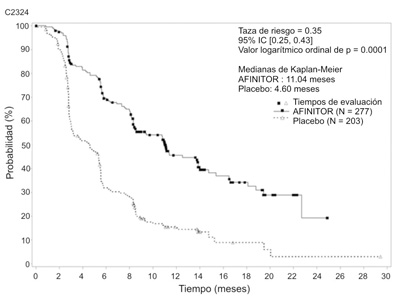
Las tasas de SLP de 18 meses de duración fueron 34.2% para la terapia con AFINITOR® comparada con 8.9% para el placebo.
Los resultados de la supervivencia total no son suficientemente avanzados y no se apreció diferencia estadísticamente significativa en la ST (HR = 0.99 (95% IC 0.68 a 1.43)). El cambio en el tratamiento de 72.9% de los pacientes (148/203) de placebo a AFINITOR® tras comprobarse la progresión de la enfermedad probablemente confundió la detección de cualquier diferencia relacionada con el tratamiento en la ST.
El estudio RADIANT-2 (estudio CRAD001C2325), un estudio de diseño aleatorizado, a doble-ciego, multicéntrico de fase III para AFINITOR® más octreotida (Sandostatina Lar®) en comparación con placebo más octreotida en pacientes con tumores neuroendocrinos en estadio avanzado (carcinomas) principalmente de origen gastrointestinal o pulmonar presentó evidencia de beneficio clínico de AFINITOR® sobre el placebo por una prolongación de 5.1 meses en el promedio de SLP (16.43 meses en comparación con 11.33 meses; HR 0.77, 95% IC: 0.59 a 1.00, p = 0.026) resultando en una reducción del riesgo de 23% en la SLP primaria (ver tabla 4 y figura 7). Aunque no se alcanzó significancia estadística para el análisis primario (intervalo de confianza fue de p = 0.0246), los análisis que se ajustan a la censura informativa y los desequilibrios entre los dos grupos de tratamiento mostraron un efecto del tratamiento en favor de everolimus.
El estudio RADIANT-2 reclutó pacientes con tumores neuroendocrinos en estadio avanzado (carcinoides), principalmente de origen gastrointestinal o pulmonar en los cuales la enfermedad había progresado en los 12 meses previos y que presentaron antecedentes de síntomas secretorios. El 80.1% de los pacientes en el grupo de AFINITOR® recibieron terapia análoga con somatostatina previa al inicio del estudio en comparación de 77.9% de los pacientes en el grupo del placebo.
La variable primaria del estudio fue la SLP evaluada por RECIST. Después de documentar la progresión radiológica los pacientes podrían ser informados por el investigador: los asignados a placebo fueron habilitados para recibir AFINITOR® no enmascarado.
Las variables secundarlas incluyen la seguridad, la tasa de respuesta objetiva, duración de la respuesta y de supervivencia total.
En total, 429 pacientes fueron aleatorizados con una relación 1:1 para recibir ya sea 10 mg/día AFINITOR® (n = 216) o placebo (n = 213) en conjunto con octreotida (Sandostatina Lar®, administrada por vía intramuscular) 30 mg cada 28 días. Se evidenciaron desequilibrios notables para varios factores de referencia pronosticados de importancia, sobre todo en favor del grupo del placebo.
Tabla 4. RADIANT-2 - Resultados para la supervivencia libre de progresión.
|
Análisis |
n |
AFINITOR® |
Placebo |
Taza de riesgos, (Hazard Ratio) (95% IC) |
Valor de p |
|
429 |
Promedio de la supervivencia libre de progresión (meses) (IC de 95%) |
||||
|
Revisión radiológica independiente* |
16.43 (13.67 a 21.19) |
11.33 (8.44 a 14.59) |
0.77 (0.59 a 1.00) |
0.026 |
|
|
Revisión radiológica del investigador |
11.99 (10.61 a 16.13) |
8.61 (8.08 a 11.14) |
0.78 (0.62 a 0.98) |
0.018 |
|
* Incluye la adjudicación para las discrepancias entre las evaluaciones radiológicas obtenidas por la revisión del investigador y la revisión central.
Los análisis adicionales para la revisión radiológica independiente que se ajustan a la censura informativa y a los desequilibrios en los dos grupos de tratamiento mostraron un efecto del tratamiento en favor de everolimus. Los resultados de un nuevo análisis de variables múltiples, el cual ajusta los desequilibrios entre los grupos de tratamiento generó como resultado un HR de 0.73 (IC 95%: 0.56 a 0.97). Un modelo de Cox con Probabilidad Inversa de Pesos Sesgados (PIPS) fue utilizado para tratar y corregir la censura informativa y los desequilibrios en las características iniciales entre los dos grupos del estudio. El HR estimado (95% IC) a partir del análisis PIPS fue de 0.60 (0.44 a 0.84), con un valor de p unilateral r 0.0014 a favor de everolimus.
Figura 7. Estudio RADIANT-2 - Curvas de supervivencia libre de progresión de Kaplan-Meier.
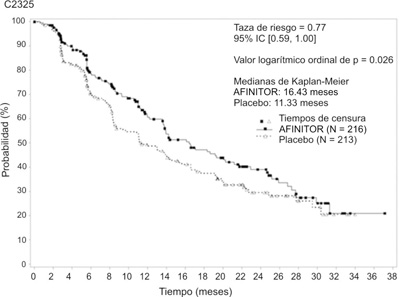
Las tasas de SLP a 18 meses fueron de 47.2% para la terapia de everolimus mas octreotida (Sandostatina Lar®) comparado con 37.4% para el placebo más octreotida (Sandostatina Lar®).
Los resultados de la supervivencia total no son suficientemente avanzados y no se apreció diferencia estadísticamente significativa en la ST [para los análisis ajustados preespecificados la HR = 1.00 (95% IC 0.76 a 1.33)]. El cambio en el tratamiento del 58.2% de los pacientes (124/213) de placebo a AFINITOR® tras comprobarse la progresión de la enfermedad probablemente confundió la detección de cualquier diferencia relacionada con el tratamiento en la ST.
Carcinomas de células renales en estadio avanzado: El estudio RECORD-1 (CRAD001C2240), un estudio clínico de fase III, internacional, multicéntrico, aleatorizado, doble-ciego y comparativo de AFINITOR® (10 mg/día) contra placebo (más un tratamiento óptimo de apoyo en ambos brazos) fue llevado a cabo en pacientes con carcinoma de células renales metastásicos y cuya enfermedad había presentado progresión tumoral a pesar del tratamiento previo con un VEGFR-TKI (inhibidor de tirosin cinasa y del receptor del factor de crecimiento del endotelio vascular), como sunitinib o sorafenib (o ambos fármacos). El tratamiento previo con bevacizumab e interferón alfa también fue permitido. Se clasificaron a los pacientes en estratos o grupos según la puntuación pronostica del MSKCC [Memorial Sloan-Ketiering Cancer Center) (grupos de riesgo pronóstico favorable, versus intermedio versus pobre) y el tratamiento antineoplásico anterior (1 o 2 VEGFR-TKI previos).
El criterio principal de valoración fue la supervivencia libre de progresión, documentada según los criterios RECIST (Response Evaluation Criteria in Solid Tumors) y evaluada mediante un examen centralizado, independiente y ciego. Los criterios secundarios de valoración fueron la seguridad, la tasa objetiva de remisión tumoral, la supervivencia global, los síntomas relacionados con la enfermedad y la calidad de vida. Cuando los estudios radiológicos indicaban que había progresión, el investigador podía revelar a los pacientes el tratamiento recibido, entonces los del grupo del placebo podían recibir AFINITOR® (10 mg/día) sin cegar. El Comité Independiente de Vigilancia de Datos (CIVD) recomendó la finalización de este estudio al momento de realizar el segundo análisis intermedio por haberse satisfecho el criterio principal de valoración.
En total, 416 pacientes fueron aleatorizados 2:1 para recibir AFINITOR® (n = 277) o placebo (n = 139). La demografía estuvo bien equilibrada (agrupados mediana edad 61 años [rango de 27 a 85], 77% hombres, 88% caucásicos, y 74% tenía previamente una terapia con un ITK-VEGFR).
Los resultados del análisis interino planificado indicaron que AFINITOR® era superior al placebo en lo referente al criterio principal de valoración de supervivencia libre de progresión al producir una reducción estadísticamente significativa del 67% en el riesgo de progresión o muerte (ver la tabla 5 y la figura 8).
Tabla 5. RECORD-1 - Resultados de la supervivencia libre de progresión
|
Población |
n |
AFINITOR® |
Placebo |
Hazard Ratio |
Valor de pa |
|
Mediana de la supervivencia libre de progresión (meses) (IC de 95%) |
|||||
|
Análisis principal |
|||||
|
Total de pacientes (examen centralizado, independiente) |
416 |
4.9 (4.0 a 5.5) |
1.9 (1.8 a 1.9) |
0.33 (0.25 a 0.43) |
< 0.001a |
|
Análisis complementarios o de sensibilidad |
|||||
|
Total de pacientes (examen local realizado por el investigador) |
416 |
5.5 (4.6 a 5.8) |
1.9 (1.8 a 2.2) |
0.32 (0.25 a 0.41) |
< 0.001a |
|
Puntuación pronóstica del MSKCC |
|||||
|
Riesgo favorable |
120 |
5.8 (4.0 a 7.4) |
1.9 (1.9 a 2.8) |
0.31 (0.19 a 0.50) |
< 0.001b |
|
Riesgo intermedio |
235 |
4.5 (3.8 a 5.5) |
1.8 (1.8 a 1.9) |
0.32 (0.22 a 0.44) |
< 0.001b |
|
Riesgo pobre (adverso) |
61 |
3.6 (1.9 a 4.6) |
1.8 (1.8 a 3.6) |
0.44 (0.22 a 0.85) |
< 0.007b |
|
Tratamiento previo con ITK-VEGFR |
|||||
|
Sunitinib |
184 |
3.9 (3.6 a 5.6) |
1.8 (1.8 a 1.9) |
0.34 (0.23 a 0.51) |
< 0.001b |
|
Sorafenib |
124 |
5.9 (4.9 a 11.4) |
2.8 (1.9 a 3.6) |
0.25 (0.16 a 0.42) |
< 0.001b |
|
Sunitinib + sorafenib |
108 |
4.0 (3.6 a 5.4) |
1.8 (1.8 a 2.0) |
0.32 (0.19 a 0.54) |
< 0.001b |
a Prueba logarítmico-ordinal estratificada por pronóstico de puntuación.
b Prueba no estratificada unilateral log-rank.
Figura 8. RECORD-1 - Curvas de supervivencia libre de progresión de Kaplan-Meier.
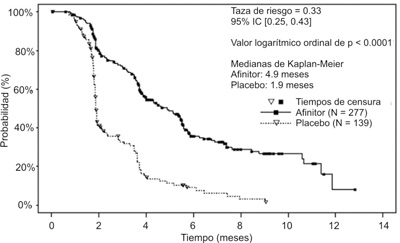
Las tasas de supervivencia libre de progresión a 6 meses fueron de 36% para el tratamiento con AFINITOR® en comparación con 9% para el placebo.
Se observaron remisiones tumorales, objetivas y confirmadas en 5 pacientes (2%) del grupo de AFINITOR® y en ninguno del grupo del placebo. Por tanto, la supervivencia sin progresión es un reflejo de lo que ocurre en la población cuya enfermedad se ha vuelto estable (que viene a ser 67% del grupo tratado con AFINITOR®).
No se observó ninguna diferencia estadísticamente significativa con relación al tratamiento en la supervivencia total, pero sí una tendencia a favor de AFINITOR® (HR 0.82; 95% IC: 0.57 a 1.17; p = 0.137). El entrecruzamiento a AFINITOR® sin cegar, tras comprobarse la progresión de la enfermedad en el grupo del placebo confundió la detección de cualquier posible diferencia vinculada al tratamiento en la supervivencia global.
Una fuerte tendencia es evidente y ésta soporta una mejor calidad de vida entre los pacientes que recibieron AFINITOR® como fue medida por los síntomas relacionados con la enfermedad (HR 0.75, IC 95%: 0.53 a 1.06; p = 0.053).
Astrocitomas subependimarios de células gigantes (SEGA) asociados con esclerosis tuberosa.
Se realizó un estudio prospectivo, abierto, de fase II para evaluar la seguridad y la eficacia de AFINITOR® en pacientes con SEGA. Como criterio de inclusión, se requirió evidencia radiológica en serie del crecimiento del SEGA.
El cambio en el volumen del SEGA durante la fase principal del tratamiento de 6 meses de duración fue la variable de eficacia primaria, conforme a lo evaluado en la revisión radiológica independiente principal. Después de la fase principal del tratamiento, los pacientes pueden entrar a la fase de extensión del tratamiento donde el volumen del SEGA fue evaluado cada 6 meses.
En total, los datos de la población de 28 pacientes tratados con AFINITOR® son; la edad promedio fue de 11 años, (entre 3 y 34 años), 61% varones, 86% caucásicos. Trece pacientes (46%) tenían un astrocitoma más pequeño secundario al SEGA principal incluyendo a 12 pacientes con SEGA en el ventrículo contralateral.
AFINITOR® fue asociado con una reducción clínicamente relevante y estadísticamente significativa en el volumen del SEGA principal desde el inicio hasta los seis meses de tratamiento (p < 0.001). La reducción del tumor fue más rápida durante los primeros 3 meses de tratamiento y se evidencia que existía una respuesta continua en los puntos de evaluación posterior (ver tabla 6). Ningún paciente desarrolló nuevas lesiones, empeoramiento de la hidrocefalia o aumento de la presión intracraneal, además de que ninguno requirió intervención quirúrgica u otro tratamiento contra el SEGA.
Tabla 6: Respuesta de la lesión generada por el SEGA principal al tratamiento con AFINITOI®.
|
Volumen del SEGA (cm3) |
Valor Inicial n = 28 |
3 meses n = 26 |
6 meses n = 27 |
12 meses n = 26 |
18 meses n = 18 |
24 meses n = 8 |
|
Promedio |
2.45 |
1.47 |
1.33 |
1.26 |
1.45 |
1.05 |
|
Mediana |
1.74 |
0.84 |
0.93 |
0.84 |
0.90 |
0.57 |
|
Intervalo |
0.49-14.23 |
0.25-8.32 |
0.31-7.98 |
0.29-8.18 |
0.33-5.20 |
0.33-3.66 |
|
Reducción a partir del valor inicial |
||||||
|
Promedio |
1.08 |
1.19 |
1.07 |
1.46 |
1.01 |
|
|
Mediana |
0.63 |
0.83 |
0.85 |
0.74 |
0.46 |
|
|
Intervalo |
-0.12-5.91 |
0.06-6.25 |
0.02-6.05 |
-0.24-9.03 |
0.12-3.79 |
|
|
Porcentaje de reducción a partir del valor inicial, n (%) |
||||||
|
³ 50% |
10 (38) |
9 (33) |
9 (35) |
8 (44) |
3 (38) |
|
|
³ 30% |
17 (65) |
21 (78) |
20 (77) |
12 (67) |
6 (75) |
|
|
> 0% |
25 (96) |
27 (100) |
26 (100) |
16 (89) |
8 (100) |
|
|
Sin cambio |
0 |
0 |
0 |
1 (6) |
0 |
|
|
Crecimiento tumoral relativo al valor inicial |
1 (4) |
0 |
0 |
1 (6) |
0 |
|
El análisis primario fue avalado por:
• El cambio en el volumen del SEGA principal conforme a la evaluación del investigador (p < 0.001), con 75 y 39% de los pacientes que tuvieron reducciones mayores a 30 y 50% respectivamente.
• El cambio en el volumen del SEGA total, (principal y astrocitomas secundarios) conforme a la revisión central independiente (p < 0.001) o a la evaluación local del investigador (p < 0.001).
Un paciente presentó los requisitos predefinidos para el éxito del tratamiento (reducción en el SEGA mayor a 75%) y le fue retirada la terapia temporalmente en el estudio clínico; sin embargo, el rebrote del SEGA fue evidente durante los siguientes 3 meses y el tratamiento fue reiniciado.
El everolimus fue asociado con una reducción clínicamente relevante en la frecuencia general de los ataques a los seis meses con respecto al valor inicial (mediana-1.0; p = 0.022).
CONTRAINDICACIONES: Hipersensibilidad al principio activo, a otros derivados de la rapamicina o a cualquiera de los excipientes (ver Precauciones generales).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Mujeres en edad reproductiva: Debe aconsejarse a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con AFINITOR® y en las 8 semanas siguientes a la finalización de la terapia.
Embarazo: No se dispone de datos suficientes sobre el uso de AFINITOR® en mujeres embarazadas. Los estudios en animales han evidenciado efectos tóxicos reproductivos como embriotoxicidad y fetotoxicidad (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Se desconoce el riesgo potencial para el humano. AFINITOR® no debe administrarse a mujeres embarazadas a menos que los posibles beneficios justifiquen el riesgo para el feto.
Lactancia: No se sabe si el everolimus pasa a la leche humana. Sin embargo, en los estudios con animales, tanto el everolimus como sus metabolitos pasan fácilmente a la leche de las ratas lactantes. Por consiguiente, las mujeres que toman AFINITOR® no deben amamantar.
Fertilidad: A juzgar por los hallazgos preclínicos el tratamiento con AFINITOR® puede disminuir la fertilidad masculina (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
REACCIONES SECUNDARIAS Y ADVERSAS:
Cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos en estadio avanzado de origen gastrointestinal, pulmonar o pancreático y carcinoma de células renales en estadio avanzado:
Resumen del perfil de seguridad: La información sobre las reacciones adversas esta basada principalmente en datos obtenidos en los cuatro estudios clínicos de fase III aleatorizados, a doble-ciego, controlados por placebo:
• BOLERO-2 (CRAD001Y2301): AFINITOR® en combinación con exemestano en el tratamiento de mujeres posmenopáusicas con cáncer de mama con receptores positivos de estrógeno localmente avanzado o metastásico, que son refractarios a letrozol o anastrozol. A partir de los datos de la fecha de corte del análisis interno (11-Feb-2011), la duración media del tratamiento fue de 14.6 semanas para los pacientes que recibían AFINITOR® y 12.0 semanas para los que recibieron placebo más exemestano.
• RADIANT-3 (CRAD001C2324): AFINITOR® más el mejor cuidado de apoyo en pacientes con tumores neuroendocrinos pancreáticos en estadio avanzado. La duración promedio del tratamiento de estudio ciego fue 37.8 semanas para pacientes que recibían AFINITOR® y 16.1 semanas para pacientes que recibían placebo.
• RADIANT-2 (CRAD001C2325): AFINITOR® más octreotida en pacientes con tumores neuroendocrinos (tumores carcinoides) principalmente de origen gastrointestinal o pulmonar. La duración promedio del tratamiento de estudio ciego fue de 37.0 semanas para los pacientes que recibieron AFINITOR® y 36.6 semanas para los que recibieron placebo.
• RECORD-1 (CRAD001C2240): AFINITOR® más el mejor cuidado de apoyo en pacientes con carcinoma de células renales en estadio avanzado. La duración promedio del tratamiento de estudio ciego fue de 141 días para los pacientes que estaban recibiendo AFINITOR® y 60 días para los que recibieron placebo.
Las reacciones adversas más frecuentes (incidencia igual o superior a 10% en al menos uno de los estudios fase III y que se sospeche a estar relacionada al tratamiento por el investigador) fueron (en orden decreciente): estomatitis, exantema, diarrea, fatiga, infecciones, astenia, náuseas, edema periférico, disminución del apetito, cefalea, disgeusia, epitaxis, inflamación de mucosa, neumonitis, disminución de peso, vómito, anorexia, prurito, tos, disnea, sequedad cutánea, trastorno de las uñas y pirexia. Las reacciones adversas de grado 3 o 4 más frecuentes (incidencia igual o superior a 2% en al menos uno de los estudios) fueron: estomatitis, fatiga, diarrea, infecciones, estomatitis, neumonitis y diabetes mellitus.
Resumen tabulado de las reacciones adversas de los estudios clínicos: En la tabla 7 presenta la incidencia de reacciones adversas notificadas con una incidencia igual o superior a 5% en los pacientes tratados con AFINITOR® (10 mg/día) en al menos uno de los estudios clínicos; todos los términos incluidos están basados en el porcentaje más alto reportado en el estudio clínico.
Las reacciones adversas de la tabla 5 están enlistadas de acuerdo a la clase de órgano, aparato o sistema afectado del MedDRA. Dentro de cada clase de órgano, aparato o sistema, las reacciones adversas se han clasificado por orden de frecuencia, las más frecuentes aparecen en primer lugar. Además, para clasificar cada reacción adversa en la correspondiente categoría de frecuencia se ha seguido la convención siguiente (CIOMS III): muy frecuente (³ 1/10); frecuente (³ 1/100, < 1/10) infrecuente (³ 1/1,000, < 1/100); rara (³ 1/10 000, < 1/1,000); muy rara (< 1/10,000), incluidas las notificaciones aisladas.
Tabla 7. Reacciones adversas registradas en al menos un estudio clínico con por lo menos 5% de los pacientes y en una mayor proporción en el grupo de AFINITOR® que en el del placebo
|
Clase órgano-sistema |
Muy frecuente |
Frecuente |
|
Infecciones e infestaciones |
Infecciones1 |
|
|
Trastornos del metabolismo y nutricionales |
Disminución del apetito2 |
Diabetes mellitus |
|
Trastornos vasculares |
Hipertensión |
|
|
Trastornos del sistema nervioso |
Disgeusia, cefalea |
|
|
Trastornos respiratorios, torácicos y del mediastino |
Tos, neumonitis3, epitaxia, disnea |
|
|
Trastornos gastrointestinales |
Estomatitis4, diarrea, náuseas, vómito |
Sequedad bucal |
|
Trastornos de la piel y del tejido subcutáneo |
Rash, piel seca, prurito, desórdenes de las uñas |
Acné |
|
Trastornos generales y del sitio de aplicación |
Fatiga, astenia, inflamación de las mucosas, edema periférico, pirexia |
|
|
Investigaciones |
Pérdida de peso |
1 Incluye todos los eventos reportados dentro de la clase órgano-sistema y los casos aislados de infecciones oportunistas, incluyendo la reactivación de la hepatitis B (< 1%),
2 Reportado como anorexia en el estudio RECORD-1 conforme a MedDRA v11.0
3 Incluye alveolitis, enfermedad pulmonar intersticial, infiltración pulmonar, neumonitis, hemorragia pulmonar alveolar y toxicidad pulmonar.
4 Incluye estomatitis aftosa y ulceración de la boca y lengua.
Otras reacciones adversas notorias, que ocurrieron en al menos un estudio clínico con mayor asiduidad en el grupo de AFINITOR® que en el del placebo, aunque con una incidencia inferior a 5% se enumeran abajo. Todos los términos incluidos están basados en el porcentaje más alto reportado en el estudio clínico.
Trastornos hematolóqicos y del sistema linfático:
Infrecuente: aplasia de células rojas puras.
Trastornos del metabolismo y de la nutrición:
Frecuente: deshidrataron (2.5%), exacerbación preexistente de la diabetes mellitus (1.1%).
Infrecuente: nueva aparición de la diabetes mellitus (< 1%).
Trastornos psiquiátricos:
Frecuente: insomnio (3.3%).
Trastornos del sistema nervioso:
Infrecuente: ageusia (< 1%).
Trastornos vasculares:
Frecuente: hemorragias (4.7%, en diferentes lugares).
Infrecuentes: trombosis venosa profunda (< 1%).
Trastornos cardiacos:
Infrecuente: insuficiencia cardiaca congestiva (< 1%).
Trastornos respiratorios, torácicos y del mediastino:
Frecuente: embolia pulmonar (1.5%), hemoptisis (1.1%).
Infrecuente: síndrome de agotamiento respiratorio agudo (< 1%).
Trastornos gastrointestinales:
Frecuente: dolor bucal (3.7%), dolor abdominal (3.6%), dispepsia (2.9%), disfagia (2.6%), dispepsia (2.6%).
Trastornos de la piel y del tejido subcutáneo:
Frecuente: síndrome mano pie (4.7%) eritema (3.7%).
Trastornos musculoesqueléticos y del tejido conjuntivo:
Frecuente: artralgia (2.8%).
Trastornos renales y urinarios:
Frecuente: proteinuria (2.5%), insuficiencia renal (2.3%, incluyendo insuficiencia renal aguda), micción diurna elevada (1.8%).
Trastornos generales y afecciones en el sitio de administración:
Frecuente: dolor torácico (1.1%).
Infrecuente: impedimento de cicatrización de las heridas (< 1%).
Anomalías de laboratorio de importancia reportadas en al menos uno de los estudios clínicos en los cuales se presentaron en proporciones mayores en el grupo de AFINITOR® que en el grupo del placebo: En los cuatro estudios clínicos de fase III, la mayoría de las anomalías de laboratorio de importancia fueron reportadas con una incidencia menor a 10% (enlistadas en frecuencia decreciente).
Decremento en los parámetros hematológicos, como hemoglobina, linfocitos, plaquetas y neutrófilos (o pancitopenia, cuando se acumulan todas). Incremento de los parámetros de química clínica como el colesterol, triglicéridos, glucosa, aspartato-transaminasas, creatinina, alanino-transaminasas y la bilirrubina. Decremento de los parámetros de químico clínica como el fosfato y el potasio.
La mayoría de las anomalías observadas fueron leves (grado 1) o moderadas (grado 2). Las anomalías de grado 4 incluían reducciones en los linfocitos (22%), hemoglobina (2%), y potasio (2%), neutrófilos, plaquetas y fosfatos (cada uno menor al 1%) e incrementos en la creatinina (1%), colesterol, ASAT, ALAT, bilirrubina y glucosa (cada uno menor a 1%) y reducciones en linfocitos (2.2%), hemoglobina (2%), neutrófilos, plaquetas y fosfato (cada uno menor a 1%).
SEGA: Los datos descritos a continuación reflejan la exposición a AFINITOR® (n = 28) en un estudio de fase II para el tratamiento de SEGA. En total, 16 de los 28 pacientes fueron tratados con AFINITOR® por más de 21 meses.
La exposición total fue 49.0 pacientes-años. La edad promedio de los pacientes fue de 11 años (rango entre 3 y 34 años).
Los eventos adversos más frecuentes (incidencia mayor o igual a 10% y posiblemente atribuida al tratamiento del investigador) fueron infecciones, estomatitis, pirexia, dermatitis acneiforme, diarrea, acné, tos, triglicéridos elevados, y disminución en el conteo de glóbulos blancos. Los únicos eventos adversos de grado 3 fueron infecciones (casos individuales de sinusitis, neumonía, infecciones dentales y bronquitis viral), y casos individuales de estomatitis y disminución en el conteo de glóbulos blancos. No se reportaron reacciones adversas de grado 4.
Resumen tabulado de las reacciones adversas del estudio clínico: La tabla 8 resume la incidencia de reacciones adversas relacionadas con el tratamiento, reportadas con una incidencia mayor o igual a 5%. Las reacciones adversas se enlistan de acuerdo a la clasificación órgano-sistema de MedDRA. En cada clasificación, las reacciones adversas están agrupadas por frecuencia, mencionando las más frecuentes al principio.
Tabla 8. Reacciones adversas reportadas en al menos 5% de los pacientes
|
Frecuencia |
AFINITOR® |
|||
|
Todos los grados |
Grado 3 |
Grado 4 |
||
|
Cualquier reacción adversa |
100 |
18 |
0 |
|
|
Infecciones e infestaciones |
||||
|
Infeccionesa |
Muy frecuente |
89 |
14 |
0 |
|
Trastornos nutricionales y metabólicos |
||||
|
Hipertrigliceremiab |
Muy frecuente |
11 |
0 |
0 |
|
Trastornos respiratorios, torácicos y del mediastino |
||||
|
Tos |
Muy frecuente |
11 |
0 |
0 |
|
Inflamación faríngea |
Frecuente |
7.1 |
0 |
0 |
|
Trastornos gastrointestinales |
||||
|
Estomatitis |
Muy frecuente |
79 |
3.6 |
0 |
|
Diarrea |
Muy frecuente |
21 |
0 |
0 |
|
Gastritis |
Frecuente |
7.1 |
0 |
0 |
|
Vómito |
Frecuente |
7.1 |
0 |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Dermatitis acneiforme |
Muy frecuente |
25 |
0 |
0 |
|
Acné |
Muy frecuente |
11 |
0 |
0 |
|
Trastornos generales y del sitio de administración |
||||
|
Pirexia |
Muy frecuente |
29 |
0 |
0 |
|
Inflamación de las mucosas |
Frecuente |
7.1 |
0 |
0 |
|
Investigaciones |
||||
|
Decremento en el conteo de glóbulos blancosc |
Muy frecuente |
11 |
3.6 |
0 |
|
Incremento de los triglicéridos en sangreb |
Frecuente |
7.1 |
0 |
0 |
CTCAE Versión 3.0.
a Para todas las infecciones reportadas, el protocolo estipula que todas las infecciones serán clasificadas como reacciones adversas (las infecciones reportadas incluyen infecciones del tracto respiratorio superior, sinusitis y otitis media).
b Reportado como una anomalía de laboratorio en 43% de los pacientes.
c Reportado como una anomalía de laboratorio en 54% de los pacientes.
Otras reacciones adversas de importancia, (con una incidencia menor a 5%) son:
Trastornos psiquiátricos:
Frecuentes: ansiedad (3.6%).
Trastornos del sistema nervioso:
Frecuentes: somnolencia (3.6%).
Trastornos vasculares:
Frecuentes: hipertensión (3.6%).
Trastornos respiratorios, torácicos y del mediastino:
Frecuentes: trastorno respiratorio (3.6%).
Trastornos de la piel y del tejido subcutáneo:
Frecuentes: Piel seca (3.6%), pitiriasis rosada (3.6%).
Trastornos renales y urinarios:
Frecuentes: proteinuria (3.6%).
Trastornos generales y del sitio de administración:
Frecuentes: fatiga (3.6%), edema periférico (3.6%).
No se incluyen las reacciones aisladas de grado 1.
Reacciones adversas de especial importancia: En estudios clínicos, everolimus se ha asociado a casos serios de reactivación de hepatitis B, incluyendo desenlace fatal. La reactivación de infecciones es un evento esperado durante los periodos de inmunosupresión.
En los estudios clínicos y en los reportes espontáneos posteriores a la comercialización, el everolimus ha sido asociado con eventos de insuficiencia renal (incluyendo casos mortales) y proteinuria. Se recomienda vigilar la función renal (ver Precauciones generales).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: La toxicidad preclínica de everolimus se estudió en ratones, ratas, cerdos de raza minipig, monos y conejos. Los órganos más afectados fueron los del aparato reproductor femenino y masculino de diversas especies (degeneración tubular testicular, reducción del contenido de esperma en los epidídimos y atrofia uterina), así como los pulmones de los ratones y las ratas (aumento de macrófagos alveolares) y los ojos de las ratas (opacidades en la línea de sutura cristalinianas anteriores). Se observaron alteraciones renales mínimas en las ratas (incremento de lipofuscina en el epitelio tubular vinculada a la edad del animal, incremento en la hidronefrosis) y los ratones (agravamiento de lesiones subyacentes). No hubo signos de toxicidad renal en los monos o los cerdos minipig.
El everolimus pareció exacerbar espontáneamente las enfermedades subyacentes (miocarditis crónica en las ratas, infección del plasma y el corazón por el virus de Coxsackie en los monos, infestación del tubo digestivo por coccidios en los cerdos minipig, lesiones cutáneas en los ratones y los monos). Estos efectos se observaron generalmente con una exposición sistémica incluida dentro del intervalo terapéutico o superior al mismo, salvo en las ratas, en las que ocurrieron con una exposición inferior al intervalo terapéutico debido a la elevada distribución en los tejidos.
En un estudio sobre fecundidad de ratas machos, se observó una alteración de la morfología testicular con la dosis de 0.5 mg/kg o mayor, así como una reducción de la motilidad de los espermatozoides, del número de cabezas de los espermatozoides y de las concentraciones plasmáticas de testosterona con la dosis de 5 mg/kg, que se sitúa dentro del intervalo terapéutico (52 ng.h/ml y 414 ng.h/ml respectivamente comparado con la exposición en humanos de 560 ng.h/ml con 10 mg/día) y menoscabó la fecundidad masculina. Hubo signos de reversibilidad.
El everolimus no alteró la fecundidad de las hembras, pero atravesó la barrera placentaria y fue tóxico para el producto de la concepción. Causó embriotoxicidad y fetotoxicidad por debajo del nivel terapéutico de exposición sistémica en las ratas, lo cual se manifestó como mortalidad y un menor peso fetal. Con 0.3 y 0.9 mg/kg se observó una mayor incidencia de malformaciones y anomalías óseas (por ejemplo, hendidura esternal). En los conejos, el aumento de resorciones tardías fue una señal de embriotoxicidad.
En estudios de toxicidad en ratas jóvenes a dosis bajas como 0.15 mg/kg/día, la toxicidad sistémica presentada incluye la disminución del peso corporal y el consumo de alimentos, y en el retraso de algunos signos del desarrollo en todas las dosis, con recuperación total o parcial después de la interrupción de la administración. Con la posible excepción de un hallazgo específico a encontrar en la rata (donde los animales jóvenes parecen ser más susceptibles), parece que no hay diferencias significativas en la sensibilidad de los animales jóvenes a los efectos adversos de everolimus en comparación con los animales adultos a dosis de 0.5 a 5 mg/kg/día. No hubo toxicidad de importancia evidente en monos jóvenes en dosis de hasta 0.5 mg/kg/día durante 4 semanas.
Los estudios de genotoxicidad en los que se investigaron las variables pertinentes de genotoxicidad no evidenciaron actividad clastógena ni mutágena alguna. La administración de everolimus a ratones y ratas durante dos años tampoco reveló poder cancerígeno alguno, incluso cuando se usaron las dosis más elevadas, que son unas 4.3 y 0.2 veces mayores que la exposición clínica prevista para una dosis diaria de 10 mg.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: El everolimus es un sustrato del CYP3A4 y asimismo, un sustrato e inhibidor moderado de la bomba de expulsión de fármacos conocida como glicoproteína P, por consiguiente, los fármacos que afectan al CYP3A4 o la glicoproteína P pueden alterar la absorción y la eliminación posterior de everolimus.
In vitro, el everolimus es un inhibidor competitivo del CYP3A4 y un inhibidor mixto del CYP2D6.
Agentes que pueden aumentar las concentraciones sanguíneas de everolimus: Las sustancias que inhiben la actividad del CYP3A4 (y que por eso mismo reducen el metabolismo de everolimus) pueden incrementar las concentraciones sanguíneas de everolimus.
Los inhibidores de la glicoproteína P (capaces de reducir la expulsión de everolimus de las células intestinales) pueden aumentar las concentraciones sanguíneas de everolimus.
Si las circunstancias lo permiten debe evitarse el tratamiento recurrente con inhibidores potentes del CYP3A4 o de la glicoproteína P (por ejemplo: ketoconazol e itraconazol, voriconazol, ritonavir, claritromicina y telitromicina).
Se apreció un significativo incremento de exposición a everolimus (la Cmáx y el ABC aumentaron unas 3.9 y 15 veces respectivamente) en sujetos sanos que habían recibido everolimus junto con ketoconazol (inhibidor potente del CYP3A4 e inhibidor de la glicoproteína P).
El tratamiento concomitante con inhibidores potentes del CYP3A4 o PgP (incluyendo, pero no limitado a, el ketoconazol, itraconazol, ritonavir, claritromicina y telitromicina) debe evitarse.
Hubo un incremento significativo en la exposición a everolimus (Cmáx y ABC incrementaron 3.9 y 15 veces respectivamente) en pacientes sanos cuando everolimus fue coadministrado con ketoconazol (un potente inhibidor de CYP3A4 y de PgP).
El tratamiento concomitante con inhibidores moderados de CYP3A4, pero no limitado a eritromicina, verapamil, ciclosporina, fluconazol, diltiazem, amprenavir, fosamprenvair, o aprepitant y los inhibidores de PgP requiere precaución. Se debe reducir la dosis de AFINITOR® si se coadministra con inhibidores moderados de CYP3A4/PgP (ver Dosis y vía de administración y Precauciones generales).
Hubo un aumento de exposición a everolimus en sujetos sanos que habían recibido everolimus junto con:
– Eritromicina (inhibidor moderado del CYP3A4 e inhibidor de la glicoproteína P; Cmáx 2.0 veces mayor y ABC 4.4 veces mayor).
– Verapamil (inhibidor moderado del CYP3A4 e inhibidor de la glicoproteína P; Cmáx 2.3 veces mayor y ABC 3.5 veces mayor).
– Ciclosporina (sustrato del CYP3A4 e inhibidor de la glicoproteína P; Cmáx 1.8 veces mayor y el ABC 2.7 veces mayor).
Algunos antimicóticos, como el fluconazol, y bloqueantes de los canales de calcio como el diltiazem, son inhibidores moderados del CYP3A4 y de la glicoproteína P que pueden aumentar las concentraciones sanguíneas de everolimus.
Durante el tratamiento con AFINITOR® se debe evitar el consumo de toronjas, jugo de toronja y cualquier otro alimento que pueda alterar la actividad de la glicoproteína P y del citocromo P-450.
No hubo diferencia aparente en la Cmín de everolimus cuando se administra en presencia o ausencia de sustratos de la CYP3A4 y/o de la glicoproteína P en dosis diarias de 10 y 5 mg.
La administración conjunta de inhibidores débiles de la CYP3A4 con o sin inhibidores de la glicoproteína P no tuvo impacto aparente en la Cmín de everolimus en dosis diarias de 10 y 5 mg.
Agentes que pueden reducir las concentraciones sanguíneas de everolimus: Las sustancias que son inductoras del CYP3A4 o de la glicoproteína P pueden reducir las concentraciones sanguíneas de everolimus mediante un aumento del metabolismo de everolimus o la expulsión de everolimus de las células intestinales.
El tratamiento concomitante con inductores fuertes de CYP3A4 o PgP debe evitarse. Si AFINITOR® debe ser coadministrado con un inductor fuerte de CYP3A4 o PGP (por ejemplo, rifabutina y rifampicina), puede ser necesario ajustar la dosis (ver Dosis y vía de administración y Precauciones generales).
El tratamiento preliminar de sujetos sanos con dosis múltiples de 600 mg de rifampicina (un inductor de CYP3A4 y de la glicoproteína P) al día durante 8 días y, luego, con una dosis única de everolimus prácticamente triplica la depuración de everolimus y reduce la Cmáx en 58% así como el ABC, en 63%.
Entre los inductores del CYP3A4 que pueden aumentar el metabolismo de everolimus y reducir sus concentraciones sanguíneas figuran asimismo la hierba de San Juan (Hypericum perforatum), corticoesteroides (por ejemplo, dexametosona, prednisona, prednisolona), anticonvulsivos como la carbamazepina, el fenobarbital y fenitoína, y antirretrovíricos como el efavirenz y la nevirapina.
Agentes cuyas concentraciones sanguíneas pueden verse alteradas por everolimus: Los estudios en sujetos sanos indican que no existen interacciones farmacocinéticas clínicamente significativas entre AFINITOR® y los inhibidores de la HMG-CoA reductasa como la atrovastatina (un sustrato del CYP3A4) y la pravastatina (la cual no es un sustrato del CYP3A4) así como la simvastatina (un sustrato del CYP3A4), de la cual no se detectó en los análisis de farmacocinética poblacional influencia alguna que afecte de alguna forma la depuración de everolimus (AFINITOR®).
In vitro, el everolimus es un inhibidor competitivo del metabolismo de la ciclosporina (un sustrato del CYP3A4) y un inhibidor mixto del dextrometorfano (un sustrato del CYP2D6). Tras la administración de una dosis oral de 10 mg al día o de 70 mg a la semana, la media del estado estable de la concentración máxima de everolimus en el estado estable se encuentra de 12 o 36 veces por debajo de los valores de Ki de la inhibición in vitro. Por consiguiente, no cabe esperar que everolimus afecte el metabolismo de los sustratos de CYP3A4 o CYP2D6.
Un estudio realizado en sujetos sanos demostró que la administración de una dosis oral de midazolam con everolimus resultó en un aumento de 25% de la Cmáx de midazolam y un aumento de 30% en el ABC(0-inf) de midazolam, mientras que la relación del ABC(0-inf) de sus metabolitos (1-hidroxi-midazolam/midazolam) y la vida media terminal (t½) de midazolam no se vieron afectados. Esto sugiere que el incremento en la exposición de midazolam es debido a los efectos del everolimus en el sistema gastrointestinal, cuando ambos medicamentos se administran al mismo tiempo. Por tanto, el everolimus puede afectar la biodisponibilidad oral de los fármacos administrados conjuntamente que son sustratos del CYP3A4.Es poco probable que el everolimus afecte la exposición de otros fármacos que son sustratos del CYP3A4 que se administran por vías no orales como por las vías de administración intravenosa, subcutánea y transdérmica (ver Precauciones generales).
La coadministración de everolimus y octreotida incrementó la Cmín de la octreotida con una relación geométrica promedio (everolimus/placebo) de 1.47 (90% IC: 1.32-1.64) lo cual no se espera que tenga relevancia clínica en la eficacia de la respuesta al tratamiento con everolimus en los pacientes que padezcan tumores neuroendócrinos en estado avanzado.
La administración concomitante de everolimus y exemestano aumentó la Cmín y la C2h en 45 y 71%, respectivamente. Sin embargo, los niveles correspondientes de estradiol en el estado de equilibrio (4 semanas) no fueron diferentes entre los dos grupos de tratamiento. No se observó un aumento de los eventos adversos relacionados con exemestano en pacientes con cáncer de mama avanzado con receptores hormonales positivos que recibieron esta combinación. Es poco probable que el aumento en los niveles de exemestano tenga un impacto en la eficacia o la seguridad.
Vacunas: Los inmunosupresores pueden alterar la reacción a una vacuna y ésta puede ser menos eficaz durante el tratamiento con AFINITOR®. Es necesario evitar el uso de vacunas vivas atenuadas durante el tratamiento con AFINITOR® (ver Precauciones generales). Ejemplos son la influenza intranasal, sarampión, parotiditis, varicela, rubéola, polio oral, BCG, fiebre amarilla, tifoidea elaborada con la cepa TY21a de S. Typha.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No se han reportado.
PRECAUCIONES GENERALES:
Neumonitis no infecciosa: La neumonitis no infecciosa es un efecto de clase de los derivados de rapamicina. También se han descrito casos de neumonitis no infecciosa (incluida la enfermedad pulmonar intersticial) en pacientes que toman AFINITOR® (ver Reacciones secundarias y adversas). Algunos de éstos han sido graves y, en raras ocasiones, se ha observado desenlace fatal.
Se debe considerar el diagnóstico de neumonitis no infecciosa en los pacientes que presentan signos y síntomas respiratorios inespecíficos, como hipoxia, derrame pleural, tos o disnea, y en quienes se han descartado las causas infecciosas o neoplásicas y otras causas no medicinales por medio de estudios apropiados. Se ha de aconsejar al paciente que comunique de inmediato cualquier síntoma respiratorio ya sea nuevo o agravado.
Los pacientes en quienes se descubren signos radiológicos indicativos de neumonitis no infecciosa, pero con escasos síntomas o ninguno, pueden continuar su tratamiento con AFINITOR® sin modificar la dosis.
Si los síntomas son moderados, cabe la posibilidad de interrumpir la terapia hasta que los síntomas mejoren. Puede ser necesario el uso de corticoesteroides.
• En pacientes con cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos en estadio avanzado de origen gastrointestinal, pulmonar o pancreático o en pacientes con carcinoma de células renales en estadio avanzado, el tratamiento con AFINITOR® se puede reanudar a una dosis de 5 mg diarios.
• En pacientes con SEGA, el tratamiento con AFINITOR® deberá reanudarse a una dosis diaria aproximadamente un 50% menor a la dosis administrada previamente.
Si los síntomas de neumonitis infecciosa son graves, se debe interrumpir el tratamiento con AFINITOR® y puede indicarse el uso de corticoesteroides hasta que desaparezcan los síntomas clínicos.
• En pacientes con cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos en estadio avanzado de origen gastrointestinal, pulmonar o pancreático o en pacientes con carcinoma de células renales en estadio avanzado, el tratamiento con AFINITOR® se puede reanudar a una dosis reducida de 5 mg diarios según las circunstancias clínicas del paciente.
• En pacientes con SEGA, el tratamiento con AFINITOR® deberá reanudarse a una dosis diaria aproximadamente 50% menor a la dosis administrada previamente dependiendo de las circunstancias clínicas individuales.
Infecciones: AFINITOR® tiene propiedades inmunosupresoras y puede hacer que los pacientes sean más propensos a contraer infecciones bacterianas, fúngicas, virales y parasíticas, especialmente las atribuidas a patógenos oportunistas (ver Reacciones secundarias y adversas). En pacientes tratados con AFINITOR®, se han descrito infecciones localizadas y generalizadas, incluidas las neumonías, otras infecciones bacterianas, infecciones fúngicas invasivas como la aspergillosis o la candidiasis e infecciones virales incluyendo la reactivación de hepatitis B. Algunas han sido graves (por ejemplo, produjeron insuficiencia respiratoria o hepática) y en ocasiones tuvieron un desenlace fatal. Los médicos y los pacientes deben ser concientes del mayor riesgo de infección asociado a AFINITOR®. Deberán estar atentos a los síntomas y signos de infección; si se diagnostica una infección, se debe emprender un tratamiento adecuado sin demora además de considerar la interrupción o retiro del tratamiento con AFINITOR®.
Se debe tratar las infecciones fúngicas invasivas preexistentes antes de Iniciar el tratamiento con AFINITOR®. Si se realiza el diagnóstico de infección fúngica sistémica invasiva, se debe suspender AFINITOR® y tratar esta infección con una terapia antifúngica apropiada.
Reacciones de hipersensibilidad: Las reacciones de hipersensibilidad que se han observado con everolimus se manifiestan por síntomas que incluyen, pero no se limitan a, anafilaxis, disnea, enrojecimiento, dolor torácico o angioedema (por ejemplo, edema de las vías respiratorias o la lengua, con o sin deterioro respiratorio) (ver Contraindicaciones).
Úlceras bucales: Se han observado úlceras, estomatitis y mucositis bucales en pacientes tratados con AFINITOR® (ver Reacciones secundarias y adversas). En dichas situaciones se recomiendan los tratamientos tópicos, pero no los colutorios a base de alcohol o peróxido, que pueden exacerbar la afección. No se deben utilizar agentes antimicóticos, salvo si se ha diagnosticado una infección fúngica (ver Interacciones medicamentosas y de otro género).
Eventos de insuficiencia renal: Se han observado casos de insuficiencia renal (incluyendo insuficiencia renal aguda), con desenlace fatal en algunos casos, en pacientes tratados con AFINITOR® (ver Reacciones secundarias y adversas así como Vigilancia y análisis de laboratorio).
Vigilancia y análisis de laboratorio:
Función renal: Se han notificado elevaciones de creatinina sérica, usualmente leves, y proteinuria en los estudios clínicos (ver Reacciones secundarias y adversas). Se recomienda la vigilancia de la función renal, que incluye la determinación de urea (BUN), proteínas urinarias o de creatinína sérica, antes del inicio del tratamiento con AFINITOR® y periódicamente durante el mismo.
Glucemia: Se ha notificado hiperglucemia en los estudios clínicos (ver Reacciones secundarias y adversas). Se aconseja la vigilancia de la glucemia en ayunas antes de comenzar el tratamiento con AFINITOR® y periódicamente durante el mismo. La glucemia debe estar perfectamente regulada antes de instaurar un tratamiento con AFINITOR® en el paciente.
Parámetros hematológicos: Se han registrado cifras reducidas de hemoglobina, linfocitos, neutrófilos y plaquetas en los estudios clínicos (ver Reacciones secundarias y adversas). Se aconseja la supervisión de la citometría hemática antes de comenzar el tratamiento con AFINITOR® y periódicamente durante el mismo.
Interacciones farmacológicas: Debe evitarse la coadministración con inhibidores potentes de CYP3A4 o de la P-glicoproteína (PgP) (ver Interacciones medicamentos y de otro género).
Se debe tener precaución cuando se administra AFINITOR® en combinación con inhibidores moderados del CYP3A4 o los inhibidores de PgP. Si AFINITOR® debe ser coadministrado con un inhibidor moderado de CYP3A4 o de PgP, el paciente debe ser cuidadosamente vigilado debido a los efectos adversos y si es necesario, se deberá realizar una reducción de la dosis (ver Dosis y vía de administración e Interacciones medicamentosas y de otro género).
En la medida de lo posible, también debe evitarse la coadministración de inductores potentes del CYP3A4 o de la glicoproteína P (ver Interacciones medicamentos y de otro género). En caso de que sea necesario administrar AFINITOR® con un inductor potente del CYP3A4 o de la glicoproteína P, se deberá vigilar atentamente la respuesta clínica del paciente. Considere la posibilidad de un aumento de la dosis de AFINITOR® cuando se administre con inductores fuertes de CYP3A4 o PGP si el tratamiento alternativo no es posible (ver Dosis y vía de administración e Interacciones medicamentosas y de otro género).
Se debe tener cuidado cuando se administre AFINITOR® en combinación con sustratos del CYP3A4 orales con un estrecho margen terapéutico, debido al potencial de interacciones farmacológicas. En caso de que se tome AFINITOR® administrado con sustratos orales del CYP3A4 con un estrecho margen terapéutico, el paciente debe ser vigilado en caso de que se presenten las reacciones adversas descritas en la información para prescribir dicho sustrato del CYP3A4 (ver Interacciones medicamentos y de otro género).
Insuficiencia hepática: La exposición a everolimus se incrementó en los pacientes con insuficiencia hepática leve (Child-Pugh A), moderada (Child-Pugh B) y severa (Child-Pugh C) (ver Farmaocinética y farmacodinamia).
El everolimus no está recomendado para su uso en mujeres posmenopáusicas con cáncer de mama avanzado con receptores hormonales positivos, o en pacientes con tumores neuroendocrinos de origen gastrointestinal, pulmonar o pancreático o carcinomas renales en estadio avanzado con insuficiencia hepática severa (Child-Pugh C) a menos que el beneficio potencial supere el riesgo (ver Dosis y vía de administración y Farmacocinética y farmacodinamia).
El everolimus no está recomendado para su uso en pacientes con SEGA que padezcan insuficiencia hepática severa (Child-Pugh C) (ver Dosis y vía de administración y Farmacocinética y farmacodinamia).
Vacunas: Durante el tratamiento con AFINITOR® es preciso evitar la aplicación de vacunas vivas atenuadas, así como el contacto íntimo con personas que han recibido tales vacunas (ver Interacciones medicamentosas y de otro género).
DOSIS Y VÍA DE ADMINISTRACIÓN: Oral.
Dosis: AFINITOR® debe administrarse por vía oral una vez al día, a la misma hora todos los días (preferentemente por la mañana), consistentemente en ayunas o consistentemente después de un refrigerio exento de grasas (ver Farmacocinética y farmacodinamia).
Los comprimidos se deben tragar enteros con un vaso de agua. No se deben masticar ni triturar. Para los pacientes que no pueden tragar los comprimidos, la tableta de AFINITOR® deberá ser dispersada por completo en un vaso de agua (aproximadamente 30 ml) por agitación suave, para después consumirla inmediatamente. El vaso deberá ser enjuagado con el mismo volumen de agua y ésta deberá ser ingerida para garantizar que toda la dosis fue administrada (ver Farmacología clínica).
Es necesario continuar con el tratamiento mientras se observen beneficios clínicos y no surjan reacciones adversas intolerables.
Pacientes adultos:
Población destinataria en general:
Dosificación para cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos en estadio avanzado de origen gastrointestinal, pulmonar o pancreático y en el carcinoma de células renales en estadio avanzado.
El tratamiento con AFINITOR® ha de instaurarlo un médico experimentado en el uso de terapias antineoplásicas.
La dosis recomendada AFINITOR® para el tratamiento carcinomas del adulto es de 10 mg una vez al día.
El tratamiento de presuntas reacciones adversas graves o intolerables puede exigir la reducción momentánea de la dosis a la interrupción del tratamiento con AFINITOR®. Si fuera necesario reducir la dosis, se sugiere una dosis de 5 mg diarios (ver Precauciones generales).
Inhibidores moderados de CYP3A4 o PgP: Tenga precaución cuando se administra en combinación con inhibidores del CYP3A4 moderados o los inhibidores de PgP. Si los pacientes requieren la coadministración de un volumen moderado de CYP3A4 o inhibidor de PgP, se debe reducir la dosis a 5 mg diarios. Podrán requerirse más reducciones de dosis como la de 5 mg cada dos días o 2.5 mg diarios para el manejo de reacciones adversas (ver Advertencias y precauciones e Interacciones medicamentosas y de otro género).
Pacientes pediátricos:
Dosificación para astrocitomas subependimarios de células gigantes (SEGA) asociados con esclerosis tuberosa (ET): El tratamiento con AFINITOR® debe iniciarse por un médico con experiencia en el tratamiento de pacientes con ET y con acceso a servicios terapéuticos de monitoreo para everolimus. El monitoreo terapéutico de las concentraciones sanguíneas de everolimus es necesario para los pacientes tratados con SEGA (ver Monitoreo terapéutico de fármacos en el texto más abajo).
Para obtener el efecto terapéutico óptimo puede ser necesario un reajuste posológico. La tolerancia y eficacia de las dosis varían entre los pacientes. La terapia concomitante con antiepilépticos puede afectar el metabolismo de everolimus y pueden contribuir a esta variación (ver Interacciones medicamentosas y de otro género).
Para asegurar que sea deglutido adecuadamente, sobre todo en niños muy pequeños, el comprimido deberá disolverse en 30 ml de agua y posteriormente dar 30 ml adicionales en el mismo recipiente para asegurar que se administró la dosis completa.
La dosis inicial recomendada de AFINITOR® para el tratamiento de pacientes con SEGA se describe en la siguiente tabla:
Tabla 9. Dosis inicial recomendada de AFINITOR® para el tratamiento de pacientes con SEGA
|
Superficie corporal |
Dosis inicial diaria |
|
£ 1.2 m2 |
2.5 mg |
|
1.3 m2 a 2.1 m2 |
5 mg |
|
³ 2.2 m2 |
7.5 mg |
Las concentraciones totales de everolimus en sangre deberán ser evaluadas aproximadamente 2 semanas después de comenzar el tratamiento. La dosis se ajustará para alcanzar las concentraciones mínimas de 5 a 15 ng/ml. Si las concentraciones están por debajo de 5 ng/ml, la dosis diaria puede incrementarse en 2.5 mg cada 2 semanas, sujeto a la tolerabilidad (ver Farmacología clínica).
El volumen de SEGA deberá ser evaluado aproximadamente 3 meses después de comenzar el tratamiento con AFINITOR®, con ajustes de dosis subsecuentes teniendo en cuenta los cambios en el volumen de SEGA, la concentración sanguínea correspondiente, y la tolerabilidad (ver Farmacología clínica). Se ha observado respuesta a concentraciones mínimas tan bajas como 2 ng/ml. En tal caso, al alcanzar una eficacia aceptable, podría no ser necesario realizar incrementos adicionales en la dosis.
El manejo de las reacciones adversas graves o intolerables podrán requerir la reducción temporal de la dosis y/o la interrupción del tratamiento (ver Advertencias y precauciones). Si se requiere una reducción de la dosis en los pacientes que reciben 2.5 mg al día, se deberá considerar la administración en días alternos.
Inhibidores moderados del CYP3A4 o de la PqP: Use con cautela cuando se administre AFINITOR® en combinación con inhibidores moderados del CYP3A4 o inhibidores de la PgP. Si los pacientes requieren la administración de un inhibidor moderado del CYP3A4 o de la PgP, se debe reducir la dosis diaria en aproximadamente 50%. Otras reducciones adicionales en la dosis pueden ser necesarias para el manejo de las reacciones adversas (ver Advertencias y precauciones e Interacciones medicamentosas y de otro género). Las concentraciones totales de everolimus en sangre deberán ser evaluadas aproximadamente 2 semanas después de la adición de un inhibidor moderado del CYP3A4 o PgP. Si la administración del inhibidor moderado se interrumpe la dosis de AFINITOR® deberá ser devuelta a la dosis utilizada antes de la iniciación del tratamiento con los inhibidores moderados del CYP3A4 o de PgP y la concentración total de everolimus en sangre deberá ser reevaluada aproximadamente 2 semanas después (ver Advertencias y precauciones e Interacciones medicamentosas y de otro género).
Inductores potentes del CYP3A4: Evítase el uso concomitante de inductores potentes de CYP3A4. Los pacientes que reciben concomitantemente inductores fuertes del CYP3A4 (por ejemplo, inductores enzimáticos de fármacos antiepilépticos) pueden requerir una dosis mayor de AFINITOR® para alcanzar las concentraciones sanguíneas de 5 a 15 ng/ml. Si las concentraciones están por debajo de 5 ng/ml, la dosis diaria puede incrementarse en 25 mg cada 2 semanas, considerando antes de aumentar la dosis las concentraciones sanguíneas y la tolerancia.
Si la administración del inductor potente se interrumpe la dosis de AFINITOR® deberá ser devuelta a la dosis utilizada antes de la iniciación del tratamiento con el inductor potente del CYP3A4 y la concentración total de everolimus en sangre deberá ser reevaluada aproximadamente 2 semanas después (ver Advertencias y Precauciones e Interacciones).
Monitoreo terapéutico para el tratamiento de pacientes con SEGA: El monitoreo terapéutico de las concentraciones sanguíneas de everolimus es necesario para los pacientes tratados para SEGA con un método bioanalítico LC/MS validado. Las concentraciones totales deberán ser evaluadas aproximadamente 2 semanas después de la dosis inicial, después de cualquier cambio en la dosis, o después de una iniciación o cambio en la administración conjunta de inductores o inhibidores del CYP3A4 (ver Advertencias y precauciones e Interacciones medicamentosas y de otro género), o después de que se observe cualquier cambio hepático (Child-Pugh) (ver Dosis y vía de administración y Farmacocinética y farmacodinamia). Se ajustará con el propósito de alcanzar las concentraciones sanguíneas de everolimus de 5 a 15 ng/ml, teniendo en cuenta la tolerabilidad (ver Farmacología clínica).
Posología en poblaciones especiales:
Niños y adolescentes:
• No se recomienda el uso de AFINITOR® en pacientes oncológicos pediátricos.
• Las recomendaciones de dosificación para pacientes pediátricos con SEGA son equiparables con las de los adultos con SEGA. AFINITOR® no ha sido estudiado en pacientes pediátricos con SEGA, menores a 3 años y actualmente no está recomendado para su uso en este grupo de edad.
Pacientes de edad avanzada (de 65 años como mínimo): No es necesario reajustar la dosis (ver Farmacología clínica).
Pacientes con insuficiencia renal: No es necesario reajustar la dosis (ver Farmacología clínica).
Pacientes con insuficiencia hepática:
Cáncer de mama avanzado con receptores hormonales positivos, tumores neuroendocrinos en estadio avanzado de origen gastrointestinal, pulmonar o pancreático y para el carcinoma de células renales en estadio avanzado:
• Insuficiencia hepática leve (Chíld-Pugh A)- La dosis recomendada es de 7.5 mg diariamente.
• Insuficiencia hepática moderada (Child-Pugh B)- La dosis recomendada es de 2.5 mg diariamente.
• Insuficiencia hepática grave (Child-Pugh C)- Su uso no está recomendado.
Si el beneficio supera al riesgo, no deberá de excederse de la dosis de 2.5 mg diarios.
• Los ajustes en la dosis deberán realizarse si el paciente presenta alteraciones de su estado hepático (Child-Pugh) durante el tratamiento.
Astrocitomas subependimarios de células gigantes (SEGA, por sus siglas en inglés) asociados con esclerosis tuberosa (TS): La dosis inicial recomendada en los pacientes con SEGA que presentan insuficiencia hepática se indica en la tabla 10.
AFINITOR® no está recomendado en pacientes con SEGA que presentan insuficiencia hepática severa (Child-Pugh C).
Tabla 10: Dosis inicial recomendada de AFINITOR® para el tratamiento de pacientes de SEGA con daño hepático.
|
Área de superficie corporal (ASC) |
Insuficiencia hepática leve (Child-Pugh A) |
Insuficiencia hepática moderada (Child-Pugh B) |
|
£ 1.2 m2 |
2.5 mg cada dos días |
No se recomienda |
|
1.3 m2 a 2.1 m2 |
2.5 mg diarios |
2.5 mg cada dos días |
|
³ 2.2 m2 |
5 mg diarios |
2.5 mg diarios |
Las concentraciones sanguíneas de everolimus deben ser evaluadas aproximadamente 2 semanas después de comenzar el tratamiento o después de cualquier cambio en el estado hepático (Child-Pugh). La dosis se ajustará para alcanzar concentraciones sanguíneas de 5 a 15 ng/ml. Si las concentraciones están por debajo de 5 ng/ml, la dosis diaria puede incrementarse en 2.5 mg cada 2 semanas, dependiendo de la tolerabilidad (ver Farmacocinética y farmacodinamia).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: En los estudios en animales, el everolimus reveló tener un bajo potencial de toxicidad aguda. No se observó letalidad ni toxicidad grave en las ratas o ratones que recibieron dosis orales únicas de 2,000 mg/kg (ensayo límite).
Se conocen muy pocos casos de sobredosis en humanos. Se han administrado dosis únicas de hasta 70 mg con una aceptable tolerabilidad aguda.
En todos los casos de sobredosis se deben tomar medidas generales de apoyo.
PRESENTACIONES:
Caja de cartón con 30 comprimidos con 2.5, 5 o 10 mg, en envase de burbuja (PA-AL-PVC)-AI.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a temperatura ambiente a no más de 30°C y en lugar seco. Protéjase de la luz.
LEYENDAS DE PROTECCIÓN:
No se deje al alcance de los niños. Su venta requiere receta médica. No se use en el embarazo ni en la lactancia.
Para mayor información comuníquese al Centro de Atención a Clientes de Novartis Farmacéutica, S.A. de C.V., Calzada de Tlalpan No. 1779, Col. San Diego Churubusco, Coyoacán, C.P. 04120, teléfono 5420-8685, en el Interior de la República 01 800 718 54 59.
Hecho en Suiza por:
Novartis Pharma Stein AG
Distribuido por:
NOVARTIS FARMACÉUTICA, S. A. de C. V
Reg. Núm. 028M2010, SSA IV
CDS: 14-Oct-2011
NPI:28-Oct-2011

