

CADUET
AMLODIPINO, ATORVASTATINA
Tabletas
1 Caja, 10 Tabletas, 5/10 mg/mg
1 Caja, 10 Tabletas, 5/20 mg/mg
1 Caja, 10 Tabletas, 5/40 mg/mg
1 Caja, 10 Tabletas, 5/80 mg/mg
1 Caja, 30 Tabletas, 5/10 mg/mg
1 Caja, 30 Tabletas, 5/20 mg/mg
1 Caja, 30 Tabletas, 5/40 mg/mg
1 Caja, 30 Tabletas, 5/80 mg/mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Besilato de amlodipino equivalente a 5 mg de amlodipino
Atorvastatina cálcica equivalente a 10 mg, 20 mg, 40 mg, 80 mg
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
CADUET® (amlodipino/atorvastatina) está indicado en las siguientes poblaciones de pacientes:
1. Pacientes con mayor riesgo cardiovascular por la presencia de dos factores de riesgo modificables, hipertensión y dislipidemia; y/o
2. Pacientes con aumento de riesgo cardiovascular por la presencia enfermedad cardiaca coronaria (ECC) sintomática expresada como angina con factor de riesgo modificable adicional de dislipidemia; y/o
3. Prevención de complicaciones cardiovasculares en pacientes hipertensos (ver a continuación - Prevención de Complicaciones cardiovasculares).
En estos pacientes con factores de riesgo cardiovascular múltiple, CADUET® (amlodipino/atorvastatina) está indicado para:
Hipertensión: El componente de amlodipino está indicado para tratamiento de primera línea de hipertensión y puede emplearse como agente único para controlar la presión arterial (PA) en la mayoría de los pacientes. Aquellos pacientes no controlados adecuadamente con un solo agente antihipertensivo (distinto de amlodipino) quizá se beneficien de la adición del componente de amlodipino de CADUET® (amlodipino/atorvastatina), del mismo modo que se beneficiarían por la adición de amlodipino solo.
amlodipino también está indicado para reducir el riesgo de ECC mortal e infarto del miocardio (IM) no mortal y reducir el riesgo de accidente cerebrovascular.
Enfermedad arterial coronaria: El componente amlodipino está indicado para reducir el riesgo de procedimientos de revascularización coronaria y la necesidad de hospitalización por angina en pacientes con enfermedad coronaria (EAC).
Angina estable crónica: El componente amlodipino está indicado para tratamiento de primera línea de isquemia del miocardio ya sea causa de obstrucción fija (angina estable) y/o vasoespasmo/vasoconstricción (angina de Prinzmetal o variable) de la vasculatura coronaria. CADUET® (amlodipino/atorvastatina) puede emplearse cuando la presentación clínica sugiere posible componente vasoespástico/vasoconstrictor pero no se ha confirmado que exista vasoespasmo/vasoconstrucción. CADUET® (amlodipino/atorvastatina) puede emplearse solo o combinado con otros fármacos antianginales en pacientes con angina refractaria a nitratos y/o dosis adecuadas de beta bloqueadores.
Dislipidemia: El componente de atorvastatina está indicado como coadyuvante a la dieta para tratamiento de pacientes con valores elevados de colesterol total (C-total), colesterol de lipoproteínas de baja densidad (C-C-LDL), apolipoproteína B (apo B) y triglicéridos (TG) y para aumentar el colesterol de lipoproteínas de alta densidad (C-C-HDL) en pacientes con hipercolesterolemia primaria (hipercolesterolemia familiar y no familiar heterocigota), hiperlipidemia combinada (mixta) (tipos IIa y IIb de Fredrickson), niveles altos de TG séricos (tipo IV de Fredrickson) y en pacientes con disbetalipoproteinemia (tipo III de Fredrickson) que no respondan adecuadamente a la dieta.
El componente atorvastatina también está indicado para reducir colesterol total y C-LDL en pacientes con hipercolesterolemia familiar homocigoto (FH).
Prevención de complicaciones cardiovasculares (CV):
En pacientes sin enfermedad cardiovascular clínicamente evidente (ECV) y con o sin dislipidemia pero con múltiples factores de riesgo para ECC como tabaquismo, hipertensión, diabetes C-HDL bajo o historia familiar de ECC temprana, atorvastatina está indicada para:
– Reducir el riesgo de ECC mortal e IM no mortal.
– Reducir el riesgo de accidente cerebrovascular.
– Reducir el riesgo de procedimientos de revascularización y angina de pecho.
En pacientes con ECC clínicamente evidente, atorvastatina está indicado para:
– Reducir el riesgo de IM no mortal.
– Reducir el riesgo de accidente cerebrovascular mortal y no mortal.
– Reducir el riesgo en procedimientos de revascularización.
– Reducir el riesgo de hospitalización por insuficiencia cardiaca congestiva (ICC).
– Reducir el riesgo de angina.
Pacientes pediátricos (de 10 a 17 años): Atorvastatina está indicada como coadyuvante de la dieta para reducir los niveles de colesterol total, C-LDL y apo B en niños y niñas después de la menarca, de 10 a 17 años con FH heterocigota si están presentes las siguientes observaciones tras una prueba adecuada con terapia dietética:
a. C-LDL permanece > 190 mg/dL o
b. C-LDL permanece > 160 mg/dL y:
• Existen antecedentes de ECV prematura o
• Están presentes dos más factores de riesgo ECV distintos en el paciente pediátrico.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades Farmacocinéticas:
Absorción:
En estudios con CADUET® (amlodipino/atorvastatina): Después de la administración oral de CADUET® (amlodipino/atorvastatina) se observan dos concentraciones plasmáticas máximas distintas. La primera dentro de 1 a 2 horas de la administración es atribuible a atorvastatina, la segunda entre 6 y 12 horas después de la dosificación es atribuible a amlodipino. La tasa y el grado de absorción (biodisponibilidad) de amlodipino y atorvastatina de CADUET® (amlodipino/atorvastatina) no son significativamente diferentes de la biodisponibilidad de la coadministración de tabletas de amlodipino y de atorvastatina evaluadas por la Cmáx. 101% (IC a 90%: 98, 104) y ABC (área bajo la curva): 100% (IC al 90%: 97, 103) para el componente amlodipino y Cmáx.: 94% (IC a 90%: 85, 104) y ABC: 105% (IC a 90%: 99, 111) para el componente atorvastatina, respectivamente.
La biodisponibilidad de amlodipino de CADUET® (amlodipino/atorvastatina) no se vio afectada en la administración con alimentos, evaluada por la Cmáx. 105% (IC al 90%: 99, 111) y ABC: 101% (IC al 90%: 97, 105). Aunque los alimentos disminuyen la velocidad y el grado de absorción de atorvastatina de CADUET® (amlodipino/atorvastatina) en aproximadamente 32% y 11%, respectivamente, evaluada por Cmáx. 68% (IC al 90%: 60, 79) y ABC: 89% (IC al 90%: 83, 95), se han observado reducciones similares en las concentraciones plasmáticas en la administración con alimentos con atorvastatina sin una reducción en el efecto del C-LDL.
En estudios con amlodipino: Después de la administración oral de dosis terapéuticas, amlodipino es bien absorbido con niveles máximos en sangre entre 6 a 12 horas después de la dosis. La biodisponibilidad absoluta se ha estimado que está entre 64 y 80%. El volumen de distribución es aproximadamente de 21 L/kg. Estudios in vitro han demostrado que aproximadamente 97. 5% del amlodipino circulante está ligado a las proteínas del plasma.
La absorción de amlodipino no se ve afectada por el consumo de alimento.
En estudios con atorvastatina:
Atorvastatina se absorbe rápidamente tras la administración oral; las concentraciones plasmáticas máximas se producen dentro de una o dos horas después. El grado de absorción y la concentración de atorvastatina en plasma aumentan en proporción a la dosis de atorvastatina. La biodisponibilidad de las tabletas de atorvastatina es de 95% a 99% en comparación con las soluciones. La biodisponibilidad absoluta de atorvastatina es aproximadamente de 14% y la disponibilidad sistémica de la actividad inhibidora de la HMG-CoA reductasa es de aproximadamente 30%. La baja disponibilidad sistémica se atribuye a un aclaramiento pre-sistémico en la mucosa gastrointestinal y/o el metabolismo de primer paso hepático. Aunque la comida disminuye la velocidad y grado de absorción del fármaco en aproximadamente un 25% y 9% respectivamente, según la evaluación de Cmáx. y ABC, la reducción de C-LDL es similar si atorvastatina se administra con o sin alimentos. Las concentraciones de atorvastatina son más bajas (aproximadamente del 30% para Cmáx. y ABC) después de la administración del fármaco por las noches en comparación con la mañana. Sin embargo, la reducción de C-LDL es el mismo, independientemente de la hora del día en que se administre el fármaco (ver sección Dosis y vía de administración).
Distribución:
En estudios con atorvastatina:
El volumen de distribución promedio de atorvastatina es aproximadamente 381 L. Atorvastatina está ≥ 98% ligada a las proteínas del plasma. Una proporción eritrocito/plasta de aproximadamente 0.25 indica penetración deficiente del medicamento en los glóbulos rojos.
Metabolismo y excreción:
En estudios con amlodpino:
La vida media de eliminación terminal en el plasma es aproximadamente 35 a 50 horas y es consistente con la dosis de una vez al día. Niveles en el plasma en estado estable son alcanzados después de 7 a 8 días de administración consecutiva. amlodipino es metabolizado ampliamente por el hígado a metabolitos inactivos con 10% del compuesto original (parent) y 60% de metabolitos excretados en la orina.
En estudios con atorvastatina: Atorvastatina es metabolizada de manera extensa a derivados orto - y para - hidroxilados y diversos productos de beta-oxidación. La inhibición in vitro de HMG-CoA reductasa por metabolitos orto - y para - hidroxilados equivale a la atorvastatina. Aproximadamente 70% de la actividad inhibitoria en circulación para HMG-CoA reductasa es atribuible a los metabolitos activos. Estudios in vitro sugieren la importancia del metabolismo de atorvastatina por citocromo P450 3A4 hepático congruente con aumento de concentraciones plasmáticas de atorvastatina en humanos tras coadministración con eritromicina, inhibidor conocido de esta isozima. Estudios in vitro también indican que atorvastatina es inhibidor débil de citocromo P450 3A4. La coadministración de atorvastatina no produjo efecto clínicamente significativo en concentraciones plasmáticas de terfenadina, compuesto metabolizado principalmente por citocromo P450 3A4; por lo tanto, es poco probable que atorvastatina altere significativamente la farmacocinética de otros sustratos de citocromo P450 3A4 (ver Interacciones medicamentosas y de otro género). En animales, el metabolito orto-hidroxilado experimenta glucuronidación posterior.
Atorvastatina y sus metabolitos son eliminados principalmente en bilis tras metabolismo hepático; sin embargo, el fármaco no sufre recirculación enterohepática. La media de la vida media de eliminación plasmática de atorvastatina en humanos es aproximadamente 14 horas, pero la vida media de actividad inhibitoria para HMG-CoA reductasa es de 20 a 30 horas debido a la contribución de los metabolitos activos. Menos del 2% de la dosis de atorvastatina se recupera en orina tras administración oral.
La atorvastatina es un sustrato de los transportadores hepáticos, transportador OATP1B1 y OATP1B3. Los metabolitos de la atorvastatina son sustratos de OATP1B1. La atorvastatina también es definida como un sustrato de los transportadores de salida MDR1 y BCRP, los cuales pueden limitar la absorción intestinal y el aclaramiento biliar de la atorvastatina.
Poblaciones especiales:
Insuficiencia hepática:
En estudios con atorvastatina: Las concentraciones plasmáticas de atorvastatina se incrementa notablemente (aproximadamente 16 veces para Cmáx. y 11 veces para ABC) en pacientes con enfermedad hepática alcohólica crónica (Child-Pugh B) (ver sección Contraindicaciones).
Insuficiencia renal (ver sección Dosis y vía de administración):
En estudios con amlodipino: Los cambios de concentración plasmática de amlodipino no se correlacionan con el grado de afección renal. amlodipino no es dializable.
En estudios con atorvastatina: La enfermedad renal no influye en las concentraciones plasmáticas o efectos de atorvastatina sobre los lípidos. Por tanto, no se requiere ajuste de dosis en pacientes con disfunción renal.
Género:
En estudios con atorvastatina: Las concentraciones plasmáticas de atorvastatina en mujeres difieren (Cmáx. es aproximadamente 20% más alta y ABC es 10% más baja) respecto a la de varones. Sin embargo, no hubo diferencias clínicamente significativas en efectos sobre lípidos entre varones y mujeres.
Personas de edad avanzada:
En estudios con amlodipino: El tiempo para alcanzar concentraciones plasmáticas máximas de amlodipino es similar en sujetos de edad avanzada y más jóvenes. La depuración de amlodipino tiende a disminuir produciéndose aumentos resultantes en ABC y vida media de eliminación en pacientes de edad avanzada. Los incrementos en ABC y vida media de eliminación en pacientes con ICC fueron según se esperaba para el grupo estudiado de pacientes de esa edad. Amlodipino empleado a dosis similares en pacientes de edad avanzada o más jóvenes es igual de bien tolerada.
En estudios con atorvastatina: Las concentraciones plasmáticas de atorvastatina son más altas (Cmáx. aproximadamente 40% y ABC 30%) en sujetos saludables de edad avanzada (≥ 65 años) que en adultos jóvenes. El estudio ACCES evaluó de manera específica 65 pacientes de edad avanzada con respecto a alcanzar sus metas de tratamiento ENCP. El estudio incluyó 1087 pacientes de menos de 65 años, 815 de más de 65 años y 185 de más de 75 años. No se observaron diferencias en seguridad, eficacia o metas de tratamientos para lípidos entre pacientes de edad avanzada y la población general.
Pediatría:
En estudios con amlodipino: En un estudio clínico de exposición crónica, se administró amlodipino a 73 pacientes pediátricos hipertensos, con edades de 12 meses hasta 17 años o menores a una dosis diaria promedio de 0.17 mg/kg. La depuración para sujetos con peso mediano de 45 kg fue 23.7 L/h y 17.6 L/h para varones y mujeres respectivamente. Esto se encuentra en un rango similar a las estimaciones publicadas de 24.8 L/h en un adulto de 70 kg. La estimación promedio para el volumen de distribución para un paciente de 45 kg fue 1130 L (25.11 L/kg). La preservación del efecto sobre la PA durante el intervalo de dosificación de 24 horas se observó con poca diferencia en efecto de variación desde el máximo hasta el mínimo. Al comparar con la farmacocinética histórica en adultos los parámetros observados en este estudio indican que la dosificación una vez al día es adecuada.
Interacciones medicamentosas:
En estudios con atorvastatina: A continuación se resume el efecto de los medicamentos administrados conjuntamente sobre la farmacocinética de la atorvastatina, así como el efecto de la atorvastatina sobre la farmacocinética de los medicamentos administrados conjuntamente (consulte la sección Precauciones generales y la sección Interacciones medicamentosas y de otro género).
Tabla 1. Efecto de los medicamentos administrados conjuntamente sobre la farmacocinética de la atorvastatina
|
Medicamento administrado conjuntamente y régimen de dosificación |
Atorvastatina |
||
|
Dosis (mg) |
Proporción en ABC& |
Proporción en Cmáx.& |
|
|
#Ciclosporina 5.2 mg/kg/día, dosis estable |
10 mg QDa durante 28 días |
8.7 |
10.7 |
|
#Tipranavir 500 mg BID/ritonavir 200 mg BIDb, 7 días |
10 mg, SDc |
9.4 |
8.6 |
|
#Glecaprevir 400 mg QDª/Pibrentasvir 120 mg QDª, 7 días |
10 mg QDa durante 7 días |
8.3 |
22.0 |
|
#Telaprevir 750 mg c 8 hf, 10 días |
20 mg, SDc |
7.9 |
10.6 |
|
#Elbasvir 50 mg QDª/grazoprevir 200 mg QDª, 13 días |
10 mg, SDc |
1.95 |
4.3 |
|
#Boceprevir 800 mg TIDd, 7 días |
40 mg, SDC |
2.3 |
2.7 |
|
#Simeprevir 150 me QDª, 10 días |
40 mg SDC |
2.12 |
1.7 |
|
#Lopinavir 400 mg BIDb/ritonavir 100 mg BIDb, 14 días |
20 mg QDa durante 4 días |
5.9 |
4.7 |
|
#‡Saquinavir 400 mg BIDb/ritonavir 400 mg BIDb, 15 días |
40 mg QDa durante 4 días |
3.9 |
4.3 |
|
#Claritromicina 500 mg BIDb, 9 días |
80 mg QDa durante 8 días |
4.5 |
5.4 |
|
#Darunavir 300 mg BIDb/ritonavir 100 mg BIDb, 9 días |
10 mg QDa durante 4 días |
3.4 |
2.2 |
|
#Itraconazol 200 mg QDª, 4 días |
40 mg SDC |
3.3 |
1.20 |
|
#Letermovir 480 mg QD, 10 díasª |
20 mg SDC |
3.29 |
2.17 |
|
#Fosamprenavir 700 mg BIDb/ritonavir 100 mg BIDb, 14 días |
10 mg QD durante 4 días |
2.5 |
2.8 |
|
#Fosamprenavir 1400 mg BIDb, 14 días |
10 mg QD durante 4 días |
2.3 |
4.0 |
|
#Nelfinavir 1250 mg BIDb, 14 días |
10 mg QDa durante 28 días |
1.74 |
2.2 |
|
#Jugo de toronja, 240 mL QDª* |
40 mg, SDC |
1.37 |
1.16 |
|
Diltiazem 240 mg QDª, 28 días |
40 mg, SDC |
1.51 |
1.00 |
|
Eritromicina 500 mg QIDe, 7 días |
10 mg, SDC |
1.33 |
1.38 |
|
Amlodipino 10 mg, dosis única |
80 mg, SDC |
1.18 |
0.91 |
|
Cimetidina 300 mg QIDe, 2 semanas |
10 mg QDa durante 2 semanas |
1.00 |
0.89 |
|
Colestipol 10 g BIDb, 24 semanas |
40 mg QDa durante 8 semanas |
No se determinó |
0.74** |
|
Maalox TC® 30 mL QIDe 17 días |
10 mg QDa durante 15 días |
0.66 |
0.67 |
|
Efavirenz 600 mg QDª, 14 días |
10 mg durante 3 días |
0.59 |
1.01 |
|
#Rifampicina 600 mg QDª, 7 días (administrada conjuntamente)t |
40 mg SDC |
1.12 |
2.9 |
|
#Rifampicina 600 mg QDª, 5 días (dosis separadas)t |
40 mg SDC |
0.20 |
0.60 |
|
#Gemfibrozilo 600 mg BIDb, 7 días |
40 mg SDC |
1.35 |
1.00 |
|
#Fenofibrato 160 mg QDª, 7 días |
40 mg, SDC |
1.03 |
1.02 |
& Representa la proporción de los tratamientos (fármaco coadministrado más atorvastatina versus atorvastatina sola).
# Puede ver secciones Precauciones generales e Interacciones medicamentosas y de otro género, para determinar su significado clínico.
* Se ha informado de mayores aumentos del ABC (proporción de hasta 2.5 veces) y/o la Cmáx. (hasta 1.71 veces) con el consumo excesivo de jugo de toronja (≥ 750 mL-1.2 L/día).
** Se tomó una muestra única 8-16 h después de la dosis.
† Debido al mecanismo de interacción dual de rifampicina, se recomienda la administración conjunta simultánea de atorvastatina con rifampicina, ya que la demora de la administración de atorvastatina después de la administración de rifampicina se ha asociado con una reducción significativa de las concentraciones plasmáticas de atorvastatina.
‡ La dosis de saquinavir/ritonavir en este estudio no es la dosis usada clínicamente. El incremento a la exposición de atorvastatina cuando se usa clínicamente es ligeramente más alto que el observado en este estudio. Por ende debe ejercer con cautela utilizando la dosis menor necesaria.
a Una vez al día.
b Dos veces al día.
c Una sola dosis.
d Tres veces al día.
e Cuatro veces al día.
f Cada 8 horas.
Tabla 2. Efecto de la atorvastatina sobre la farmacocinética de los medicamentos administrados conjuntamente
|
Atorvastatina |
Medicamento administrado conjuntamente y régimen de dosificación |
||
|
Medicamento/dosis (mg) |
Cambio en ABC& |
Cambio en Cmáx.& |
|
|
80 mg QDa durante 15 días |
Antipirina, 600 mg SDc |
1.03 |
0.89 |
|
80 mg QDa durante 10 días |
#Digoxina 0.25 mg QDa, 20 días |
1.15 |
1.20 |
|
40 mg QDa durante 22 días |
Anticonceptivo oral QDa, 2 meses - Noretindrona 1 mg - Etinilestradiol 35 μg |
1.28 1.19 |
1.23 1.30 |
|
10 mg, SDC |
Tripanavir 500 mg BIDb/ritonavir 200 mg BIDb, 7 días |
1.08 |
0.96 |
|
10 mg, QDa durante 4 días |
Fosamorenavir 1400 mg BIDb, 14 días |
0.73 |
0.82 |
|
10 mg QDa durante 4 días |
Fosamprenavir 700 mg BIDb/ritonavir 100 mg BIDb, 14 días |
0.99 |
0.94 |
& Representa la proporción de los tratamientos (fármaco coadministrado más atorvastatina versus atorvastatina sola).
# Para fines clínicos ver sección Interacciones medicamentosas y de otro género.
a Una vez al día.
b Dos veces al día.
c Una sola dosis.
Propiedades Farmacodinámicas:
Farmacodinamia de CADUET® (amlodipino/atorvastatina): El componente de besilato de amlodipino en CADUET® (amlodipino/atorvastatina) se describe químicamente como bencenosulfonato de (R.S.) 3-etil-5-metil-2-(2-aminoetoximetil)-4-(-2-clorofenil)-1,4-dihidro-6-metil-3,5-piridinodicarboxilato. Su fórmula empírica es C20H25CIN2O5• C6H6O3S. El componente de atorvastatina cálcica en CADUET® (amlodipino/atorvastatina) se describe químicamente como sal cálcica trihidratada de [R-(R*, R*)-2-(4-fluorofenil)-β,δ-dihidroxi-5-(1-metiletil)-3-fenil-4(fenilamino)carbonil)]-1H-pirrol-1-ácido heptanoico. La fórmula empírica de atorvastatina cálcica es: (C33H34 FN2O5)2 Ca• 3H2O. Las fórmulas estructurales se muestran a continuación:
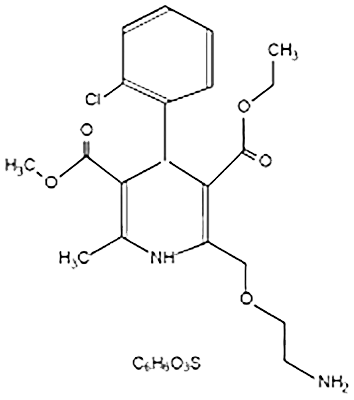
Besilato de amlodipino
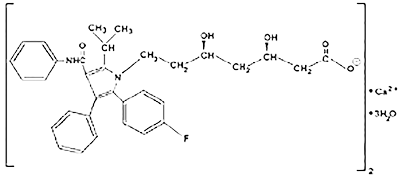
Atorvastatina
Mecanismo de acción de amlodipino/atorvastatina: CADUET® (amlodipino/atorvastatina) combina dos mecanismos de acción: La acción antagonista del calcio de dihidropiridina (antagonista del ion calcio o bloqueador de canal lento) de amlodipino y la inhibición de HMG-CoA reductasa de atorvastatina. El componente amlodipino en CADUET® (amlodipino/atorvastatina) inhibe la entrada tras membrana de iones de calcio al músculo liso vascular y el músculo cardiaco. El componente de atorvastatina en CADUET® (amlodipino/atorvastatina) es un inhibidor selectivo y competitivo de la HMG-CoA reductasa enzima limitante de la velocidad que transforma la HMG-CoA a mevalonato un precursor de esteroles incluyendo colesterol.
Estudios clínicos de la combinación de CADUET® (amlodipino/atorvastatina) en pacientes con hipertensión y dislipidemia:
En un estudio doble ciego controlado por placebo en 1660 pacientes con hipertensión comórbida y dislipidemia el tratamiento una vez al día con ocho combinaciones de dosificación de CADUET® (amlodipino/atorvastatina) (5/10 mg, 10/10 mg, 5/20 mg, 10/20 mg, 5/40 mg, 10/40 mg, 5/80 mg, o 10/80 mg) se comparó contra amlodipino solo (5 mg o 10 mg), atorvastatina sola (10 mg, 20 mg, 40 mg u 80 mg) y placebo. Además de hipertensión concomitante y dislipidemia, 15% de los pacientes tenía diabetes mellitus 22% eran fumadores y 14% tenían historia familiar de ECV. A las 8 semanas los ocho grupos de tratamiento combinado demostraron reducción estadísticamente significativa relacionada con la dosis en presión arterial sistólica (PAS), presión arterial diastólica (PAD) y C-LDL en comparación con placebo sin modificación general de efecto de cualquiera de los componentes sobre PAS, PAD y C-LDL (ver la siguiente tabla).
Eficacia en términos de reducción de presión arterial y C-LDL:
Tabla 3. Eficacia de los tratamientos combinados para reducción de la presión arterial sistólica BPa
|
Parámetro/análisis |
ATOb 0 mg |
ATO 10 mg |
ATO 20 mg |
ATO 40 mg |
ATO 80 mg |
|
|
AMLc 0 mg |
Cambio promedio (mm Hg) |
-3.0 |
-4.5 |
-6.2 |
-6.2 |
-6.4 |
|
Diferencia vs. placebo (mm Hg) |
- |
-1.5 |
-3.2 |
-3.2 |
-3.4 |
|
|
AML 5 mg |
Cambio promedio (mm Hg) |
-12.8 |
-13.7 |
-15.3 |
-12.7 |
-12.2 |
|
Diferencia vs. placebo (mm Hg) |
-9.8 |
-10.7 |
-12.3 |
-9.7 |
-9.2 |
|
|
AML 10 mg |
Cambio promedio (mm Hg) |
-16.2 |
-15.9 |
-16.1 |
-16.3 |
-17.6 |
|
Diferencia vs. placebo (mm Hg) |
-13.2 |
-12.9 |
-13.1 |
-13.3 |
-14.6 |
|
Tabla 4. Eficacia de los tratamientos combinados para reducir la presión arterial diastólicaa
|
Parámetro/análisis |
ATOb 0 mg |
ATO 10 mg |
ATO 20 mg |
ATO 40 mg |
ATO 80 mg |
|
|
AMLc 0 mg |
Cambio promedio (mm Hg) |
-3.3 |
-4.1 |
-3.9 |
-5.1 |
-4.1 |
|
Diferencia vs. placebo (mm Hg) |
- |
-0.8 |
-0.6 |
-1.8 |
-0.8 |
|
|
AML 5 mg |
Cambio promedio (mm Hg) |
-7.6 |
-8.2 |
-9.4 |
-7.3 |
-8.4 |
|
Diferencia vs. placebo (mm Hg) |
-4.3 |
-4.9 |
-6.1 |
-4.0 |
-5.1 |
|
|
AML 10 mg |
Cambio promedio (mm Hg) |
-10.4 |
-9.1 |
-10.6 |
-9.8 |
-11.1 |
|
Diferencia vs. placebo (mm Hg) |
-7.1 |
-5.8 |
-7.3 |
-6.5 |
-7.8 |
|
Tabla 5. Eficacia de los tratamientos combinados para reducir C-LDLa (cambio porcentual)
|
Parámetro/análisis |
ATOb 0 mg |
ATO 10 mg |
ATO 20 mg |
ATO 40 mg |
ATO 80 mg |
|
|
AMLc 0 mg |
% de cambio promedio |
-1.1 |
-33.4 |
-39.5 |
-43.1 |
-47.2 |
|
AML 5 mg |
% de cambio promedio |
-0.1 |
-38.7 |
-42.3 |
-44.9 |
-48.4 |
|
AML 10 mg |
% de cambio promedio |
-2.5 |
-36.6 |
-38.6 |
-43.2 |
-49.1 |
a Colesterol de lipoproteínas de baja densidad.
b Atorvastatina.
c Amlodipino.
En un estudio abierto 1220 pacientes con hipertensión comórbida y dislipidemia recibieron titulación de dosis selectiva con amlodipino/atorvastatina en un periodo de 14 semanas. Se requirió que los pacientes tuvieran PA descontrolada al entrar el estudio (estuvieran o no usando antihipertensivos en la inscripción; se permitió que los pacientes continuaran con los antihipertensivos previos distintos de bloqueadores de canal del calcio durante el periodo de titulación de dosis de 14 semanas), pero pudieron ser inscritos con C-LDL controlado o sin control. Como resultado ningún paciente entró al estudio teniendo controlados tanto la PA como C-LDL y ninguno de éstos estuvo bajo control en 62% de los pacientes. El tratamiento con amlodipino/atorvastatina redujo la PA media menos 17.1 mm Hg sistólica y menos 9.6 mm Hg diastólica y redujo C-LDL medio en -32.7% produciendo control tanto de PA como C-LDL para 58% de estos pacientes (presión arterial y C-LDL controlados se definieron respectivamente como < 140/90 mm Hg y < 160 mg/dL para pacientes con hipertensión comórbida y dislipidemia únicamente 140/90 mm Hg y < 130 mg/dL para pacientes con hipertensión comórbida y dislipidemia más 1 factor de riesgo cardiovascular adicional excluyendo ECC conocida o diabetes mellitus; y 130/85 mm Hg y < 100 mg/dL en pacientes con hipertensión comórbida y dislipidemia más ECC conocida diabetes mellitus u otra enfermedad ateroesclerócica). Sólo el 13% de los pacientes del estudio usaron amlodipino/atorvastatina como terapia inicial para hipertensión comórbida y dislipidemia mientras que el componente de amlodipino del compuesto amlodipino/atorvastatina constituyó terapia adicional para hipertensión de 56% de los pacientes, incluyendo aquellos en quienes el componente de atorvastatina del compuesto amlodipino/atorvastatina constituyó terapia inicial para dislipidemia (20%), una sustitución para atorvastatina tomada previamente (18%) o un cambio de otra estatina (18%). Al ser evaluados de acuerdo con el uso de medicamentos antihipertensivos y para reducción de lípidos en la inscripción los resultados demostraron que tanto la PA como el C-LDL quedaron bajo control para 65% de los pacientes que usaron como terapia inicial amlodipino/atorvastatina para hipertensión comórbida y dislipidemia, y para 55% a 64% de los pacientes en quienes el componente de amlodipino el compuesto amlodipino/atorvastatina constituyó terapia adicional para hipertensión (55% para aquellos pacientes que previamente usaban medicamentos para reducción de lípidos distintos de atorvastatina, 58% para aquellos pacientes que previamente usaban atorvastatina y 64% para aquellos pacientes que no habían usado previamente medicamentos para reducción de lípidos).
Estudio anglo-escandinavo sobre resultados cardiacos: El estudio Anglo-Escandinavo sobre Resultados Cardiacos (ASCOT) es un estudio de diseño factorial aleatorizado 2x2 que comparó dos regímenes anti-hipertensivos en un total de 19,342 pacientes (Grupo de Disminución de Presión Sanguínea – ASCOT-BPLA), así como el efecto de la adición de 10 mg de atorvastatina contra placebo en 10,305 pacientes (grupo de Disminución de Lípidos – ASCOT-LLA) en eventos coronarios fatales y no fatales. Existen 19,257 y 10,240 pacientes para evaluación de eficacia en ASCOT-BPLA y ASCOT-LLA, respectivamente.
En el Grupo de disminución de la presión arterial del estudio Anglo-escandinavo de resultados cardiacos: El efecto de los regímenes de tratamiento basado en amlodipino (5-10 mg) (n = 9,681) o atenolol (50-100 mg) (n = 9,661) fue comparado en un estudio prospectivo, aleatorizado, abierto y con evaluación ciega de resultados finales, (PROBE) en 19,342 pacientes hipertensos, ≥40 a < 80 años de edad sin IM previo ni tratamiento de angina, al menos tres de los siguientes factores de riesgo cardiovascular predefinidos: Hombres, edad ≥ 55 años), fumadores, diabetes tipo 2, historia de evento de EAC (enfermedad coronaria, CAD) en un familiar en primer grado antes de la edad de 55 (hombres) o 60 años (mujeres), total-C: HDL ≥ 6, enfermedad vascular periférica, hipertrofia ventricular izquierda, evento cerebro vascular previo, anormalidades de electrocardiogramas (ECG) específicas, proteinuria/albuminuria.
Para alcanzar los objetivos de presión sanguínea (PA) (< 140/90 mm Hg para pacientes no-diabéticos, < 130/80 mm Hg para pacientes diabéticos), perindopril (4-8 mg) podía ser agregado al grupo de amlodipino y bendroflumetiazida potasio (1.25-2.5 mg) al grupo con atenolol. El tratamiento de tercera línea fue doxazosina sistema clínico gastrointestinal (GITS) (4-8 mg) en ambos grupos.
El estudio ASCOT-BPLA fue suspendido prematuramente después de 903 eventos primarios (IM no fatal y ECC fatal) con 5.5 años promedio de seguimiento debido al beneficio significativo del régimen basado en amlodipino en los siguientes resultados finales secundarios: Mortalidad por cualquier causa, mortalidad cardiovascular (CV) y accidente cerebrovascular. Se había planeado requerir al menos 1150 resultados finales primarios en el estudio.
El resultado final primario de IM no fatal + fatal ECC no alcanzó significancia estadística al comparar el grupo con amlodipino vs. el grupo con atenolol. Los resultados finales secundarios de todos los eventos coronarios, la mortalidad por todas las causas, los accidentes cerebro vasculares fatales y no fatales fueron reducidos de manera estadísticamente significativa al comparar el grupo con amlodipino con el grupo de atenolol.
Incidencia de los resultados finales primario y secundario en los 19,257 pacientes evaluables en términos de eficacia:
|
Evento |
Tratamiento basado en |
Disminución de riesgo (%) |
Valor-p rango logarítmico |
|
|
Amlodipino N = 9,639 n (%) |
Atenolol N = 9,618 n (%) |
|||
|
IMa no fatal + ECC fatal (resultado final primario) |
429 (4.5) |
474 (4.9) |
10 |
0.105 |
|
Total Eventos y Procedimientos CVb |
1362 (14.1) |
1602 (16.7) |
16 |
< 0.001 |
|
Total Eventos Coronariosc |
753 (7.8) |
852 (8.9) |
13 |
0.007 |
|
IM no fatal (excluyendo IM silencioso) + ECC fatal |
390 (4.0) |
444 (4.6) |
13 |
0.046 |
|
Todas las causas de mortalidad |
738 (7.7) |
820 (8.5) |
11 |
0.025 |
|
Mortalidad cardiovasculard |
263 (2.7) |
342 (3.6) |
24 |
< 0.001 |
|
ACV (accidente cerebrovascular) fatal y no fatal |
327 (3.4) |
422 (4.4) |
23 |
< 0.001 |
|
Insuficiencia cardiaca fatal y no fatal |
134 (1.4) |
159 (1.7) |
16 |
0.126 |
a Infarto del miocardio.
b Mortalidad cardiovascular, IM no-fatal (sintomático y silencioso), angina inestable, Angina crónica estable, Arritmia letal, Insuficiencia cardiaca no fatal, Accidente cerebrovascular no fatal, ataque isquémico transitorio (AIT), déficit neurológico isquémico reversible (RIND, por sus siglas en inglés: reversible ischemic neurologic deficit), trombosis vascular de la retina, enfermedad arterial periférica y procedimientos de revascularización.
c ECC fatal, IM no-fatal (sintomático y silencioso). Angina Crónica Estable, angina inestable, Insuficiencia Cardiaca fatal y no fatal.
d Incluye RIND.
La presión sanguínea (PAS/PAD) disminuyó significativamente en ambos regímenes de tratamiento al compararse con el inicio (valores-p < 0.001).
Las disminuciones de PAS/PAD desde el inicio fueron significativamente más numerosas con el régimen basado en amlodipino que con el régimen basado en atenolol (-27.5/-17.7 mm Hg vs. -25.7/-15.6 mm Hg, respectivamente) y los valores-p en cuanto a diferencias entre los dos grupos fueron en ambos < 0.001 para PAS y PAD.
En el Grupo de disminución de lípidos del estudio Anglo-escandinavo de resultados cardiacos:
En el Grupo de disminución de lípidos del estudio Anglo-escandinavo de resultados cardiacos (ASCOT-LLA), el efecto de la atorvastatina en la ECC fatal y no fatal fue evaluado en 10,305 pacientes hipertensos de 40 a 80 años de edad (promedio de 63 años), sin IM previo y con niveles de colesterol total CT < 6.5 mmol/L (251 mg/dL). Adicionalmente, todos los pacientes presentaban al menos tres de los siguientes factores de riesgo cardiovascular: Hombres, edad > 55 años, fumadores, diabetes, historia de ECC en un pariente en primer grado, CT: HDL >6, enfermedad vascular periférica, hipertrofia ventricular izquierda evento cerebrovascular previo, anormalidad ECG específica, proteinuria/albuminuria. En este estudio doble-ciego controlado con placebo los pacientes fueron tratados con terapia anti-hipertensiva (PA Objetivo < 140/90 mm Hg para pacientes no diabéticos, < 130/80 mm Hg para pacientes diabéticos) y asignados a 10 mg al día de atorvastatina (n = 5168) o a un placebo (n = 5137). Debido a que el efecto del tratamiento con atorvastatina en comparación con el placebo excedió el umbral de significancia durante un análisis temporal, ASCOT-LLA fue terminado anticipadamente a los 3.3 años en lugar de a los 5 años. Adicionalmente, la PA fue bien controlada y similar en los pacientes asignados a atorvastatina y al placebo. Estos cambios continuaron durante todo el periodo de tratamiento.
Atorvastatina redujo la tasa de los siguientes eventos:
|
Evento |
Disminución de riesgo (%) |
No. de eventos (atorvastatina vs. placebo) |
Valor-p |
|
Eventos coronarios (ECCa fatal más IMb no fatal) |
36% |
100 vs. 154 |
0.0005 |
|
Eventos cardiovasculares totales y procedimientos de revascularización |
20% |
389 vs. 483 |
0.0008 |
|
Eventos coronarios totales |
29% |
178 vs. 247 |
0.0006 |
|
Accidente cerebrovascular fatal y no-fatal* |
26% |
89 vs. 119 |
0.0332 |
a Enfermedad coronaria.
b Infarto del miocardio.
* Aunque la disminución de accidentes cerebrovasculares fatales y no fatales no alcanzó el nivel de significancia predefinido (p = 0.01), se observó una tendencia favorable con una disminución del 26% del riesgo relativo.
La mortalidad total y mortalidad cardiovascular no han sido disminuidas significativamente aunque se observó una tendencia favorable.
En el estudio Anglo-escandinavo de resultados cardiacos 2×2: El análisis factorial pre-especificado ASCOT 2x2 investigó el efecto potencial diferencial (interacción) de agregar atorvastatina al amlodipino vs. el grupo con atenolol en ASCOT-LLA.
En los 10,305 pacientes reclutados en ASCOT-LLA, hubo 5,168 pacientes en el grupo con atorvastatina (2,584 pacientes recibieron amlodipino y 2,584 pacientes recibieron atenolol) y 5,137 en el grupo con placebo (2,554 pacientes recibieron amlodipino y 2,583 pacientes recibieron atenolol).
Las disminuciones de riesgo en el resultado final compuesto de IM no-fatal y ECC fatal se basaron en 10,240 pacientes evaluables en términos de eficacia.
La combinación de amlodipino con atorvastatina produjo una disminución de riesgo significativa en el resultado compuesto primario de ECC fatal e IM no fatal por:
• 53% (95% IC 31% a 68%, p < 0.0001) comparado con amlodipino + placebo,
• 39% (95% IC 8% a 59%, p < 0.016) comparado con atenolol + atorvastatina.
El valor-p de la interacción fue 0.027 el cual no fue estadísticamente significativo en el nivel 0.01 preespecificado.
La presión sanguínea (PAS/PAD) disminuyó significativamente en los cuatro regímenes de tratamiento en comparación con el inicio (valores-p < 0.001). Las disminuciones de PAS/PAD desde el inicio fueron significativamente más numerosas con el régimen basado en amlodipino que con los regímenes basados en atenolol (-26.5/-15.6 mm Hg versus -24.7/-13.6 mm Hg para amlodipino/atorvastatina vs. atenolol/atorvastatina, y -27.1/-15.8 mm Hg vs. -24.1/-13.6 mm Hg para amlodipino/placebo vs. atenolol/placebo, respectivamente). Los valores-p en las diferencias entre los dos grupos fueron todos < 0.01 para PAS y PAD.
Farmacodinamia de amlodipino: Amlodipino es un inhibidor de la afluencia ion calcio (bloqueador del canal lento o antagonista del ion calcio) e inhibe el influjo de los iones calcio a músculo liso cardiaco y vascular.
El mecanismo de acción antihipertensiva de amlodipino se debe al efecto relajante directo sobre músculo liso vascular. El mecanismo preciso por el cual amlodipino alivia la angina no ha sido determinado en su totalidad, pero amlodipino reduce la carga isquémica total por las dos acciones siguientes:
1) Amlodipino dilata las arteriolas periféricas y por lo tanto reduce la resistencia periférica total (carga posterior) contra la cual trabaja el corazón. Como la frecuencia cardiaca permanece estable este alivio de carga del corazón reduce el consumo energético miocardial y los requisitos de oxígeno.
2) El mecanismo de acción de amlodipino también incluye probablemente dilatación de las arterias coronarias principales y arteriolas coronarias tanto en regiones normales como isquémicas. Esta dilatación aumenta el suministro de oxígeno al miocardio en pacientes con espasmo de la arteria coronaria (angina de prinzmetal o variantes) y amortigua la vasoconstricción coronaria inducida por tabaquismo.
En pacientes con hipertensión la dosificación una vez al día suministra reducción clínicamente significativa de PA en posición supina y de pie durante el intervalo de 24 horas. Debido al inicio de acción lento la hipotensión aguda no es una característica para administración de amlodipino.
En pacientes con angina la administración una vez al día de amlodipino aumenta el tiempo total de ejercicio, el tiempo hasta el inicio de angina y el tiempo hasta depresión de 1 mm en el segmento ST y reduce tanto la frecuencia de ataques de angina como el consumo de tabletas de nitroglicerina.
Amlodipino no se asocia con ningún efecto metabólico adverso ni cambio en lípidos plasmáticos si es adecuada para uso en pacientes con asma, diabetes y gota.
Uso en pacientes con EAC: Los efectos de amlodipino sobre morbilidad y mortalidad cardiovascular, la progresión de arteroesclerosis coronaria y la arteroesclerosis de la carótida fueron estudiados en El Estudio Evaluación Prospectiva Aleatoria de Efectos Vasculares de NORVASC (PREVENT). Este estudio multicéntrico aleatorizado doble ciego controlado con placebo dio seguimiento a 825 pacientes con EAC angiográficamente definida durante tres años. La población incluyó pacientes con IM previo (45%), angioplastia coronaria trasluminal percutánea (ACTP) en la visita basal (42%) o historia de angina (69%). La severidad de EAC fue desde enfermedad en un vaso (45%) hasta enfermedad de más de 3 vasos (21% de los pacientes). Los pacientes con hipertensión no controlada (PAD > 95 mm Hg) fueron excluidos del estudio. Los eventos cardiovasculares mayores (MCVE, por sus siglas en inglés Major Cardio Vascular Events) fueron adjudicados por un comité ciego al criterio de valoración. Aunque no hubo efectos demostrables sobre la tasa de progresión de lesiones coronarias, amlodipino detuvo la progresión de engrosamiento de la media íntima de la carótida. Se observó una reducción significativa (-31%) en pacientes tratados con amlodipino respecto al criterio de valoración combinado de muerte cardiovascular, IM, accidente cerebrovascular, ACTP, injerto de derivación coronaria (CABG, por sus siglas en inglés Coronary Arlery Bypass Graff), hospitalizaciones por angina inestable y empeoramiento de ICC. También se observó una reducción significativa (-42%) en procedimientos de revascularización (ACTP y CABG) en pacientes tratados con amlodipino. Se observaron menos hospitalizaciones (-33%) por angina inestable en pacientes con amlodipino que en el grupo con placebo.
La eficacia con amlodipino para prevenir eventos clínicos en pacientes con EAC ha sido evaluada en un estudio independiente multicéntrico aleatorio doble ciego controlado con placebo en 1997 pacientes: La Comparación de amlodipino contra Enalapril para Limitar Ocurrencia de Trombosis (CAMELOT). De ellos, 663 fueron tratados con 5 mg-10 mg de amlodipino y 655 fueron tratados con placebo además de cuidado estándar con estatinas, bloqueadores beta, diuréticos y aspirina por dos años. Los resultados clave de eficacia se presentan en la Tabla 6. Los resultados indican que el tratamiento con amlodipino se asoció con menos hospitalizaciones por angina y procedimientos de revascularización en pacientes con EAC.
Tabla 6. Incidencia de resultados clínicamente significativos en el estudio CAMELOT
|
Estudio CAMELOTa |
|||
|
Resultados clínicos N (%) |
Amlodipino (n = 663) |
Placebo (n = 655) |
Reducción de riesgo (valor p) |
|
Criterio de valoración* CVb Compuesto |
110 |
151 (23.1) |
31% (0.003) |
|
Hospitalización por angina |
51 (7.7) |
84 (12.8) |
42% (0.002) |
|
Revascularización coronaria |
78 (11.8) |
103 (15.7) |
27% (0.033) |
* 1) Definido en el Estudio CAMELOT como muerte cardiovascular, IM no mortal, paro cardiaco con reanimación, revascularización coronaria, hospitalización por angina de pecho, hospitalización por ICC, accidente cerebro vascular o AIT mortal o no mortal, cualquier diagnóstico de enfermedad arterial periférica (EAP) en un sujeto a quien no se hubiera diagnosticado previamente EAP o cualquier ingreso para procedimiento para tratamiento de EAP.
2) El criterio de valoración (CV) compuesto por el Criterio de valoración de eficacia primaria del Estudio CAMELOT.
a Comparación de amlodipino versus enalapril para limitar las ocurrencias de trombosis.
b Cardiovascular.
Estudio de Tratamiento para Prevenir Ataque Cardiaco: Se realizó un estudio aleatorio doble ciego de morbilidad-mortalidad llamado Tratamiento con Antihipertensivo y Reductor de Lípido para Prevenir Ataque Cardiaco (ALLHAT) con el fin de comparar terapias farmacológicas novedosas: amlodipino 2.5-10 mg/día (bloqueador del canal del calcio) o lisinopril 10-40 mg/día (inhibidor ECA, enzima convertidora de angiotensina) como terapia de primera línea contra el diurético tiazídico, clortalidona 12.5-25 mg/día en hipertensión de ligera a moderada. Un total de 33,357 pacientes hipertensos de 55 años o más fueron asignados aleatoriamente y se les dio seguimiento una media de 4.9 años. Los pacientes tenían por lo menos un factor de riesgo adicional de ECC incluyendo IM o accidente cerebro vascular > 6 meses o documentación de otros ECV (eventos cardiovasculares) ateroescleróticos (en total 51.5%), diabetes tipo 2, (36.1), C-HDL < 35 mg/dL (11.6%), hipertrofia del ventrículo izquierdo diagnosticado por electrocardiograma o ecocardiograma (20.9%) tabaquismo actual (21.9%).
El Criterio de valoración primario fue una combinación de Enfermedad Cardiaca Coronaria fatal o IM no fatal. No hubo diferencia significativa en el criterio de valoración primario entre terapia a base de amlodipino y terapia a base de clortalidona: RR 0.98, 95% IC 0.90-1.07; p = 0.65. Además, no hubo diferencia significativa en mortalidad por cualquier causa entre el tratamiento basado en amlodipino y el tratamiento basado en clortalidona: RR 0.96; 95% IC 0.89-1.02 p = 0.20.
Uso en pacientes con insuficiencia cardiaca: Estudios hemodinámicos y estudios clínicos controlados basados en ejercicio en pacientes con insuficiencia cardiaca Clase II-IV de NYHA (New York Heart Association) han demostrado que el amlodipino no condujo a deterioro clínico medido por tolerancia al ejercicio, fracción de expulsión de ventrículo izquierdo y sintomatología clínica.
Un estudio controlado con placebo (PRAISE) diseñado para valorar pacientes con insuficiencia cardiaca clase III-IV de NYHA que reciben digoxina diuréticos de inhibidores de la ECA ha demostrado que amlodipino no condujo a un aumento en el riesgo de mortalidad o mortalidad combinada y morbilidad en pacientes con insuficiencia cardiaca.
Un estudio de seguimiento a largo plazo controlado con placebo (PRAISE-2) de amlodipino en pacientes con insuficiencia cardiaca Clase III y IV de NYHA sin síntomas clínicos u observaciones objetivas que sugirieran enfermedad isquémica subyacente, tomando dosis estables de inhibidores ECA, digital y diuréticos, indicó que amlodipino no producía efectos sobre la mortalidad total o cardiovascular. En esta misma población amlodipino se asoció con aumento de reportes de edema pulmonar a pesar de que no hubo diferencia significativa en la incidencia de empeoramiento de insuficiencia cardiaca en comparación con placebo.
Uso en pacientes pediátricos (de 6 a 17 años de edad): La eficacia de amlodipino en pacientes pediátricos hipertensos de 6 a 17 años de edad fue demostrada en un estudio doble ciego aleatorio controlado con placebo de supresión con duración de 8 semanas en 268 pacientes con hipertensión. Todos los pacientes fueron asignados aleatoriamente a los grupos de tratamiento de 2.5 mg o 5 mg y se les dio seguimiento 4 semanas después de lo cual fueron asignados aleatoriamente a continuar con 2.5 mg o 5 mg de amlodipino o placebo por 4 semanas adicionales. En comparación con la línea basal al tratamiento una vez al día con amlodipino 5 mg produjo reducción estadísticamente significativa en presión arterial sistólica y PAD. La reducción media ajustada por placebo en PAS en posición sentada se estimó como 5.0 mm Hg para la dosis de 5 mg de amlodipino y 3.3 mm Hg para la dosis de 2.5 mg de amlodipino. Los análisis subgrupales indicaron que los pacientes pediátricos más jóvenes de 6 a 13 años de edad tuvieron resultados de eficacia comparables con los de los pacientes pediátricos mayores de 14 a 17 años.
Farmacodinamia de atorvastatina: Atorvastatina es un inhibidor selectivo y competitivo de HMG-CoA reductasa, la enzima limitante de velocidad que convierte la HMG-CoA a mevalonato, un precursor de los esteroles, incluyendo el colesterol. En pacientes con HF homocigota y heterocigota, formas no familiares de hipercolesterolemia y dislipidemia mixta atorvastatina reduce el C-total, el C-LDL y la apo B. Atorvastatina también reduce el colesterol de lipoproteínas de muy baja densidad (C-VLDL) y TG y produce incrementos variables en C-HDL.
Atorvastatina reduce el colesterol plasmático y los niveles de lipoproteína inhibiendo las HMG-CoA reductasa y la síntesis de colesterol en hígado y aumentando el número de receptores LDL hepáticos en la superficie celular para aumento de la captación y catabolismo de LDL.
Atorvastatina reduce la producción de LDL y el número de partículas LDL. Atorvastatina produce un incremento profundo y sostenido en actividad de receptor LDL acoplada con cambio benéfico en la calidad de partículas LDL en circulación. Atorvastatina es eficaz para reducir LDL en pacientes con HF homocigoto una población que normalmente no responde a medicamentos para reducción de lípidos.
Atorvastatina y algunos de sus metabolitos son farmacológicamente activos en humanos. El sitio primario de acción de atorvastatina es el hígado que es el principal sitio de síntesis de colesterol y depuración de LDL. La reducción de C-LDL se correlaciona mejor con la dosis de fármaco que con la concentración sistémica de fármaco. Debe usarse individualización de dosificación de fármaco basándose en respuesta terapéutica (ver sección Dosis y vía de administración).
En un estudio de dosis respuesta, atorvastatina (10 mg a a 80 mg) redujo el colesterol total (30% a 46%), el C-LDL (41%-61%), apo B (34% a 50%) y TG (14%-33%). Estos resultados son congruentes en pacientes con HF heterocigoto, formas no familiares de hipercolesterolemia e hiperlipidemia mixta incluyendo pacientes con diabetes mellitus no dependiente de insulina.
En pacientes con hipertrigliceridemia aislada atorvastatina reduce el colesterol total, C-LDL, C-VLDL apo B, TG, y no C-HDL, e incrementa C-HDL en pacientes con disbetalipoproteinemia, atorvastatina reduce el colesterol de lipoproteínas de densidad intermedia (C-IDL).
En pacientes con hiperlipoproteinemia Fredrickson Tipo IIa y IIb acumulados de 24 estudios controlados los incrementos porcentuales medianos desde la línea basal en C-HDL para atorvastatina (10-80 mg) fueron 5.1-8.7% de manera no relacionada con la dosis. Además, el análisis de estos datos acumulados demostró reducciones significativas relacionadas con la dosis en proporciones de colesterol total C/C-HDL y LDL-C/HDL-C, que va de -29% a -44% y -37% a -55%, respectivamente.
Los efectos de atorvastatina sobre eventos isquémicos y mortalidad total fueron estudiados en el Estudio de Reducción de Isquemia del Miocardio con Reducción Agresiva del Colesterol (MIRACL). Este estudio multicéntrico aleatorio doble ciego controlado con placebo dio seguimiento a 3086 pacientes con síndromes coronarios agudos, angina inestable o IM de ondas no Q. Los pacientes fueron tratados con cuidados estándar incluyendo dieta y 80 mg al día de atorvastatina o placebo con una duración mediana de 16 semanas. Los niveles finales de LDL, colesterol total, C-HDL y TG fueron 72 mg/dL, 147 mg/dL, 48 mg/dL, 139 mg/dL en el grupo con atorvastatina, respectivamente y 135 mg/dL, 217 mg/dL, 46 mg/dL y 187 mg/dL, en el grupo con placebo, respectivamente. Atorvastatina redujo significativamente el riesgo de eventos isquémicos y muerte en 16%. El riesgo de experimentar rehospitalización por angina de pecho con evidencia documentada de isquemia del miocardio se redujo significativamente en 26%. Atorvastatina redujo el riesgo de eventos isquémicos y muerte en un grado similar a través del rango de C-LDL basal.
Además la atorvastatina redujo el riesgo de eventos isquémicos y muerte en grado similar en pacientes con infarto del miocardio de ondas no Q y angina inestable y también en varones y mujeres en pacientes ≤ 65 años y > 65 años de edad.
Prevención de complicaciones cardiovasculares: El efecto de la atorvastatina en ECC fatal y no fatal se discute en esta sección bajo el título Estudios Clínicos de amlodipino y atorvastatina Combinadas en pacientes con Hipertensión y Dislipidemia, Estudio Anglo-Escandinavo de Resultados Cardiacos.
En el Estudio Colaborativo de Atorvastatina y Diabetes (CARDS), el efecto de la atorvastatina en la ECV (enfermedad cardiovascular, ECV) fatal y no fatal fue evaluado en 2,838 pacientes con diabetes tipo 2 de 40-75 años de edad, sin historia previa de ECV y con LDL ≤ 4.14 mmol/L (160 mg/dL) y TG ≤ 6.78 mmol/L (600 mg/dL). Adicionalmente, todos los pacientes reportaron al menos uno de los siguientes factores de riesgo: Hipertensión, fumadores actuales, retinopatía, microalbuminuria o macroalbuminuria.
En este estudio aleatorizado, doble-ciego, multicéntrico, controlado con placebo, los pacientes fueron tratados con 10 mg al día de atorvastatina (n = 1428) o con un placebo (n = 1410) en una mediana de seguimiento de 3.9 años. Debido a que el efecto del tratamiento con atorvastatina en el resultado final primario alcanzó las reglas de suspensión predefinidas de eficacia, CARDS fue terminado 2 años antes de lo programado.
El efecto de reducción de riesgo absoluto y relativo de atorvastatina es el siguiente:
|
Evento |
Disminución de riesgo relativo (%) |
Núm. de eventos (atorvastatina vs. placebo) |
Valor-p |
|
Eventos cardiovasculares mayores (IAM fatal y no fatal, IM silencioso, muerte por ECC angina inestable, CABG, ACTP, revascularización, evento vascular cerebral) |
37% |
83 vs. 127 |
0.0010 |
|
IM (IAM fatal y no-fatal, IM silencioso) |
42% |
38 vs. 64 |
0.0070 |
|
Evento vascular cerebral (fatal y no fatal) |
48% |
21 vs. 39 |
0.0163 |
IAM = infarto agudo del miocardio; CABG = injerto de derivación de arteria coronaria; ECC = enfermedad coronaria; IM = infarto del miocardio; ACTP = angioplastia coronaria transluminal percutánea.
No hubo evidencia de diferencia en el efecto del tratamiento por género, edad o nivel C-LDL basal del paciente.
Se observó una reducción de riesgo relativo de muerte del 27% (82 muertes en el grupo de placebo en comparación con 61 muertes en el grupo de tratamiento) con significancia estadística limitante (p = 0.0592).
La incidencia total de eventos adversos o eventos adversos graves fue similar entre los grupos de tratamiento.
Aterosclerosis: El Estudio para Inversión de Aterosclerosis con Reducción Agresiva de Lípidos (REVERSAL), se evalúa el efecto de 80 mg de atorvastatina y de 40 mg de pravastatina sobre aterosclerosis coronaria por ultrasonido intra-vascular (IVUS, por sus siglas en inglés, IntraVascular UltraSound), durante angiografía en pacientes con ECC. En este estudio aleatorio doble ciego multicéntrico clínico controlado, el IVUS fue realizado en la visita basal y a los 18 meses en 502 pacientes. En grupo con atorvastatina (n = 253), el cambio mediano porcentual respecto a la línea basal en volumen total de ateroma (criterio primario del estudio) fue -0.4% (p = 0.98) en el grupo de atorvastatina y + 2.7% (p = 0.001) en grupo de pravastatina (n = 249). En comparación con pravastatina los efectos de atorvastatina fueron estadísticamente significativos (p = 0.02).
En el grupo con atorvastatina C-LDL se redujo a una media de 2.04 mmol/L ± 0.8 (78.9 mg/dL ± 30) respecto a la línea basal de 3.89 mmol/L ± 0.7 (150 mg/dL ± 28) y en el grupo de pravastatina C-LDL se redujo a una media de 2.85 mmol/L ± 0.7 (110 mg/dL ± 26) respecto a la línea basal 3.89 mmol/L ± 0.7 (150 mg/dL ± 26) (p < 0.0001). Atorvastatina también redujo significativamente el CT medio en 34.1% (pravastatina -18.4%, p < 0.0001), niveles medios de TG en 20% (-6.8%, p = < 0.0009), y media de apo B en 39.1% (pravastatina: -22.0%, p < 0.0001). Atorvastatina aumentó C-HDL medio en 2.9% (pravastatina: + 5.6%, p = NS). Hubo una reducción media de 36.4% en PCR (Proteína C Reactiva) en el grupo de atorvastatina en comparación con una reducción de 5.2% en el grupo de pravastatina (p < 0.0001).
Los perfiles de seguridad y tolerabilidad de los dos grupos del tratamiento fueron comparables.
Evento vascular cerebral recurrente: En el estudio prevención de accidentes cerebro vascular por reducción agresiva de niveles de colesterol (SPARCL) se evalúa el efecto de atorvastatina, 80 mg al día o placebo sobre accidentes cerebrovascular en 4,731 pacientes que padecieron accidentes cerebro vascular o AIT en los 6 meses anteriores y no tenían historia de ECC. Los pacientes fueron un 60% varones de 21 a 92 años de edad (la edad promedio fue 63 años) y tenían LDL basal promedio de 133 mg/dL (3.4 mmol/L). El C-LDL medio fue 73 mg/dL (1.9 mmol/L) durante el tratamiento con atorvastatina y 129 mg/dL (3.3 mmol/L) durante el tratamiento con placebo. La mediana de seguimiento fue 4.9 años.
80 mg de atorvastatina redujeron el riesgo del criterio de valoración primario de accidente cerebro vascular mortal o no mortal en 15% de tasa de riesgo [HR] 0.85; 95% IC, 0.72-1.00; p = 0.05 o HR 0.84; 95% IC, 0.71-0.99; p = 0.03 tras ajuste tomando en cuenta factores basales) en comparación con placebo. 80 mg de atorvastatina redujeron significativamente el riesgo de eventos coronarios mayores (HR 0.67; 95% IC, 0.51-0.89; p = 0.006) cualquier evento de ECC (HR 0.60; 95% IC, 0.48-0.74; p < 0.001), y procedimientos de revascularización (HR 0.57; 95% IC, 0.44; p < 0.001).
En un análisis post hoc, 80 mg de atorvastatina redujeron la incidencia de accidente cerebrovascular isquémico (218/2365, 9.2% vs. 274/2366, 11.6%, p = 0.01) y aumentaron la incidencia de accidente cerebrovascular hemorrágico (55/2365, 2.3% vs. 33/2366, 1.4%, p = 0.02) en comparación con placebo. La incidencia de accidente cerebrovascular hemorrágico mortal fue similar entre grupos (17 con atorvastatina vs. 18 con placebo). La reducción en el riesgo de eventos cardiovasculares con 80 mg de atorvastatina se demostró en todos los grupos de pacientes excepto en aquellos que entraron al estudio con accidente cerebro vascular hemorrágico y tuvieron recaída del mismo (7 con atorvastatina vs. 2 con placebo), en cuyo caso el número de eventos fue demasiado pequeño para discernir el riesgo o beneficio.
En pacientes tratados con 80 mg de atorvastatina hubo menos accidentes cerebrovasculares de cualquier tipo (265 con atorvastatina vs. 311 con placebo) y menos eventos ECC (123 con atorvastatina vs. 204 con placebo). La mortalidad total fue similar a través de grupos de tratamiento (216 con atorvastatina vs. 211 con placebo). La incidencia total de eventos adversos y eventos adversos graves fue similar entre grupos de tratamiento.
Prevención secundaria de eventos cardiovasculares: En el Estudio de Tratamiento Hasta Nuevas Metas (TNT), se evalúa el efecto de 80 mg/día de atorvastatina vs. 10 mg/día de atorvastatina sobre la reducción de eventos cardiovasculares en 10,001 sujetos (94% blancos, 81% varones, 38% > 65 años) con ECC clínicamente evidente que habían alcanzado un nivel meta de C-LDL < 130 mg/dL tras completar un periodo de inducción abierto de 8 semanas con 10 mg/día de atorvastatina. Los sujetos fueron asignados aleatoriamente a 10 mg/día u 80 mg/día de atorvastatina y se les dio seguimiento por una duración mediana de 4.9 años. El promedio de los niveles de C-LDL, CT, TG, colesterol no HDL y HDL a las 12 semanas fueron 73 mg/dL, 145 mg/dL, 128 mg/dL, 98 mg/dL y 47 mg/dL respectivamente, durante el tratamiento con 80 mg de atorvastatina y 99 mg/dL, 177 mg/dL, 152 mg/dL, 129 mg/dL y 48 mg/dL respectivamente durante el tratamiento con 10 mg de atorvastatina.
El tratamiento con 80 mg/día de atorvastatina redujo significativamente la tasa de ECM (434 eventos en el grupo de 80 mg/día vs. 548 eventos en el grupo de 10 mg/día) con reducción de riesgo relativo de 22%.
80 mg de atorvastatina redujeron significativamente el riesgo de lo siguiente:
|
Resultado final significativo |
Atorvastatina 10 mg (N = 5006) |
Atorvastatina 80 mg (N = 4995) |
HRa (95% IC) |
||
|
Criterio de valoración primario* |
n |
(%) |
n |
(%) |
|
|
Primer criterio de valoración cardiovascular mayor |
548 |
10.9 |
434 |
8.7 |
0.78 (0.69, 0.89) |
|
Componentes del Criterio de valoración primario |
|||||
|
IM No-fatal, no relacionado con procedimientos |
308 |
6.2 |
243 |
4.9 |
0.78 (0.66, 0.93) |
|
Accidente cerebrovascular (fatal y no-fatal) |
155 |
3.1 |
117 |
2.3 |
0.75 (0.59, 0.96) |
|
Criterios de valoración secundarios** |
n |
(%) |
n |
(%) |
|
|
Primer episodio ICC con hospitalización |
164 |
3.3 |
122 |
2.4 |
0.74 (0.59, 0.94) |
|
Primer CABG u otros procedimientos de revascularización coronaria |
904 |
18.1 |
667 |
13.4 |
0.72 (0.65, 0.80) |
|
Primer criterio de valoración de angina documentado |
615 |
12.3 |
545 |
10.9 |
0.88 (0.79, 0.99) |
a Atorvastatina 80 mg: atorvastatina 10 mg.
b Componente de otros Criterios de valoración secundarios.
* ECM = muerte por ECC, IM no mortal, paro cardiaco con reanimación y accidente cerebrovascular mortal y no mortal.
** Criterio de valoración secundarios no incluidos en el Criterio de valoración primario.
HR = proporción de riesgos; IC = intervalo de confianza; IM = infarto del miocardio; ICC = insuficiencia cardiaca congestiva; CABG = injerto de derivación de la arteria coronaria.
Intervalos de confianza para los Criterios de valoración secundarios no fueron ajustados para comparaciones múltiples.
No hubo diferencia significativa entre los grupos de tratamiento para mortalidad por todas las causas: 282 (5.6%) en el grupo de 10 mg/día de atorvastatina vs. 284 (5.7%) en el grupo de 80 mg/día. La proporción de sujetos que experimentó muerte cardiovascular incluyendo los componentes de muerte por ECC y accidente cerebrovascular mortal fueron numéricamente más bajos en el grupo de 80 mg de atorvastatina que en el de 10 mg de atorvastatina. Las proporciones de sujetos que experimentaron muerte no cardiovascular fueron numéricamente mayores en el grupo de 80 mg de atorvastatina que en el grupo de 10 mg de atorvastatina.
En el Estudio de Reducción Incremental de Criterios de valoración a través de Reducción Agresiva de Lípido (IDEAL) el tratamiento con 80 mg/día de atorvastatina se comparó con tratamiento con 20 mg a 40 mg/día de simvastatina en 8,888 sujetos hasta de 80 años de edad con historia de ECC para evaluar si era posible lograr reducir el riesgo CV (cardiovascular). Los pacientes eran principalmente varones (81%), blancos (99%), con edad promedio de 61.7 años y un C-LDL promedio de 121.5 mg/dL en la asignación aleatoria; 76% estaban recibiendo terapia con estatinas. Este estudio prospectivo aleatorio abierto con Criterio de valoración ciego (PROBE) sin periodo de inducción, realizó seguimiento de los sujetos durante una mediana de 4.8 años. La media de los niveles de C-LDL, CT, TG, colesterol HDL y no HDL en la semana 12 fue 78 mg/dL, 145 mg/dL, 115 mg/dL, 45 mg/dL y 100 mg/dL respectivamente durante el tratamiento con 80 mg de atorvastatina y 105 mg/dL, 179 mg/dL, 142 mg/dL, 47 mg/dL y 132 mg/dL respectivamente durante el tratamiento con 20 mg a 40 mg de simvastatina.
No hubo diferencia significativa entre grupos de tratamiento para el Criterio de valoración primario, la tasa de primer evento coronario mayor (ECC mortal, IM no mortal y paro cardiaco con reanimación): 411 (9.3%) del grupo de 80 mg/día de atorvastatina vs. 463 (10.4%) en el grupo de 20 mg a 40 mg/día de simvastatina, HR 0.89, 95% IC 0.78, 1.01, p = 0.07.
No hubo diferencia significativa entre grupos de tratamiento para mortalidad por todas las causas: 366 (8.2%) en el grupo de 80 mg/día de atorvastatina vs. 374 (8.4%) en el grupo de 20 mg/día a 40 mg/día de simvastatina. La proporción de sujetos que experimentó muerte CV o no CV fue similar para el grupo de 80 mg de atorvastatina y el grupo de 20 mg a 40 mg de simvastatina.
Atorvastatina para hiperlipidemia y dislipidemia mezclada:
Atorvastatina reduce el colesterol total, C-LDL, lipoproteínas de muy baja densidad (C-VLDL), apo B y TG y aumenta el C-HDL en pacientes con hiperlipidemia (heterocigótica familiar y no familiar) y dislipidemia mixta (Fredrickson tipos IIa y IIb). La respuesta terapéutica se observa dentro de las 2 semanas siguientes y la respuesta máxima se alcanza usualmente en 4 semanas y se mantiene durante la terapia crónica.
Atorvastatina es eficaz en una amplia variedad de poblaciones de pacientes con hiperlipidemia, con y sin hipertrigliceridemia, en hombres y mujeres y en ancianos.
En dos estudios multicéntricos, controlados con placebo, de respuesta a la dosis, en pacientes con hiperlipidemia, atorvastatina administrada como dosis única durante 6 semanas, redujo de manera significativa el colesterol total, C-LDL, apo B y TG. (Los resultados agrupados se presentan en la Tabla 7).
Tabla 7. Respuesta de dosis en pacientes con hiperlipidemia primaria (cambio medio ajustado % a partir de la línea de referencia)ª
|
Dosis |
N |
TC |
C-LDL |
Apo B |
TG |
C-HDL |
C No-HDL/C-HDL |
|
Placebo |
21 |
4 |
4 |
3 |
10 |
-3 |
7 |
|
10 |
22 |
-29 |
-39 |
-32 |
-19 |
6 |
-34 |
|
20 |
20 |
-33 |
-43 |
-35 |
-26 |
9 |
-41 |
|
40 |
21 |
-37 |
-50 |
-42 |
-29 |
6 |
-45 |
|
80 |
23 |
-45 |
-60 |
-50 |
-37 |
5 |
-53 |
* Los resultados están agrupados a partir de los estudios de respuesta a la dosis.
En pacientes con Fredrickson Tipos IIa y IIb la hiperlipoproteinemia agrupada a partir de 24 estudios controlados, los cambios medios en porcentaje (Percentil 25 y 75) a partir de la línea de referencia en C-HDL para atorvastatina de 10, 20, 40 y 80 mg fueron de 6.4 (-1.4, 14), 8.7 (0, 17), 7.8 (0, 16) y 5.1 (-2.7, 15), respectivamente. Adicionalmente, los análisis de los datos agrupados demostraron disminuciones consistentes y significativas en el Colesterol total, C-LDL, TG, C-Total/C-HDL y C-LDL/C-HDL.
En tres estudios multicéntricos, doble ciego, en pacientes con hiperlipidemia, atorvastatina se comparó con otras estatinas. Después de la asignación aleatoria, se trató a los pacientes por 16 semanas con atorvastatina 10 mg por día o una dosis fija del agente comparador (Tabla 8).
Tabla 8. Cambio medio en porcentaje a partir de la línea de referencia en el resultado final (estudios doble ciego, aleatorios a 2, con control activo)
|
Tratamiento (dosis diaria) |
N |
Colesterol total |
C-LDL |
Apo B |
TG |
C-HDL |
C No-HDL/ |
|
Estudio 1 Atorvastatina 10 mg Lovastatina 20 mg 95% IC para Dif.1 |
707 191 |
-27a -19 -9.2, -6.5 |
-36a -27 -10.7, -7.1 |
-28a -20 -10.0, -6.5 |
-17a -6 -15.2, -7.1 |
+7 +7 -1.7, 2.0 |
-37a -28 -11.1, -7.1 |
|
Estudio 2 Atorvastatina 10 mg Pravastatina 20 mg 95% IC para Dif.1 |
222 77 |
-25b -17 -10.8, -6.1 |
-35b -23 -14.5, -8.2 |
-27b -17 -13.4, -7.4 |
-17b -9 -14.1, -0.7 |
+6 +8 -4.9, 1.6 |
-36b -28 -11.5, -4.1 |
|
Estudio 3 Atorvastatina 10 mg Simvastatina 10 mg 95% IC para Dif.1 |
132 45 |
-29c -24 -8.7, -2.7 |
-37c -30 -10.1, -2.6 |
-34c -30 -8.0, -1.1 |
-23c -15 -15.1, -0.7 |
+7 +7 -4.3, 3.9 |
-39c -33 -9.6, -1.9 |
1 Un valor negativo para el 95% de IC para la diferencia entre los tratamientos favorece a atorvastatina en todo, excepto para C-HDL, para la cual un valor positivo favorece a atorvastatina. Si el rango no incluye 0, esto indica una diferencia estadísticamente significativa.
a Significativamente diferente de lovastatina, ANCOVA, p ≤ 0.05.
b Significativamente diferente de pravastatina, ANCOVA, p ≤ 0.05.
c Significativamente diferente de simvastatina, ANCOVA, p ≤ 0.05.
Se desconoce el impacto sobre los resultados clínicos de las diferencias en los efectos de alteración de los lípidos entre los tratamientos que se muestran en la Tabla 8. La Tabla 8 no contiene datos que comparen los efectos de atorvastatina 10 mg y dosis mayores de lovastatina, pravastatina y simvastatina. Los medicamentos comparados en los estudios que se resumen en la tabla no son necesariamente intercambiables.
Atorvastatina para hipertrigliceridemia:
La respuesta a atorvastatina en 64 pacientes con hipertrigliceridemia aislada (Fredrickson Tipo IV) tratados a través de varios estudios clínicos se muestra en la tabla a continuación (Tabla 9). Para los pacientes tratados con atorvastatina, el nivel medio de líneas de referencia TG (min, máx) fue de 565 (267-1502).
Tabla 9. Pacientes combinados con hipertrigliceridemia aislada: Cambio medio en porcentaje (min, máx) a partir de la línea de referencia
|
Placebo (N=12) |
Atorvastatina 10 mg (N=37) |
Atorvastatina 20 mg (N=13) |
Atorvastatina 80 mg (N=14) |
|
|
TG |
-12.4 (-36.6, 82.7) |
-41.0 (-76.2, 49.4) |
-38.7 (-62.7, 29.5) |
-51.8 (-82.8, 41.3) |
|
Colesterol total |
-2.3 (-15.5, 24.4) |
-28.2 (-44.9, -6.8) |
-34.9 (-49.6, -15.2) |
-44.4 (-63.5, -3.8) |
|
C-LDL |
3.6 (-31.3, 31.6) |
-26.5 (-57.7, 9.8) |
-30.4 (-53.9, 0.3) |
-40.5 (-60.6, -13.8) |
|
C-HDL |
3.8 (-18.6, 13.4) |
13.8 (-9.7, 61.5) |
11.0 (-3.2, 25.2) |
7.5 (-10.8, 37.2) |
|
C-VLDL |
-1.0 (-31.9, 53.2) |
-48.8 (-85.8, 57.3) |
-44.6 (-62.2, -10.8) |
-62.0 (-88.2, 37.6) |
|
C No-HDL |
-2.8 (-17.6, 30.0) |
-33.0 (-52.1, -13.3) |
-42.7 (-53.7, -17.4) |
-51.5 (-72.9, -4.3) |
Atorvastatina para disbetalipoproteinemia:
Los resultados de un estudio transversal abierto de 16 pacientes (genotipos: 14 apo E2/E2 y 2 apo E3/E2) con disbetalipoproteinemia (Fredrickson Tipo III) se muestran en la tabla a continuación (Tabla 10).
Tabla 10. Estudio transversal abierto de 16 pacientes con disbetalipoproteinemia (Fredrickson Tipo III
|
Mediana (min, máx) a la línea de referencia (mg/dL) |
Cambio medio en % (min, máx) |
||
|
Atorvastatina 10 mg |
Atorvastatina 80 mg |
||
|
Colesterol total |
442 (225, 1320) |
-37 (-85, 17) |
-58 (-90, -31) |
|
TG |
678 (273, 5990) |
-39 (-92, -8) |
-53 (-95, -30) |
|
Colesterol lipoproteico de densidad intermedia (C-IDL) +C-VLDL |
215 (111, 613) |
-32 (-76, 9) |
-63 (-90, -8) |
|
C No-HDL |
411 (218, 1272) |
-43 (-87, -19) |
-64 (-92, -36) |
Atorvastatina para hipercolesterolemia homocigótica familiar:
En un estudio sin un grupo de control concurrente, 29 pacientes en edades entre 6 años y 37 años con HoFH recibieron dosis diarias máximas de 20 a 80 mg de atorvastatina. La reducción media del C-LDL en este estudio fue del 18%. Veinticinco pacientes con una reducción en el C-LDL tuvieron una respuesta media del 20% (rango de 7% a 53%, mediana de 24%); Los 4 pacientes restantes tuvieron aumentos del 7% al 24% en el C-LDL. Cinco de los 29 pacientes tuvieron una función de receptor ausente. De éstos, 2 pacientes también tuvieron derivación portocava y no tuvieron una reducción significativa en el C-LDL. Los 3 pacientes restantes negativos para receptor tuvieron una reducción media del C-LDL de un 22%.
Hipercolesterolemia familiar heterocigoto en pacientes pediátricos:
Los siguientes estudios exclusivos para niños se completaron con atorvastatina.
En un estudio abierto, de un solo grupo, se inscribieron 271 niños y niñas de 6 a 15 años de edad con Hipercolesterolemia Familiar Heterocigota (HeFH) y se trataron con atorvastatina durante un máximo de 3 años. La inclusión al estudio requirió HeFH confirmada y un nivel inicial de C-LDL ≥ 4 mmol/L (aproximadamente 152 mg/dL). El estudio incluyó a 139 niños en etapa de desarrollo de Tanner 1 (por lo general de 6 a 10 años de edad). La dosis de atorvastatina (una vez al día) se inició con 5 mg (tabletas masticables) en niños menores de 10 años. Los niños de ≥ 10 años se iniciaron con 10 mg de atorvastatina (una vez al día). A todos los niños se les podían ajustar a dosis más altas para lograr un objetivo de C-LDL < 3.35 mmol/L. La dosis media ponderada para los niños de 6 a 9 años fue de 19.6 mg y la dosis media ponderada para los niños de 10 años o más fue de 23.9 mg.
El valor inicial medio de C-LDL (+/- SD) fue de 6.12 (1.26) mmol/L, que fue aproximadamente 233 (48) mg/dL Consulte la tabla 6 a continuación para conocer los resultados finales.
Los datos no demostraron ningún efecto del medicamento sobre ninguno de los parámetros de crecimiento y desarrollo (es decir, altura, peso, índice de masa corporal, etapa de Tanner, evaluación del investigador de la maduración y el desarrollo general) en niños y adolescentes con HeFH que recibieron tratamiento con atorvastatina durante el estudio de 3 años. El investigador no señaló ningún efecto del medicamento sobre la altura, peso, índice de masa corporal por edad o por sexo por visita.
Tabla 11. Efectos hipolipemiantes de la atorvastatina en niños y niñas adolescentes con hipercolesterolemia familiar heterocigota (mmol/L)
|
Punto temporal |
N |
CT (SD) |
C-LDL (SD) |
C-HDL (SD) |
TG (SD) |
Apo B (SD)# |
|
Valor inicial |
271 |
7.86 (1.30) |
6.12 (1.26) |
1.314 (0.2663) |
0.93 (0.47) |
1.42 (0.28)** |
|
Mes 30 |
206 |
4.95 (0.77)* |
3.25 (0.67) |
1.327 (0.2796) |
0.79 (0.38)* |
0.90 (0.17)* |
|
Mes 36/ET |
240 |
5.12 (0.86) |
3.45 (0.81) |
1.308 (0.2739) |
0.78 (0.41) |
0.93 (0.20)*** |
CT=colesterol total; C-LDL=colesterol de lipoproteína de baja densidad; C-HDL=colesterol de lipoproteína de alta densidad; TG=triglicéridos; Apo B=apolipoproteína B; SD=desviación estándar; el "Mes 36/ET" incluyó los datos de la última visita de los sujetos que terminaron el tratamiento antes del punto temporal programado de 36 meses, así como los datos completos de 36 meses de los sujetos que completaron el tratamiento en 36 meses; "*"=N del mes 30 para este parámetro fue de 207; "**"=N del valor inicial para este parámetro fue de 270; "***" =N del mes 36/ET para este parámetro fue de 243: "#"=g/L para apo B.
En un estudio doble ciego controlado con placebo seguido por una fase de marbete abierto, 187 muchachos y muchachas postmenarca de 10 a 17 años de edad (edad media 14.1 años) con HF heterocigoto o hipercolesterolemia severa fueron asignados aleatoriamente a atorvastatina (n=140) o placebo (n=47) por 26 semanas y después todos recibieron atorvastatina por 26 semanas. Para la inclusión en el estudio se requirió 1) nivel C-LDL basal ≥ 190 mg/dL o 2) C-LDL basal ≥ 160 mg/dL e historia familiar positiva de HF (hipercolesterolemia familiar) o ECC prematura documentada en paciente de primer o de segundo grado.
El valor pasal medio de C-LDL fue 218.6 mg/dL (rango: 138.5-385.0 mg/dL) en el grupo de atorvastatina en comparación con 230.0 mg/dL (rango: 160.0-324.5 mg/dL) en el grupo de placebo. La dosis de atorvastatina una vez al día) fue 10 mg durante las 4 primeras semanas y se tituló a 20 mg cuando el nivel de C-LDL fue > 130 mg/dL. El número de pacientes tratados con atorvastatina que requiere una titulación ascendente hasta 20 mg tras la semana 4 durante la fase doble ciego fue 78 (55.7%).
Atorvastatina redujo significativamente los niveles plasmáticos de colesterol total, C-LDL, TG y apo B durante la fase doble ciego de 26 semanas (ver Tabla 12).
Tabla 12. Efectos de Reducción de Lípidos y Atorvastatina en muchachos y muchachas adolescentes con hipercolesterolemia familiar heterocigoto o hipercolesterolemia severa (cambio medio porcentual respecto a la línea basal en criterio de valoración en la población de intención de tratamiento)
|
Dosis |
N |
C-totalª |
C-LDLb |
C-HDLc |
TGd |
Apo Be |
|
Placebo |
47 |
-1.5 |
-0.4 |
-1.9 |
1.0 |
0.7 |
|
Atorvastatina |
140 |
-31.4 |
-39.6 |
2.8 |
-12.0 |
-34.0 |
a Colesterol total.
b Colesterol de lipoproteínas de baja densidad.
c Colesterol de lipoproteínas de aIta densidad.
d Total de glicéridos.
e Apolipoproteína B.
El valor medio alcanzado de C-LDL fue 130.7 mg/dL (rango: 70.0-242.0 mg/dL) en el grupo de atorvastatina en comparación con 228.5 mg/dL (rango: 152.0-385.0 mg/dL) en el grupo con placebo durante la fase doble ciego de 26 semanas. En este estudio de 1 año, no hubo efecto detectable sobre el crecimiento o la maduración sexual en niños o sobre la duración del periodo menstrual en niñas.
La eficacia a largo plazo de terapia con atorvastatina en la niñez para reducir la morbilidad y la mortalidad en etapa adulta no se ha establecido.
CONTRAINDICACIONES: CADUET® (amlodipino/atorvastatina) está contraindicada en pacientes que:
1. Presentan hipersensibilidad conocida a dihidropiridinas,* amlodipino, atorvastatina o cualquier componente de este medicamento.
2. Presentan enfermedad hepática activa o elevaciones persistentes no explicadas de transaminasas séricas > 3 veces el límite superior normal (LSN).
3. Están embarazadas, en etapa de lactancia o tienen potencial reproductivo y no están usando medidas anticonceptivas adecuadas. Amlodipino/atorvastatina debe administrarse a mujeres en edad reproductiva sólo cuando tengan muy pocas probabilidades de concebir y se les haya informado sobre los riesgos potenciales para el feto.
* Amlodipino es un antagonista de la dihidropiridina cálcica.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: CADUET® (amlodipino/atorvastatina) está contraindicada en el embarazo debido al componente de atorvastatina. Las mujeres con potencial reproductivo deben usar medidas anticonceptivas adecuadas.
CADUET® (amlodipino/atorvastatina) debe administrarse a mujeres en edad reproductiva sólo cuando tengan muy pocas probabilidades de concebir y ya hayan sido informadas del riesgo potencial para el feto.
CADUET® (amlodipino/atorvastatina) está contraindicado en mujeres en periodo de lactancia debido al componente de atorvastatina. Se desconoce si atorvastatina se excreta en leche materna. Debido al potencial de reacciones adversas en lactantes las mujeres que tomen CADUET® (amlodipino/atorvastatina) no deben lactar.
No se ha establecido la seguridad de amlodipino en embarazo humano o lactancia. El amlodipino no demuestra toxicidad en estudios reproductivos en animales distintos de retraso de parto y prolongación del trabajo de parto en ratas a nivel de dosificación cincuenta veces la dosis máxima recomendada en humanos. No hubo efecto alguno sobre la fertilidad de las ratas tratadas con amlodipino (ver sección Precauciones en relación con efecto de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
La experiencia en seres humanos indica que el amlodipino se transfiere a la leche materna humana. La mediana de la relación de concentración de amlodipino en leche/plasma en 31 mujeres en periodo de lactancia con hipertensión inducida por el embarazo fue de 0.85 después de la administración de amlodipino con una dosis inicial de 5 mg una vez al día que se ajustó según lo requerido (dosis diaria promedio y dosis diaria ajustada según, peso: 6 mg y 98.7 mcg/kg, respectivamente). La dosis diaria estimada de amlodipino en el lactante mediante la leche materna fue de 4.17 mcg/kg.
REACCIONES SECUNDARIAS Y ADVERSAS: La terapia combinada de CADUET® (amlodipino con atorvastatina) ha sido evaluada para seguridad de 1092 pacientes en estudios doble ciego controlados con placebo, como tratamiento para hipertensión concomitante y dislipidemia. En estudios clínicos no se observaron eventos adversos peculiares a la terapia combinada de amlodipino y atorvastatina. Los eventos adversos se han limitado a aquéllos reportados previamente con amlodipino y/o atorvastatina (favor de ver las experiencias respectivas de evento adverso a continuación).
En general la terapia combinada de amlodipino y atorvastatina fue bien tolerada. En su mayoría los eventos adversos fueron de severidad leve o moderada. En estudios clínicos controlados la descontinuación de terapia por eventos adversos o anomalías de laboratorio se requirió en 5.1% de los pacientes tratados tanto con amlodipino y atorvastatina en comparación con 4.0% de los pacientes que recibieron placebo.
La siguiente información se basa en estudios clínicos y experiencia poscomercialización con CADUET® (amlodipino/atorvastatina).
Experiencia con amlodipino: Amlodipino es bien tolerado. En estudios clínicos controlados con placebo que incluyen pacientes con hipertensión o angina, los efectos secundarios observados de manera más común fueron:
|
Clase de órgano MedDRA |
Efectos no deseados |
|
Trastornos del sistema nervioso |
Dolor de cabeza, mareo, somnolencia |
|
Trastornos cardiacos |
Palpitaciones |
|
Trastornos vasculares |
Sofoco |
|
Trastornos gastrointestinales |
Dolor abdominal, náusea |
|
Trastornos generales y condiciones del sitio de administración |
Edema, fatiga |
En estos estudios clínicos no se ha observado ningún patrón de anomalías clínicamente significativas en pruebas de laboratorio relacionados con amlodipino.
Los efectos secundarios observados menos frecuentemente en la experiencia de venta con amlodipino incluyen:
|
Clase de órgano MedDRA |
Efectos no deseados |
|
Trastornos de Sangre y Sistema linfático |
Leucopenia, trombocitopenia |
|
Trastornos de Metabolismo y Nutrición |
Hiperglicemia |
|
Trastornos Psiquiátricos |
Insomnio, alteración del humor |
|
Trastornos del Sistema nervioso |
Hipertonía, hipoestesia/parestesia, neuropatía periférica, síncope, disgeusia, temblor, trastorno extrapiramidal |
|
Trastornos Oculares |
Trastornos visuales |
|
Trastornos de Oído y laberinto |
Tinnitus |
|
Trastornos Vasculares |
Hipotensión, vasculitis |
|
Trastornos Respiratorios, Torácicos y del Mediastino |
Tos, disnea, rinitis |
|
Trastornos Gastrointestinales |
Cambios en los hábitos intestinales, boca seca, dispepsia (incluyendo gastritis), hiperplasia gingival, pancreatitis, vómito |
|
Trastornos de Piel y Tejido Subcutáneo |
Alopecia, hiperhidrosis, púrpura, decoloración de piel, urticaria |
|
Trastornos Músculo esqueléticos y de Tejido conectivo |
Artralgia, dolor de espalda, espasmos musculares, mialgia |
|
Trastornos Renales y Urinarios |
Polaquiuria, trastorno en la micción, nicturia |
|
Trastornos del Sistema Reproductor y Senos |
Ginecomastia, disfunción eréctil |
|
Trastornos Generales y condiciones del Sitio de administración |
Astenia, malestar, dolor |
|
Investigaciones |
Peso aumentado/disminuido |
Rara vez se han reportado reacciones alérgicas incluyendo prurito, salpullido, angioedema y eritema multiforme.
Hepatitis, ictericia y elevaciones de la enzima hepática también han sido reportados muy infrecuentemente (casi siempre consistentes con colestasis). Se han reportado algunos casos asociados con el uso de amlodipino que requieren hospitalización por su severidad. En muchos casos, la asociación causal es incierta.
Como con otros bloqueadores del canal de calcio, los siguientes eventos adversos han sido reportados raramente y no pueden ser distinguidos de la historia natural de la enfermedad subyacente: IM, arritmia (incluyendo bradicardia, taquicardia ventricular y fibrilación atrial) y dolor de pecho.
Pacientes pediátricos (de 6 a 17 años): Amlodipino es bien tolerado en niños. Los eventos adversos fueron similares a los que se observan, en adultos. En un estudio en 268 niños, los eventos adversos reportados con mayor frecuencia fueron:
|
Clase de órgano MedDRA |
Efectos no deseados |
|
Trastornos del Sistema nervioso |
Dolor de cabeza, mareo |
|
Trastornos Vasculares |
Vasodilatación |
|
Trastornos Respiratorios, Torácicos y del Mediastino |
Epistaxis |
|
Trastornos Gastrointestinales |
Dolor abdominal |
|
Trastornos Generales y Condiciones del Sitio de administración |
Astenia |
La mayoría de los eventos adversos fueron leves o moderados. Eventos adversos severos (principalmente cefalea) fueron experimentados por 7.2% con amlodipino 2.5 mg, 4.5% con amlodipino 5 mg y 4.6% con placebo. La causa más común para descontinuar el estudio fue hipertensión no controlada. No hubo descontinuaciones a causa de anomalías de laboratorio. No hubo cambio significativo en frecuencia cardiaca.
Experiencia con atorvastatina: Atorvastatina es bien tolerada en general. Las reacciones adversas han generalmente sido leves y transitorias. En la base de datos del estudio clínico controlado con placebo de atorvastatina de 16,066 (8,755 atorvastatina vs. 7,311 placebo) pacientes tratados por un periodo promedio de 53 semanas, 5.2% de los pacientes que tomaban atorvastatina la descontinuaron debido a reacciones adversas en comparación con 4.0% de los pacientes con placebo.
Los efectos adversos más frecuentes (≥ 1%) asociados con terapia con atorvastatina en pacientes que participaron en estudios clínicos controlados fueron:
Infecciones e infestaciones: Nasofaringitis.
Trastornos de metabolismo y nutrición: Hiperglicemia.
Trastornos respiratorios, torácicos y del mediastino: Dolor faríngeo, laríngeo, epistaxis.
Trastornos gastrointestinales: Diarrea, dispepsia, náuseas, flatulencias.
Trastornos músculo esqueléticos y de tejido conectivo: Artralgia, dolor en extremidades, dolor músculo-esquelético, espasmos musculares, mialgia, inflamación de articulaciones.
Investigaciones: Prueba de función hepática anormal, creatina fosfoquinasa en sangre elevada.
Los efectos adversos adicionales reportados en los estudios clínicos controlados con placebo con atorvastatina incluyen:
Trastornos psiquiátricos: Pesadillas.
Trastornos oculares: Visión borrosa.
Trastornos de oído y laberinto: Tinnitus.
Trastornos gastrointestinales: Incomodidad abdominal, eructos.
Trastornos hepatobiliares: Hepatitis, colestasis.
Trastornos de piel y tejido subcutáneo: Urticaria.
Trastornos músculo esqueléticos y de tejido conectivo: Fatiga muscular, dolor de cuello.
Trastornos generales y condiciones del sitio de administración: Malestar, pirexia.
Investigaciones: Leucocitos en orina positivos.
No todos los efectos listados con anterioridad han sido asociados causalmente con la terapia con atorvastatina.
Pacientes pediátricos (de 10 a 17 años): Los pacientes tratados con atorvastatina presentaron un perfil de experiencias adversas generalmente similar al de pacientes tratados con placebo y las experiencias adversas más comunes observadas en ambos grupos sin importar la evaluación causal fueron infecciones.
Experiencia poscomercialización:
En experiencia poscomercialización se han reportado los siguientes efectos indeseables adicionales no deseados adicionales con atorvastatina:
Trastornos de la sangre y el sistema linfático: Trombocitopenia.
Trombocitopenia, trastornos del sistema inmune: Reacciones alérgicas (incluyendo anafilaxia).
Lesiones, intoxicación y complicaciones de procedimientos: Ruptura del tendón.
Trastornos del metabolismo y la nutrición: Aumento de peso.
Trastornos del sistema nervioso: Hipoestesia, amnesia, mareo, disgeusia.
Trastornos gastrointestinales: Pancreatitis.
Trastornos de la piel y el tejido subcutáneo: Síndrome de Stevens Johnson, necrólisis epidérmica tóxica, angioedema, eritema multiforme, erupción bullosa.
Trastornos músculo esquelético y del tejido conectivo: Radiomiólisis, miopatía necrosante inmunomediada, miositis, dolor de la espalda.
Trastornos generales y afecciones en el sitio de administración: Dolor toráxico, edema periférico, fatiga.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis:
En estudios con amlodipino: Ratas y ratones tratados con amlodipino en la dieta durante dos años en concentraciones que se calcula suministra niveles diarios de dosificación de 0.5, 1.25 y 2.5 mg/kg/día no demostraron evidencia de carcinogénesis. La dosis más alta (en ratones, que es similar y para ratas el doble* de la dosis clínica máxima recomendada de 10 mg con base en mg/m2) fue cercana a la dosis máxima tolerada en ratones, pero no en ratas.
En estudios con atorvastatina: Atorvastatina no fue carcinogénica en ratas. La dosis máxima usada fue 63 veces más alta que la dosis más alta en humanos (80 mg/día) con base en mg/kg de peso del cuerpo y de 8 a 16 veces más alta basándose en valores de ABC (0-24). En un estudio a 2 años en ratón las incidencias de adenoma hepatocelular en machos y carcinomas hepatocelulares en hembras incrementaron con la dosis máxima empleada que fue 250 veces más alta que la dosis más alta en humanos con base en mg/kg de peso del cuerpo. La exposición sistémica fue de 6 a 11 veces más alta basada en ABC (0-24).
Todos los demás fármacos químicamente similares en esta clase han inducido a tumores tanto en ratón como en rata a múltiplos de 12 a 125 veces su dosis clínica más alta recomendada con base en mg/kg de peso del cuerpo.
Mutagénesis:
En estudios con amlodipino: Los estudios de mutagénesis no revelaron efectos relacionados con el fármaco a nivel de genes o cromosomas.
En estudios con atorvastatina: Atorvastatina no demostró potencial mutagénico o clastogénico en cuatro pruebas in vitro con y sin activación metabólica ni en un ensayo in vivo. Fue negativa en la prueba de Ames con Salmonella typhimurium y Escherichia coli y en el ensayo de mutación anterógrada hipoxantina guanina fosforibosil transferasa (HGPRT) in vitro en células de pulmón de hámster chino. Atorvastatina no produjo incremento significativo de aberraciones cromosomales en el ensayo de células pulmonares de hámster chino in vitro y fue negativo en la prueba de micronúcleo de ratón in vivo.
Alteración de la fertilidad:
En estudios con amlodipino: No hubo efecto sobre la fertilidad de ratas tratadas con amlodipino (machos durante 64 días y hembras 14 días antes del apareamiento) a dosis hasta de 10 mg/kg/día (8 veces* la dosis máxima recomendada en humanos de 10 mg con base en mg/m2).
En estudios con atorvastatina: No se observaron efectos adversos sobre la fertilidad o la reproducción en ratas macho que recibieron dosis de atorvastatina hasta de 175 mg/kg/día en ratas hembra que recibieron dosis hasta de 225 mg/kg/día. Estas dosis son de 100 a 140 veces la dosis máxima recomendada en humanos con base en mg/kg. Atorvastatina no provocó efectos adversos sobre parámetros de espermatozoides o semen ni sobre histopatología de órganos reproductivos en perros que recibieron dosis de 10 mg/kg, 40 mg/kg, o 120 mg/kg por 2 años.
* Basado en peso de paciente de 50 kg.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Los datos de un estudio de interacción farmacológica respecto a 10 mg de amlodipino y 80 mg atorvastatina en sujetos saludables indican que la farmacocinética de amlodipino no se altera cuando estos fármacos se coadministran. El efecto de amlodipino sobre la farmacocinética de atorvastatina no demostró efectos sobre Cmáx.: 91% (intervalo de confianza del 90%: 80% a 103%), pero ABC de atorvastatina aumentó en 18% (IC del 90%: 109% a 127%) en presencia de amlodipino.
No se han realizado estudios de interacción farmacológica con CADUET® (amlodipino/atorvastatina) y otros fármacos aunque se han realizado estudios usando los componentes individuales amlodipino y atorvastatina como se describe a continuación.
Interacciones de amlodipino: Amlodipino ha sido administrado de manera segura con diuréticos tiazídicos, bloqueadores alfa, bloqueadores beta inhibidores de la ECA, nitratos de larga duración, nitroglicerina sublingual, antiinflamatorios no esteroides, antibióticos e hipoglucemiantes orales.
Inhibidores CYP 3A4: La administración concomitante de la dosis de 180 mg diaria de diltiazen con 5 mg de amlodipino en pacientes de edad avanzada hipertensos (69 a 87 años) resultó en un incremento de 57% en la exposición sistémica de amlodipino. La administración concomitante con eritromicina en voluntaios sanos (18 a 43 años) no cambió significativamente la exposición sistémica de amlodipino (incremento del 22% ABC). La importancia clínica de este hallazgo es incierta, las variaciones farmacocinéticas podrían ser más pronunciadas en personas con edad avanzada.
Inhibidores más potentes de CYP 3A4 (por ejemplo: ketoconazol, itraconazol, ritonavir) pueden aumentar las concentraciones de amlodipino en el plasma en mayor medida que diltiazem. Amlodipino debe ser usado con precaución junto con inhibidores de CYP 3A4.
Claritromicina: La claritromicina es un inhibidor de CYP 3A4. Existe un mayor riesgo de hipotensión en, pacientes bajo claritromicina con amlodipino. Se recomienda la observación minuciosa de los pacientes cuando se coadministra amlodipino con claritromicina.
Inductores CYP 3A4: No existe información disponible sobre el efecto de los inductores de CYP 3A4 en amlodipino. El uso concomitante de inductores de CYP 3A4 (por ejemplo: rifampicina, Hypericum perforatum) puede producir una menor concentración de amlodipino en el plasma. Amlodipino debe ser usado con precaución junto con inductores de CYP 3A4.
Jugo de toronja: La administración concomitante de 240 mL de jugo de toronja con una dosis oral, única de 10 mg de amlodipino en 20 voluntarios no tuvo reacciones significativas sobre la farmacocinética de amlodipino. El estudio no permite la examinación del efecto del polimorfismo genético en CYP 3A4, la enzima primaria responsable del metabolismo de amlodipino, por lo tanto, la administración de amlodipino con toronja o jugo de toronja no es recomendable porque la biodisponibilidad puede aumentar en los pacientes resultando en mayores efectos de disminución de la PA.
Datos in vitro de estudios en plasma humano indican que amlodipino no produce efectos sobre el enlace proteico de los fármacos probados (digoxina, fenitoína, warfarina o indometacina).
En los estudios que se mencionan a continuación no hubo cambios significativos en la farmacocinética, de amlodipino u otro fármaco dentro del estudio cuando se coadministraron.
Estudios especiales: Efectos de otros agentes sobre amlodipino.
Cimetidina: La coadministración de amlodipino con cimetidina no modificó la farmacocinética de amlodipino.
Aluminio/magnesio (antiácido): La coadministración de un antiácido con aluminio/magnesio y una sola dosis de amlodipino no produjo efecto significativo sobre la farmacocinética de amlodipino.
Sildenafil: Una dosis de 100 mg de sildenafil en sujetos con hipertensión esencial no produjo efectos sobre los parámetros farmacocinéticos de amlodipino cuando se usaron amlodipino y sildenafil en combinación cada agente de manera independiente ejerció su propio efecto de reducción en la PA.
Estudios especiales: Efecto de amlodipino sobre otros agentes.
Digoxina: La coadministración de amlodipino con digoxina no modificó los niveles séricos de digoxina ni la depuración renal de digoxina en voluntarios normales.
Etanol (alcohol): Dosis únicas y múltiples de 10 mg de amlodipino no produjeron efectos significativos sobre la farmacocinética de etanol.
Warfarina: La coadministración de amlodipino con warfarina no modificó el tiempo de respuesta de protombina a la warfarina.
Ciclosporina: No se han llevado a cabo estudios de interacciones medicamentosas con ciclosporina y amlodipino en voluntarios sanos ni en otras poblaciones, con excepción de pacientes de trasplante renal. Varios estudios en pacientes de trasplante renal informan que la coadministración de amlodipino con ciclosporina afecta las concentraciones pico de la ciclosporina desde ningún cambio hasta un aumento promedio del 40%. Debe considerarse el moritoreo de los niveles de ciclosporina en pacientes de trasplante renal con amlodipino.
Tacrolimus: Hay riesgo de un aumento de tacrolimus en los niveles sanguíneos cuando se lo coadministra con amlodipino. A fin de evitar la toxicidad del tacrolimus, para la administración de amlodipino en un paciente bajo tacrolimus, es necesario monitorear los niveles sanguíneos de tacrolimus y el ajuste de la dosis de tacrolimus cuando se estime apropiado.
lnhibidores de la molécula diana de rapamicina en células de mamíferos (mTOR):
Los inhibidores de la mTOR, como el sirolimus, el temsirolimus y el everolimus, son sustratos de CYP3A. La amlodipina es un inhibidor débil de CYP3A. Con el uso concomitante de los inhibidores de la mTOR el amlodipino puede aumentar la exposición de los inhibidores de la mTOR.
Interacciones entre el fármaco y las pruebas de laboratorio: Ninguna conocida.
Interacciones de atorvastatina: El riesgo de miopatía durante tratamiento con inhibidores de la HMG-CoA reductasa se incrementa con la administración concurrente de ciclosporina derivados del ácido fíbrico, dosis modificadoras de lípidos de niacina o inhibidores del transportador de citocromo P450 3A4 (por ejemplo eritromicina y funguicidas azólicos) (ver sección Dosis y vía de administración y Precauciones generales).
Inhibidores de citocromo P450 3A4: Atorvastatina es metabolizada por citocromo P450 3A4. La administración concomitante de atorvastatina e inhibidores de citocromo P450 3A4 puede conducir incrementos en las concentraciones plasmáticas de atorvastatina. El grado de interacción y la potenciación de los efectos depende de la variabilidad del efecto sobre el citocromo P450 3A4.
Eritromicina/claritromicina: La coadministración de atorvastatina y eritromicina (500 mg cuatro veces al día) o claritromicina (500 mg dos veces al día), inhibidores conocidos de citocromo P450 3A4, se asoció con concentraciones plasmáticas más altas de atorvastatina (ver sección Precauciones generales, Efectos en el músculo esquelético y Farmacocinética y farmacodinamia, Propiedades Farmacocinéticas).
Inhibidores de proteasa: La coadministración de atorvastatina con inhibidores de proteasa, inhibidores conocidos de citocromo P450 3A4 se asoció con incremento de concentraciones plasmáticas de atorvastatina (Ver sección Farmacocinética y farmacodinamia, Propiedades farmacocinéticas).
Clorhidrato de diltiazem: La coadministración de atorvastatina (40 mg) con diltiazem (240 mg) se asocian con concentraciones plasmáticas más altas de atorvastatina (ver sección Farmacocinética y farmacodinamia, Propiedades farmacocinéticas).
Cimetidina: Se realizó un estudio de interacción de atorvastatina con cimetidina y no se detectaron interacciones clínicamente significativas (ver sección Farmacocinética y Farmacodinamia, Propiedades farmacocinéticas).
Itraconazol: La administración concomitante de atorvastatina (20 mg a 40 mg) e itraconazol (200 mg) se asoció con un aumento de ABC de atorvastatina (ver sección Farmacocinética y Farmacodinamia, Propiedades farmacocinéticas).
Jugo de toronja: Contiene uno o más componentes que inhiben a CYP 3A4 y puede elevar las concentraciones plasmáticas de atorvastatina en particular cuando se consume jugo de toronja en exceso (> 1.2 L/día) (ver sección Farmacocinética y Farmacodinamia, Propiedades farmacocinéticas).
lnhibidores de transportadores:
Atorvastatina es un sustrato de los transportadores hepáticos (ver sección Propiedades Farmacocinéticas).
La administración concomitante de atorvastatina 10 mg y ciclosporina 5.2 mg/kg/día resultó en un incremento en la exposición a la atorvastatina (relación de ABC: 8.7 ver sección Propiedades Farmacocinéticas). La ciclosporina es un inhibidor del polipéptido transportador de aniones orgánicos 1B1 (OATP1B1), OATP1B3, proteína de resistencia a múltiples medicamentos 1 (MDR1), y la proteína de resistencia del cáncer de mama (BCRP, por sus siglas en inglés), así como del CYP 3A4, de esta manera éste incrementa su exposición a la atorvastatina. No exceder los 10 mg de atorvastatina por día (ver sección Dosis y vía de administración, Uso en combinación con otros compuestos medicinales).
Glecaprevir y pibrentasvir son inhibidores de OATP1B1, OATP1B3, MDR1 y BCRP, de esta manera éstos incrementan la exposición a la atorvastatina. No exceder los 10 mg de atorvastatina por día (ver sección Dosis y vía de administración, Uso en combinación con otros compuestos medicinales).
La administración concomitante de atorvastatina 20 mg y letermovir 480 mg diaria incrementó la exposición a la atorvastatina (cociente del ABC: 3.29; ver sección Propiedades Farmacocinéticas). Letermovir inhibe los transportadores de eflujo P-gp, BCRP, MRP2, OAT2 y el transportador hepático OATP1B1/1B3, por lo tanto, aumenta la exposición a la atorvastatina. No exceda la administración de 20 mg de atorvastatina al día (ver sección Dosis y vía de administración, Uso en combinación con otros compuestos medicinales).
La magnitud de las interacciones medicamentosas mediadas por CYP3A y OATP1B1/1B3 sobre los medicamentos concomitantes puede ser diferente cuando se administra de manera simultánea letermovir con ciclosporina. No se recomienda la administración de atorvastatina a pacientes que se encuentran en tratamiento con letermovir y ciclosporina de manera simultánea.
Elbasvir y grazoprevir son inhibidores de OATP1B1, OATP1B3, MDR1 y BCRP, de esta manera éstos incrementan la exposición a la atorvastatina. Use con precaución y en la menor dosis necesaria (ver sección Dosis y vía de administración, Uso en combinación con otros compuestos medicinales).
Inductores de citocromo P450 3A4: La administración concomitante de atorvastatina e inductores de citocromo P450 3A4 (por ejemplo efavirenz, rifampina) puede conducir a reducciones variables de concentraciones plasmáticas de atorvastatina. Debido al mecanismo de interacción dual de rifampicina (inducción de citocromo P450 3A4 e inhibición del transportador de captación de hepatocitos OATP1B1), se recomienda coadministración simultánea de atorvastatina con rifampicina ya que el retraso de administración de atorvastatina tras administrar rifampicina se ha asociado por reducción significativa de concentraciones plasmáticas de atorvastatina (ver sección Farmacocinética y Farmacodinamia, Propiedades farmacocinéticas).
Antiácidos: La coadministración de atorvastatina con antiácidos orales en suspensión, que contengan hidróxidos de magnesio y aluminio redujo las concentraciones plasmáticas de atorvastatina (proporción de ABC: 0.66). Sin embargo, la reducción de C-LDL no se alteró.
Antipirina: Como atorvastatina no afecta la farmacocinética de antipirina no se esperan interacciones con otros fármacos metabolizados a través de las mismas isoenzimas de citocromo.
Colestipol: Las concentraciones plasmáticas de atorvastatina fueron más bajas (relación de concentración: 0.74) al administrar colestipol con atorvastatina. Sin embargo, los efectos en lípidos fueron mayores cuando atorvastatina y colestipol se coadministraron que cuando cualquiera de estos fármacos se administró solo.
Digoxina: Cuando se coadministraron dosis múltiples de digoxina y 10 mg de atorvastatina las concentraciones plasmáticas de digoxina en estado estable no se vieron afectadas. Sin embargo, las concentraciones de digoxina aumentaron (relación de ABC: 1.15) tras la administración de digoxina con 80 mg de atorvastatina al día. Los pacientes que tomen digoxina deben ser monitoreados de manera adecuada.
Azitromicina: La coadministración de atorvastatina (10 mg una vez al día) con azitromicina (500 mg una vez al día) no modificó las concentraciones plasmáticas de atorvastatina.
Anticonceptivos orales: La coadministración de atorvastatina con un anticonceptivo oral que contiene noretindrona y etinil estradiol aumentó los valores del área bajo la curva de la concentración versus el tiempo (ABC) de noretindrona (relación de ABC: 1.28) y etinilestradiol (relación de ABC: 1.19) respectivamente. Estos incrementos deben tomarse en cuenta al elegir un anticonceptivo oral para mujeres que tomen atorvastatina.
Warfarina: Se realizó un estudio de interacción de atorvastatina con warfarina y no se observaron interacciones clínicamente significativas.
Ácido fusídico: Aunque no se han realizado estudios de interacción con atorvastatina y ácido fusídico, existe un mayor riesgo de rabdomiólisis en pacientes que reciben una combinación de estatinas, incluyendo atorvastatina y ácido fusídico. El mecanismo de interacción se desconoce. En pacientes donde el uso de ácido fusídico sistémico es considerado esencial, el tratamiento con estatinas debe interrumpirse durante el tratamiento con ácido fusídico. La terapia con estatinas puede ser reiniciada siete días después de la última dosis de ácido fusídico.
En circunstancias excepcionales cuando se requiere un uso prolongado de ácido fusídico, como por ejemplo para el tratamiento de infecciones graves, la necesidad de administrar conjuntamente atorvastatina y ácido fusídico debe considerarse caso por caso y bajo estrecha supervisión médica. Se debe informar al paciente de acudir inmediatamente al médico en caso de experimentar algún síntoma de debilidad muscular, dolor o sensibilidad.
Colchicina: Aunque no se han llevado a cabo estudios de interacción con atorvastatina y colchicina, se han reportado casos de miopatía con administración concomitante de atorvastatina con colchicina y la prescripción de atorvastatina con colchicina deberá ser ejercida con precaución.
Otra terapia concomitante: En estudios clínicos atorvastatina se empleó concomitantemente con agentes antihipertensivos y terapia de reemplazo de estrógenos sin evidencia de interacciones adversas clínicamente significativas. No se han realizado estudios de interacción con agentes específicos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Elevación de transaminasas. Elevación de los valores de la creatinina fosfocinasa (ver sección Precauciones generales).
PRECAUCIONES GENERALES:
Uso en pacientes con insuficiencia cardiaca: En un estudio a largo plazo controlado con placebo (PRAISE-2) de pacientes tratados con amlodipino con insuficiencia cardiaca clases III y IV de New York Heart Association (NYHA) de etiología no isquémica, amlodipino se asoció con aumento de reportes de edema pulmonar a pesar de que no hubo diferencia significativa en la incidencia de empeoramiento de insuficiencia cardiaca en comparación con placebo (ver sección Farmacocinética y farmacodinamia).
Uso en pacientes con alteración del funcionamiento hepático (ver sección Contraindicaciones):
Efectos hepáticos: Como ocurre con otros agentes reductores de lípidos de la clase de inhibidores de HMG-CoA reductasa se han reportado elevaciones moderadas (> 3 x LSN) de transaminasas séricas tras terapia con atorvastatina. El funcionamiento hepático se monitoreó antes de que saliera el producto al mercado y también se han realizado estudios clínicos poscomercialización con atorvastatina a dosis de 10 mg, 20 mg, 40 mg y 80 mg.
Ocurrieron incrementos persistentes de transaminasas séricas (> 3 x LSN en dos o más ocasiones) en 0.7% de los pacientes que recibieron atorvastatina en estos estudios clínicos. La incidencia de estas anomalías fue de 0.2%, 0.2%, 0.6% y 2.3% para 10 mg, 20 mg, 40 mg y 80 mg respectivamente. En general los incrementos no se asociaron con ictericia ni otros signos o síntomas clínicos. Al reducir la dosis de atorvastatina o interrumpir o descontinuar el tratamiento farmacológico los niveles de transaminasa regresaron a niveles pretratamiento. La mayoría de los pacientes continuaron con el tratamiento con dosis reducida de atorvastatina sin secuelas.
Deben realizarse pruebas de funcionamiento hepático antes de iniciar el tratamiento y periódicamente después de ello. Los pacientes que desarrollen cualquier signo o síntoma que sugiera lesión hepática deben someterse a pruebas de funcionamiento hepático. Los pacientes que desarrollen incremento a niveles de transaminasa deben ser monitoreados hasta que la(s) anomalía(s) se resuelva(n). En caso de incremento de alanina transaminasa (ALT) o aspartato transaminasa (AST) >3 veces LSN persistente, se recomienda reducción de la dosis o supresión de amlodipino/atorvastatina. Atorvastatina puede provocar elevación de transaminasas (ver sección Reacciones secundarias y adversas).
CADUET® (amlodipino/atorvastatina) debe usarse con cautela en pacientes que consuman cantidades sustanciales de alcohol y/o tengan historia de enfermedad hepática. La enfermedad hepática activa o las elevaciones persistentes o inexplicables de transaminasa son contraindicaciones para el uso de amlodipino/atorvastatina (ver sección Contraindicaciones).
Efectos en el músculo esquelético: Se ha reportado mialgia en pacientes tratados con atorvastatina (ver sección Reacciones secundarias y adversas). La miopatía, que se define como dolores musculares o debilidad muscular junto con elevación de valores de creatina fosfocinasa (CPK) > 10 x LSN, debe considerarse en cualquier paciente con mialgias difusas, sensibilidad o debilidad muscular y/o elevación notable de CPK. Se debe aconsejar al paciente que reporte con prontitud dolor muscular inexplicable, sensibilidad o debilidad en particular si va acompañado de malestar general o fiebre. La terapia con amlodipino/atorvastatina debe descontinuarse si ocurren niveles marcadamente altos de CPK o se diagnostica o se sospecha miopatía. El riesgo de miopatía se incrementa con la administración concurrente de medicamentos que incrementan la concentración sistémica de atorvastatina (ver sección Interacciones medicamentosas y de otro género y Propiedades farmacocinéticas). Muchos de estos fármacos inhiben el metabolismo del citocromo P450 3A4 y/o el transporte del fármaco. CYP 3A4 son las isozimas hepáticas primarias que se sabe participan en la biotransformación de atorvastatina. Los médicos que consideran una terapia combinada con atorvastatina y derivados del ácido fíbrico, eritromicina, fármacos inmunosupresores, fungicidas azólicos inhibidores de la proteasa VIH/VHC, inhibidores VHC NS5A/NS5B, letermovir o dosis modificadoras de lípidos de niacina, deben sopesar cuidadosamente los beneficios y riesgos potenciales y monitorear con cuidado a los pacientes para cualquier signo y síntoma de dolor muscular, sensibilidad o debilidad en particular durante los meses iniciales de la terapia y durante cualquier periodo de titulación ascendente de la dosis de cualquiera de los fármacos. Por lo tanto, también deben considerarse dosis iniciales y de mantenimiento más bajas del componente atorvastatina cuando se tome concomitantemente con los fármacos anteriormente mencionados (ver sección Dosis y vía de administración). No se recomienda el uso concomitante de atorvastatina y ácido fusídico, por lo tanto, se aconseja la suspensión temporal de atorvastatina durante la terapia de ácido fusídico (ver sección Interacciones medicamentosas y de otro género). Las determinaciones periódicas de CPK pueden considerarse en estos casos, pero no hay seguridad de que dicho monitoreo prevenga la ocurrencia de miopatía severa. Amlodipino/atorvastatina puede causar una elevación de CPK debido al componente de atorvastatina (ver sección Reacciones secundarias y adversas).
Ha habido informes muy raros de miopatía necrotizante inmunomediada (MNIM) durante o después del tratamiento con algunas estatinas (ver sección Reacciones secundarias y adversas). La MNIM se caracteriza clínicamente por debilidad muscular proximal persistente y creatina cinasa sérica elevada, que persisten a pesar de la interrupción del tratamiento con estatinas, anticuerpos anti-HMG CoA reductasa positivos y mejoría con agentes inmunosupresores.
Igual que sucede con otros medicamentos en la clase de inhibidores de la HMG-CoA reductasa se ha reportado casos raros de rabdomiólisis con insuficiencia renal aguda secundaria a mioglobinuria. Los antecedentes de deterioro de la función renal pueden constituir un factor de riesgo para el desarrollo de la rabdomiólisis. Estos pacientes ameritan una vigilancia más estrecha de los efectos sobre el músculo esquelético. La terapia con CADUET® (amlodipino/atorvastatina) deberá pausarse temporalmente o descontinuarse en cualquier paciente con afección aguda grave que sugiera miopatía o que tenga un factor de riesgo que le predisponga a desarrollar insuficiencia renal secundaria a rabdomiólisis, (por ejemplo, infección aguda severa, hipotensión, cirugía mayor, traumatismo, trastornos severos de tipo metabólico, endocrino y de electrólitos y convulsiones descontroladas). El control de la hipertensión puede continuarse con la dosis adecuada de amlodipino.
Accidente cerebrovascular hemorrágico: Un análisis post-hoc de un estudio clínico en 4,731 pacientes sin ECC que presentaron accidente cerebrovascular o ataque isquémico transitorio (AIT) en los 6 meses anteriores y en quienes se inició atorvastatina 80 mg, revelaron mayor incidencia de accidente cerebrovascular hemorrágico en el grupo de atorvastatina 80 mg en comparación con placebo (55 atorvastatina vs 33 placebo). Los pacientes con accidente cerebrovascular hemorrágico al comenzar el estudio aparentemente corrieron mayor riesgo de accidente cerebrovascular hemorrágico recurrente (7 atorvastatina contra 2 placebo). Sin embargo, en pacientes tratados con atorvastatina 80 mg hubo menos accidentes cerebrovasculares de cualquier tipo (265 atorvastatina contra 311 placebo) y menos eventos de ECC (123 atorvastatina vs. 204 placebo) (ver sección Propiedades farmacodinámicas - Accidente cerebrovascular recurrente).
Función endocrina:
Se han reportado aumentos en la hemoglobina glicada (HbA 1c) y en las concentraciones de glucosa en ayuno en suero con los inhibidores de la HMG-CoA reductasa, incluyendo atorvastatina. Sin embargo, el beneficio en la reducción de eventos cardiovasculares sobrepasa al riesgo de hiperglucemia.
Efectos en la habilidad para manejar y usar máquinas: Con base en información disponible de amlodipino y atorvastatina, es improbable que este medicamento altere la habilidad del paciente para manejar o usar maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Consideraciones generales: CADUET® (amlodipino/atorvastatina) es un producto combinado que tiene como blanco afecciones cardiovasculares concomitantes/hipertensión/angina y dislipidemia.
El rango de dosificación para amlodipino/atorvastatina es 5 mg/10 mg hasta dosis máxima de 10 mg/80 mg una vez al día. La dosis inicial y la dosis de mantenimiento deben individualizarse basándose tanto en la eficacia como en la tolerancia para cada componente individual en el tratamiento de hipertensión/angina y dislipidemia. Las directrices de tratamiento actualizadas deben consultarse para establecer metas de tratamiento para pacientes basándose en sus características basales. Las dosis pueden tomarse en cualquier hora del día con o sin alimentos.
Como componente de intervención por factores múltiples de riesgo, amlodipino/atorvastatina debe usarse en complemento de medidas no farmacológicas, incluyendo dieta adecuada, ejercicio y reducción de peso en pacientes obesos, sensación de tabaquismo y para tratar problemas médicos subyacentes cuando la respuesta a estas medidas haya sido inadecuada.
Después del inicio y/o titulación de amlodipino/atorvastatina los niveles de lípidos deben analizarse y la PA debe medirse en 2 a 4 semanas, para ajustar en consecuencia la dosis de los componentes amlodipino/atorvastatina. La titulación de respuesta de PA puede realizarse más rápido si es clínicamente necesario.
Tratamiento inicial: Amlodipino/atorvastatina puede emplearse para iniciar tratamiento en pacientes con hiperlipidemia e hipertensión o angina. La dosis inicial recomendada de amlodipino/atorvastatina debe basarse la combinación adecuada de recomendaciones para los componentes amlodipino y atorvastatina consideradas por separado. La dosis máxima de componente amlodipino del producto amlodipino/atorvastatina es 10 mg una vez al día. La dosis máxima del componente atorvastatina del producto amlodipino/atorvastatina es 80 mg una vez al día.
Terapia de reemplazo: Amlodipino/atorvastatina puede sustituirse por sus componentes titulados individualmente. Los pacientes pueden recibir la dosis equivalente de amlodipino/atorvastatina o una dosis de amlodipino/atorvastatina con mayores cantidades de amlodipino, atorvastatina o ambas para efectos anti anginosos adicionales, reducción de PA o efecto de reducción de lípidos.
Amlodipino/atorvastatina puede emplearse como terapia adicional en pacientes que ya estén tomando uno de sus componentes. Como terapia inicial para una indicación y continuación de tratamiento de otra, la dosis inicial recomendada de amlodipino/atorvastatina debe elegirse basándose en continuación de los componentes usados previamente y en dosis inicial recomendada para el componente que se agrega.
Medicamento concomitante (ver Interacciones medicamentosas y de otro género): El componente del producto amlodipino/atorvastatina ha sido coadministrado de manera segura con diuréticos tiazídicos, alfa bloqueadores, beta bloqueadores, inhibidores de la enzima convertidora de la angiotensina (ECA), nitratos de larga duración y nitroglicerina sublingual. Amlodipino/atorvastatina también se ha administrado de manera segura con los medicamentos anteriormente mencionados.
El componente atorvastatina de amlodipino/atorvastatina puede usarse combinado con una resina secuestradora de ácido biliar para efectos aditivos de reducción de lípidos. La combinación de inhibidores de 3-hidroxi-3-metilglutanil-coenzima A (HMG-CoA) reductasa y fibratos en general debe evitarse (ver sección Precauciones generales e Interacciones medicamentosas y de otro género).
Poblaciones especiales y condiciones especiales para dosificación:
Enfermedad arterial coronaria (estudios de amlodipino): En pacientes con EAC el rango de dosificación recomendado es de 5 a 10 mg de amlodipino una vez al día. En estudios clínicos la mayoría de los pacientes requirieron 10 mg una vez al día (ver sección Farmacodinamia de amlodipino/atorvastatina, Uso en pacientes con EAC).
Hipercolesterolemia primaria e hiperlipidemia combinada (mixta) (estudios con atorvastatina): La mayoría de los pacientes se controlan con 10 mg de atorvastatina una vez al día. Es evidente una respuesta terapéutica en un lapso de 2 semanas y la respuesta máxima suele alcanzarse en 4 semanas. La respuesta se mantiene durante la terapia crónica.
Hipercolesterolemia familiar homocigota (estudios con atorvastatina): En un estudio de uso compasivo en pacientes con HF homocigoto la mayoría de los pacientes respondieron a 80 mg de atorvastatina con reducción mayor de 15% de C-LDL (18-45%).
Uso en pacientes con alteración de funcionamiento hepático: Amlodipino/atorvastatina no debe emplearse en pacientes con afección hepática (ver secciones Contraindicaciones y Precauciones generales).
Uso en pacientes con deterioro de la función renal: No se requiere ajuste de dosis en pacientes con deterioro de la función renal (ver secciones Contraindicaciones y Precauciones generales).
Uso en personas de edad avanzada: No se requiere ajuste de dosis en pacientes de edad avanzada.
Uso en niños: No se han realizado estudios para determinar la seguridad o eficacia de amlodipino/atorvastatina (producto combinado) en poblaciones pediátricas. Sin embargo, se han realizado estudios en poblaciones pediátricas con amlodipino solo y atorvastatina sola (ver a continuación).
Estudios con amlodipino: La dosis oral de antihipertensivo recomendada en pacientes pediátricos de 6 a 17 años es 2.5 a 5 mg una vez al día. No se han estudiado dosis en exceso de 5 mg al día en pacientes pediátricos (ver secciones Propiedades Farmacodinámicas y Propiedades Farmacocinéticas).
El efecto de amlodipino sobre la PA en pacientes de menos de 6 años se desconoce.
Estudios con atorvastatina:
Uso en pacientes pediátricos con dislipidemias severas:
Para pacientes de 10 años o más la dosis inicial recomendada de atorvastatina es 10 mg/día. La dosis puede aumentarse a 80 mg al día según la respuesta y la tolerancia. Las dosis deben individualizarse de acuerdo con la meta de terapia recomendada (ver secciones Indicaciones terapéuticas, Propiedades Farmacodinámicas). Deben realizarse ajustes e intervalos de 4 semanas o más.
La experiencia en pacientes pediátricos menores de 10 años se deriva de estudios abiertos (ver secciones Reacciones secundarias y adversas y Farmacocinética y Farmacodinamia).
Uso en combinación con otros compuestos medicinales:
Estudios con atorvastatina:
En casos en que es necesario coadministrar atorvastatina con ciclosporina, telaprevir la combinación tipranavir/ritonavir o glecaprevir/pibrentasvir, la dosis de atorvastatina no debe exceder 10 mg.
No se recomienda el uso de atorvastatina en pacientes que reciben letermovir de manera simultánea con ciclosporina.
También se han observado interacciones medicamentosas farmacocinéticas que resultan en un aumento de la concentración sistémica de atorvastina con otros inhibidores de la proteasa del virus de la inmunodeficiencia humana (VIH) (lopinavir/ritonavir, saquinavir/ritonavir, darunavir/ritonavir, fosamprenavir, fosamprenavir/ ritonavir y nelfinavir), inhibidor de la proteasa de la hepatitis C (VHC) (boceprevir, elbasvir/grazoprevir, simeprevir), claritromicina, itraconazol y letermovir. Se debe tener precaución al co-prescribir atorvastatina y se recomienda realizar la evaluación clínica apropiada para garantizar que se emplea la menor dosis necesaria de atorvastatina (ver secciones Precauciones generales, Efectos músculo esqueléticos e lnteracciones medicamentosas y de otro género).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No se tiene información sobre sobredosis con CADUET® (amlodipino/atorvastatina) en humanos.
Debido al extenso enlace del fármaco CADUET® (amlodipino/atorvastatina) con las proteínas plasmáticas no se espera que la hemodiálisis aumente significativamente la depuración de amlodipino/atorvastatina (ver sección Propiedades Farmacocinéticas, Insuficiencia renal).
Datos adicionales sobre ingestión de amlodipino sugieren que la sobredosificación exagerada podría provocar vasodilatación periférica excesiva y posiblemente taquicardia refleja. La hipotensión sistémica marcada y prolongada que quizá llegue hasta un choque con resultado mortal ha sido reportada. La administración de carbón activado en voluntarios saludables inmediatamente o hasta 2 horas tras la ingestión de amlodipino 10 mg se ha demostrado reduce significativamente la absorción de amlodipino. En algunos casos podría ser conveniente el lavado gástrico. La hipotensión clínicamente significativa por sobredosis de amlodipino requiere de apoyo cardiovascular activo incluyendo monitoreo frecuente de funcionamiento cardiaco y respiratorio, elevación de extremidades y atención al volumen de líquido en circulación y la producción de orina. Un vasoconstrictor podría ser útil para restaurar el tono vascular y la PA, siempre y cuando no haya contraindicaciones para su uso. El gluconato de calcio intravenoso podría ser benéfico al invertir los efectos de bloqueo del canal del calcio.
Datos adicionales sobre ingestión de atorvastatina sugieren que no hay tratamiento específico para sobredosis de atorvastatina. En caso de ocurrir sobredosis el paciente deberá ser tratado sintomáticamente instituyendo las medidas de apoyo necesarias.
PRESENTACIONES:
Cajas con 10 y 30 tabletas.
Tabletas de 5/10 mg: (5 mg de amlodipino y 10 mg de atorvastatina).
Tabletas de 5/20 mg: (5 mg de amlodipino y 20 mg de atorvastatina).
Tabletas de 5/40 mg: (5 mg de amlodipino y 40 mg de atorvastatina).
Tabletas de 5/80 mg: (5 mg de amlodipino y 80 mg de atorvastatina).
RECOMENDACIONES SOBRE ALMACENAMIENTO: Conserve a no más de 30 °C.
Protéjase de la luz.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para el profesional de la salud. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo y la lactancia.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
MEX.AEReporting@pfizer.com o a la línea 800 401 2002
PFIZER, S.A. de C.V.
Km. 63 Carretera México Toluca,
Zona Industrial, C.P. 50140,
Toluca, México, México.
Reg. Núm. 204M2004, SSA IV
®Marca Registrada


