
CAPRELSA
VANDETANIB
Tabletas recubiertas
1 Caja , 30 Tabletas , 100 Miligramos
1 Caja , 30 Tabletas , 300 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Vandetanib 100 y 300 mg
Excipiente, c.b.p. 1 tableta.
INDICACIONES TERAPÉUTICAS: CAPRELSA® está indicado en el tratamiento de pacientes con cáncer medular de tiroides no resecable localmente avanzado o metastásico.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética: La farmacocinética de vandetanib con la dosis de 300 mg en pacientes con MTC se caracteriza por una depuración de ~13.2 L/h, un volumen de distribución de ~7,450 L y una vida media plasmática de ~19 días.
Absorción: Después de la administración oral, la absorción de vandetanib es lenta, con concentraciones plasmáticas pico que se alcanzan por lo general a una mediana de tiempo de 6 horas, con un intervalo de 4-10 horas, después de la administración. Vandetanib se acumula ~8 veces con administración múltiple y se alcanza el estado estacionario desde los ~2 meses.
Distribución: Vandetanib se une a la albúmina sérica humana y a la glucoproteína ácida a1, con una unión in vitro a proteínas de ~90%. En muestras de plasma ex vivo de pacientes con cáncer colorrectal con exposición en estado estacionario después de 300 mg una vez al día, el porcentaje medio de unión a proteínas fue de 93.7% (intervalo de 92.2 a 95.7%).
Metabolismo: Después de la administración oral de 14C-vandetanib, se detectó vandetanib original y los metabolitos de vandetanib N-óxido de vandetanib y N-desmetil-vandetanib en plasma, orina y heces. El conjugado glucurónido se observó como un metabolito secundario únicamente en excrementos. N-desmetil-vandetanib es producido principalmente por CYP3A4 y N-óxido de vandetanib por las enzimas monooxigenasas que contienen flavina FMO1 y FMO3. N-desmetil-vandetanib y N-óxido de vandetanib circulan a concentraciones de ~11% y 1.4% de la concentración de vandetanib.
Excreción: En un periodo de recolección de 21 días después de una dosis única de 14C-vandetanib, se recuperó ~69%, 44% en heces y 25% en orina. La excreción de la dosis fue lenta y se esperaba que hubiera excreción adicional después de los 21 días, de acuerdo con la vida media plasmática.
Vandetanib no fue un sustrato de hOCT2 expresado en células HEK293. Vandetanib fue un inhibidor de OCT2, al inhibir la absorción del sustrato marcador selectivo de OCT2, 14C-creatinina, por células HEK-OCT2, con una IC50 media de aproximadamente 2.1 µg/mL. Esto es más elevado que las concentraciones plasmáticas de vandetanib (~0.81 y 0.32 µg/mL) observadas después de la administración de varias dosis de 300 y 100 mg. La inhibición de la excreción renal de creatinina por vandetanib ofrece una explicación de los incrementos en la creatinina plasmática observados en sujetos humanos que recibían vandetanib.
Farmacodinamia: Vandetanib es un inhibidor selectivo de la tirosina cinasa que inhibe la actividad tirosina cinasa del receptor 2 de VEGF estimulada por el factor de crecimiento endotelial vascular (VEGF) en células endoteliales. Vandetanib inhibe la migración, proliferación, supervivencia y formación de nuevos vasos sanguíneos en modelos in vitro de angiogénesis. In vivo, la administración de vandetanib redujo la angiogénesis inducida por células tumorales, la permeabilidad de vasos tumorales y la densidad de microvasos tumorales, además inhibió el crecimiento tumoral y la metástasis en modelos de xenoinjertos humanos de cáncer de pulmón en ratones atímicos.
Además, vandetanib inhibe a la tirosina cinasa receptora EGF estimulada por el factor de crecimiento epidérmico (EGF) en células tumorales y en células endoteliales. Vandetanib inhibe la proliferación de células estimuladas por EGFR y la supervivencia celular in vitro.
En los estudios in vitro se ha mostrado que vandetanib también inhibe la actividad de otras tirosina cinasas, entre ellas, la codificada por el gen rearreglado durante la transfección (RET) y el receptor 3 de VEGF (Flt-4).
Datos clínicos de cáncer medular de tiroides: Se llevó a cabo un estudio aleatorizado, doble ciego, controlado con placebo (Estudio 58) para demostrar la seguridad y eficacia de CAPRELSA® 300 mg frente a placebo en 331 pacientes con cáncer medular de tiroides (MTC) no resecable, localmente avanzado o metastásico.
El objetivo principal de este estudio fue demostrar una mejoría en la supervivencia sin progresión (PFS) con CAPRELSA® en comparación con placebo. Los criterios de valoración secundarios fueron la evaluación de la tasa de respuesta objetiva global (ORR), la tasa de control de la enfermedad (DCR), definida como enfermedad estable (SD), respuesta parcial (RP) o respuesta completa (CR) que se prolongue por 12 semanas, duración de la respuesta (DOR) y supervivencia global (OS). La respuesta bioquímica con CAPRELSA® en comparación con placebo, de acuerdo con la medición de calcitonina (CTN) y antígeno carcinoembrionario (CEA), también se evaluó como criterio de valoración secundario.
Los pacientes se trataron con CAPRELSA® o placebo hasta que llegaran a la progresión objetiva de la enfermedad. Una vez en progresión objetiva de la enfermedad, basada en la evaluación del investigador, los pacientes se suspendían del tratamiento de estudio ciego y se les daba la opción de recibir CAPRELSA® de manera abierta.
El resultado del análisis primario de PFS mostró una mejora estadísticamente significativa en PFS para pacientes aleatorizados a vandetanib en comparación con placebo (Cociente de riesgos instantáneos (HR) = 0.46; intervalo de confianza del 95% (IC) = 0.31-0.69; p = 0.0001).
La mediana de PFS para pacientes aleatorizados a placebo fue 19.3 meses. La mediana de PFS para pacientes aleatorizados a CAPRELSA® no se ha alcanzado, sin embargo, con base en la modelación estadística de los datos observados hasta el 43.º percentil, el valor pronosticado de la mediana de PFS es de 30.5 meses con un intervalo de confianza de 95%, de 25.5 a 36.5 meses. A los 12 meses, la proporción de pacientes vivos y sin progresión fue de 63 (63%) pacientes aleatorizados a placebo y de 192 (83%) pacientes aleatorizados a vandetanib. Para vandetanib, un total de 73 (32%) pacientes habían progresado; 64 (28%) por progresión RECIST y 9 (4%) por muerte en ausencia de progresión. Los restantes 158 pacientes (68%) se censuraron en el análisis de PFS. Para placebo, un total de 51 (51%) pacientes habían progresado; 46 (46%) por progresión RECIST y 5 (5%) por muerte en ausencia de progresión. Los restantes 49 pacientes (49%) se censuraron en el análisis de PFS.
Fig. 1. Gráfica de Kaplan Meier de supervivencia sin progresión
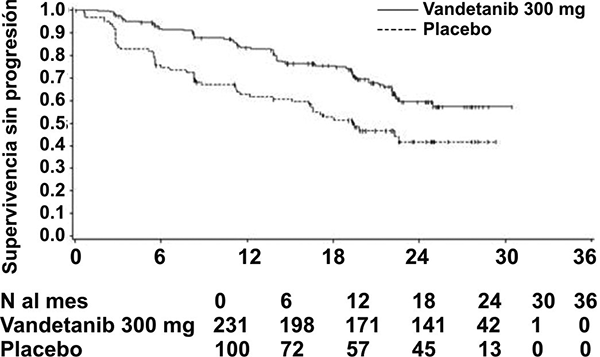
Al momento del análisis primario de PFS (fecha de corte de datos 31 de julio de 2009), 48 (15%) pacientes habían muerto, y no hubo diferencia significativa en la supervivencia global entre grupos de tratamiento (Cociente de riesgos instantáneos = 0.89, IC 99.98% = 0.28 - 2.85; p = 0.712). Al momento de este análisis, 32 pacientes (14%) en el grupo de vandetanib y 16 pacientes (16%) en el grupo de placebo habían muerto.
También se observaron ventajas estadísticamente significativas para vandetanib en los criterios de valoración secundarios de tasa de respuesta, tasa de control de la enfermedad, respuesta bioquímica y tiempo hasta el empeoramiento del dolor (TWP), como se muestra en la tabla 3. Los resultados en la tasa de respuesta y en la tasa de control de la enfermedad provienen del análisis de intención de tratamiento, en el que se incluyen pacientes que cruzaron del tratamiento ciego al tratamiento abierto con vandetanib antes de la progresión, de acuerdo con la evaluación de la lectura central. De los 13 pacientes que experimentaron una respuesta después de la aleatorización a placebo, 12 pacientes experimentaron la respuesta sólo después de recibir vandetanib en forma abierta. Los datos para calcitonina y respuesta de CEA, así como el tiempo hasta el empeoramiento del dolor, provienen únicamente de la fase aleatorizada del estudio.
Tabla 3. Resumen de resultados clave de eficacia en el estudio 58
|
Supervivencia sin progresión |
n |
PFS mediana |
HRa |
IC 95% |
Valor de p |
|
Vandetanib 300 mg |
73/231 (32%) |
No se alcanzó (pronosticado 30.5 meses) |
0.46 |
0.31, 0.69 |
0.0001 |
|
Placebo |
51/100 (51%) |
19.3 meses |
|||
|
Tasa de respuesta objetivab |
n |
Tasa de respuesta |
ORd |
IC 95% |
Valor de p |
|
Vandetanib 300 mg |
104/231 |
45% |
5.48 |
2.99, 10.79 |
< 0.0001 |
|
Placebo |
13/100 |
13% |
|||
|
Tasa de control de la enfermedadc |
n |
Tasa de respuesta |
ORd |
IC 95% |
Valor de p |
|
Vandetanib 300 mg |
200/231 |
87% |
2.64 |
1.48, 4.69 |
0.001 |
|
Placebo |
71/100 |
71% |
|||
|
Respuesta de CTN (calcitonina) |
n |
Tasa de respuesta |
ORd |
IC 95% |
Valor de p |
|
Vandetanib 300 mg |
160/231 |
69% |
72.9 |
26.2, 303.2 |
< 0.0001 |
|
Placebo |
3/100 |
3% |
|||
|
Respuesta de CEA (antígeno carcinoembrionario) |
n |
Tasa de respuesta |
ORd |
IC 95% |
Valor de p |
|
Vandetanib 300 mg |
119/231 |
52% |
52.0 |
16.0, 320.3 |
< 0.0001 |
|
Placebo |
2/100 |
2% |
|||
|
Supervivencia global |
n |
OS mediana |
HRa |
IC 99.98% |
Valor de p |
|
Vandetanib 300 mg |
32/231 (14%) |
No se alcanzó |
0.89 |
0.28, 2.85 |
0.712 |
|
Placebo |
16/100 (16%) |
No se alcanzó |
|||
|
Tiempo hasta el empeoramiento del dolore |
n |
Mediana de TWP |
HR |
IC 97.5% |
Valor de p |
|
Vandetanib 300 mg |
114/231 (49%) |
7.85 meses |
0.61 |
0.43, 0.87 |
0.006 |
|
Placebo |
57/100 (57%) |
3.25 meses |
a HR = Cociente de riesgos instantáneos. Un valor < 1 favorece a CAPRELSA®. El análisis se realizó mediante una prueba del orden logarítmico con el tratamiento como el único factor.
b La tasa de respuesta objetiva es la proporción de pacientes con la mejor respuesta objetiva de respuesta completa (CR) o respuesta parcial (PR).
c La tasa de control de la enfermedad es la proporción de pacientes con la mejor respuesta objetiva de respuesta completa, respuesta parcial o Enfermedad Estable (SD) a las 24 semanas.
d OR = Cociente de probabilidades. Un valor >1 favorece a vandetanib. El análisis se realizó mediante modelo de regresión logístico con el tratamiento como el único factor.
e TWP (Tiempo hasta el empeoramiento del dolor) fue un criterio de valoración compuesto, derivado del uso de analgésicos opioides y del inciso sobre el peor dolor en el cuestionario del Índice Breve del Dolor (BPI).
N, número de eventos/número de pacientes aleatorizados; OS, supervivencia global; PFS, supervivencia sin progresión; IC, intervalo de confianza.
No hay evidencia de una relación entre el estatus de mutación en RET y la eficacia de vandetanib.
Con diferentes duraciones de exposición, los niveles medianos de hemoglobina en pacientes tratados con vandetanib se incrementaron en 5-15 g/L en comparación con los valores iniciales. Los datos de animales sugieren que esto puede deberse al incremento en la producción hepática de eritropoyetina en pacientes que toman vandetanib.
CONTRAINDICACIONES: CAPRELSA® no se debe administrar en pacientes con hipersensibilidad conocida a la sustancia activa, vandetanib, ni a cualquiera de sus excipientes.
CAPRELSA® no se debe administrar en pacientes con síndrome de intervalo QT prolongado congénito.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No hay estudios adecuados y bien controlados en mujeres embarazadas que tomen CAPRELSA®. Con base en datos preclínicos, CAPRELSA® puede causar daño fetal cuando se administra en mujeres embarazadas, ya que el riesgo de que vandetanib se asocie con anormalidades del desarrollo se predice como elevado. Como se esperaba a partir de sus acciones farmacológicas, vandetanib ha mostrado importantes efectos en todas las etapas de reproducción femenina en ratas (véase Datos de seguridad preclínica).
Si CAPRELSA® se usa durante el embarazo o si una paciente se embaraza mientras toma CAPRELSA®, se le debería alertar sobre el riesgo potencial para el feto o el riesgo potencial de pérdida del embarazo. El tratamiento se debería continuar en mujeres embarazadas sólo si el beneficio potencial para la madre sobrepasa el riesgo para el feto. Las mujeres con capacidad reproductiva deberán usar un método anticonceptivo eficaz durante la terapia y durante al menos tres meses después de la última dosis de CAPRELSA®.
Lactancia: No hay datos sobre el uso de CAPRELSA® en mujeres en lactancia. Se aconseja a las madres lactantes suspender la lactancia mientras reciben terapia de CAPRELSA®. En ratas, vandetanib se excretó en la leche y se encontró en el plasma de crías después de la administración en ratas lactantes (véase Datos de seguridad preclínica).
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen global de reacciones adversas al medicamento: Las reacciones adversas al medicamento (RAM) reportadas más comúnmente han sido diarrea, exantema, náusea, hipertensión y cefalea.
Reacciones adversas al medicamento durante estudios clínicos: Se han identificado las siguientes reacciones adversas en estudios clínicos con pacientes que reciben CAPRELSA® como tratamiento para el cáncer medular de tiroides. Su frecuencia se presenta en la tabla 1 Reacciones adversas al medicamento, mediante la clasificación de frecuencia de CIOMS III y enlistadas posteriormente por SOC de MedDRA y con el nivel de término preferente. Las frecuencias de incidencia de efectos indeseables se definen como muy común (³ 1/10), común (> 1/100, < 1/10), poco común (³ 1/1,000, < 1/100), raro (³ 1/10,000, < 1/1,000), muy raro (<1/10,000) incluidos reportes aislados. En esta sección se incluyen sólo datos derivados de estudios concluidos en donde se conoce la exposición del paciente.
Tabla 1. Reacciones adversas al medicamento
|
SOC de MedDRA |
Término de MedDRA |
Descriptor CIOMS/frecuencia |
|
Trastornos cardiacos |
Prolongación del intervalo QT en el ECG |
Común |
|
Insuficiencia cardiaca, insuficiencia cardiaca aguda |
Poco común |
|
|
Trastornos gastrointestinales |
Diarrea |
Muy común |
|
Náusea |
Muy común |
|
|
Vómito |
Muy común |
|
|
Dolor abdominal |
Muy común |
|
|
Estomatitis |
Común |
|
|
Boca seca |
Común |
|
|
Pancreatitis |
Poco común |
|
|
Trastornos generales |
Fatiga |
Muy común |
|
Astenia |
Muy común |
|
|
Exploraciones complementarias |
Disminución de peso |
Común |
|
Incremento de ALT y AST séricas |
Común |
|
|
Incremento de hemoglobina |
Poco común |
|
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito |
Muy común |
|
Hipocalemia |
Muy común |
|
|
Deshidratación |
Común |
|
|
Trastornos endocrinos |
Hipotiroidismo |
Común |
|
Trastornos psiquiátricos |
Insomnio |
Muy común |
|
Depresión |
Común |
|
|
Trastornos renales y urinarios |
Proteinuria |
Común |
|
Nefrolitiasis |
Común |
|
|
Hematuria |
Común |
|
|
Trastornos respiratorios |
Epistaxis |
Común |
|
Trastornos de la piel y del tejido subcutáneo |
Exantema u otras reacciones cutáneas (incluido acné, piel seca, dermatitis, pruritis) |
Muy común |
|
Reacción de fotosensiblidad |
Muy común |
|
|
Síndrome de eritrodisestesia palmo-plantar |
Común |
|
|
Trastornos oculares |
Visión borrosa |
Común |
|
Opacidad córnea |
Común |
|
|
Conjuntivitis |
Común |
|
|
Ojos secos |
Común |
|
|
Deficiencia visual |
Común |
|
|
Trastornos del sistema nervioso |
Cefalea |
Muy común |
|
Disgeusia |
Común |
|
|
Trastornos vasculares |
Hipertensión |
Muy común |
|
Afecciones isquémicas cerebrovasculares |
Común |
|
|
Crisis hipertensiva |
Común |
Eventos como Torsade de pointes, síndrome de Stevens-Johnson, eritema multiforme y síndrome de leucoencefalopatía posterior reversible han ocurrido en pacientes tratados con monoterapia de vandetanib. Se espera que éstos sean eventos poco comunes en pacientes que reciben vandetanib para cáncer medular de tiroides.
Los eventos oculares como visión borrosa son comunes en pacientes que recibieron CAPRELSA® para cáncer medular de tiroides. Las exploraciones programadas con lámpara de hendidura han revelado opacidades corneales (queropatías en vórtex) en pacientes tratados; sin embargo, no se requiere de exploraciones de rutina con lámpara de hendidura para pacientes que reciben CAPRELSA®.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Vandetanib no ha mostrado potencial mutagénico o clastogénico.
En un modelo animal de cicatrización, el límite de rotura dérmica se redujo en ratones tratados con vandetanib en comparación con los controles. Esto sugiere que vandetanib hace más lenta la cicatrización, pero no la impide. No se ha determinado el intervalo apropiado entre la suspensión de CAPRELSA® y la posterior cirugía programada para evitar los riesgos de una cicatrización deficiente. En estudios clínicos de CAPRELSA®, un pequeño número de pacientes se sometieron a cirugía mientras tomaban CAPRELSA® y no hubo reportes de complicaciones en la cicatrización.
Toxicología reproductiva: Vandetanib no tuvo efecto en la fertilidad de ratas macho. En un estudio de fertilidad femenina hubo una tendencia hacia el incremento en la irregularidad del ciclo estral, ligera reducción en la incidencia de embarazos y aumento de las pérdidas de implantación. En un estudio de toxicidad de dosis repetidas en ratas, hubo una disminución en el número de cuerpos lúteos en los ovarios de ratas tratadas con vandetanib durante 1 mes.
En ratas, la toxicidad embriofetal fue evidente por la pérdida fetal, retardo del desarrollo fetal, anormalidades de los vasos cardiacos y osificación precoz de algunos huesos craneales. En un estudio de desarrollo pre y postnatal en ratas, con dosis que producen toxicidad materna durante la gestación y/o lactancia, vandetanib incrementó las pérdidas previas al nacimiento y redujo el crecimiento postnatal de las crías. En ratas, vandetanib se excretó en la leche y se encontró en el plasma de crías después de la administración en ratas lactantes.
No se han llevado a cabo estudios de carcinogenicidad con vandetanib.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: En sujetos sanos, no se ha mostrado interacción clínicamente significativa entre vandetanib e itraconazol, inhibidor potente de CYP3A4. En sujetos de sexo masculino, la exposición a vandetanib se redujo en 40% cuando se administró de manera conjunta con rifampicina, inductor potente de CYP3A4. Se debe evitar la administración de vandetanib con inductores potentes de CYP3A4.
La exposición a CAPRELSA® no se ve afectada por la comida.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Los resultados de laboratorio se muestran en la tabla 2.
Tabla 2. Resultados de laboratorio
|
Resultado de laboratorio |
|
|
Proteína en orina por tira reactiva1 |
Muy común |
|
Sangre en orina por tira reactiva1 |
Muy común |
|
Incremento de TSH sérica |
Muy común |
|
Incremento de amilasa sérica |
Muy común |
|
Incremento de lipasa sérica |
Muy común |
|
Incremento de hemoglobina |
Muy común |
|
Incremento de creatinina sérica2 |
Muy común |
1 En la tabla 2 se representa la incidencia de resultados de laboratorio en un estudio clínico aleatorizado de cáncer medular de tiroides, no de eventos adversos reportados.
2 Los incrementos en creatinina sérica fueron de grado CTCAE 1-2 y podrían estar relacionados con la inhibición de la proteína transportadora humana OCT2 (véase Propiedades farmacocinéticas).
PRECAUCIONES GENERALES:
Prolongación del segmento QTc: Se ha observado la prolongación del intervalo QTc en el electrocardiograma en pacientes que reciben CAPRELSA® (véase Efectos indeseables). Con una dosis de 300 mg por día en cáncer medular de tiroides, se observó prolongación confirmada del intervalo QTc en el electrocardiograma, en un estudio de fase III en 8% de los pacientes. La prolongación del electrocardiograma QTc parece ser dependiente de la dosis y se puede manejar con monitoreo adecuado, interrupción o reducción de la dosis según sea apropiado.
Se ha reportado de forma poco común Torsade de pointes y taquicardia ventricular en pacientes que recibieron CAPRELSA® 300 mg.
El tratamiento con CAPRELSA® no se debería iniciar en pacientes cuyo intervalo QT en electrocardiograma corregido se confirme como superior a 480 ms. CAPRELSA® no se debería administrar en pacientes que tienen antecedentes de Torsade de pointes, a menos que todos los factores de riesgo que contribuyeron al Torsade se hayan corregido. CAPRELSA® no se ha estudiado en pacientes con arritmias ventriculares ni infarto de miocardio reciente.
Se debería obtener un electrocardiograma, así como los niveles séricos de potasio, calcio, magnesio y TSH al inicio, a las 2-4 semanas y a las 8-12 semanas de iniciar tratamiento con CAPRELSA®, así como cada 3 meses durante por lo menos un año después. También se deberían obtener ECG y pruebas sanguíneas, según indicación clínica durante este periodo y posteriormente. El nivel sérico de potasio se debería mantener en 4 mEq/L o más y el magnesio y calcio séricos se deberían mantener dentro de los intervalos normales para reducir el riesgo de prolongación de QT en electrocardiograma.
CAPRELSA® se puede administrar con medicamentos que prolonguen el intervalo QT en el electrocardiograma si no existe terapia alterna adecuada. Si se administran tales medicamentos a pacientes que ya están recibiendo CAPRELSA®, se debería realizar el monitoreo del intervalo QT en el ECG según sea apropiado para la farmacocinética del medicamento añadido.
Los pacientes que desarrollan un solo valor de intervalo QT en electrocardiograma corregido de por lo menos 550 ms o un valor repetido de por lo menos 500 ms, deberían suspender la toma de CAPRELSA®. La administración de CAPRELSA® se puede reanudar con una dosis reducida después de que se haya confirmado que el intervalo QTc en el electrocardiograma ha regresado al estado inicial.
Reacciones cutáneas: Se ha observado exantema y otras reacciones cutáneas (entre ellas, reacciones de fotosensibilidad y síndrome de eritrodisaestesia palmar-plantar) en pacientes que han recibido CAPRELSA®.
Las reacciones cutáneas leves a moderadas pueden generalmente manejarse con tratamiento sintomático o por reducción de la dosis. Las reacciones cutáneas más severas (como el síndrome de Stevens-Johnson) pueden requerir de glucocorticoides sistémicos y suspensión permanente de CAPRELSA®.
Debe tenerse precaución con la exposición solar mediante el uso de ropa protectora y/o bloqueadores solares.
Diarrea: Se ha observado diarrea en pacientes que reciben CAPRELSA®. Se recomiendan antidiarreicos de rutina para el tratamiento de la diarrea. Se deben monitorear los electrólitos séricos según sea adecuado. Si se desarrolla diarrea severa (grado CTCAE 3-4) se debería suspender CAPRELSA® hasta que la diarrea mejore. Después de mejorar, se debe reanudar el tratamiento con CAPRELSA® a una dosis reducida (véase Posología y método de administración, y Efectos indeseables).
Hipertensión: Se ha observado hipertensión, incluida crisis hipertensiva, en pacientes tratados con CAPRELSA®; los pacientes se deben monitorear para detectar hipertensión y controlarla de manera adecuada. Si no se puede controlar la presión arterial elevada con manejo médico, no se deberá reanudar CAPRELSA®, sino hasta que la presión arterial se controle médicamente. Podría ser necesario reducir la dosis (véase Efectos indeseables).
Insuficiencia cardiaca: Se ha observado diarrea en pacientes que reciben CAPRELSA®. La suspensión temporal o permanente de CAPRELSA® puede ser necesaria en pacientes con insuficiencia cardiaca. Podría no ser reversible con la suspensión de CAPRELSA®. Algunos casos han sido fatales.
Elevaciones de alanina aminotransferasa: En pacientes tratados con CAPRELSA® ocurren comúnmente elevaciones de alanina aminotransferasa. La mayoría de las elevaciones se resolverán mientras se continúa con el tratamiento con CAPRELSA®, otras se resuelven por lo general después de 1-2 semanas de interrupción de la terapia. Se recomienda el monitoreo periódico de la alanina aminotransferasa en pacientes que toman CAPRELSA®.
Enfermedad pulmonar intersticial: Se ha observado enfermedad pulmonar intersticial (ILD) en pacientes que reciben CAPRELSA® y en algunos casos ha sido fatal. Si un paciente presenta síntomas respiratorios como disnea, tos o fiebre, se debe interrumpir CAPRELSA® e iniciarse una investigación inmediata. Si se confirma que es ILD, se ha de suspender CAPRELSA® en forma permanente y tratar al paciente de manera adecuada.
Síndrome de leucoencefalopatía posterior reversible: El síndrome de leucoencefalopatía posterior reversible (RPLS), un síndrome de edema vasogénico subcortical diagnosticado mediante una IRM del cerebro, se ha observado de manera poco frecuente en pacientes que reciben tratamiento con CAPRELSA® en combinación con quimioterapia o en pacientes pediátricos con tumores cerebrales que reciben CAPRELSA® como monoterapia. Se ha observado RPLS en pacientes que reciben vandetanib. Este síndrome se debe considerar en cualquier paciente que presente convulsiones, cefalea, alteraciones visuales, confusión o función mental alterada.
Insuficiencia renal: La dosis inicial se debería reducir a 200 mg en pacientes con insuficiencia renal moderada (depuración de creatinina ³ 30 a < 50 mL/min).
DOSIS Y VÍA DE ADMINISTRACIÓN:
Administración en adultos: CAPRELSA® 300 mg tabletas orales una vez al día. La administración también puede ser de 3 tabletas por 100 mg una vez al día.
Las tabletas de CAPRELSA® se pueden tomar con o sin alimentos.
Las tabletas de CAPRELSA® también se pueden dispersar en medio vaso (2 onzas fluidas o 50 mL) de agua para beber no carbonatada. No se debe emplear ningún otro líquido. La tableta se pone en el agua, sin triturarla, se agita hasta que se disperse (aproximadamente 10 minutos) y la dispersión resultante se toma inmediatamente. Cualquier residuo en el vaso se ha de mezclar con medio vaso de agua y tomarse. El líquido también puede administrarse a través de sondas nasogástricas o tubos de gastrostomía.
Duración: CAPRELSA® se puede administrar hasta que los pacientes con cáncer medular de tiroides ya no se beneficien del tratamiento.
Omisión de dosis: Si un paciente omite una dosis, deberá tomar la siguiente dosis diaria según la prescripción.
Ajustes de la dosis: En caso de toxicidad o prolongación del intervalo QT en el electrocardiograma de grado CTCAE 3 o mayor, se deberá suspender temporalmente la administración de vandetanib y reanudarse con una dosis inferior cuando la toxicidad se haya resuelto o haya mejorado hasta grado CTCAE 1 (véase también Advertencias especiales y precauciones especiales de uso). La dosis diaria de 300 mg se puede reducir a 200 mg (dos tabletas de 100 mg) y luego hasta 100 mg si es necesario.
Poblaciones de pacientes especiales:
Niños o adolescentes: CAPRELSA® no está indicado para su uso en pacientes pediátricos, ya que no se ha establecido la seguridad y eficacia de CAPRELSA® en niños.
Pacientes de edad avanzada (> 65 años): No se requiere del ajuste de la dosis para pacientes de edad avanzada. Existen datos clínicos limitados en pacientes con edades superiores a 75 años.
Insuficiencia renal: Existen datos clínicos limitados en pacientes con insuficiencia renal moderada. Sin embargo, los datos indican que los pacientes con insuficiencia renal leve tienen un perfil de seguridad similar al de pacientes con función renal normal. Estos datos clínicos junto con los datos farmacocinéticos de voluntarios sugieren que no se requiere que haya cambios en la dosis inicial en pacientes con insuficiencia renal leve. La dosis inicial se debería reducir a 200 mg en pacientes con insuficiencia renal moderada (depuración de creatinina ³ 30 a < 50 mL/min). El uso de vandetanib no se recomienda en pacientes con insuficiencia renal severa (depuración inferior a 30 mL/min), puesto que existen datos limitados en pacientes con insuficiencia renal severa y no se ha establecido la seguridad y eficacia. Un estudio farmacocinético sugiere que en voluntarios con insuficiencia renal severa, la exposición a vandetanib se puede incrementar hasta 2 veces.
Insuficiencia hepática: Los datos farmacocinéticos de voluntarios sugieren que no se requiere que haya cambios en la dosis inicial en pacientes con insuficiencia hepática leve, moderada o severa. Existen datos limitados en pacientes con insuficiencia hepática (bilirrubina sérica superior a 1.5 veces el límite superior de lo normal). CAPRELSA® no está indicado para su uso en pacientes con insuficiencia hepática, puesto que no se ha establecido la seguridad ni la eficacia.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No hay tratamiento específico en el caso de sobredosis con CAPRELSA® y no se han establecido los posibles síntomas de sobredosis. Con dosis múltiples y con dosis superiores a 300 mg se observó un incremento en la frecuencia y severidad de algunas reacciones adversas como exantema, diarrea e hipertensión tanto en estudios con voluntarios sanos como en pacientes. Además, se debe considerar la posibilidad de prolongación del segmento QT y de Torsade de pointes.
Las reacciones adversas asociadas con sobredosis se han de tratar de manera sintomática, en particular, la diarrea severa se debe manejar adecuadamente. En caso de sobredosis, se deben interrumpir las dosis posteriores de CAPRELSA® y se han de tomar medidas apropiadas para asegurar que no haya ocurrido un evento adverso, es decir, registrar un ECG en un periodo posterior de 24 horas para determinar si hay prolongación del segmento QTc.
PRESENTACIONES:
Caja con 30 tabletas con 100 mg.
Caja con 30 tabletas con 300 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a temperatura ambiente a no más de 30°C.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos. No se use en el embarazo ni en la lactancia. Este medicamento deberá ser administrado únicamente por médicos especialistas en oncología y con experiencia en quimioterapia antineoplásica.

ASTRAZENECA, S. A. de C. V.
Súper Av. Lomas Verdes No. 67
Fracc. Lomas Verdes
C.P. 53120, Naucalpan de Juárez, México
Reg. Núm. 113300EL870026
133300EL870007RM/2013


