
COLMESDANT
COLISTINA
Solución inyectable
1 Caja, 1 Frasco ámpula con liofilizado,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Hecha la mezcla, el frasco ámpula contiene:
Colistimetato de sodio equivalente a 150 mg de colistina base activa
Vehículo cbp 2 mL
Forma farmacéutica: Solución.
INDICACIONES TERAPÉUTICAS: COLMESDANT® está indicado en el tratamiento de infecciones agudas o crónicas causadas por variedades susceptibles de ciertos bacilos gram negativos, tales como Enterobacter aerogenes, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa y otros bacilos gram negativos no fermentadores.
COLMESDANT® podrá usarse en el tratamiento de infecciones producidas por bacterias gram negativas susceptibles solamente cuando otros antibióticos potencialmente efectivos y menos tóxicos estén contraindicados o sean ineficaces. No obstante, COLMESDANT® puede ser de utilidad en el tratamiento de infecciones producidas por bacterias gram negativas multirresistentes, tales como las infecciones de vías respiratorias bajas producidas por Pseudomonas aeruginosa en pacientes que padecen de fibrosis quística.
COLMESDANT® no está indicado en infecciones producidas por bacterias de los géneros Proteus, Providencia, Serratia, Neisseria o Bacteroides.
FARMACOCINÉTICA Y FARMACODINAMIA:
Descripción: El colistimetato sódico es un producto antibiótico parenteral estéril que, al ser reconstituido, es adecuado para su administración intramuscular o intravenosa. Cada frasco ámpula contiene el equivalente a 150 mg de colistina base activa. El colistimetato sódico es un antibiótico polipéptido del grupo de las polimixinas obtenido de cultivos de Bacillus polymoxa var. Colistinus.
La colistina, también conocida como polimixina E, está relacionada estructural y farmacológicamente a la polimixina B.
La colistina se encuentra disponible comercialmente para su aplicación parenteral en forma de colistimetato sódico (derivado sulfametilo de la colistina). El colistimetato sódico es un polvo fino, blanco o ligeramente amarillento, ampliamente soluble en agua. Tras su reconstitución con agua inyectable estéril, la solución de colistimetato sódico tiene un pH de 7-8.
Absorción: El colistimetato sódico no se absorbe en el tracto digestivo, por lo que debe ser aplicado por vía parenteral.
Las concentraciones séricas y urinarias típicas después de una dosis única de 150 mg de colistimetato inyectable IV o IM en sujetos adultos normales se muestran en la figura 1.
Figura 1
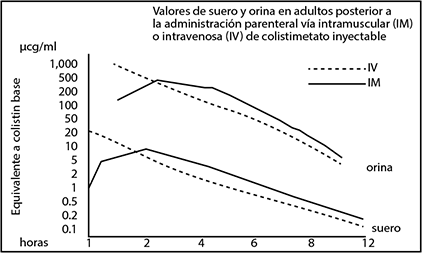
La concentración pico se obtiene a los 10 minutos después de la administración IV y antes de dos horas con la administración IM. Los niveles pico promedian 30 mcg/mL tras la administración IV y entre 5 y 7.5 mcg/mL tras la administración IM. Hay concentraciones séricas antimicrobianas detectables hasta 12 horas después de la administración. Estudios en pacientes críticamente enfermos mostraron que se requiere una dosis de carga para alcanzar las concentraciones deseadas en las primeras 24 horas de tratamiento. Los regímenes de dosificación que emplean intervalos de tiempo más cortos pueden ser favorecidos.
Distribución: Tras la administración IV o IM, el colistimetato se distribuye ampliamente en los tejidos corporales; las concentraciones del antibiótico obtenidas en el líquido sinovial, pleural o pericárdico son insignificantes. Los estudios en animales revelan que, al igual que la polimixina B, la colistina se une reversiblemente y persiste en tejidos tales como el hígado, riñones, pulmones, corazón y músculos. El colistimetato sódico se une en más del 50% a proteínas.
Las concentraciones de colistimetato obtenidas en el líquido cefalorraquídeo son mínimas aún con inflamación de las meninges. El colistimetato cruza la barrera placentaria; se obtienen concentraciones sanguíneas de alrededor de 1 mcg/mL en el feto después de la administración intravenosa de 150 mg a la madre.
Eliminación: La vida media plasmática del colistimetato tras su administración IV o IM es de 1.5 a 8 horas en adultos con función renal normal. Las concentraciones séricas tienden a declinar más rápidamente en población pediátrica que en los adultos. Las concentraciones séricas llegan a ser más altas y la vida media del medicamento es más prolongada en personas con alteraciones en la función renal. En personas con depuración de creatinina por debajo de los 20 mL/min, la vida media del colistimetato es de entre 10 y 20 horas. En pacientes anúricos la vida media puede extenderse hasta 48 a 72 horas.
El colistimetato sódico es hidrolizado in vivo a colistina y posiblemente a otros metabolitos con menos substituciones en los grupos amino. El grado y extensión de la hidrólisis, así como los metabolitos específicos resultantes de la misma y su actividad antibacteriana no han sido determinados a la fecha.
El colistimetato sódico y sus metabolitos son excretados principalmente por vía renal mediante filtración glomerular. La actividad antimicrobiana en la orina es habitualmente mayor que en el suero. Después de la administración IV o IM de 150 mg de colistimetato sódico a personas con función renal normal en una única dosis, las concentraciones del medicamento en orina tras dos horas de la administración van de 200 a 270 mcg/mL, y de 15 a 25 mcg/mL tras 8 horas. Se desconoce si el colistimetato sódico es removido por hemodiálisis o diálisis peritoneal.
Mecanismo de acción: La colistina es bactericida, mediante la unión al lipopolisacárido de la membrana de los bacilos gram negativos, provocando lisis celular por el desplazamiento de cationes.
Microbiología: La actividad antimicrobiana se asocia con mejor relación de las áreas bajo la curva (AUC)/concentración mínima inhibitoria (CMI). El Instituto de Estándares Clínicos y de Laboratorio (CLSI) recomienda los siguientes puntos de corte:
|
Susceptible |
Intermedio |
Resistente |
|
|
P. aeuruginosa |
≤ 2 mcg/mL |
4 mcg/mL |
8 mcg/mL |
|
Acinetobacter |
≤ 2 mcg/mL |
≥ 4 mcg/mL |
|
|
Enterobacterias |
Sin recomendación |
La colistina se usa mejor como parte de una combinación altamente activa, especialmente cuando se trata de una infección causada por un organismo con una CMI > 0.5 mcg/mL.
El efecto post-antibiótico alcanza concentraciones séricas antimicrobianas detectables hasta 12 horas después de la administración.
Estudios de toxicidad en animales:
Toxicidad aguda: La dosis letal (LD50) intravenosa fue de 41.5 mg/kg en pruebas realizadas con perros y 739 mg/kg en pruebas realizadas con ratones; la toxicidad intramuscular fue de 42 mg/kg en pruebas realizadas con perros y 267 mg/kg en pruebas realizadas con ratones.
Toxicidad subaguda: En conejos albinos y perros Beagle, dosis intravenosas de 5, 10 y 20 mg/kg/día durante 28 días resultaron en BUN (nitrógeno de urea sanguínea) elevado en pruebas realizadas con perros (grupos de dosis de 10 mg/kg/día) y en ambos grupos de dosis de 20 mg/kg.
CONTRAINDICACIONES: El uso de COLMESDANT® está contraindicado en pacientes con historia de hipersensibilidad al colistimetato sódico, aminoglucósidos o a las polimixinas, en embarazo, lactancia y menores de 12 años.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Categoría C: No se ha establecido la seguridad del uso de COLMESDANT® en mujeres embarazadas.
Recordando que el medicamento cruza parcialmente la barrera placentaria, se recomienda que el medicamento se utilice solamente cuando los beneficios potenciales justifiquen el posible riesgo al feto.
Se desconoce si el colistimetato sódico se distribuye en la leche materna; sin embargo, se ha detectado sulfato de colistina en la leche materna, por lo que se recomienda ejercer precaución al indicar COLMESDANT® en mujeres lactantes.
REACCIONES SECUNDARIAS Y ADVERSAS: Los efectos adversos reportados con el colistimetato sódico son similares a los reportados con el sulfato de polimixina B. La neurotoxicidad y la nefrotoxicidad son los efectos adversos potencialmente más serios; la probabilidad de que ocurran es mayor cuando el medicamento se administra a dosis superiores a las recomendadas o en pacientes con alteraciones de la función renal.
Toxicidad renal: Se ha reportado nefrotoxicidad, manifestada por reducción en el gasto urinario, concentraciones séricas elevadas de BUN y creatinina, proteinuria, hematuria y cilindros en la orina en pacientes sometidos a tratamiento con dosis convencionales de COLMESDANT®. Se han reportado casos de necrosis tubular aguda no necesariamente precedida por reducción progresiva de la función renal. La nefrotoxicidad es habitualmente reversible al suspender la administración del medicamento; sin embargo, se han reportado cifras elevadas de azoados en suero hasta 1 a 2 semanas después de la suspensión del medicamento. La administración de dosis excesivas del medicamento de acuerdo a la función renal puede resultar en mayor deterioro de la misma.
Efectos gastrointestinales: Se han reportado alteraciones gastrointestinales en pacientes que reciben COLMESDANT®. Dado que la colitis y diarrea asociadas con Clostridium difficile (colitis pseudomembranosa) se han asociado con el uso de diversos antibióticos de amplio espectro, deberá ser considerado como parte del diagnóstico diferencial en un paciente que recibe COLMESDANT® y desarrolla diarrea.
Neurotoxicidad: Pueden ocurrir alteraciones neurológicas transitorias. Éstas incluyen parestesias circumorales, adormecimiento u hormigueo de las extremidades o lengua, mareo, vértigo, ataxia, visión borrosa y dislalia. Estas manifestaciones generalmente aparecen dentro de los primeros cuatro días de tratamiento y desaparecen al suspenderse el mismo. De aparecer este tipo de efectos, el tratamiento no necesariamente deberá discontinuarse, pero sí deberá vigilarse estrechamente al paciente. En algunas ocasiones los síntomas mejoran al reducir la dosis del medicamento.
Algunos efectos neurotóxicos más severos pueden presentarse, incluyendo confusión, coma, psicosis y convulsiones, particularmente en pacientes que están recibiendo dosis altas o en pacientes con disfunción renal.
El bloqueo neuromuscular, que puede resultar en paro respiratorio, puede presentarse en pacientes que reciben COLMESDANT®, particularmente si el medicamento es utilizado en pacientes que sufren de padecimientos neuromusculares, tales como Miastenia gravis o en personas que reciben bloqueadores musculares, anestésicos y otros medicamentos que pueden producir o exacerbar un bloqueo neuromuscular (ver Interacciones medicamentosas y de otro género). La apnea y el bloqueo neuromuscular asociados a la administración de COLMESDANT® se han reportado principalmente en pacientes con disfunción renal previa en los que la dosis del medicamento no se ajustó convenientemente.
Otros efectos secundarios: Se han reportado prurito generalizado, urticaria, rash y fiebre medicamentosa en pacientes bajo tratamiento con COLMESDANT®. También se han reportado disfonía y dolor en el sitio de aplicación IM del medicamento.
Aunque no se ha establecido una relación causal definitiva, se han reportado ocasionalmente casos de leucopenia, granulocitopenia y hepatotoxicidad en pacientes bajo tratamiento con COLMESDANT®.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: No se han realizado estudios a largo plazo de los efectos carcinogenéricos o mutagénicos del colistimetato sódico.
No se observaron efectos adversos sobre la fertilidad o la capacidad reproductiva en ratas a las que se administraron dosis de 9.3 mg/kg/día de colistimetato sódico, equivalentes al 30% de la dosis máxima en humanos calculada en mg/m2.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Dado que los efectos nefrotóxico y neurotóxicos pueden ser aditivos, el uso concurrente o secuencial de COLMESDANT® con otros medicamentos con un perfil de toxicidad similar (aminoglucósidos, anfotericina B, capreomicina, metoxiflurano, polimixina B, vancomicina) debe ser evitado en la medida de lo posible.
Los agentes que producen bloqueo neuromuscular (tubocurarina, succinilcolina, decametonio) potencian el efecto bloqueante del colistimetato sódico, por lo que deberán utilizarse con precaución.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Se han reportado las siguientes alteraciones de laboratorio:
Pruebas de función renal: Elevación de BUN y creatinina sérica, reducción en la depuración de creatinina, proteinuria y cilindruria.
Pruebas de función hepáticas: Elevación de transaminasas y fosfatasa alcalina.
Hematológicas: Leucopenia y granulocitopenia.
PRECAUCIONES GENERALES: La dosis máxima diaria no debe exceder de 5 mg/Kg/día de colistina base activa en personas con función renal normal.
Debido a que el colistimetato inyectable es eliminado principalmente por excreción renal, debe ser usado con precaución cuando existe la posibilidad de insuficiencia renal. La disminución de la función renal en edad avanzada debe ser considerada.
Cuando se presenta insuficiencia renal, puede usarse el colistimetato inyectable, pero debe tenerse la mayor precaución y la dosis debe reducirse en proporción al nivel de insuficiencia (ver Dosis y vía de administración). La administración de cantidades de colistimetato inyectable en exceso de la capacidad excretoria renal llevará a concentraciones séricas elevadas y puede resultar en insuficiencia renal, y la posterior concentración del antibiótico a niveles tóxicos en el cuerpo. En este punto puede ocurrir bloqueo neuromuscular resultando en debilidad muscular y apnea. Si se presenta apnea, puede ser manejada con respiración asistida, oxígeno e inyecciones de cloruro de calcio. El bloqueo neuromuscular inducido por el colistimetato sódico es no competitivo y no responde a la neostigmina.
Algunas señales fácilmente reconocibles del desarrollo de insuficiencia renal son: disminución de la emisión de orina, incremento en BUN y creatinina sérica. Si se presentan, la terapia con colistimetato inyectable debe descontinuarse inmediatamente. Ahora bien, si existe una infección que pone en peligro la vida y para la cual no hay alternativas de tratamiento, la terapia puede ser retomada a una dosis menor de acuerdo a la depuración de creatinina.
Precauciones en población pediátrica: Se ha administrado COLMESDANT® a neonatos, lactantes, niños y adolescentes. El perfil de efectos adversos en estas poblaciones parece ser similar al observado en adultos. Dado que los síntomas de toxicidad no van a ser reportados por pacientes de corta edad, se recomienda un monitoreo estrecho de la función renal.
Precauciones en población geriátrica: Los estudios clínicos con colistimetato no han incluido una población suficientemente grande de personas con edades por arriba de los 65 años que permitan determinar si tienen una respuesta diferente a la de personas más jóvenes. La evidencia con la que se cuenta sugiere que éste no es el caso.
En general, la dosificación en personas de avanzada edad deberá ser cuidadosa e iniciar en los rangos bajos del esquema recomendado, tomando en cuenta la mayor frecuencia de disfunción renal, hepática o cardiaca, de comorbilidad y uso de otros medicamentos que se observa en esta población.
Dado que los ancianos tienen grados variables de disfunción renal, deberá tenerse precaución con la dosificación, y monitorear estrechamente la función renal a lo largo del tratamiento.
DOSIS Y VÍA DE ADMINISTRACIÓN: COLMESDANT® es suministrado en frascos ámpula que contienen colistimetato sódico equivalente a 150 mg de colistina base activa por frasco.
Reconstitución y administración: COLMESDANT® puede administrarse por vía IV o IM.
El frasco ámpula de 150 mg debe ser reconstituido con 2 mL de agua esterilizada para inyección. La solución resultante proporciona colistimetato sódico en una concentración de 75 mg/mL.
Durante la reconstitución, agitar el frasco suavemente para evitar la formación de espuma.
Administración intravenosa: La administración intravenosa podrá hacerse mediante bolos intermitentes o infusión continua:
a. Administración intravenosa: Inyectar directo en la vena, lentamente, una mitad de la dosis total diaria por un periodo de 3 a 5 minutos, cada 12 horas.
b. Infusión continua:
a Inicialmente, se aplicará un bolo directo en la vena por 3 a 5 minutos consistente en la mitad de la dosis total diaria.
b La mitad restante de la dosis total diaria de colistimetato sódico será diluido con una de las siguientes soluciones:
NaCI al 0.9% (solución fisiológica o salina isotónica).
Dextrosa al 5.0% con NaCI al 0.9% (solución mixta).
Dextrosa al 5% (solución glucosada).
5% dextrosa en 0.45 NaCI.
5% dextrosa en 0.225% NaCI.
Solución lactada de Ringer.
10% solución de azúcar invertida.
c No hay suficiente información para recomendar el uso del colistimetato sódico con alguna solución distinta de las listadas arriba.
d La infusión de la segunda mitad de la dosis total diaria ya diluida deberá iniciarse 1 a 2 horas después del bolo inicial, y mantenerse por las siguientes 22 a 23 horas.
e La elección de la solución intravenosa y el volumen a ser empleado deberá definirse en función de los requisitos hídricos del paciente.
f Cualquier infusión que contenga colistimetato sódico estéril deberá ser utilizada no más de 24 horas después de haberse preparado.
Administración intramuscular: Para la inyección intramuscular, inyecte profundamente en una masa muscular grande (como los músculos de los glúteos o la región lateral del muslo).
Guía de dosificación: La dosis de colistimetato sódico (CMS) es expresa como colistina base activa (CBA). Cada vial contiene 150 mg de colistina base activa.
Cálculo de la dosis de carga (todos los pacientes):
Dosis de carga de CBA (mg) = 5.0 x Kg de peso.
*** Use el peso menor ideal o actual.
*** No exceda los 300 mg.
Dosis de mantenimiento: La primera dosis puede ser administrada 24 horas después de la dosis de carga. Cálculo de la dosis de mantenimiento en pacientes sin terapia de reemplazo renal.
Dosis diaria de colistina base activa (CBA) (mg) = 2.5 x (1.5 x Crcl + 30).
La dosis diaria se administrará de manera dividida de acuerdo al intervalo ajustado a depuración de creatinina (mL/min/1.73 m2) que se describe a continuación:
|
Depuración de creatinina (CrCL) |
Frecuencia de la administración |
|
< 10 mL/min/1.73 m2 |
cada 12 horas |
|
10-70 mL/min/1.73 m2 |
cada 8-12 horas |
|
> 70 mL/min/1.73 m2 |
cada 8 horas |
Dosis en pacientes en hemodiálisis intermitente: Los días en los que no se realiza la hemodiálisis, la dosis diaria es de 75 mg, administrados como 37.5 mg cada 12 horas.
En los días que se realiza la hemodiálisis, se administra un 30% adicional de la dosis diaria de mantenimiento después de hemodiálisis administrando, primero 37.5 mg y 12 horas después del procedimiento, una segunda dosis de 60 mg.
Dosis de mantenimiento en pacientes con terapia de reemplazo renal continuo: La dosis diaria es de 480 mg administrados como 160 mg cada 8 horas.
Pacientes pediátricos: La dosis para niños que pesan menos de 40 Kg es de 2.5 a 5 mg/Kg/día dividido en 2 a 3 dosis.
Para los niños que pesen más de 40 Kg, se deberá considerar la dosis de adulto.
No hay ninguna recomendación de dosis de carga en niños.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: La sobredosis de colistimetato sódico puede provocar bloqueo neuromuscular caracterizado por parestesia, letargo, confusión, mareo, ataxia, nistagmus, trastornos del habla y apnea.
La parálisis de los músculos respiratorios puede causar apnea, paro respiratorio y muerte.
La sobredosis también puede producir insuficiencia renal aguda, manifestada por disminución del volumen urinario e incremento de las concentraciones de creatinina y nitrógeno en urea (BUN) séricos.
Cuando ocurra sobredosis, debe ser descontinuada y se deben implementar medidas de soporte. No se sabe si el colistimetato sódico puede ser removido por hemodiálisis y diálisis peritoneal.
Ver la sección de Interacciones medicamentosas y de otro género para uso junto con medicamentos curariformes y la sección de Dosis y vía de administración para el uso en insuficiencia renal.
PRESENTACIÓN: Caja de cartón con un frasco ámpula con liofilizado e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: El liofilizado sin reconstituir deberá almacenarse a no más de 30°C y en lugar seco.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos. Dilúyase con 2 ml de agua estéril para uso inyectable. No se use durante el embarazo o lactancia. Si no se administra todo el producto, deséchese el sobrante. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se administre si el cierre ha sido violado. No exceder las dosis recomendadas. ANTIBIÓTICO: El uso incorrecto de este producto puede causar resistencia bacteriana.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
ESPECÍFICOS STENDHAL, S.A. de C.V.
Escorpión Lote 10, Fraccionamiento Industrial San Isidro,
C.P. 56506, La Paz, México.
Reg. Núm. 152M2015, SSA IV


