
COZAAR - Comprimidos
Sustancia(s):
- Losartán
Presentaciones:
- 1 Caja, 21 Comprimidos, 12.5 mg
- 1 Caja, 15 Comprimidos, 50 mg
- 1 Caja, 15 Comprimidos, 100 mg
- 1 Caja, 30 Comprimidos, 50 mg
- 1 Caja, 30 Comprimidos, 100 mg
III. FORMA FARMACÉUTICA Y FORMULACIÓN
Cada COMPRIMIDO contiene como ingrediente activo:
Losartán potásico 12.5 mg, 50 mg, 100 mg
Excipiente, c.b.p. 1 comprimido
IV. INDICACIONES TERAPÉUTICAS
COZAAR® (losartán potásico), el primero de una nueva clase de agentes para el tratamiento de la hipertensión, en un antagonista del receptor de angiotensina tipo II (tipo AT1). COZAAR® también brinda una reducción en el riesgo combinado de muerte cardiovascular, apoplejía e infarto al miocardio en pacientes hipertensos con hipertrofia ventricular izquierda y protección renal para pacientes con diabetes tipo 2 y proteinuria.
Hipertensión
COZAAR® está indicado para el tratamiento de la hipertensión.
Reducción del riesgo de morbilidad y mortalidad cardiovascular en pacientes hipertensos con hipertrofia ventricular izquierda
Protección renal en pacientes diabéticos tipo 2 con proteinuria: COZAAR® está indicado para retardar la progresión de enfermedad renal determinada por la reducción en la incidencia combinada de duplicación de creatinina sérica, enfermedad renal terminal (necesidad de diálisis o trasplante de riñón) o muerte; y para reducir la proteinuria.
Insuficiencia cardiaca
COZAAR® está indicado para el tratamiento de la insuficiencia cardiaca, cuando el tratamiento con un inhibidor de la ECA ya no se considera apropiado. No se recomienda cambiar a tratamiento con COZAAR® a pacientes con insuficiencia cardiaca que estén estables con un inhibidor de la ECA.
V. FARMACOCINÉTICA Y FARMACODINAMIA
MECANISMO DE ACCIÓN
La angiotensina II, un potente vasoconstrictor, es la hormona activa principal del sistema renina-angiotensina y un importante factor determinante de la fisiopatología de la hipertensión. Se une a los receptores AT1 existentes en muchos tejidos (p. ej., músculo liso vascular, glándulas suprarrenales, riñones y corazón) e induce varias acciones biológicas importantes, como vasoconstricción y liberación de aldosterona. También estimula la proliferación de las células musculares lisas. Se ha identificado un segundo receptor de angiotensina II, el subtipo AT2, pero no tiene ningún papel conocido en la homeostasis cardiovascular.
El losartán es un compuesto sintético potente, activo por vía oral. Los bioensayos de fijación y farmacológicos han mostrado que se une selectivamente a los receptores AT1. In vitro e in vivo, tanto el losartán como su metabolito ácido carboxílico farmacológicamente activo (E-3174) bloquean todas las acciones de importancia fisiológica de la angiotensina II, independientemente del origen o de la vía de síntesis de ésta. En contraste con algunos péptidos antagonistas de la angiotensina II, el losartán no tiene ningún efecto agonista.
El losartán se une selectivamente a los receptores AT1, y no se une ni bloquea a otros receptores hormonales o canales de iones importantes en la regulación cardiovascular. Además, no inhibe la ECA (cininasa II), la enzima que degrada la bradicinina. Por consiguiente, el losartán no tiene efectos que no estén directamente relacionados con el bloqueo de los receptores AT1, como la potenciación de los efectos mediados por la bradicinina o la generación de edema (losartán, 1.7%; placebo, 1.9%).
FARMACOCINÉTICA
ABSORCIÓN
El losartán administrado por vía oral se absorbe bien y sufre un metabolismo de primer paso en el que se forman un metabolito ácido carboxílico activo y otros metabolitos inactivos. La biodisponibilidad sistémica del losartán administrado en comprimidos es de 33% aproximadamente. El losartán alcanza concentraciones máximas promedio en el plasma en una hora, y su metabolito activo en tres a cuatro horas. Cuando se administró losartán con una comida estandarizada, no hubo ningún cambio clínicamente significativo en su curva de concentración plasmática.
DISTRIBUCIÓN
El 99% o más del losartán y de su metabolito activo se unen a las proteínas plasmáticas, principalmente a la albúmina. El volumen de distribución del losartán es de 34 litros. Los estudios en ratas indican que el losartán atraviesa poco o nada la barrera hematoencefálica.
METABOLISMO
Alrededor de 14% de una dosis intravenosa u oral de losartán es convertida en su metabolito activo. Tras la administración oral o intravenosa de losartán potásico marcado con 14C, la radiactividad del plasma es atribuida principalmente al losartán y a su metabolito activo. En 1% aproximadamente de los sujetos estudiados, la conversión de losartán en su metabolito activo fue mínima. Además del metabolito activo, se forman metabolitos inactivos, que incluyen dos metabolitos principales por hidroxilación de la cadena lateral butílica y un metabolito menor, un glucurónido N-2-tetrazólico.
ELIMINACIÓN
La depuración plasmática del losartán es de unos 600 mL/min, y la de su metabolito activo, de unos 50 mL/min. Sus depuraciones renales son, respectivamente, de unos 74 mL/min y 26 mL/min. Aproximadamente 4% de una dosis de losartán administrada por vía oral es excretada sin cambio con la orina y 6% en forma de su metabolito activo. Con las dosis orales de losartán de hasta 200 mg, la farmacocinética del losartán y de su metabolito activo es lineal.
Tras su administración por vía oral, las concentraciones plasmáticas de losartán y de su metabolito activo van disminuyendo de manera poliexponencial, con una semivida terminal de unas dos horas y de seis a nueve horas, respectivamente. Durante la administración de 100 mg una vez al día ni el losartán ni su metabolito activo se acumulan significativamente en el plasma.
El losartán y sus metabolitos se eliminan principalmente por las vías biliar y urinaria. En el hombre, después de una dosis oral de losartán marcado con 14C alrededor de 35% de la radiactividad se recupera en la orina y 58% en las heces, y después de una dosis intravenosa aproximadamente 43% de la radiactividad se recupera en la orina y 50% en las heces.
FARMACODINAMIA
El losartán inhibe las respuestas presoras sistólica y diastólica a la administración de angiotensina II por goteo intravenoso. En el momento de su efecto máximo, 100 mg de losartán inhiben esas respuestas un 85% aproximadamente y 24 horas después de la administración de dosis únicas o múltiples la inhibición es de 26-39%.
Durante la administración de losartán, la supresión de la retroalimentación negativa de la angiotensina II sobre la secreción de renina causa un aumento de la actividad de la renina plasmática, lo cual, a su vez, aumenta la angiotensina II en el plasma. Durante el tratamiento prolongado (seis semanas) de pacientes hipertensos con 100 mg diarios de losartán, la concentración plasmática de angiotensina II aumentó aproximadamente al doble o al triple en el momento de la concentración máxima del medicamento en el plasma. En algunos pacientes se observaron aumentos mayores, en particular durante el tratamiento a corto plazo (dos semanas). Sin embargo, la actividad antihipertensiva y la supresión de la concentración plasmática de aldosterona eran apreciables a las dos y a las seis semanas, lo cual indica un bloqueo efectivo de los receptores de angiotensina II. Después de suspender la administración de losartán la actividad de la renina plasmática y las concentraciones de angiotensina II disminuyeron hasta sus niveles anteriores al tratamiento en un término de tres días.
Como el losartán es un antagonista específico de los receptores de angiotensina II del tipo AT1, no inhibe la ECA (cininasa II), la enzima que degrada la bradicinina. En un estudio en el que se compararon los efectos de 20 mg y 100 mg de losartán potásico y de un inhibidor de la ECA sobre las respuestas a la angiotensina I, la angiotensina II y la bradicinina, el losartán bloqueó las respuestas a la angiotensina I y a la angiotensina II sin afectar las respuestas a la bradicinina, lo cual concuerda con su mecanismo de acción específico. En contraste, el inhibidor de la ECA bloqueó las respuestas a la angiotensina I y aumentó las respuestas a la bradicinina sin alterar la respuesta a la angiotensina II, lo cual proporciona una distinción farmacodinámica entre el losartán y los inhibidores de la ECA.
Las concentraciones plasmáticas de losartán y de su metabolito activo y el efecto antihipertensivo del losartán son mayores a medida que se aumenta la dosis. Como tanto el losartán como su metabolito activo son antagonistas de los receptores de angiotensina II, ambos contribuyen al efecto antihipertensivo.
En un estudio en hombres sanos, la administración de dosis únicas de 100 mg de losartán potásico con dietas altas y bajas en sal no alteró la tasa de filtración glomerular, el flujo plasmático renal efectivo ni la fracción de filtración. El losartán tuvo un efecto natriurético que fue más pronunciado con una dieta baja en sal y no pareció estar relacionado con una inhibición de la reabsorción temprana de sodio en el túbulo proximal y causó también un aumento transitorio de la excreción de ácido úrico en la orina.
En pacientes hipertensos no diabéticos con proteinuria (≥ 2 g/24 horas) tratados durante ocho semanas, la administración de 50 mg diarios de losartán potásico, aumentados hasta 100 mg diarios, disminuyó significativamente la proteinuria (42%) y la excreción fraccional de albúmina y de IgG, mantuvo la tasa de filtración glomerular y disminuyó la fracción de filtración.
En mujeres posmenopáusicas hipertensas tratadas durante cuatro semanas, la administración de 50 mg diarios de losartán potásico no tuvo ningún efecto sobre las concentraciones renales o sistémicas de prostaglandinas.
El losartán no tiene ningún efecto sobre los reflejos autonómicos ni ningún efecto sostenido sobre la noradrenalina plasmática.
A dosis de hasta 150 mg una vez al día, el losartán potásico no causó cambios clínicamente importantes en las concentraciones en ayunas de triglicéridos, colesterol total, colesterol de HDL y glucosa en pacientes hipertensos. Las mismas dosis de losartán no tuvieron efecto sobre los niveles de glucosa plasmática.
En general, el losartán causó una disminución del ácido úrico en el suero (usualmente < 0.4 mg/dL), que persistió durante el tratamiento prolongado. En los estudios clínicos controlados en pacientes hipertensos, en ningún caso se suspendió el tratamiento por aumento de la creatinina o del potasio séricos.
En un estudio de diseño paralelo de 12 semanas de duración en pacientes con insuficiencia ventricular izquierda (clases funcionales II a IV de la New York Heart Association), la mayoría de los cuales estaban recibiendo diuréticos y/o digitálicos, se comparó el losartán potásico a dosis de 2.5, 10, 25 y 50 mg una vez al día con un placebo. Las dosis de 25 mg y 50 mg tuvieron efectos hemodinámicos y neurohormonales positivos que se mantuvieron durante todo el estudio. Las respuestas hemodinámicas se caracterizaron por un aumento del índice cardiaco y disminuciones de la presión capilar pulmonar en cuña, la resistencia vascular sistémica, la presión arterial sistémica promedio y la frecuencia cardiaca. En esos pacientes con insuficiencia cardiaca, la aparición de hipotensión estuvo relacionada con la dosis. Los efectos neurohormonales se caracterizaron por una disminución de las concentraciones circulantes de aldosterona y de noradrenalina.
ESTUDIOS CLÍNICOS
ESTUDIOS EN HIPERTENSIÓN
La eficacia antihipertensiva de COZAAR® fue demostrada en 11 estudios controlados que incluyeron 1 679 pacientes tratados con COZAAR®, 471 tratados con un placebo y 488 tratados con diversos medicamentos de comparación. En los pacientes con hipertensión esencial leve a moderada, la administración de COZAAR® una vez al día produjo disminuciones estadísticamente significativas de las presiones sistólica y diastólica. El efecto antihipertensivo se mantuvo en los estudios clínicos hasta por un año.
La comparación de las presiones arteriales en el momento de la concentración mínima del medicamento en el plasma (24 horas después de la dosis) y durante su concentración máxima (5-6 horas después de la dosis) demostró una reducción relativamente suave de la presión en el transcurso de 24 horas. El efecto antihipertensivo fue paralelo al ritmo diario natural. La reducción de la presión al final del intervalo entre las dosis fue aproximadamente 70-80% de la observada 5-6 horas después de la dosis. El efecto antihipertensivo máximo se alcanzó tres a seis semanas después de iniciar el tratamiento. A pesar de la significativa disminución de la presión arterial, COZAAR® no tuvo ningún efecto clínicamente significativo sobre la frecuencia cardiaca. La suspensión de la administración de losartán en los pacientes hipertensos no causó un aumento brusco de la presión.
La administración de 50-100 mg de COZAAR® una vez al día tuvo un efecto antihipertensivo significativamente mayor que la de 50-100 mg de captopril una vez al día. El efecto antihipertensivo de 50 mg de COZAAR® una vez al día fue similar al de 20 mg de enalapril una vez al día. El de 50-100 mg de COZAAR® una vez al día fue similar al de 50-100 mg de atenolol una vez al día, y en pacientes hipertensos de edad avanzada (≥ 65 años) fue equivalente al de 5-10 mg de felodipina de liberación prolongada después de 12 semanas de tratamiento.
COZAAR® tiene la misma eficacia en los pacientes hipertensos de uno y otro sexo, y en más jóvenes (< 65 años) o más viejos (≥ 65 años). Aunque COZAAR® tuvo efecto antihipertensivo en todas las razas estudiadas, como sucede con otros medicamentos que afectan el sistema renina-angiotensina el promedio de respuesta a la monoterapia con losartán fue menor en los hipertensos de raza negra que en los de otras razas.
Cuando se asocia COZAAR® con un diurético tiacídico, sus efectos antihipertensivos son aproximadamente aditivos.
Como COZAAR® bloquea selectivamente los receptores de AII, es de esperarse que no provoque tos. En un estudio controlado de ocho semanas en pacientes hipertensos con antecedentes de tos durante el tratamiento con un inhibidor de la ECA, la incidencia de la tos fue similar en los que recibieron COZAAR® o un medicamento no inhibidor de la ECA (hidroclorotiazida), y significativamente menor que en los que volvieron a tomar un inhibidor de la ECA. Además, en un análisis global de 16 estudios clínicos por el método doble ciego en 4 131 pacientes, la incidencia de tos reportada espontáneamente por los pacientes fue similar con COZAAR® (3.1%), con placebo (2.6%) o con la hidroclorotiazida (4.1%), mientras que con inhibidores de la ECA la incidencia de tos fue de 8.8%.
Estudio LIFE
LIFE (Losartan Intervention For Endpoint Reduction in Hipertension) fue un estudio grande, multicéntrico, multinacional, con distribución al azar, triple ciego, controlado con principio activo y realizado en 9 193 pacientes hipertensos de 55 a 80 años de edad (promedio 67 años), con hipertrofia ventricular izquierda confirmada por electrocardiograma. De los pacientes incluidos al inicio, 1 195 (13%) tenían diabetes; 1 326 (14%) hipertensión sistólica aislada; 1 468 (17%) cardiopatía coronaria y 728 (8%) enfermedad vascular cerebral. El objetivo del estudio fue demostrar los efectos cardioprotectores de COZAAR® comparado con atenolol, independientemente de los beneficios de controlar solamente la presión arterial (cuyas mediciones se hacían durante la concentración mínima [valle] del medicamento). Para alcanzar este objetivo, el estudio se diseñó para que, con ambos grupos de tratamiento, se alcanzase una presión arterial similar. Los pacientes fueron distribuidos al azar para recibir una vez al día COZAAR® 50 mg o atenolol 50 mg. Si el objetivo de la presión arterial no se lograba (< 140/90 mm Hg), primero se añadía hidroclorotiazida (12.5 mg) y, si era necesario, entonces la dosis de COZAAR® o de atenolol se incrementaba a 100 mg una vez al día. Si era necesario, para alcanzar el objetivo de la presión arterial podía añadirse a cada régimen de tratamiento otros tratamientos antihipertensivos (p. ej., aumentar la dosis de hidroclorotiazida a 25 mg o añadir otro tratamiento diurético, antagonistas de los canales de calcio, bloqueadores alfa o agentes de acción central, pero no inhibidores de la ECA, antagonistas de angiotensina II o bloqueadores beta).
En ambos grupos de tratamiento la presión arterial se redujo significativamente a niveles similares y una proporción similar de pacientes alcanzó el objetivo de la presión arterial. La duración promedio de seguimiento fue de 4.8 años.
El objetivo primario fue el compuesto de morbilidad y mortalidad cardiovasculares por la reducción en la incidencia combinada de muerte cardiovascular, apoplejía e infarto del miocardio. Los resultados mostraron que el tratamiento con COZAAR® resultó en una reducción de 13% en el riesgo (p=0.021) comparado con atenolol en los pacientes que alcanzaron el objetivo primario compuesto (véase figura 1).
Figura 1. Estimados de Kaplan-Meier del objetivo primario compuesto de muerte cardiovascular, apoplejía o infarto del miocardio en los grupos tratados con COZAAR® y atenolol, ajustado por la puntuación basal de riesgo de Framingham y el nivel de hipertrofia ventricular izquierda según el electrocardiograma.
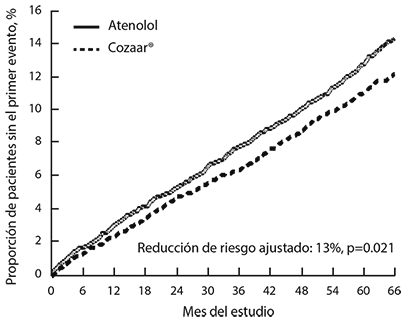
El tratamiento con COZAAR® redujo el riesgo de apoplejía en 25% en relación con atenolol (p=0.001). Los porcentajes de muerte cardiovascular e infarto del miocardio no fueron significativamente diferentes entre los grupos de tratamiento. El efecto de COZAAR® en el objetivo primario compuesto fue independiente de los beneficios de controlar solamente la presión arterial (véase la siguiente tabla).
PUNTOS FINALES DEL ESTUDIO LIFE
|
Desenlace |
COZAAR® (N=4 605) n (%) |
Tasa* |
Atenolol (N=4 588) n (%) |
Tasa* |
Reducción del riesgo** |
Valor de p |
|
Punto final primario compuesto |
508 (11%) |
23.8 |
588 (13%) |
27.9 |
13% |
0.021 |
|
Componentes del punto final primario compuesto |
||||||
|
Mortalidad CV |
204 (4%) |
9.2 |
234 (5%) |
10.6 |
11% |
0.206 |
|
Apoplejía |
232 (5%) |
10.8 |
309 (7%) |
14.5 |
25% |
0.001 |
|
Infarto al miocardio |
198 (4%) |
9.2 |
188 (4%) |
8.7 |
-7% |
0.491 |
* Por 1 000 pacientes-años de seguimiento.
** Ajustado para.
Otros objetivos clínicos del estudio LIFE fueron: Mortalidad total, hospitalización por insuficiencia cardiaca o angina de pecho, procedimientos de revascularización coronaria o periférica y resucitación por paro cardiaco. No hubo diferencias significativas en los porcentajes de estos puntos finales entre los grupos de tratamiento. Los pacientes que recibieron COZAAR® tuvieron una reducción significativamente mayor en la incidencia de hipertrofia ventricular izquierda según el electrocardiograma en comparación con los pacientes tratados con atenolol.
Los efectos de COZAAR®, respecto a atenolol, en la morbilidad y la mortalidad cardiovasculares fueron examinados según el subgrupo de pacientes con antecedentes basales de diabetes mellitus (n=1 195) o hipertensión sistólica aislada (n=1 326). Para el objetivo primario compuesto, los resultados observados en estos subgrupos fueron consistentes con los beneficios que COZAAR® brindó a la población general del estudio: En los pacientes diabéticos se observó una reducción de riesgo de 24% (p=0.03) y, en los pacientes con hipertensión sistólica aislada, se observó una reducción de riesgo de 25% (p=0.06). Consistente con los resultados observados en la población general, la reducción en el riesgo de apoplejía fue un importante factor que contribuyó a los beneficios observados en los pacientes con diabetes o hipertensión sistólica aislada.
Raza: Con base en el estudio LIFE los beneficios de COZAAR® comparado con atenolol en la morbilidad y mortalidad cardiovasculares en pacientes hipertensos con hipertrofia ventricular izquierda no se observaron en pacientes de raza negra, aun cuando ambos tratamientos redujeron eficazmente la presión arterial en los pacientes de raza negra. En el estudio LIFE, comparado con atenolol, COZAAR® disminuyó el riesgo de morbilidad y mortalidad en todos los pacientes hipertensos con hipertrofia ventricular izquierda de raza no negra (n=8 660) de acuerdo al punto final primario combinado de incidencia de muerte cardiovascular, apoplejía e infarto del miocardio (p=0.003). En este estudio, sin embargo, los pacientes de raza negra tratados con atenolol tenían un menor riesgo de presentar algún evento del punto final primario compuesto que los mismos pacientes tratados con COZAAR® (p=0.03). En el subgrupo de pacientes de raza negra (n=533, 6% de los pacientes del estudio LIFE), ocurrieron 29 eventos del punto final primario en los 263 pacientes con atenolol (11%, 25.9 por 1 000 años-paciente) y 46 en los 270 pacientes tratados con COZAAR® (17%, 41.8 por 1 000 años-paciente).
En este estudio, COZAAR® generalmente fue bien tolerado, y el perfil de tolerabilidad de COZAAR® fue superior al de atenolol, según se evidenció por una incidencia significativamente menor de abandonos debidos a efectos colaterales.
Estudio RENAAL
El estudio RENAAL (Reduction of Endpoints in NIDDM with the Angiotensin II Receptor Antagonist Losartan), fue un estudio grande, multicéntrico, con distribución al azar, controlado con placebo, doble ciego, realizado a nivel mundial en 1 513 pacientes diabéticos tipo 2 con proteinuria (751 tratados con COZAAR®) con o sin hipertensión. El objetivo del estudio fue demostrar los efectos de protección renal de COZAAR® más allá de los beneficios de solamente controlar la presión arterial. Para lograr este objetivo, el estudio fue diseñado para que en ambos grupos de tratamiento se lograra el control de la presión arterial. Los pacientes con proteinuria y creatinina sérica de 1.3-3.0 mg/dL fueron distribuidos al azar para recibir COZAAR® 50 mg una vez al día ajustado de acuerdo a la respuesta de la presión arterial o placebo, con un tratamiento antihipertensivo convencional de fondo que excluía inhibidores de la ECA y antagonistas de angiotensina II. Los investigadores fueron instruidos para ajustar el medicamento en estudio a 100 mg al día según fuera apropiado; 72% de los pacientes estaban tomando la dosis diaria de 100 mg la mayoría del tiempo en que estuvieron con el medicamento en estudio. A ambos grupos, se podían añadir otros agentes antihipertensivos (diuréticos, bloqueadores de los canales de calcio, bloqueadores alfa o beta y agentes de acción central). Los pacientes fueron seguidos por hasta 4.6 años (promedio 3.4 años).
El punto final primario del estudio fue el punto final compuesto de duplicación de la creatinina sérica, enfermedad renal terminal (necesidad de diálisis o trasplante de riñón) o muerte. Los resultados mostraron que, comparado con placebo (359 eventos), el tratamiento con COZAAR® (327 eventos) resultó en una reducción de riesgo de 16.1% (P=0.022) para los pacientes que alcanzaron el punto final primario compuesto. Para los siguientes puntos finales primarios, solos y combinados, los resultados también mostraron reducciones significativas en el grupo tratado con COZAAR®: Reducción de 25.3% en el riesgo de duplicación de creatinina sérica (P = 0.006); reducción de 28.6% en el riesgo de enfermedad renal terminal (P = 0.002); reducción de 19.9% en el riesgo de enfermedad renal terminal o muerte (P = 0.009); reducción de 21.0% en el riesgo de duplicación de creatinina sérica o enfermedad renal terminal (P = 0.010). La tasa del componente de muerte por todas las causas no fue significativamente diferente entre los dos grupos de tratamiento.
Los puntos finales secundarios del estudio fueron: Cambio en la proteinuria, índice de progresión de enfermedad renal; y el compuesto de morbilidad y mortalidad por causas cardiovasculares (hospitalización por insuficiencia cardiaca, infarto del miocardio, revascularización, apoplejía, hospitalización por angina inestable, o muerte cardiovascular). Los resultados mostraron una reducción promedio de 34.3% en los niveles de proteinuria en el grupo tratado con COZAAR® (P < 0.001). Durante la fase crónica del estudio, el tratamiento con COZAAR® redujo el índice de disminución de la función renal en 13.9%, p =0.003 (tasa mediana de disminución media de 18.5%, p = 0.01), según se midió con la concentración recíproca de creatinina sérica. No hubo diferencias significativas entre el grupo tratado con COZAAR® (247 eventos) y el grupo con placebo (268 eventos) en el punto final compuesto de morbilidad y mortalidad cardiovasculares, si bien el estudio no tuvo la potencia para detectar ese efecto.
En este estudio, COZAAR® fue generalmente bien tolerado, como se evidencia por el índice similar de abandonos por efectos adversos comparado con placebo.
Estudio HEAAL
El estudio Heart Failure Endpoint Evaluation of Angiotensin II Antagonist Losartan (HEAAL) fue un estudio clínico controlado realizado a nivel mundial en 3 834 pacientes con insuficiencia cardiaca (NYHA clases II-IV) intolerantes al tratamiento con un inhibidor de la ECA. Los pacientes fueron seguidos por más de 4 años (mediana 4.7 años) para comparar los efectos de losartán 50 mg para reducir la mortalidad por todas las causas o la hospitalización por insuficiencia cardiaca. En comparación con losartán 50 mg, losartán 150 mg redujo significativamente el riesgo de muerte por todas las causas o la hospitalización por insuficiencia cardiaca (probabilidad de riesgo 0.899, p = 0.027).
Estudios ELITE I y ELITE II
En el estudio ELITE de 48 semanas en 722 pacientes con insuficiencia cardiaca (de las clases II-IV de la NYHA) no se encontró diferencia significativa en el punto final primario de disfunción renal persistente entre los pacientes tratados con COZAAR® y los tratados con captopril. La observación inesperada del estudio ELITE, respecto a la superioridad de COZAAR® sobre captopril para reducir el riesgo de muerte, no se confirmó en el definitivo estudio ELITE II, descrito a continuación.
En un estudio con diseño prospectivo en pacientes con insuficiencia cardiaca que evaluó la mortalidad (ELITE II), se comparó el tratamiento con 50 mg de COZAAR® una vez al día (dosis inicial de 12.5 mg, incremento progresivo a 25 mg y a 50 mg una vez al día) con 50 mg de captopril tres veces al día (dosis inicial de 12.5 mg, incremento progresivo a 25 mg y 50 mg tres veces al día). En este estudio se siguió a 3 152 pacientes con insuficiencia cardiaca (de las clases II-IV de la NYHA) durante aproximadamente dos años (mediana de seguimiento 1.5 años) para evaluar si COZAAR® era superior a captopril para reducir la mortalidad total. El punto final primario no mostró diferencia estadísticamente significativa entre COZAAR® y captopril en la reducción de muerte total (17.7% para COZAAR® y 15.9% para captopril, p = 0.16). El punto final secundario no mostró diferencia estadísticamente significativa en la reducción de muerte cardiaca súbita y/o resucitación por paro cardiaco (9.0% para COZAAR® y 7.3% para captopril, p=0.08). El punto final terciario de mortalidad y/o de hospitalización (ambas por todas las causas) no mostró diferencia estadísticamente significativa entre COZAAR® y captopril (47.7% para COZAAR® y 44.9% para captopril, p = 0.18). En general, no hubo otras diferencias entre grupos en lo que respecta a otros puntos finales de morbilidad y mortalidad, incluyendo mejoría en la clase funcional de la NYHA.
En estos dos estudios clínicos controlados en pacientes con insuficiencia cardiaca, COZAAR® fue generalmente bien tolerado, y el perfil de tolerabilidad de COZAAR® fue superior al de captopril, como lo muestra la incidencia significativamente menor de abandonos de tratamiento por efectos adversos y la incidencia significativamente menor de tos.
IV. CONTRAINDICACIONES
COZAAR® está contraindicado en pacientes hipersensibles a cualquiera de los componentes de este producto.
COZAAR® no debe ser coadministrado con aliskiren en pacientes con diabetes (véase Interacciones medicamentosas y de otro género).
V. PRECAUCIONES GENERALES
Toxicidad Fetal
El uso de fármacos que actúan en el sistema renina-angiotensina durante el segundo y tercer trimestres de gestación reduce la función renal fetal y aumenta la morbilidad y muerte fetal y neonatal. El oligohidramnios resultante puede asociarse con hipoplasia pulmonar fetal y deformaciones esqueléticas. Entre los efectos adversos neonatales potenciales se encuentran hipoplasia del cráneo, anuria, hipotensión, insuficiencia renal y muerte. Cuando se detecte la gestación, suspender COZAAR® tan pronto como sea posible. Véase Restricciones de uso durante el embarazo y la lactancia.
Hipersensibilidad: Angioedema (véase Reacciones secundarias y adversas).
Hipotensión y desequilibrio hidroelectrolítico
Los pacientes que tienen disminuido el volumen intravascular (p. ej., los tratados con dosis altas de diuréticos) pueden presentar síntomas de hipotensión. Se deben corregir esos trastornos antes de administrar COZAAR®, o se debe utilizar una dosificación inicial menor (véase Dosis y vía de administración).
El desequilibrio de electrólitos es común en pacientes con daño renal, con o sin diabetes, y debe ser controlado. En un estudio clínico realizado en pacientes con diabetes tipo 2 con proteinuria, la incidencia de hiperpotasemia fue mayor en el grupo tratado con COZAAR® respecto al grupo placebo; sin embargo, pocos pacientes suspendieron el tratamiento debido a hiperpotasemia (véase Reacciones secundarias y adversas y Alteraciones en los resultados de pruebas de laboratorio).
El uso concomitante de otros medicamentos que puedan incrementar el potasio sérico puede producir hiperpotasemia (véase Interacciones medicamentosas y de otro género).
Deterioro de la función hepática
Basándose en los datos farmacocinéticos que demuestran un aumento significativo de las concentraciones plasmáticas de losartán en pacientes cirróticos, se debe considerar el empleo de una dosificación menor en los pacientes con antecedentes de deterioro hepático (véase Dosis y vía de administración y Farmacocinética y farmacodinamia, Farmacocinética).
Deterioro de la función renal
Como consecuencia de la inhibición del sistema renina-angiotensina, en sujetos susceptibles se han reportado cambios en la función renal, incluyendo insuficiencia renal; estos cambios pueden ser reversibles al suspender el tratamiento.
Otros medicamentos que afectan el sistema renina-angiotensina pueden aumentar la urea sanguínea y la creatinina sérica en pacientes con estenosis bilateral de las arterias renales o de la arteria de un riñón único. Se han reportado efectos similares con COZAAR®, los cuales pueden ser reversibles al suspender el tratamiento.
Empleo en pacientes de edad avanzada
En los estudios clínicos, no hubo ninguna diferencia relacionada con la edad en la eficacia o la seguridad del losartán.
Empleo en niños
Neonatos con antecedentes de exposición in utero a COZAAR®: Si ocurre oliguria o hipotensión, dirigir la atención a respaldar la presión sanguínea y la perfusión renal. Pueden requerirse exanguinotransfusiones o diálisis como medio para revertir la hipotensión y reemplazar la función renal trastornada.
Los efectos antihipertensivos de COZAAR® han sido establecidos en pacientes pediátricos hipertensos de un ≥ 1 mes de edad a 16 años de edad. El uso de COZAAR® en esos grupos de edad está sustentado en la evidencia obtenida en estudios adecuados y bien controlados de COZAAR® en pacientes pediátricos y en pacientes adultos, así como en la literatura referente a pacientes pediátricos.
La farmacocinética de losartán ha sido investigada en 50 pacientes pediátricos hipertensos > 1 mes de edad a < 16 años de edad, después de la administración de una dosis diaria oral de aproximadamente 0.54 a 0.77 mg/kg de losartán (dosis promedio). El metabolito activo está formado a partir de losartán en todos los grupos de edad. La farmacocinética de losartán y de su metabolito activo es generalmente similar a lo largo de todos los grupos de edad estudiados y es consistente con los datos históricos de la farmacocinética en adultos.
En un estudio clínico que incluyó 177 pacientes pediátricos hipertensos de 6 a 16 años de edad, los pacientes que pesaban ≥ 20 kg a < 50 kg recibieron 2.5, 25 o 50 mg diarios de losartán y los pacientes que pesaban ≥ 50 kg recibieron 5, 50 o 100 mg diarios de losartán. La administración de losartán una vez al día disminuyó la presión arterial valle en forma dependiente de la dosis. La respuesta a la dosis de losartán se observó en todos los subgrupos (p. ej., edad, escala de Tanner, sexo, raza). Sin embargo, la menor dosis estudiada, 2.5 mg y 5 mg, correspondientes a una dosis diaria promedio de 0.07 mg/kg, no mostró eficacia antihipertensiva consistente. En ese estudio, COZAAR® fue generalmente bien tolerado.
Para pacientes que pueden tragar comprimidos, la dosis recomendada es 25 mg una vez al día en pacientes que pesan ≥ 20 kg a < 50 kg. La dosis puede ser incrementada a un máximo de 50 mg una vez al día. En pacientes que pesan ≥ 50 kg, la dosis inicial es de 50 mg una vez al día. Esta dosis puede incrementarse hasta 100 mg una vez al día.
En pacientes pediátricos que tienen disminuido el volumen intravascular se deben corregir esos trastornos antes de administrar COZAAR®.
El perfil de efectos adversos en pacientes pediátricos ha sido similar al observado en pacientes adultos.
No se recomienda el uso de COZAAR® en pacientes pediátricos con índice de filtración glomerular < 30 mL/min/1.73 m2. No hay datos disponibles en pacientes pediátricos con función hepática comprometida ni en pacientes neonatos.
VIII. RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA
EMBARAZO
Los medicamentos que actúan directamente sobre el sistema renina-angiotensina pueden causar lesiones y muerte al feto en desarrollo. En cuanto se detecte la gestación se debe suspender la administración de COZAAR® tan pronto como sea posible.
Aunque no hay experiencia con el uso de COZAAR® en mujeres embarazadas, los estudios con losartán potásico en animales han demostrado lesiones y muertes fetales y neonatales, que al parecer son mediadas farmacológicamente por los efectos sobre el sistema renina-angiotensina. En el feto humano la perfusión renal, que depende del desarrollo del sistema renina-angiotensina, se inicia en el segundo trimestre, por lo que el riesgo para el feto aumenta si COZAAR® se administra durante el segundo o el tercer trimestre del embarazo.
El uso de fármacos que actúan sobre el sistema de renina-angiotensina durante el segundo y tercer trimestres de gestación reduce la función renal fetal y aumenta la morbilidad y muerte fetal y neonatal. El oligohidramnios resultante se puede asociar con hipoplasia pulmonar fetal y deformaciones esqueléticas. Entre los efectos neonatales adversos potenciales se encuentran hipoplasia del cráneo, anuria, hipotensión, insuficiencia renal y muerte. Cuando se detecte la gestación, suspender COZAAR® tan pronto como sea posible.
Por lo general, estos resultados adversos se asocian con el uso de estos fármacos en el segundo y tercer trimestres de gestación. La mayoría de los estudios epidemiológicos que examinan las anomalías fetales después de exposición al uso de antihipertensivos en el primer trimestre no han distinguido los fármacos que afectan el sistema renina-angiotensina de otros agentes antihipertensivos. Es importante el manejo apropiado de la hipertensión materna durante la gestación para optimizar los resultados tanto para la madre como para el feto.
En el caso poco común en que no existe alternativa apropiada para la terapia con fármacos que afectan el sistema renina-angiotensina para un paciente particular, notificar a la madre del riesgo potencial para el feto. Realizar ultrasonidos seriales para valorar el medio ambiente intraamniótico. Si se observa oligohidramnios, suspender COZAAR®, a menos que se considere que salva la vida de la madre. Puede ser apropiado hacer pruebas fetales, basadas en la semana de gestación. Sin embargo, pacientes y médicos deben ser conscientes de que el oligohidramnios puede no aparecer hasta después de que el feto tiene daño irreversible. Observar con cuidado a lactantes con antecedentes de exposición in utero a COZAAR® para hipotensión, oliguria e hipercaliemia.
Lactancia
No se sabe si el losartán es excretado en la leche humana. Como muchos medicamentos sí son excretados por esa vía, y debido al riesgo de efectos adversos en el lactante, se debe decidir si se suspende la lactancia o no se administra el medicamento, teniendo en cuenta la importancia de éste para la madre.
IX. REACCIONES SECUNDARIAS Y ADVERSAS
COZAAR® ha sido generalmente bien tolerado en los estudios clínicos controlados en pacientes hipertensos. Usualmente, los efectos colaterales han sido leves y pasajeros y no han hecho necesario suspender el tratamiento. La incidencia total de efectos colaterales reportados con COZAAR® fue similar a la observada con un placebo.
En los estudios clínicos controlados en pacientes con hipertensión esencial, el mareo fue el único efecto colateral reportado como relacionado con el medicamento que ocurrió con una incidencia mayor que con placebo en 1% o más de los pacientes tratados con COZAAR®. Además, se observaron efectos ortostáticos relacionados con la dosis en menos de 1% de los pacientes. Hubo raros casos de erupción cutánea, aunque en los estudios clínicos controlados su incidencia fue menor que con el placebo.
En esos estudios clínicos controlados, doble-ciego, en pacientes con hipertensión esencial, ocurrieron las siguientes reacciones adversas en 1% o más de los pacientes tratados con COZAAR®, relacionadas o no con el medicamento:
|
COZAAR® (n = 2 085) |
Placebo (n = 535) |
|
|
Todo el organismo |
||
|
Dolor abdominal |
1.7 |
1.7 |
|
Astenia/fatiga |
3.8 |
3.9 |
|
Dolor de pecho |
1.1 |
2.6 |
|
Edema/hinchazón |
1.7 |
1.9 |
|
Cardiovasculares |
||
|
Palpitación |
1.0 |
0.4 |
|
Taquicardia |
1.0 |
1.7 |
|
Digestivo |
||
|
Diarrea |
1.9 |
1.9 |
|
Dispepsia |
1.1 |
1.5 |
|
Náuseas |
1.8 |
2.8 |
|
Musculoesqueléticas |
||
|
Dolor de espalda |
1.6 |
1.1 |
|
Calambres musculares |
1.0 |
1.1 |
|
Nervioso/psiquiátrico |
||
|
Mareo |
4.1 |
2.4 |
|
Cefalea |
14.1 |
17.2 |
|
Insomnio |
1.1 |
0.7 |
|
Respiratorio |
||
|
Tos |
3.1 |
2.6 |
|
Congestión nasal |
1.3 |
1.1 |
|
Faringitis |
1.5 |
2.6 |
|
Trastorno de los senos paranasales |
1.0 |
1.3 |
|
Infección del tracto respiratorio superior |
6.5 |
5.6 |
COZAAR® fue generalmente bien tolerado en un estudio clínico controlado en pacientes hipertensos con hipertrofia ventricular izquierda. Los efectos colaterales más comunes relacionados con el medicamento fueron mareo, astenia/fatiga y vértigo.
En el estudio LIFE, entre los pacientes que al inicio no tenían diabetes, hubo una menor incidencia de diabetes mellitus de nueva aparición en el grupo con COZAAR® comparado con el grupo con atenolol (242 contra 320 pacientes, respectivamente, p < 0.001). Debido a que en este estudio no hubo un grupo con placebo, no se puede determinar si esto representa un efecto benéfico de COZAAR® o un efecto colateral del atenolol.
COZAAR® fue generalmente bien tolerado en un estudio clínico controlado en pacientes diabéticos tipo 2 con proteinuria. Los efectos colaterales más comunes relacionados con el medicamento fueron astenia/fatiga, mareo, hipotensión e hiperpotasemia (véase Precauciones generales, Hipotensión y desequilibrio hidroelectrolítico).
COZAAR® ha sido generalmente bien tolerado en los estudios clínicos en pacientes con insuficiencia cardiaca. Los efectos colaterales han sido típicos de esa población. Los efectos colaterales más frecuentes relacionados con el medicamento fueron mareo e hipotensión.
En el estudio HEAAL (Heart Failure Endpoint Evaluation of Angiotensin II Antagonist Losartan) (véase estudio HEAAL en Estudios clínicos) las reacciones adversas clínicamente importantes relacionadas con el medicamento que ocurrieron más frecuentemente en los pacientes que recibieron COZAAR® 150 mg que en los que recibieron COZAAR® 50 mg fueron hipercaliemia, deterioro renal, insuficiencia renal, hipotensión e incrementos en la creatinina, potasio y urea sanguíneos. Estas reacciones adversas no condujeron a significativamente más suspensiones del tratamiento en los pacientes que recibieron COZAAR® 150 mg.
Después de la salida del producto al mercado se han reportado las siguientes reacciones adversas adicionales:
Hipersensibilidad: Reacciones anafilácticas, se han reportado raros casos de angioedema incluyendo tumefacción de la laringe y la glotis provocando obstrucción de la vía aérea y/o tumefacción de la cara, los labios, la faringe y/o lengua en pacientes tratados con losartán; algunos de estos pacientes previamente experimentaron angioedema con otros medicamentos incluyendo inhibidores de la ECA. Rara vez se ha reportado, vasculitis, incluyendo púrpura de Schönlein-Henoch.
Gastrointestinales: Hepatitis (reportada en raros casos), trastornos de la función hepática, vómito.
Trastornos generales y condiciones en el sitio de administración: Malestar.
Hematológicas: Anemia, trombocitopenia (reportada en raros casos).
Musculoesqueléticas: Mialgia, artralgia.
Neurológicas/psiquiátricas: Migraña, disgeusia.
Sistema reproductivo y trastornos de las mamas: Disfunción eréctil/impotencia.
Respiratorias: Tos.
Piel: Urticaria, prurito, eritroderma, fotosensibilidad.
X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO
En estudios de farmacocinética no se ha identificado ninguna interacción farmacológica de importancia clínica con hidroclorotiazida, digoxina, warfarina, cimetidina, fenobarbital, ketoconazol y eritromicina. Se ha reportado que la rifampicina y el fluconazol reducen los niveles del metabolito activo. No se han evaluado las consecuencias clínicas de estas interacciones.
Como ocurre con otros medicamentos que bloquean la angiotensina II o sus efectos, el uso concomitante con diuréticos ahorradores de potasio (p. ej. espironolactona, triamtereno, amilorida), suplementos de potasio, substitutos de sal que contienen potasio u otros medicamentos que puedan incrementar el potasio sérico (p. ej., medicamentos que contengan trimetoprima), puede producir incrementos del potasio sérico.
Como ocurre con otros medicamentos que afectan la excreción de sodio, la excreción de litio puede disminuir. Por lo tanto, los niveles de litio sérico deben vigilarse cuidadosamente, si se coadministran sales de litio con antagonistas de los receptores de angiotensina II.
Los antiinflamatorios no esteroides (AINEs), incluyendo los inhibidores selectivos de la ciclooxigenasa 2 (inhibidores de COX-2), pueden reducir el efecto de los diuréticos y de otros medicamentos antihipertensivos. Por ello, el efecto antihipertensivo de los antagonistas de receptores de angiotensina II o de los inhibidores de la ECA puede ser atenuado por los AINEs, incluyendo los inhibidores selectivos de COX-2.
En algunos pacientes con función renal comprometida (p. ej., pacientes de edad avanzada o pacientes que tienen disminución del volumen, incluyendo aquellos que reciben diuréticos) que están siendo tratados con antiinflamatorios no esteroides, incluyendo inhibidores selectivos de COX-2, la coadministración con antagonistas de receptores de angiotensina II o de inhibidores de la ECA puede resultar en un deterioro ulterior de la función renal, incluyendo posible falla renal aguda. Estos efectos usualmente son reversibles. Por ello, la coadministración debe administrarse con precaución en pacientes con función renal comprometida.
El bloqueo dual del sistema renina-angiotensina-aldosterona (RAAS por sus siglas en inglés) con bloqueadores de los receptores de angiotensina, inhibidores de la ECA o aliskiren está asociado con un mayor riesgo de hipotensión, síncope, hiperpotasemia y cambios en la función renal (incluyendo insuficiencia renal aguda) comparado con la monoterapia. Monitorear de cerca la presión arterial, función renal y electrólitos en los pacientes con COZAAR® y otros agentes que afecten al RAAS. No coadministre aliskiren con COZAAR® en pacientes con diabetes. Evite el uso de aliskiren con COZAAR® en pacientes con insuficiencia renal (tasa de filtración glomerular < 60 mL/min).
XI. ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO
En los estudios clínicos controlados en pacientes con hipertensión esencial, rara vez se asociaron con la administración de COZAAR® cambios clínicamente importantes en los parámetros de laboratorio usuales. En los estudios clínicos en pacientes con hipertensión ocurrió hiperpotasemia (potasio sérico > 5.5 mEq/L) en 1.5% de los pacientes. En un estudio realizado en pacientes diabéticos tipo 2 con proteinuria, 9.9% de los pacientes tratados con COZAAR® y 3.4% de los tratados con placebo desarrollaron hiperpotasemia (véase Precauciones generales, Hipotensión y desequilibrio hidroelectrolítico). Hubo raros casos de aumento de la alanina-aminotransferasa, que generalmente cesaron al suspender el tratamiento.
XIII. DOSIS Y VÍA DE ADMINISTRACIÓN
COZAAR® se puede administrar con o sin alimentos.
COZAAR® se puede administrar con otros agentes antihipertensivos.
Hipertensión
La dosificación inicial y de mantenimiento usual para la mayoría de los pacientes es de 50 mg una vez al día. El efecto antihipertensivo máximo se alcanza tres a seis semanas después de iniciar el tratamiento. Algunos pacientes pueden obtener un beneficio adicional aumentando la dosis a 100 mg una vez al día.
En los pacientes que tienen disminuido el volumen intravascular (p. ej., los tratados con dosis altas de diuréticos) se debe considerar una dosificación inicial de 25 mg una vez al día (véase Precauciones generales).
No es necesario hacer ningún ajuste inicial de la dosificación en los pacientes de edad avanzada o con deterioro renal, incluyendo los que están en diálisis. Se debe considerar una dosificación más baja en los pacientes con antecedentes de deterioro hepático (véase Precauciones generales).
Reducción del riesgo de morbilidad y mortalidad cardiovascular en pacientes hipertensos con hipertrofia ventricular izquierda
La dosificación inicial usual es de 50 mg de COZAAR® una vez al día. De acuerdo a la respuesta de la presión arterial, se debe añadir una dosis baja de hidroclorotiazida o aumentar la dosis de COZAAR® a 100 mg una vez al día.
Protección renal en pacientes diabéticos tipo 2 con proteinuria
La dosificación inicial usual es de 50 mg una vez al día. La dosis puede ser incrementada a 100 mg una vez al día con base en la respuesta de la presión arterial. COZAAR® puede ser administrado con otros agentes antihipertensivos (p. ej., diuréticos, bloqueadores de los canales de calcio, bloqueadores alfa o beta y agentes de acción central), así como con insulina y otros agentes hipoglucemiantes comúnmente utilizados (p. ej., sulfonilureas, glitazonas e inhibidores de la glucosidasa).
Insuficiencia cardiaca
La dosificación inicial de COZAAR® en pacientes con insuficiencia cardiaca es de 12.5 mg una vez al día. Generalmente, esa dosis se debe ajustar a intervalos de una semana (a 12.5 mg diarios, 25 mg diarios, 50 mg diarios, 100 mg diarios, hasta una dosis máxima de 150 mg una vez al día), según lo vaya tolerando el paciente.
XIV. MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL
Los datos relativos a la sobredosificación en seres humanos son limitados. Las manifestaciones más probables de la sobredosificación serían hipotensión y taquicardia; podría ocurrir bradicardia por estimulación parasimpática (vagal). Si ocurre hipotensión sintomática, se debe establecer tratamiento de sostén.
Ni el losartán ni su metabolito activo se pueden extraer por hemodiálisis.
XII. PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD
Toxicidad animal
Carcinogenicidad
El losartán potásico no fue carcinogénico cuando se administró a ratas durante 105 semanas y a ratones durante 92 semanas a las dosificaciones máximas toleradas. Estas dosificaciones produjeron exposiciones sistémicas al losartán y a su metabolito farmacológicamente activo que fueron, respectivamente, 270 y 150 veces mayores en las ratas y 45 y 27 veces mayores en los ratones que en los seres humanos tratados con 50 mg diarios de losartán.
Mutagenicidad
El losartán potásico fue negativo en los ensayos de mutagénesis microbiana y de mutagénesis de células de mamífero V-79. Además, no mostró ningún indicio de genotoxicidad directa en los ensayos in vitro de elución alcalina y de aberración cromosómica a concentraciones aproximadamente 1 700 veces mayores que la concentración plasmática máxima producida en el hombre a las dosificaciones terapéuticas recomendadas. Tampoco indujo aberraciones cromosómicas en células de médula ósea de ratones machos o hembras a dosis tóxicas por vía oral de hasta 1 500 mg/kg (4 500 mg/m2) (750 veces más que la dosis diaria máxima recomendada en seres humanos). El metabolito activo tampoco mostró ningún indicio de genotoxicidad en los ensayos de mutagénesis microbiana, de elución alcalina in vitro y de aberración cromosómica in vitro.
Reproducción
El losartán potásico no afectó la fertilidad ni la conducta reproductiva de ratas machos y hembras que recibieron dosificaciones orales de hasta 150 y 300 mg/kg/día, respectivamente. Estas dosificaciones producen exposiciones sistémicas al losartán y a su metabolito farmacológicamente activo aproximadamente 150/125 veces mayores en las ratas machos y 300/170 veces mayores en las ratas hembras que en el hombre tratado con la dosis diaria recomendada.
Desarrollo
Se ha demostrado que el losartán potásico tiene efectos adversos en los fetos y las crías de las ratas, que incluyen menor peso corporal, mortalidad y/o toxicidad renal. Además, se encontraron concentraciones significativas de losartán y de su metabolito activo en la leche de las ratas. Basándose en los datos farmacocinéticos, esos resultados son atribuidos a la exposición al medicamento durante la gestación avanzada y la lactancia.
XV. PRESENTACIONES
Caja con 21 comprimidos de 12.5 mg.
Caja con 15 y 30 comprimidos de 50 mg.
Caja con 15 y 30 comprimidos de 100 mg.
XVI. RECOMENDACIONES SOBRE ALMACENAMIENTO Consérvese a no más de 30 °C y en lugar seco. Protéjase de la luz.
XVII. LEYENDAS DE PROTECCIÓN
Su venta requiere receta médica. Mantener fuera del alcance de los niños. No se use en el embarazo, y lactancia. Literatura exclusiva para médicos.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx y
dpocmx@merck.com
Av. 16 de Septiembre No. 301, Col. Xaltocan
C.P. 16090, Deleg. Xochimilco, Ciudad de México, México
Reg. Núm. 284M95, SSA IV
Número de Tracer: 0954-MEX-2018-017385