DARZALEX - Solución
Sustancia(s):
- Daratumumab
Presentaciones:
- 1 Caja, 1 Frasco(s) ámpula, 5 ml, 100 Miligramos
- 1 Caja, 1 Frasco(s) ámpula, 20 ml, 400 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
DARZALEX® está disponible como concentrado líquido incoloro a amarillo sin conservantes para la infusión intravenosa después de la dilución.
Cada frasco ámpula contiene:
Daratumumab 100 mg
Vehículo cbp 5 mL
Cada frasco ámpula contiene:
Daratumumab 400 mg
Vehículo cbp 20 mL
INDICACIONES TERAPÉUTICAS:
Mieloma Múltiple:
DARZALEX® está indicado para el tratamiento de pacientes con mieloma múltiple en combinación con lenalidomida y dexametasona para pacientes de diagnóstico reciente que no han recibido tratamiento previo y que no son candidatos para trasplante autólogo de células hematopoyéticas y para pacientes con mieloma múltiple recurrente o refractario que han recibido al menos un tratamiento anterior.
DARZALEX® está indicado en combinación con bortezomib, melfalán y prednisona en pacientes con Mieloma Múltiple que no han recibido tratamiento previo y que no son candidatos para trasplante autólogo de células hematopoyéticas.
DARZALEX® está indicado en combinación con bortezomib, talidomida y dexametasona en pacientes con mieloma múltiple que no han recibido tratamiento previo y que son candidatos para trasplante autólogo de células hematopoyéticas.
DARZALEX® está indicado en combinación con un agente inmunomodulador (IMiD)* y dexametasona o bortezomib y dexametasona, para el tratamiento de pacientes con mieloma múltiple que han recibido por lo menos un tratamiento previo.
*ver Farmacocinética y farmacodinamia – Estudios clínicos.
DARZALEX® está indicado para el tratamiento de los pacientes con mieloma múltiple que han recibido al menos tres líneas de tratamiento incluyendo un inhibidor de proteasoma (PI) y un agente inmunomodulador o que son doble refractario a un PI y un agente inmunomodulador.
FARMACOCINÉTICA Y FARMACODINAMIA: Daratumumab es un anticuerpo monoclonal humano de inmunoglobulina G1 kappa (IgG1k) contra el antígeno CD38, producido en líneas celulares de mamíferos (ovario de hámster chino [OHC] utilizando tecnología de ADN recombinante.
Propiedades farmacocinéticas: La farmacocinética (PK) de daratumumab después de la administración intravenosa de monoterapia de DARZALEX® fue evaluada en los pacientes con mieloma múltiple en recaída y refractario, a niveles de dosis de 0.1 mg/kg a 24 mg/kg. Se desarrolló un modelo PK de población de daratumumab para describir las características PK de daratumumab y para evaluar la influencia de las covariables en la disposición de daratumumab en los pacientes con mieloma múltiple. El análisis PK de población incluyó 223 pacientes que recibieron monoterapia de DARZALEX® en dos estudios clínicos (150 sujetos recibieron 16 mg/kg).
En las cohortes de 1 a 24 mg/kg, las concentraciones séricas pico (Cmáx) después de la primera dosis aumentaron en una proporción aproximada a la dosis y el volumen de distribución fue consistente con la distribución inicial en el compartimiento plasmático. Los aumentos en el área bajo la curva (ABC) fueron más que proporcionales a la dosis y la depuración (CL) disminuyó con el aumento de la dosis. Estas observaciones sugieren que CD38 puede saturarse a dosis más altas, después de lo cual el impacto en la depuración de unión objetivo se minimiza y la depuración de daratumumab se aproxima a la depuración lineal de IgG1 endógeno. La depuración también disminuyó con dosis múltiples, lo cual puede estar relacionado con las disminuciones de la carga tumoral.
La vida media terminal aumenta con el incremento de la dosis y con la dosificación repetida. La vida media terminal estimada promedio (desviación estándar [DE]) de daratumumab después de la primera dosis de 16 mg/kg fue de 9 (4.3) días. Con base en el análisis PK de la población, la vida media promedio (DE) asociada con la eliminación lineal no específica fue de aproximadamente 18 (9) días; ésta es la vida media terminal que puede esperarse después de la saturación completa de la depuración mediada por el objetivo y la dosificación repetida de daratumumab.
Al final de la dosificación semanal para el esquema recomendado de monoterapia y la dosis de 16 mg/kg, el valor medio (DE) de Cmáx sérico fue de 915 (410.3) microgramos/mL, aproximadamente 2.9 veces más alto que después de la primera infusión. La media de concentración sérica (DE) previa a la dosis (punto mínimo) al final de la dosificación semanal fue de 573 (331.5) microgramos/mL.
Con base en el análisis PK de la población de la monoterapia con DARZALEX®, el estado estacionario de daratumumab se alcanza aproximadamente 5 meses dentro del periodo de dosificación de cada 4 semanas (para la infusión 21), y la proporción media (DE) de Cmáx en estado estacionario a Cmáx después de la primera dosis fue de 1.6 (0.5). El volumen central de distribución medio (DE) es de 56.98 (18.07) mL/kg.
Se realizaron tres análisis PK de la población adicionales en pacientes con mieloma múltiple que recibieron daratumumab en diferentes terapias de combinación (N = 1390). Los perfiles de concentración-tiempo de daratumumab fueron similares después de la monoterapia y de las terapias de combinación. La vida media terminal estimada promedio asociada con la depuración lineal en la terapia de combinación fue de aproximadamente 15-23 días.
Con base en el análisis PK de la población, se identificó el peso corporal como una covariable estadísticamente significativa para la depuración de daratumumab. Por lo tanto, la dosificación basada en el peso corporal es una estrategia apropiada de dosificación para los pacientes con mieloma múltiple.
La simulación de la farmacocinética de daratumumab se realizó para todos los esquemas de dosificación recomendados utilizando parámetros PK individuales de pacientes con mieloma múltiple (n=1309). Los resultados de la simulación confirmaron que la dosis dividida y la dosis única para la primera dosis deberían proporcionar una PK similar, con la excepción del perfil de PK en el primer día del tratamiento.
Poblaciones especiales:
Edad y género: Con base en los análisis PK de la población en pacientes que reciben monoterapia o varias terapias de combinación, la edad (rango: 31-93 años) no tuvo un efecto clínicamente importante en la PK de daratumumab, y la exposición de daratumumab fue similar entre pacientes más jóvenes (< 65 años de edad, n = 518), y mayores (≥ 65 a < 75 años de edad, n = 761; ≥ 75 años de edad, n = 334).
El género no afectó la exposición de daratumumab a un grado clínicamente relevante en los análisis PK de la población.
Insuficiencia renal: No se han llevado a cabo estudios formales de DARZALEX® en pacientes con insuficiencia renal. Se llevaron a cabo análisis PK de la población con base en datos preexistentes de función renal en pacientes que reciben monoterapia o varias terapias de combinación de daratumumab, incluyendo 441 pacientes con función renal normal (depuración de creatinina [CRCL] ≥ 90 mL/min), 621 con insuficiencia renal leve (CRCL < 90 y ≥ 60 mL/min), 523 con insuficiencia renal moderada (CRCL < 60 y ≥ 30 mL/min), y 27 con insuficiencia renal severa o enfermedad renal en etapa terminal (CRCL < 30 mL/min). No se observaron diferencias clínicamente importantes en la exposición a daratumumab entre los pacientes con insuficiencia renal y aquellos con una función renal normal.
Insuficiencia hepática: No se han llevado a cabo estudios formales de DARZALEX® en pacientes con insuficiencia hepática.
Se llevaron a cabo análisis PK de la población en pacientes que reciben monoterapia o varias terapias de combinación de daratumumab incluyendo 1404 pacientes con función hepática normal (bilirrubina total [BT] y aspartato aminotransferasa [AST] ≤ límite superior normal [LSN]) y 189 con insuficiencia hepática leve (BT 1.0x a 1.5x LSN o AST > LSN) y 8 pacientes con insuficiencia hepática moderada (BT > 1.5x a 3.0x LSN; n = 7), o severa (BT > 3.0x LSN; n = 1). No se observaron diferencias clínicamente importantes en la exposición a daratumumab entre pacientes con insuficiencia hepática y aquellos con función hepática normal.
Raza: Con base en los análisis PK de la población en pacientes que recibieron ya sea monoterapia o varias terapias de combinación de daratumumab, la exposición a daratumumab fue similar entre sujetos blancos (n = 1371) y no blancos (n = 242).
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Agentes antineoplásicos, anticuerpos monoclonales, código ATC: L01XC24.
Mecanismo de acción: Daratumumab es un anticuerpo monoclonal humano IgG1k (mAb) que se une a la proteína CD38 expresada en un alto nivel en la superficie de las células en una variedad de neoplasias hematológicas malignas, incluyendo células tumorales de mieloma múltiple, así como en otros tipos de células y tejidos en varios niveles. La proteína CD38 tiene múltiples funciones tales como adhesión mediada por el receptor, señalización y actividad enzimática.
Daratumumab ha demostrado inhibir potentemente el crecimiento in vivo de las células tumorales que expresan CD38. Con base en los estudios in vitro, daratumumab puede utilizar múltiples funciones efectoras, resultando en muerte inmuno mediada de las células tumorales. Estos estudios sugieren que daratumumab puede inducir la lisis de las células tumorales a través de la citotoxicidad dependiente del complemento (CDC), la citotoxicidad dependiente de anticuerpos (ADCC) y la fagocitosis celular dependiente de anticuerpos (ADCP) en neoplasias malignas que expresan CD38. Un subgrupo de células supresoras derivadas de mieloides (CD38 + MDSCs), células T reguladoras (CD38 + Tregs) y células B (CD38 + Bregs) son disminuidos por daratumumab. También se sabe que las células T (CD3 +, CD4 + y CD8 +) expresan CD38 dependiendo de la etapa de desarrollo y el nivel de activación. Se han observado aumentos significativos en los recuentos absolutos de células T CD4 + y CD8 +, y en los porcentajes de linfocitos, con el tratamiento con DARZALEX® en la sangre total periférica y médula ósea. La secuenciación de ADN de los receptores de células T verificó que la clonalidad de las células T aumentó con el tratamiento con DARZALEX®, indicando efectos moduladores inmunológicos que pueden contribuir a la respuesta clínica.
Daratumumab indujo apoptosis in vitro después de la reacción cruzada mediada por Fc. Además, daratumumab moduló la actividad enzimática de CD38, inhibiendo la actividad de la enzima ciclasa y estimulando la actividad de la hidrolasa. El significado de estos efectos in vitro en un ambiente clínico y las implicaciones en el crecimiento tumoral, no son bien comprendidas.
Efectos farmacodinámicos:
Recuento de células asesinas naturales (NK) y de células T: Las células NK son conocidas por expresar niveles altos de CD38 y son susceptibles a la lisis celular mediada por daratumumab. Con el tratamiento con DARZALEX® se observaron disminuciones en los recuentos absolutos y los porcentajes de células NK totales (CD16 + CD56 +) y de células NK activadas (CD16 + CD56dim) en la sangre total periférica y la médula ósea. Sin embargo, los niveles basales de las células NK no mostraron una asociación con la respuesta clínica.
Inmunogenicidad: Los pacientes tratados con daratumumab en monoterapia (n = 199) y terapia de combinación (n = 1051) fueron evaluados para respuestas de anticuerpos antiterapéuticos a daratumumab en múltiples puntos de tiempo durante el tratamiento y hasta 8 semanas después del final del tratamiento. Después del inicio del tratamiento con DARZALEX®, ninguno de los pacientes en monoterapia y 2 de los 1051 pacientes en terapia de combinación dieron positivo para anticuerpos anti-daratumumab; 1 de los pacientes en terapia de combinación desarrolló anticuerpos neutralizantes transitorios contra daratumumab.
Los datos de inmunogenicidad dependen en gran medida de la sensibilidad y la especificidad de los métodos de pruebas usados. Adicionalmente, la incidencia observada de un resultado positivo en un método de prueba puede ser influenciada por varios factores, incluyendo el manejo de las muestras, el tiempo de recolección de las muestras, la interferencia de los fármacos, los medicamentos concomitantes y la enfermedad subyacente. Por lo tanto, la comparación de la incidencia de los anticuerpos a daratumumab con la incidencia de los anticuerpos a otros productos puede ser desorientadora.
Estudios clínicos:
Mieloma Múltiple que no han recibido tratamiento previo: Tratamiento combinado con lenalidomida y dexametasona en pacientes no aptos para el trasplante autólogo de células hematopoyéticas.
El estudio MMY3008, un estudio de fase 3 abierto, aleatorizado, controlado con activo, comparó el tratamiento con DARZALEX® 16 mg/kg en combinación con lenalidomida y dosis bajas de dexametasona (DRd) con el tratamiento con lenalidomida y dosis bajas de dexametasona (Rd) en pacientes con mieloma múltiple que no han recibido tratamiento previo. Lenalidomida (25 mg una vez al día por vía oral en los días 1-21 de ciclos repetidos de 28 días [4 semanas]) se administró con dosis bajas de dexametasona oral o intravenosa 40 mg/semana (o una dosis reducida de 20 mg/semana para los pacientes de más de 75 años o con índice de masa corporal [IMC] <18.5). En los días de infusión de DARZALEX®, la dosis de dexametasona se administró como medicamento previo a la infusión. Los ajustes de dosis para lenalidomida y dexametasona se aplicaron de acuerdo con la información para prescribir del fabricante. Se continuó el tratamiento en ambos brazos hasta la progresión de la enfermedad o una toxicidad inaceptable.
Un total de 737 pacientes fueron aleatorizados: 368 al brazo DRd y 369 al brazo Rd. Las características demográficas y de la enfermedad basales fueron similares entre los dos grupos de tratamiento. La edad mediana fue de 73 años (rango: 45-90), con un 44% de los pacientes de ≥75 años de edad. La mayoría eran blancos (92%), hombres (52%), 34% tenían una puntuación de rendimiento del Eastern Cooperative Oncology Group (ECOG) de 0, 50% tenían una puntuación de rendimiento del ECOG de 1 y 17% tenían una puntuación de rendimiento del ECOG de ≥2. Veintisiete por ciento tenía Etapa I del Sistema Internacional de Estadificación (ISS, por sus siglas en inglés), 43% tenía Etapa II del ISS y 29% tenía enfermedad en Etapa III del ISS. La eficacia se evaluó mediante la supervivencia libre de progresión (SLP) basada en los criterios del International Myeloma Working Group (IMWG).
El estudio MMY3008 demostró una mejoría en la supervivencia libre de progresión (SLP) en el brazo DRd en comparación con el brazo Rd; la mediana de la SLP no se había alcanzado en el brazo DRd y fue de 31.9 meses en el brazo Rd (cociente de riesgo [CR]=0.56; IC del 95%: 0.43, 0.73; p<0.0001), lo que representa un reducción del 44% en el riesgo de progresión de la enfermedad o muerte en los pacientes tratados con DRd.
Figura 1: Curva Kaplan-Meier de SLP en el estudio MMY3008
Los resultados adicionales de eficacia del Estudio MMY3008 se presentan en la siguiente tabla.
Tabla 1: Resultados de eficacia adicionales del estudio MMY3008a
|
DRd (n=368) |
Rd (n=369) |
|
|
Respuesta general (RCe + RC + MBRP + RP) [n(%)a |
342 (92.9%) |
300 (81.3%) |
|
Valor de pb |
<0.0001 |
|
|
Respuesta completa estricta (RCe) |
112 (30.4%) |
46 (12.5%) |
|
Respuesta completa (RC) |
63 (17.1%) |
46 (12.5%) |
|
Muy buena respuesta parcial (MBRP) |
117 (31.8%) |
104 (28.2%) |
|
Respuesta parcial (RP) |
50 (13.6%) |
104 (28.2%) |
|
RC o mejor (RCe + RC) |
175 (47.6%) |
92 (24.9%) |
|
Valor de pb |
<0.0001 |
|
|
MBRP o mejor (RCe+RC+MBRP) |
292 (79.3%) |
196 (53.1%) |
|
Valor de pb |
<0.0001 |
|
|
Tasa EMR negativaa,c n(%) |
89 (24.2%) |
27 (7.3%) |
|
IC del 95% (%) |
(19.9%, 28.9%) |
(4.9%, 10.5%) |
|
Cociente de momios con IC del 95%d |
4.04 (2.55, 6.39) |
|
|
Valor de pe |
<0.0001 |
|
DRd=daratumumab-lenalidomida-dexametasona; Rd=lenalidomida-dexametasona; EMR = enfermedad mínima residual; IC = intervalo de confianza.
a Con base en la población con intención de tratar.
b valor p de la prueba de Chi cuadrada de Cochran Mantel-Haenszel.
c Con base en el umbral de 10-5.
d Se utiliza un estimado de Mantel-Haenszel del cociente de momios para tablas no estratificadas. Un cociente de momios >1 indica una ventaja para DRd.
e valor de p de la prueba exacta de Fisher.
En los respondedores, el tiempo mediano de respuesta fue de 1.05 meses (rango: 0.2 a 12.1 meses) en el grupo DRd y de 1.05 meses (rango: 0.3 a 15.3 meses) en el grupo Rd. La duración mediana de la respuesta no se había alcanzado en el grupo con DRd y fue de 34.7 meses (IC del 95%: 30.8; no estimable) en el grupo con Rd.
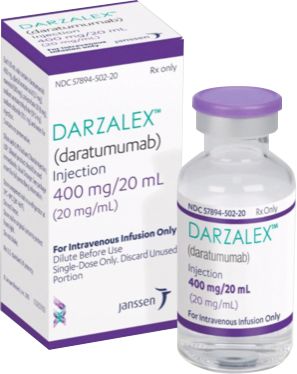
Tratamiento de combinación con bortezomib, melfalán y prednisona (VMP) en pacientes que no son candidatos para trasplante autólogo de células hematopoyéticas: En el Estudio MMY3007, un estudio abierto de Fase 3, aleatorizado, controlado con activo, se comparó el tratamiento con DARZALEX® 16 mg/kg en combinación con bortezomib, melfalán y prednisona (D-VMP), con el tratamiento con VMP en pacientes con mieloma múltiple sin tratamiento previo. Bortezomib se administró mediante inyección subcutánea (SC) a una dosis de 1.3 mg/m2 del área de superficie corporal dos veces a la semana en las semanas 1, 2, 4 y 5 para el primer ciclo de 6 semanas (Ciclo 1; 8 dosis), seguido de administraciones una vez a la semana en las Semanas 1, 2, 4 y 5 durante ocho ciclos más de 6 semanas (Ciclos 2-9; 4 dosis por ciclo). Se administraron oralmente melfalán en dosis de 9 mg/m2 y prednisona en dosis de 60 mg/m2 en los Días 1 a 4 de los nueve ciclos de 6 semanas (Ciclos 1-9). El tratamiento con DARZALEX® continuó hasta la progresión de la enfermedad o hasta observarse toxicidad inaceptable.
En total aleatorizaron 706 pacientes: 350 al grupo D-VMP y 356 al grupo VMP. Las características basales demográficas y de la enfermedad fueron similares entre los dos grupos de tratamiento. La mediana de edad fue de 71 (rango: 40-93) años, con 30% de los pacientes ≥75 años de edad. La mayoría eran de raza blanca (85%), sexo femenino (54%), 25% con una calificación de desempeño de ECOG de 0; 50% con una calificación de desempeño ECOG de 1 y 25% con una calificación de desempeño ECOG de 2. Los pacientes presentaban mieloma de IgG/IgA/cadena ligera en el 64%/22%/10% de los casos, 19% presentaba ISS en Etapa I, 42% ISS en Etapa II y 38% ISS en Etapa III. La eficacia se evaluó mediante la SLP con base en los criterios del IMWG.
En el estudio MMY3007 se demostró una mejoría en la SLP en el grupo D-VMP en comparación con el grupo VMP; no se alcanzó la mediana de la SLP en el grupo D-VMP y fue de 18.1 meses en el grupo VMP (CR=0.5; IC del 95%: 0.38, 0.65; p < 0.0001), lo que representa una reducción del 50% en el riesgo de progresión de la enfermedad o la muerte en pacientes tratados con D-VMP.
Figura 2: Curva de Kaplan-Meier de SLP en el Estudio MMY3007.
Los resultados adicionales de eficacia del Estudio MMY3008 se presentan en la siguiente tabla.
Tabla 2. Resultados adicionales de eficacia del Estudio MMY3007a
|
D-VMP (n = 350) |
VMP (n = 356) |
|
|
Respuesta general (RCe + RC + MBRP + RP) [n(%)] |
318 (90.9) |
263 (73.9) |
|
Valor de pb |
< 0.0001 |
|
|
Respuesta completa estricta (RCe) [n(%)] |
63 (18.0) |
25 (7.0) |
|
Respuesta completa (RC) [n(%)] |
86 (24.6) |
62 (17.4) |
|
Muy buena respuesta parcial (MBRP) [n(%)] |
100 (28.6) |
90 (25.3) |
|
Respuesta parcial (RP) [n(%)] |
69 (19.7) |
86 (24.2) |
|
Tasa EMR negativa (IC del 95%)c (%) |
22.3 (18.0, 27.0) |
6.2 (3.9, 9.2) |
|
Cociente de momios con IC del 95%d |
4.36 (2.64, 7.21) |
|
|
Valor de pe |
< 0.0001 |
D-VMP = daratumumab-bortezomib-melfalán-prednisona; VMP = bortezomib-melfalán-prednisona;
EMR = enfermedad mínima residual; IC = intervalo de confianza; NE = no estimable.
a Con base en la población con intención de tratar.
b Valor de p de la prueba de Chi cuadrada de Cochran Mantel-Haenszel.
c Con base en el umbral de 10-5.
d Se utiliza un estimado de Mantel-Haenszel del cociente de momios común para tablas estratificadas. Un cociente de momios > 1 indica una ventaja para D-VMP.
e Valor de p de la prueba exacta de Fischer.
En los respondedores, la mediana del tiempo de respuesta fue de 0.79 meses (rango: 0.4 a 15.5 meses) en el grupo D-VMP y de 0.82 meses (rango: 0.7 a 12.6 meses) en el grupo VMP. No se alcanzó la mediana de la duración de la respuesta en el grupo D-VMP y fue de 21.3 meses (rango: 18.4, no estimable) en el grupo VMP.
Combinación con bortezomib, talidomida y dexametasona en pacientes que son candidatos para trasplante autólogo de células hematopoyéticas (TACHP): El estudio MMY3006, un estudio abierto de fase 3, aleatorizado, controlado con activo, comparó el tratamiento de inducción y consolidación con DARZALEX® 16 mg/kg en combinación con bortezomib, talidomida y dexametasona (DVTd) con el tratamiento con bortezomib, talidomida y dexametasona (VTd) en pacientes con mieloma múltiple que no han recibido tratamiento previo y que son candidatos para TACHP. La fase de consolidación del tratamiento comenzó con un mínimo de 30 días después del TACHP, cuando el paciente se había recuperado lo suficiente, y el injerto estaba completo.
Bortezomib se administró por inyección subcutánea (SC) o intravenosa (IV) a una dosis de 1.3 mg/m2 de superficie corporal dos veces por semana durante dos semanas (días 1, 4, 8 y 11) de ciclos de tratamiento de inducción repetidos de 28 días (4 semanas) (ciclos 1-4) y dos ciclos de consolidación (ciclos 5 y 6) después del TACHP después del ciclo 4. Talidomida se administró por vía oral con una dosis de 100 mg diarios durante los seis ciclos de bortezomib. Se administró dexametasona (oral o intravenosa) con una dosis de 40 mg en los días 1, 2, 8, 9, 15, 16, 22 y 23 de los ciclos 1 y 2, y a 40 mg en los días 1-2 y 20 mg en los días de dosificación posteriores (días 8, 9, 15 y 16) de los ciclos 3 y 4. Dexametasona 20 mg se administró en los días 1, 2, 8, 9, 15 y 16 en los ciclos 5 y 6. En los días de la infusión de DARZALEX®, la dosis de dexametasona se administró por vía intravenosa como medicamento previo a la infusión. Los ajustes de dosis para bortezomib, talidomida y dexametasona se aplicaron de acuerdo con la información para prescribir del fabricante.
Un total de 1085 pacientes fueron aleatorizados: 543 al brazo del DVTd y 542 al brazo del VTd. Las características basales demográficas y de la enfermedad fueron similares entre los dos grupos de tratamiento. La edad mediana fue de 58 años (rango: 22 a 65 años). La mayoría eran hombres (59%), el 48% tenía una puntuación de rendimiento ECOG de 0, el 42% tenía una puntuación de rendimiento ECOG de 1 y el 10% tenía una puntuación de rendimiento ECOG de 2. El 40% tenía ISS Etapa I, el 45% tenía ISS Etapa II y el 15% tenía enfermedad ISS Etapa III.
La eficacia fue evaluada por la tasa de Respuesta Completa estricta (RCe) en el día 100 después del trasplante.
Tabla 3: Resultados de eficacia del estudio MMY3006a
|
DVTd (n=543) |
VTd (n=542) |
valor Pb |
|
|
Evaluación de la respuesta Día 100 después del trasplante |
|||
|
Respuesta completa estricta (RCe) |
157 (28.9%) |
110 (20.3%) |
0.0010 |
|
RC o mejor (RCe + RC) |
211 (38.9%) |
141 (26.0%) |
<0.0001 |
|
Muy buena respuesta parcial o mejor (RCe + RC + MBRP) |
453 (83.4%) |
423 (78.0%) |
|
|
Tasa EMR negativac n(%) |
346 (63.7%) |
236 (43.5%) |
<0.0001 |
|
IC del 95% (%) |
(59.5%, 67.8%) |
(39.3%, 47.8%) |
|
|
Cociente de momios con IC del 95%d |
2.27 (1.78, 2.90) |
||
|
Tasa EMR negativac n(%) |
183 (33.7%) |
108 (19.9%) |
<0.0001 |
|
IC del 95% (%) |
(29.7%, 37.9%) |
(16.6%, 23.5%) |
|
|
Cociente de momios con IC del 95%d |
2.06 (1.56, 2.72) |
||
D-VTd=daratumumab-bortezomib-talidomida-dexametasona; VTd=bortezomib-talidomida-dexametasona; EMR=enfermedad mínima residual; IC=intervalo de confianza.
a Basado en la población con intención de tratar.
b valor p de la prueba de Chi-cuadrado Cochran Mantel-Haenszel.
c Basado en el umbral de 10-5.
d Se utiliza la estimación de Mantel-Haenszel del cociente de momios común para las tablas estratificadas.
e Sólo incluye pacientes que lograron la tasa EMR negativa (umbral de 10-5) y RC o mejor.
El estudio MMY3006 demostró una mejoría en la SLP en el brazo de DVTd en comparación con el brazo de VTd; con una mediana de seguimiento de 18.8 meses, la SLP mediana no se había alcanzado en ninguno de los dos brazos. El tratamiento con DVTd produjo una reducción del riesgo de progresión o muerte del 53% en comparación con la VTd sola (CR = 0.47; IC del 95%: 0.33; 0.67; p<0.0001).
Figura 3: Curva Kaplan-Meier de SLP en el estudio MMY3006
Mieloma Múltiple Recurrente/Refractario:
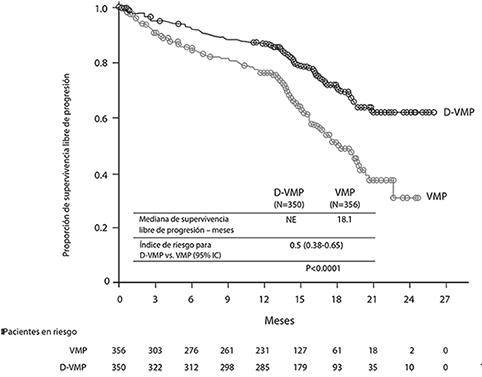
Tratamiento de combinación con lenalidomida y dexametasona: El estudio MMY3003, un estudio abierto, aleatorizado, controlado con activo, de Fase 3, comparó el tratamiento con DARZALEX® 16 mg/kg en combinación con lenalidomida y bajas dosis de dexametasona (DRd), con el tratamiento con lenalidomida y bajas dosis de dexametasona (Rd) en pacientes con mieloma múltiple que habían recibido por lo menos una terapia previa. Se administró lenalidomida (25 mg una vez al día por vía oral en los Días 1-21 de ciclos repetidos de 28 días [4 semanas]) con una dosis baja oral o intravenosa de dexametasona 40 mg/semana (o una dosis reducida de 20 mg/semana para pacientes > 75 años o un IMC < 18.5). En los días de la infusión de DARZALEX®, se administraron 20 mg de la dosis de dexametasona como medicación previa a la infusión y el resto se administró al día siguiente de la infusión. Para pacientes con una dosis reducida de dexametasona, se administró toda la dosis de 20 mg como medicación previa a la infusión de DARZALEX®. Se aplicaron ajustes de dosis para lenalidomida y dexametasona con base en la información para prescribir del fabricante. Se continuó el tratamiento en ambos grupos hasta la progresión de la enfermedad o toxicidad inaceptable.
Un total de 569 pacientes fueron aleatorizados: 286 al grupo DRd y 283 al grupo Rd. Las características demográficas y de la enfermedad basales fueron similares entre el brazo de DARZALEX® y el control. La mediana de la edad de los pacientes fue de 65 años (rango 34 a 89 años), 11% tenían ≥ 75 años, 59% eran hombres; 69% caucásicos, 18% asiáticos y 3% afroamericanos. Los pacientes habían recibido una mediana de 1 línea de terapia previa. Sesenta y tres por ciento (63%) de los pacientes había recibido previamente trasplante autólogo de células progenitoras hematopoyéticas (TACHP). La mayoría de los pacientes (86%) recibieron previamente un inhibidor de proteasoma (IP), 55% de los pacientes habían recibido previamente un agente inmunomodulador (IMiD), incluyendo 18% de los pacientes que habían recibido previamente lenalidomida, y 44% de los pacientes habían recibido previamente tanto IP como IMiD. En la basal, 27% de los pacientes fueron refractarios a la última línea de tratamiento. Dieciocho por ciento (18%) de los pacientes fueron refractarios a IP solamente, y 21% fueron refractarios a bortezomib. La eficacia se evaluó mediante la SLP con base en los criterios del IMWG.
El Estudio MMY3003 demostró una mejoría en SLP en el grupo DRd en comparación con el grupo Rd; la mediana de SLP no se había alcanzado en el grupo DRd y fue de 18.4 meses en el grupo Rd (CR=0.37; IC del 95%: 0.27, 0.52; p < 0.0001), lo que representa una reducción de 63% en el riesgo de progresión de la enfermedad o muerte en pacientes tratados con DRd.
Figura 4. Curva de Kaplan-Meier de SLP en el Estudio MMY3003

Los resultados adicionales de eficacia del estudio MMY3003 se presentan en la siguiente tabla.
Tabla 4. Resultados de eficacia adicionales del estudio MMY3003
|
Número de pacientes con respuesta evaluable |
DRd (n = 281) |
Rd (n = 276) |
|
Respuesta general (RCe + RC + MBRP + RP) n (%) |
261 (92.9) |
211 (76.4) |
|
Valor de pa |
< 0.0001 |
|
|
Respuesta completa estricta (RCe) |
51 (18.1) |
20 (7.2) |
|
Respuesta completa (RC) |
70 (24.9) |
33 (12.0) |
|
Muy buena respuesta parcial (MBRP) |
92 (32.7) |
69 (25.0) |
|
Respuesta parcial (RP) |
48 (17.1) |
89 (32.2) |
|
Mediana del tiempo a la respuesta [meses (95% IC)] |
1.0 (1.0, 1.1) |
1.3 (1.1, 1.9) |
|
Mediana de la duración de la respuesta [meses (95% IC)] |
NE (NE, NE) |
17.4 (17.4, NE) |
|
Tasa EMR negativa (95% CI)b (%) |
29.0 (23.8, 34.7) |
7.8 (4.9, 11.5) |
|
Cociente de momios con 95% ICc |
4.85 (2.93, 8.03) |
|
|
Valor de pd |
< 0.000001 |
DRd = daratumumab-lenalidomida-dexametasona; Rd = lenalidomida-dexametasona; EMR = enfermedad mínima residual; IC = intervalo de confianza; NE = no estimable.
a Valor de p de la prueba de Chi Cuadrada de Cochran Mantel-Haenszel.
b Basada en la población de intención a tratamiento y un umbral de 10-4.
c Se utiliza una estimación de Chi Cuadrada del cociente de momios común. Un cociente de momios > 1 indica una ventaja para DRd.
d El valor de p se obtiene de una prueba de Chi Cuadrada con cocientes de probabilidad.
Con una mediana general de seguimiento de 13.5 meses, el cociente de riesgo para la supervivencia global (SG) fue de 0.64 (IC del 95%: 0.40, 1.01; p = 0.0534). La tasa de SG a los 18 meses fue de 86% (IC del 95%: 79.9, 90.5) para pacientes en el grupo DRd frente a 76% (IC del 95%: 79.9, 90.5) en el grupo Rd.
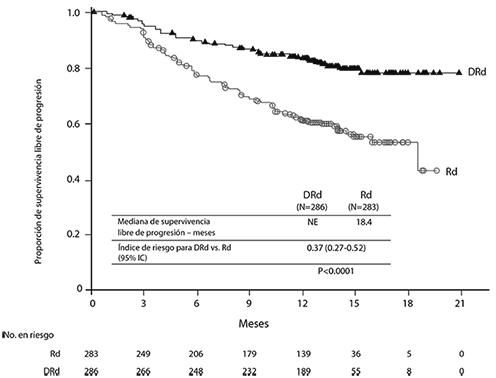
Tratamiento de combinación con bortezomib y dexametasona: El estudio MMY3004, un estudio abierto, aleatorizado, controlado con activo, de Fase 3, comparó el tratamiento con DARZALEX® 16 mg/kg en combinación con bortezomib y dexametasona (DVd), con el tratamiento con bortezomib y dexametasona (Vd) en pacientes con mieloma múltiple que han recibido por lo menos una terapia previa. Bortezomib se administró por inyección SC o infusión IV a una dosis de 1.3 mg/m2 de área de superficie corporal dos veces a la semana durante dos semanas (Días 1, 4, 8, y 11) de ciclos de tratamiento repetidos de 21 días (3 semanas), por un total de 8 ciclos. Dexametasona se administró por vía oral a una dosis de 20 mg en los Días 1, 2, 4, 5, 8, 9, 11 y 12 de los 8 ciclos de bortezomib (80 mg/semana por dos de las tres semanas de cada uno de los ciclos de bortezomib) o a una dosis reducida de 20 mg/semana para pacientes > 75 años, IMC < 18.5, diabetes mellitus con un control deficiente o intolerancia previa al tratamiento con esteroides. En los días de la infusión de DARZALEX®, se administraron 20 mg de la dosis de dexametasona como medicación previa a la infusión. Para pacientes con una dosis reducida de dexametasona, se administró la dosis completa de 20 mg como medicación previa a la infusión de DARZALEX®. Bortezomib y dexametasona se administraron por 8 ciclos de tres semanas en ambos grupos de tratamiento, mientras que DARZALEX® se administró hasta la progresión del tratamiento. Sin embargo, dexametasona 20 mg se continuó como medicación previa a la infusión de DARZALEX® en el grupo DVd. Se aplicaron ajustes de dosis para bortezomib y dexametasona de acuerdo con la información para prescribir del fabricante.
Se aleatorizaron un total de 498 pacientes; 251 al grupo DVd y 247 al grupo Vd. Las características demográficas y de enfermedad basales fueron similares entre los grupos de DARZALEX® y de control. La mediana de la edad de los pacientes fue de 64 años (rango 30 a 88 años); 12% tenían ≥ 75 años, 57% eran hombres; 87% caucásicos, 5% asiáticos y 4% afroamericanos. Los pacientes habían recibido una mediana de 2 líneas de terapia previas y 61% de los pacientes había recibido previamente trasplante autólogo de células progenitoras hematopoyéticas (TACHP). Sesenta y nueve por ciento (69%) de los pacientes habían recibido un IP previo (66% recibieron bortezomib) y 76% de los pacientes recibieron un IMiD (42% recibieron lenalidomida). En la basal, 32% de los pacientes eran refractarios a la última línea de tratamiento, y la proporción de pacientes refractarios a cualquier terapia previa específica estuvieron bien balanceados entre los grupos de tratamiento. Treinta y tres por ciento (33%) de los pacientes fueron refractarios a un IMiD solamente, y 28% fueron refractarios a la lenalidomida. La eficacia se evaluó mediante la SLP con base en los criterios del IMWG.
El estudio MMY3004 demostró una mejoría en SLP en el grupo DVd en comparación con el grupo Vd; la mediana de SLP no se había alcanzado en el grupo DVd y fue de 7.2 meses en el grupo Vd (CR [IC del 95%]: 0.39 [0.28, 0.53]; valor de p < 0.0001), lo que representa una reducción del 61% en el riesgo de progresión de la enfermedad o muerte para pacientes tratados con DVd frente a Vd.
Figura 5. Curva de Kaplan-Meier de SLP en el estudio MMY3004
Los resultados adicionales de eficacia del estudio MMY3004 se presentan en la siguiente tabla.
Tabla 5. Resultados de eficacia adicionales del Estudio MMY3004
|
Número de pacientes con respuesta evaluable |
DVd (n = 240) |
Vd (n = 234) |
|
Respuesta general (RCe + RC + MBRP + RP) n(%) |
199 (82.9) |
148 (63.2) |
|
Valor de Pa |
< 0.0001 |
|
|
Respuesta completa estricta (RCe) |
11 (4.6) |
5 (2.1) |
|
Respuesta completa (RC) |
35 (14.6) |
16 (6.8) |
|
Muy buena respuesta parcial (MBRP) |
96 (40.0) |
47 (20.1) |
|
Respuesta parcial (RP) |
57 (23.8) |
80 (34.2) |
|
Mediana del tiempo a la respuesta [meses (rango)] |
0.9 (0.8, 1.4) |
1.6 (1.5, 2.1) |
|
Mediana de la duración de la respuesta [meses (95% IC)] |
NE (11.5, NE) |
7.9 (6.7, 11.3) |
|
Tasa EMR negativa (IC del 95%)b (%) |
13.5 (9.6, 18.4) |
2.8 (1.1,5.8)) |
|
Cociente de momios con 95% ICc |
5.37 (2.33, 12.37) |
|
|
Valor de pd |
0.000006 |
DVd = daratumumab-bortezomib-dexametasona; Vd = bortezomib-dexametasona; EMR = enfermedad mínima residual; IC = intervalo de confianza; NE = no estimable.
a Valor de p de la prueba de Chi Cuadrada de Cochran Mantel-Haenszel.
b Basado en la población de intención para tratar y un umbral de 10-4.
c Se utiliza una estimación de Chi Cuadrada del cociente de momios común. Un cociente de momios > 1 indica una ventaja para DVd.
d El valor de p proviene de la prueba e Chi Cuadrada con cociente de probabilidad.
No se alcanzó la mediana de SG para ninguno de los grupos de tratamiento. Con una mediana general de seguimiento de 7.4 meses (95% IC: 0.0, 14.9), el cociente de riesgo para SG fue de 0.77 (95% IC: 0.47, 1.26; p = 0.2975).
Citogenética de alto riesgo: La eficacia fue similar en todos los subgrupos analizados, incluyendo aquellos pacientes con alteraciones citogenéticas de alto riesgo (AR), definidos como los que presentan las translocaciones t(4:14), t(14;16) o la deleción 17p (en el brazo DRd n = 28, en el brazo Rd n = 37, en el brazo DVd n = 44, en el brazo Vd n = 51). En este grupo de pacientes la SLP no fue alcanzada para aquellos del brazo DRd contra 6.58 meses en el brazo Rd (p = 0.0004) esto en el estudio MMY3003. Para el brazo de DVd fue de 9.17 meses contra 4.34 meses para el grupo Vd (p = 0.0011) esto, en el estudio MMY3004.
Tratamiento de combinación con pomalidomida y dexametasona: El estudio MMY1001 fue un estudio abierto en el que 103 pacientes con mieloma múltiple que habían recibido un IP e IMiD previo, recibieron 16 mg/kg de DARZALEX® en combinación con pomalidomina y una dosis baja de dexametasona hasta la progresión de la enfermedad. Se administró pomalidomida (4 mg una vez al día por vía oral los Días 1-21 de ciclos repetidos de 28 días [4 semanas]) con dosis bajas orales o intravenosas de dexametasona a 40 mg/semana (dosis reducida de 20 mg/semana para pacientes > 75 años o IMC < 18.5). En los días de la infusión de DARZALEX®, se administraron 20 mg de dexametasona como medicación previa a la infusión y el resto se administró al día siguiente de la infusión. Para pacientes con dosis reducida de dexametasona, se administró la dosis total de 20 mg como medicación previa a la infusión de DARZALEX®.
La mediana de edad de los pacientes fue de 64 años (rango: 35 a 86 años) con 8% de los pacientes ≥ 75 años de edad. Los pacientes en el estudio habían recibido una mediana de 4 líneas de tratamiento previas. Setenta y cuatro por ciento (74%) de los pacientes habían recibido trasplante autólogo de células progenitoras hematopoyéticas (TACHP) previo. Noventa y ocho por ciento (98%) de los pacientes recibieron tratamiento previo con bortezomib, y 33% de los pacientes recibieron previamente carfilzomib. Todos los pacientes recibieron tratamiento previo con lenalidomida, con 98% de los pacientes tratados previamente con la combinación de bortezomib y lenalidomida. Ochenta y nueve por ciento (89%) de los pacientes fueron refractarios a lenalidomida y 71% fueron refractarios a bortezomib; 64% de los pacientes fueron refractarios a bortezomib y lenalidomida.
La tasa de respuesta global fue de 59% (95% IC: 49.1%, 68.8%); se logró MBRP o mejor en 42% de los pacientes, se alcanzó RC o mejor en 14% de los pacientes y se alcanzó RCe en 8% de los pacientes. La Tasa de Beneficio Clínico (TRG + RM [Respuesta mínima]) fue del 62% (95% IC: 52.0, 71.5). La mediana del tiempo a la respuesta fue de 1 mes (rango: 0.9 a 2.8 meses). La mediana de la duración de la respuesta fue de 13.6 meses (95% IC: 10.0, no estimable). Después de una mediana de duración de seguimiento de 9.8 meses, no se alcanzó la mediana de SG. La tasa de supervivencia a los 12 meses fue del 72%.
Monoterapia: La eficacia clínica y la seguridad de la monoterapia de DARZALEX® para el tratamiento de pacientes con mieloma múltiple en recaída y refractario cuya terapia previa incluyó un inhibidor de proteasoma y un agente inmunomodulador fueron demostradas en dos estudios abiertos.
En el estudio MMY2002, 106 pacientes con mieloma múltiple en recaída y refractario recibieron 16 mg/kg de DARZALEX® hasta la progresión de la enfermedad. La mediana de la edad de los pacientes fue de 63.5 años (rango, de 31 a 84 años), 49% eran hombres y 79% eran caucásicos. Los pacientes habían recibido una mediana de 5 líneas de terapia previas. Ochenta por ciento de los pacientes había recibido un trasplante autólogo de células progenitoras hematopoyéticas (TACPH). Las terapias previas incluyeron bortezomib (99%), lenalidomida (99%), pomalidomida (63%) y carfilzomib (50%). En la basal, 97% de los pacientes eran refractarios a la última línea de tratamiento, 95% fueron refractarios a IP y IMiD, 77% fueron refractarios a agentes alquilantes, 63% fueron refractarios a pomalidomida y 48% de los pacientes fueron refractarios a carfilzomib.
Los resultados de eficacia basados en la evaluación del Comité de Revisión Independiente (CRI) se presentan en la siguiente tabla.
Tabla 6. Resultados de eficacia evaluados por el CRI para el estudio MMY2002
|
Criterio de Valoración de Eficacia |
DARZALEX® 16 mg/kg N = 106 |
|
Tasa de respuesta global1 (TRG: RCe + RC + MBRP + RP) [n (%)] 95% IC (%) |
31 (29.2) (20.8, 38.9) |
|
Respuesta completa estricta (RCe) [n (%)] |
3 (2.8) |
|
Respuesta completa (RC) [n] |
0 |
|
Muy buena respuesta parcial (MBRP) [n (%)] |
10 (9.4) |
|
Respuesta parcial (RP) [n (%)] |
18 (17.0) |
|
Tasa de Beneficios Clínicos (TRG+RM) |
36 (34.0) |
|
Mediana de Duración de Respuesta [meses (95% IC)] |
7.4 (5.5, NE) |
|
Mediana de Tiempo hasta la Respuesta [meses (rango)] |
1 (0.9; 5.6) |
1. Criterio de valoración primario de eficacia (criterios del Grupo de Trabajo Internacional de Mieloma).
IC = intervalo de confianza; NE = no estimable; RM = respuesta mínima.
La tasa de respuesta global (TRG) en MMY2002 fue similar independientemente del tipo de terapia anti-mieloma previa. Con una duración mediana de seguimiento de 9 meses, no se alcanzó la mediana de SG. La tasa de SG de 12 meses fue de 65% (IC del 95%: 51.2, 75.5).
En el estudio GEN501, 42 pacientes con mieloma múltiple en recaída y refractario recibieron 16 mg/kg de DARZALEX® hasta la progresión de la enfermedad. La mediana de edad de los pacientes fue de 64 años (rango, de 44 a 76 años), 64% eran hombres y 76% eran caucásicos. Los pacientes en el estudio habían recibido una mediana de 4 líneas de terapia previas. Setenta y cuatro por ciento de los pacientes habían recibido TACHP previo. Las terapias anteriores incluyeron bortezomib (100%), lenalidomida (95%), pomalidomida (36%) y carfilzomib (19%). En la basal, 76% de los pacientes fueron refractarios a la última línea de tratamiento, 64% fueron refractarios a IP y IMiD, 60% fueron refractarios a agentes alquilantes, 36% fueron refractarios a pomalidomida y 17% fueron refractarios a carfilzomib.
El tratamiento con 16 mg/kg de daratumumab condujo a una TRG de 36% con RC de 5% y MBRP de 5%. La mediana del tiempo hasta la respuesta fue de 1 mes (rango: de 0.5 a 3.2). La mediana de la duración de la respuesta no fue alcanzada (95% IC: 5.6 meses, no estimable). Con una mediana de la duración de seguimiento de 10 meses, no se alcanzó la mediana de SG. La tasa de SG de 12 meses fue de 77% (95% IC: 58.0, 88.2).
CONTRAINDICACIONES: Pacientes con antecedentes de hipersensibilidad severa a daratumumab o a cualquiera de los excipientes.
Embarazo y lactancia.
Cuando daratumumab es utilizado en combinación con IMiD, consultar la Información para prescribir de dichos medicamentos en relación a las contraindicaciones.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No existen datos en humanos o animales para evaluar el riesgo del uso de DARZALEX® durante el embarazo. Los anticuerpos monoclonales IgG1 son conocidos por cruzar la placenta después del primer trimestre de embarazo. Por lo tanto, no debe usarse DARZALEX® durante el embarazo a menos que se considere que el beneficio del tratamiento para la mujer rebase los riesgos potenciales para el feto. Si la paciente queda embarazada mientras está tomando este medicamento, la paciente deberá ser informada sobre el riesgo potencial para el feto.
Para evitar la exposición al feto, las mujeres con potencial reproductivo deben usar anticonceptivos efectivos durante y por 3 meses después de la interrupción del tratamiento con DARZALEX®.
Lactancia: No se sabe si daratumumab es excretado en la leche humana o animal o si afecta la producción de leche. No existen estudios para evaluar el efecto de daratumumab en el infante lactante.
El IgG materno es excretado en la leche humana, pero no ingresa a las circulaciones neonatales y de los infantes en cantidades sustanciales ya que estos son degradados en el tracto gastrointestinal y no son absorbidos. Debido a que los riesgos de DARZALEX® para el infante a partir de la ingestión oral son desconocidos, debe tomarse una decisión ya sea de descontinuar la lactancia, o descontinuar la terapia con DARZALEX®, tomando en cuenta el beneficio de la lactancia para el niño y el beneficio de la terapia para la mujer.
Fertilidad: No hay datos disponibles para determinar los efectos potenciales de daratumumab en la fertilidad de pacientes masculinos y femeninos.
REACCIONES SECUNDARIAS Y ADVERSAS: A lo largo de esta sección, se presentan las reacciones adversas. Las reacciones adversas son eventos adversos que fueron considerados estar razonablemente asociados con el uso de daratumumab, basado en una evaluación integral de la información de eventos adversos disponible. No se puede establecer una relación causal confiable con daratumumab en casos individuales. Además, debido a que los estudios clínicos se llevan a cabo bajo condiciones muy variables, las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento no se pueden comparar directamente con las tasas en los estudios clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica clínica.
Los datos de seguridad descritos a continuación reflejan la exposición a DARZALEX® (16 mg/kg) en 2066 pacientes con mieloma múltiple, incluyendo a 1910 pacientes que recibieron DARZALEX® en combinación con regímenes de fondo y 156 pacientes que recibieron DARZALEX® como monoterapia.
Mieloma Múltiple sin tratamiento previo:
Tratamiento combinado con lenalidomida y dexametasona (DRd): Las reacciones adversas descritas en la siguiente tabla reflejan la exposición a DARZALEX® para una mediana de duración del tratamiento de 25.3 meses (rango: 0.1 a 40.44 meses) para el grupo de daratumumab-lenalidomida-dexametasona (DRd) y una mediana de duración del tratamiento de 21.3 meses (rango 0.03 a 40.64 meses) para el grupo de lenalidomida-dexametasona (Rd) en un estudio fase 3 controlado con activo (Estudio MMY3008). Las reacciones adversas más frecuentes (≥20%) fueron reacciones a la infusión, diarrea, estreñimiento, náuseas, edema periférico, fatiga, dolor de espalda, astenia, pirexia, infección de las vías respiratorias superiores, bronquitis, neumonía, disminución del apetito, espasmos musculares, neuropatía sensorial periférica, disnea y tos. Las reacciones adversas graves con una incidencia 2% mayor en el brazo de DRd en comparación con el brazo de Rd fueron deshidratación (DRd 2% frente a Rd <1%), bronquitis (DRd 4% frente a Rd 2%) y neumonía (DRd 15% frente a Rd 8%).
Tabla 7: Reacciones adversas reportadas en el Estudio MMY3008*
|
Clase de Órgano/Sistema Reacción adversa |
DRd (N=364) |
Rd (N=365) |
||||
|
Cualquier grado (%) |
Grado 3 (%) |
Grado 4 (%) |
Cualquier grado (%) |
Grado 3 (%) |
Grado 4 (%) |
|
|
Reacciones a la infusióna |
41 |
2 |
<1 |
0 |
0 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
57 |
7 |
0 |
46 |
4 |
0 |
|
Estreñimiento |
41 |
1 |
<1 |
36 |
<1 |
0 |
|
Náusea |
32 |
1 |
0 |
23 |
1 |
0 |
|
Vómito |
17 |
1 |
0 |
12 |
<1 |
0 |
|
Trastornos generales y condiciones del sitio de administración |
||||||
|
Edema periféricob |
41 |
2 |
0 |
33 |
1 |
0 |
|
Fatiga |
40 |
8 |
0 |
28 |
4 |
0 |
|
Dolor de espalda |
34 |
3 |
<1 |
26 |
3 |
<1 |
|
Astenia |
32 |
4 |
0 |
25 |
3 |
<1 |
|
Pirexia |
23 |
2 |
0 |
18 |
2 |
0 |
|
Escalofríos |
13 |
0 |
0 |
2 |
0 |
0 |
|
Infecciones e infestaciones |
||||||
|
Infección de las vías respiratorias superioresc |
52 |
2 |
<1 |
36 |
2 |
<1 |
|
Bronquitisd |
29 |
3 |
0 |
21 |
1 |
0 |
|
Neumoniae |
26 |
14 |
1 |
14 |
7 |
1 |
|
Infección de vías urinarias |
18 |
2 |
0 |
10 |
2 |
0 |
|
Trastornos del metabolismo y la nutrición |
||||||
|
Disminución del apetito |
22 |
1 |
0 |
15 |
<1 |
<1 |
|
Hiperglucemia |
14 |
6 |
1 |
8 |
3 |
1 |
|
Hipocalcemia |
14 |
1 |
<1 |
9 |
1 |
1 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||||
|
Espasmos musculares |
29 |
1 |
0 |
22 |
1 |
0 |
|
Trastornos del sistema nervioso |
||||||
|
Neuropatía sensorial periférica |
24 |
1 |
0 |
15 |
0 |
0 |
|
Dolor de cabeza |
19 |
1 |
0 |
11 |
0 |
0 |
|
Parestesia |
16 |
0 |
0 |
8 |
0 |
0 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||||
|
Disneaf |
32 |
3 |
<1 |
20 |
1 |
0 |
|
Tosg |
30 |
<1 |
0 |
18 |
0 |
0 |
|
Trastornos vasculares |
||||||
|
Hipertensiónh |
13 |
6 |
<1 |
7 |
4 |
0 |
Clave: D=daratumumab, Rd=lenalidomida-dexametasona.
a Reacción a la infusión incluye términos que los investigadores han determinado que están relacionados con la infusión; ver la sección sobre reacciones a la infusión a continuación.
b Edema generalizado, edema gravitacional, edema, edema periférico, hinchazón periférica.
c Sinusitis aguda, rinitis bacteriana, laringitis, infección por metaneumovirus, nasofaringitis, candidiasis orofaríngea, faringitis, infección por virus sincitial respiratorio, infección de las vías respiratorias, infección viral de las vías respiratorias, rinitis, infección por rinovirus, sinusitis, amigdalitis, traqueítis, infección de las vías respiratorias superiores, faringitis viral, rinitis viral, infección viral de las vías respiratorias superiores.
d Bronquiolitis, bronquitis, bronquitis viral, bronquiolitis por virus sincitial respiratorio, traqueobronquitis.
e Neumonía atípica, aspergilosis broncopulmonar, infección pulmonar, infección por Pneumocystis jirovecii, neumonía por Pneumocystis jirovecii, neumonía, neumonía por aspiración, neumonía neumocócica, neumonía viral, micosis pulmonar.
f Disnea, disnea de esfuerzo.
g Tos, tos productiva.
h Aumento de la presión arterial, hipertensión.
* Nota: Las reacciones adversas que ocurrieron en ≥10% de los pacientes y con al menos un 5% de frecuencia mayor en el brazo DRd están en la lista. Las toxicidades relacionadas con el laboratorio de hematología fueron excluidas y reportadas por separado en la siguiente tabla.
Las anormalidades de laboratorio que empeoran durante el tratamiento desde la basal se enlistan en la siguiente tabla.
Tabla 8: Anormalidades hematológicas de laboratorio emergentes al tratamiento en el Estudio MMY3008
|
DRd (N=364) % |
Rd (N=365) % |
|||||
|
Cualquier grado |
Grado 3 |
Grado 4 |
Cualquier grado |
Grado 3 |
Grado 4 |
|
|
Anemia |
47 |
13 |
0 |
57 |
24 |
0 |
|
Trombocitopenia |
67 |
6 |
3 |
58 |
7 |
4 |
|
Leucopenia |
90 |
30 |
5 |
82 |
20 |
4 |
|
Neutropenia |
91 |
39 |
17 |
77 |
28 |
11 |
|
Linfopenia |
84 |
41 |
11 |
75 |
36 |
6 |
Clave: D = daratumumab, Rd = lenalidomida – dexametasona.
Tratamiento de combinación con bortezomib, melfalán y prednisona: Las reacciones adversas descritas en la siguiente tabla reflejan la exposición a DARZALEX® para una mediana de duración del tratamiento de 14.7 meses (rango: 0 a 25.8 meses) para el grupo de daratumumab-bortezomib-melfalán-prednisona (grupo D-VMP) y una mediana de la duración del tratamiento de 12 meses (rango: 0.1 a 14.9 meses) para el grupo VMP en un estudio fase 3 controlado con activo (Estudio MMY3007). Las reacciones adversas más frecuentes (≥20%) fueron reacciones a la infusión, infección de vías respiratorias superiores y edema periférico. Las reacciones adversas graves con una incidencia de por lo menos más del 2% en el grupo D-VMP, en comparación con el grupo VMP, fueron neumonía (D-VMP 11% contra VMP 4%), infección de vías respiratorias superiores (D-VMP 5% contra VMP 1%) y edema pulmonar (D-VMP 2% contra VMP 0%).
Tabla 9. Reacciones adversas reportadas en el Estudio MMY3007*
|
Clase de Órgano/Sistema Reacción adversa |
D-VMP (N = 346) |
VMP (N = 354) |
||||
|
Cualquier Grado (%) |
Grado 3 (%) |
Grado 4 (%) |
Cualquier Grado (%) |
Grado 3 (%) |
Grado 4 (%) |
|
|
Reacciones a la infusióna |
28 |
4 |
1 |
0 |
0 |
0 |
|
Trastornos generales y condiciones del sitio de administración |
||||||
|
Edema periféricob |
21 |
1 |
< 1 |
14 |
1 |
0 |
|
Infecciones e infestaciones |
||||||
|
Infección de vías respiratorias superioresb |
48 |
5 |
0 |
28 |
3 |
0 |
|
Neumoníab |
16 |
12 |
< 1 |
6 |
5 |
< 1 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||||
|
Tosb |
16 |
< 1 |
0 |
8 |
< 1 |
0 |
|
Disneab |
13 |
2 |
1 |
5 |
1 |
0 |
|
Edema pulmonarb |
2 |
1 |
< 1 |
< 1 |
< 1 |
0 |
|
Trastornos vasculares |
||||||
|
Hipertensiónb |
10 |
4 |
< 1 |
3 |
2 |
0 |
Clave: D = daratumumab, VMP = bortezomib-melfalán-prednisona.
a La reacción a la infusión incluye términos determinados por los investigadores como relacionados con la infusión, ver la sección de Reacciones a la Infusión a continuación.
b Indica el agrupamiento de términos preferidos.
* Nota: Se listan las reacciones adversas que se presentaron en ≥ 10% de los pacientes y con una frecuencia de por lo menos más del 5% en el grupo D-VMP. Además, se listan las reacciones adversas graves si su incidencia fue de por lo menos más del 2% en el grupo D-VMP en comparación con el grupo VMP.
Las toxicidades relacionadas con el laboratorio de hematología fueron excluidas y reportadas por separado en la siguiente tabla.
Las anormalidades del laboratorio que empeoraron durante el tratamiento desde la basal se enlistan en la siguiente tabla.
Tabla 10. Anormalidades del laboratorio de hematología emergentes al tratamiento en el Estudio MMY3007
|
D-VMP (N = 346) % |
VMP (N = 354) % |
|||||
|
Cualquier grado |
Grado 3 |
Grado 4 |
Cualquier grado |
Grado 3 |
Grado 4 |
|
|
Anemia |
47 |
18 |
0 |
50 |
21 |
0 |
|
Trombocitopenia |
88 |
27 |
11 |
88 |
26 |
16 |
|
Neutropenia |
86 |
34 |
10 |
87 |
32 |
11 |
|
Linfopenia |
85 |
46 |
12 |
83 |
44 |
9 |
Clave: D = daratumumab, VMP = bortezomib-melfalán-prednisona.
Tratamiento combinado con bortezomib, talidomida y dexametasona (DVTd): Las reacciones adversas descritas en la siguiente tabla reflejan la exposición a DARZALEX® hasta el día 100 después del trasplante en un estudio fase 3 controlado con activo, Estudio MMY3006 (ver Estudios clínicos). La duración mediana del tratamiento de inducción/TACHP/consolidación fue de 8.9 (rango: 7.0 a 12.0) meses para el grupo con DVTd y 8.7 (rango: 6.4 a 11.5) meses para el grupo con VTd. Las reacciones adversas más frecuentes (>20%) fueron reacciones a la infusión, náuseas, pirexia, infección de las vías respiratorias superiores y bronquitis. Las reacciones adversas graves con una incidencia 2% mayor en el brazo de DVTd en comparación con el brazo de VTd fueron bronquitis (DVTd 2% vs VTd <1%) y neumonía (DVTd 6% vs VTd 4%).
Tabla 11: Reacciones adversas reportadas en el Estudio MMY3006*
|
Clase de Órgano/Sistema Reacción adversa |
DVTd (N=536) |
VTd (N=538) |
||||
|
Cualquier grado (%) |
Grado 3 (%) |
Grado 4 (%) |
Cualquier grado (%) |
Grado 3 (%) |
Grado 4 (%) |
|
|
Reacciones a la infusióna |
35 |
3 |
<1 |
0 |
0 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Náusea |
30 |
4 |
0 |
24 |
2 |
<1 |
|
Vómito |
16 |
2 |
0 |
10 |
2 |
0 |
|
Trastornos generales y condiciones del lugar de administración |
||||||
|
Pirexia |
26 |
2 |
<1 |
21 |
2 |
0 |
|
Infecciones e infestaciones |
||||||
|
Infección de las vías respiratorias superioresb |
27 |
1 |
0 |
17 |
1 |
0 |
|
Bronquitisc |
20 |
1 |
0 |
13 |
1 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||||
|
Tosd |
17 |
0 |
0 |
9 |
0 |
0 |
|
Trastornos vasculares |
||||||
|
Hipertensión |
10 |
4 |
0 |
5 |
2 |
0 |
Clave: D=daratumumab, VTd=bortezomib-talidomida -dexametasona.
a Reacción a la infusión incluye términos determinados por los investigadores como relacionados con la infusión, ver la sección de Reacciones a la Infusión a continuación.
b Laringitis, laringitis viral, infección por metaneumovirus, nasofaringitis, candidiasis orofaríngea, faringitis, infección por virus sincitial respiratorio, infección de las vías respiratorias, infección viral de las vías respiratorias, rinitis, infección por rinovirus, sinusitis, amigdalitis, traqueítis, infección de las vías respiratorias superiores, faringitis viral, rinitis viral, infección viral de las vías respiratorias superiores.
c Bronquiolitis, bronquitis, bronquitis crónica, bronquitis por virus sincitial respiratorio, traqueobronquitis.
d Tos, tos productiva.
*Nota: Se enumeran las reacciones adversas que ocurrieron en ≥10% de los pacientes y con al menos un 5% más de frecuencia en el brazo de DVTd. Las toxicidades relacionadas con el laboratorio de hematología fueron excluidas y reportadas por separado en la siguiente tabla.
Las anormalidades de laboratorio que empeoraron durante el tratamiento desde la basal se enlistan en la siguiente tabla.
Tabla 12: Anormalidades hematológicas de laboratorio emergentes al tratamiento en el Estudio MMY3006
|
DVTd (N=536) % |
VTd (N=538) % |
|||||
|
Cualquier grado |
Grado 3 |
Grado 4 |
Cualquier grado |
Grado 3 |
Grado 4 |
|
|
Anemia |
36 |
4 |
0 |
35 |
5 |
0 |
|
Trombocitopenia |
81 |
9 |
5 |
58 |
8 |
3 |
|
Leucopenia |
82 |
14 |
10 |
57 |
6 |
9 |
|
Neutropenia |
63 |
19 |
14 |
41 |
10 |
9 |
|
Linfopenia |
95 |
44 |
15 |
91 |
37 |
10 |
Clave: D=daratumumab, VTd=bortezomib-talidomida -dexametasona.
Mieloma Múltiple recurrente/refractario:
Tratamiento de combinación con lenalidomida y dexametasona: Las reacciones adversas descritas en la siguiente tabla reflejan la exposición a DARZALEX® para una mediana de duración del tratamiento de 13.1 meses (rango: 0 a 20.7 meses) para el grupo de daratumumab-lenalidomida-dexametasona (DRd) y una mediana de duración del tratamiento de 12.3 meses (rango: 0.2 a 20.1 meses) para el grupo de lenalidomida-dexametasona (Rd) en un estudio fase 3 controlado con activo (Estudio MMY3003). Las reacciones adversas más frecuentes fueron reacciones a la infusión, diarrea, náusea, fatiga, pirexia, infección de vías respiratorias superiores, espasmos musculares, tos y disnea. Las reacciones adversas graves fueron neumonía, infección de vías respiratorias superiores, influenza y pirexia. Las reacciones adversas resultaron en descontinuación en 7% (n = 19) de los pacientes en el grupo DRd contra 8% (n = 22) en el grupo Rd.
Tabla 13. Reacciones adversas reportadas en el estudio MMY3003*
|
Clase de Órgano/Sistema Reacción adversa |
DRd (N = 283) |
Rd (N = 281) |
||||
|
Cualquier Grado (%) |
Grado 3 (%) |
Grado 4 (%) |
Cualquier Grado (%) |
Grado 3 (%) |
Grado 4 (%) |
|
|
Reacciones a la infusióna |
48 |
5 |
0 |
0 |
0 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
43 |
5 |
0 |
25 |
3 |
0 |
|
Náusea |
24 |
1 |
0 |
14 |
0 |
0 |
|
Vómito |
17 |
1 |
0 |
5 |
1 |
0 |
|
Trastornos generales y condiciones en el sitio de administración |
||||||
|
Fatiga |
35 |
6 |
< 1 |
28 |
2 |
0 |
|
Pirexia |
20 |
2 |
0 |
11 |
1 |
0 |
|
Infecciones e infestaciones |
||||||
|
Influenza |
7 |
3 |
0 |
5 |
1 |
0 |
|
Neumoníab |
19 |
10 |
2 |
15 |
7 |
2 |
|
Infección de vías respiratorias superioresb |
65 |
6 |
< 1 |
51 |
4 |
0 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||||
|
Espasmos musculares |
26 |
1 |
0 |
19 |
2 |
0 |
|
Trastornos del sistema nervioso |
||||||
|
Dolor de cabeza |
13 |
0 |
0 |
7 |
0 |
0 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||||
|
Tosb |
30 |
0 |
0 |
15 |
0 |
0 |
|
Disnea |
21 |
3 |
< 1 |
12 |
1 |
0 |
Clave: D = daratumumab, Rd = lenalidomida-dexametasona.
a Reacciones a la infusión incluye los términos determinados por los investigadores como relacionados a la infusión; consultar la sección de Reacciones a la Infusión a continuación.
b Indica el agrupamiento de los términos preferidos.
* Nota: Se enlistan las reacciones adversas que ocurrieron en ≥ 10% de los pacientes y con una frecuencia al menos 5% mayor en el grupo DRd. Adicionalmente, se enlistan los eventos adversos graves si se observó una incidencia al menos 2% mayor en el grupo DRd en comparación con el grupo Rd.
Las toxicidades relacionadas con datos de análisis hematológico fueron excluidas y se reportan por separado en la siguiente tabla.
Las anormalidades de laboratorio que empeoraron durante el tratamiento desde la basal se enlistan en la siguiente tabla .
Tabla 14. Anormalidades hematológicas de laboratorio emergentes al tratamiento
|
Estudio MMY3003 |
||||||
|
DRd (N = 283) % |
Rd (N = 281) % |
|||||
|
Cualquier Grado |
Grado 3 |
Grado 4 |
Cualquier Grado |
Grado 3 |
Grado 4 |
|
|
Anemia |
52 |
13 |
0 |
57 |
19 |
0 |
|
Trombocitopenia |
73 |
7 |
6 |
67 |
10 |
5 |
|
Neutropenia |
92 |
36 |
17 |
87 |
32 |
8 |
|
Linfopenia |
95 |
42 |
10 |
87 |
32 |
6 |
Clave: D = daratumumab, Rd = lenalidomida-dexametasona.
Tratamiento de combinación con bortezomib y dexametasona: Las reacciones adversas descritas en la tabla 15 reflejan la exposición a DARZALEX® (grupo DVd) para una mediana de duración del tratamiento de 6.5 meses (rango: 0 a 14.8 meses) para el grupo de daratumumab-bortezomib-dexametasona (DVd) y una mediana de duración del tratamiento de 5.2 meses (rango: 0.2 a 8.0 meses) para el grupo de bortezomib-dexametasona (Vd) en un estudio fase 3 controlado con activo (Estudio MMY3004).
Las reacciones adversas más frecuentes (> 20%) fueron reacciones a la infusión, diarrea, edema periférico, infección de vías respiratorias superiores, neuropatía sensorial periférica, tos y disnea. Las reacciones adversas graves incluyeron diarrea, infección de vías respiratorias superiores y fibrilación auricular. Las reacciones adversas resultaron en descontinuación en 7% (n = 18) de los pacientes en el grupo DVd contra 9% (n = 22) en el grupo Vd.
Tabla 15. Reacciones adversas reportadas en el estudio MMY3004*
|
Clase de Órgano/Sistema Reacción adversa |
DVd (N = 243) |
Vd (N = 237) |
||||
|
Cualquier Grado (%) |
Grado 3 (%) |
Grado 4 (%) |
Cualquier Grado (%) |
Grado 3 (%) |
Grado 4 (%) |
|
|
Reacciones a la infusióna |
45 |
9 |
0 |
0 |
0 |
0 |
|
Trastornos cardiacos |
||||||
|
Fibrilación auricular |
5 |
1 |
1 |
2 |
1 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
32 |
3 |
< 1 |
22 |
1 |
0 |
|
Vómito |
11 |
0 |
0 |
4 |
0 |
0 |
|
Trastornos generales y condiciones en el sitio de administración |
||||||
|
Edema periféricob |
22 |
1 |
0 |
13 |
0 |
0 |
|
Pirexia |
16 |
1 |
0 |
11 |
1 |
0 |
|
Infecciones e infestaciones |
||||||
|
Infección de vías respiratorias superioresb |
44 |
6 |
0 |
30 |
3 |
< 1 |
|
Trastornos del sistema nervioso |
||||||
|
Neuropatía sensorial periférica |
47 |
5 |
0 |
38 |
6 |
< 1 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||||
|
Tosb |
27 |
0 |
0 |
14 |
0 |
0 |
|
Disneab |
21 |
4 |
0 |
11 |
1 |
0 |
Clave: D = daratumumab, Vd = bortezomib-dexametasona.
a Reacciones a la infusión incluye los términos descritos por los investigadores como relacionados a la infusión; consultar la sección de Reacciones a la Infusión a continuación.
b Indica el agrupamiento de los términos preferidos.
* Nota: Se enlistan las reacciones adversas que ocurrieron en ≥ 10% de los pacientes y con una frecuencia al menos 5% mayor en el grupo DVd. Adicionalmente, se enlistan los eventos adversos graves si se observó una incidencia al menos 2% mayor en el grupo DVd en comparación con el grupo Vd.
Las toxicidades relacionadas con datos de análisis hematológico fueron excluidas y se reportan por separado en la siguiente tabla.
Las anormalidades de laboratorio que empeoraron durante el tratamiento se enlistan en la siguiente tabla.
Tabla 16. Anormalidades hematológicas de laboratorio emergentes al tratamiento
|
Estudio MMY3004 |
||||||
|
DVd (N = 243) % |
Vd (N = 237) % |
|||||
|
Cualquier Grado |
Grado 3 |
Grado 4 |
Cualquier Grado |
Grado 3 |
Grado 4 |
|
|
Anemia |
48 |
13 |
0 |
56 |
14 |
0 |
|
Trombocitopenia |
90 |
28 |
19 |
85 |
22 |
13 |
|
Neutropenia |
58 |
12 |
3 |
40 |
5 |
< 1 |
|
Linfopenia |
89 |
41 |
7 |
81 |
24 |
3 |
Clave: D = daratumumab, Vd = bortezomib-dexametasona.
Tratamiento combinado con pomalidomida y dexametasona: Las reacciones adversas descritas en la tabla 17 reflejan la exposición a DARZALEX® y pomalidomida (DPd) para una mediana de duración del tratamiento de 6 meses (rango: 0.03 a 16.9 meses) en el estudio MMY1001. Las reacciones adversas resultaron en descontinuación en 13% de los pacientes.
Tabla 17. Reacciones adversas reportadas en el estudio MMY1001
|
Clase de Órgano/Sistema Reacción adversa |
Estudio MMY1001 |
||
|
DPd (N = 103) |
|||
|
Cualquier Grado (%) |
Grado 3 (%) |
Grado 4 (%) |
|
|
Reacciones a la infusióna |
50 |
4 |
0 |
|
Trastornos cardiacos |
|||
|
Fibrilación auricular |
7 |
1 |
0 |
|
Trastornos gastrointestinales |
|||
|
Diarrea |
38 |
3 |
0 |
|
Náusea |
30 |
0 |
0 |
|
Vómito |
21 |
2 |
0 |
|
Trastornos generales y condiciones en el sitio de administración |
|||
|
Fatiga |
50 |
10 |
0 |
|
Edema periféricob |
17 |
4 |
0 |
|
Pirexia |
25 |
1 |
0 |
|
Infecciones e infestaciones |
|||
|
Influenza |
5 |
3 |
0 |
|
Neumoníab |
15 |
8 |
2 |
|
Infección de vías respiratorias superioresb |
50 |
4 |
1 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|||
|
Espasmos musculares |
26 |
1 |
0 |
|
Trastornos del sistema nervioso |
|||
|
Dolor de cabeza |
17 |
0 |
0 |
|
Neuropatía sensorial periférica |
8 |
2 |
0 |
|
Trastornos respiratorios, torácicos y mediastinales |
|||
|
Tosb |
43 |
1 |
0 |
|
Disneab |
33 |
6 |
1 |
Clave: D = daratumumab, Pd = pomalidomida-dexametasona.
a Reacciones a la infusión incluye los términos descritos por los investigadores como relacionados a la infusión; consultar la sección de Reacciones a la Infusión a continuación.
b Indica el agrupamiento de los términos preferidos.
Las toxicidades relacionadas con datos de análisis hematológico fueron excluidas y se reportan por separado en la siguiente tabla.
Las anormalidades de laboratorio que empeoraron durante el tratamiento se enlistan en la siguiente tabla.
Tabla 18. Anormalidades hematológicas de laboratorio emergentes al tratamiento (estudio MMY1001)
|
DPd (N = 103) |
|||
|
Cualquier Grado (%) |
Grado 3 (%) |
Grado 4 (%) |
|
|
Anemia |
57 |
30 |
0 |
|
Trombocitopenia |
75 |
10 |
10 |
|
Neutropenia |
95 |
36 |
46 |
|
Linfopenia |
94 |
45 |
26 |
Clave: D = daratumumab, Pd = pomalidomida-dexametasona.
Monoterapia: Los datos descritos a continuación reflejan la exposición a DARZALEX® en tres estudios clínicos abiertos acumulados que incluyeron a 156 pacientes con mieloma múltiple en recaída y refractario, tratados con DARZALEX® a 16 mg/kg. La mediana de duración del tratamiento de DARZALEX® fue de 3.3 meses, con duración máxima del tratamiento siendo de 14.2 meses. Las reacciones adversas que ocurrieron en una tasa ≥ 10% se presentan en la siguiente tabla. Las reacciones adversas reportadas con mayor frecuencia (≥ 20%) fueron RRIs, fatiga, náusea, dolor de espalda, anemia, neutropenia y trombocitopenia. Cuatro por ciento de los pacientes descontinuó el tratamiento con DARZALEX® debido a reacciones adversas, ninguna de las cuales fue considerada como relacionada con el fármaco.
Las frecuencias son definidas como muy común (≥ 1/10), común (≥ 1/100 a < 1/10), poco común (≥ 1/1000 a < 1/100), rara (≥ 1/10000 a < 1/1000) y muy rara (< 1/10000).
Tabla 19. Reacciones adversas en pacientes con mieloma múltiple tratados con DARZALEX® 16 mg/kg
|
Clase de Órgano/Sistema |
Reacción Adversa |
Frecuencia (todos los Grados) |
Incidencia (%) |
|
|
Todos los Grados |
Grado 3-4 |
|||
|
Infecciones e infestaciones |
Infección de las vías respiratorias superiores |
Muy Común |
17 |
1* |
|
Nasofaringitis |
12 |
0 |
||
|
Neumonía** |
10 |
6* |
||
|
Trastornos sanguíneos y del sistema linfático |
Anemia |
Muy Común |
25 |
17* |
|
Neutropenia |
22 |
12 |
||
|
Trombocitopenia |
20 |
14 |
||
|
Trastornos metabólicos y de nutrición |
Disminución del apetito |
Muy Común |
15 |
1* |
|
Trastornos respiratorios, torácicos y mediastinales |
Tos |
Muy Común |
14 |
0 |
|
Trastornos gastrointestinales |
Náuseas |
Muy Común |
21 |
0 |
|
Diarrea |
15 |
0 |
||
|
Estreñimiento |
14 |
0 |
||
|
Trastornos músculo esqueléticos y de tejidos conectivos |
Dolor de espalda |
Muy Común |
20 |
2* |
|
Artralgia |
16 |
0 |
||
|
Dolor en una extremidad |
15 |
1* |
||
|
Dolor torácico músculo esquelético |
10 |
1* |
||
|
Trastornos generales y condiciones en el sitio de administración |
Fatiga |
Muy Común |
37 |
2* |
|
Pirexia |
17 |
1* |
||
|
Lesión, envenenamiento y complicaciones de procedimientos |
Reacción relacionada con la infusión*** |
Muy Común |
51 |
4* |
* No Grado 4.
** Neumonía también incluye los términos neumonía estreptocócica y neumonía lobar.
*** Las reacciones relacionadas con la infusión incluyen, pero no están limitadas a los siguientes términos de reacciones adversas múltiples: congestión nasal, tos, escalofríos, rinitis alérgica, irritación de garganta, disnea, náusea (todas ≥5%), broncoespasmo (2.6%), hipertensión (1.9%) e hipoxia (1.3%).
Reacciones relacionadas con la infusión: En estudios clínicos (monoterapia y tratamientos combinados; N = 2066) la incidencia de las reacciones relacionadas con la infusión de cualquier grado fue de 37% con la primera (16 mg/kg, Semana 1) infusión de DARZALEX®, de 2% con la infusión de la Semana 2 y acumulativamente 6% con las infusiones subsecuentes. Menos de 1% de los pacientes presentaron una reacción a la infusión grado 3/4 con la infusión de la Semana 2 o con infusiones subsecuentes. La mediana del tiempo para el comienzo de una reacción fue 1.5 horas (rango: 0 a 72.8 horas). La incidencia de modificaciones de la infusión debidas a reacciones fue de 36%. La mediana de la duración de las infusiones de 16 mg/kg para la 1ra, 2da e infusiones subsecuentes fue aproximadamente 7, 4 y 3 horas respectivamente.
Las reacciones graves relacionadas a la infusión incluyeron broncoespasmo, disnea, edema laríngeo, edema pulmonar, hipoxia e hipertensión. Otras reacciones adversas relacionadas con la infusión incluyeron congestión nasal, tos, escalofrío, irritación en garganta, vómito y náusea.
Cuando se interrumpió la dosificación de DARZALEX® en el contexto del TACHP (Estudio MMY3006) durante una mediana de 3.75 (rango: 2.4; 6.9) meses, al reiniciar DARZALEX® la incidencia de RRIs fue de 11% en la primera infusión después del TACHP. La tasa de infusión/volumen de dilución utilizado en el momento de la reanudación fue el utilizado para la última infusión de DARZALEX® antes de la interrupción debida al TACHP. Las RRIs que ocurrieron al reiniciar DARZALEX® después del TACHP fueron consistentes en términos de síntomas y severidad (Grado 3/4: <1%) con aquellos reportados en estudios previos en la Semana 2 o infusiones subsecuentes.
En el estudio MMY1001, a los pacientes que recibieron tratamiento de combinación con daratumumab (n=97) se les administró la primera dosis de 16 mg/kg de daratumumab en la semana 1 dividida en dos días, es decir, 8 mg/kg el día 1 y el día 2, respectivamente. La incidencia de reacciones relacionadas con la infusión de cualquier grado fue del 42%, con 36% de los pacientes que experimentaron reacciones relacionadas con la infusión el día 1 de la semana 1, el 4% el día 2 de la semana 1 y el 8% con las infusiones posteriores. El tiempo mediano de inicio de una reacción fue de 1.8 horas (rango: 0.1 a 5.4 horas). La incidencia de interrupciones de la infusión debidas a reacciones fue del 30%. La duración mediana de las infusiones fue de 4.2 h para la semana 1-Día 1, 4.2 h para la semana 1-Día 2 y 3.4 horas para las infusiones posteriores.
Infecciones:
En los pacientes en terapia combinada con DARZALEX®, se reportaron infecciones grado 3 o 4 como se describe a continuación: Estudios de pacientes con recidiva/refractarios: DVd: 21%, Vd: 19%; DRd: 27%, Rd: 23%, DPd: 28%.
Estudios de pacientes recientemente diagnosticados: D-VMP: 23%, VMP: 15%; DRd: 32%, Rd: 23%; DVTd: 22%, VTd: 20%.
La neumonía fue la infección grave (grado 3 o 4) más comúnmente reportada de los estudios. En los estudios controlados con activo, las discontinuaciones del tratamiento debido a infecciones (1-4%) e infecciones fatales fueron generalmente poco frecuentes y balanceadas entre los regímenes que contienen DARZALEX® y los grupos controlados con activo. Las infecciones fatales fueron principalmente debido a neumonía y sepsis.
Datos post comercialización: Las reacciones adversas identificadas durante la experiencia post-comercialización con DARZALEX® se incluyen en la tabla 20. Las frecuencias se proveen de acuerdo a la siguiente convención:
Muy común ≥ 1/10
Común ≥ 1/100 a < 1/10
Poco común ≥ 1/1000 a < 1/100
Raro ≥ 1/10000 a < 1/1000
Muy raro < 1/10000, incluyendo reportes aislados
Desconocido la frecuencia no puede ser estimada a partir de los datos disponibles.
En la tabla 20, las reacciones adversas se presentan por categoría de frecuencia con base en la tasa de reportes espontáneos.
Tabla 20. Reacciones adversas post-comercialización
|
Clase de Sistema/Órgano Reacción adversa |
Categoría de frecuencia con base en la tasa de reportes espontáneos |
|
Trastornos del sistema inmune |
|
|
Reacción anafiláctica |
Raro |
|
Infecciones e infestaciones |
|
|
Reactivación del virus de la Hepatitis B |
Raro |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogenicidad y mutagenicidad: No se han llevado a cabo estudios en animales para establecer el potencial carcinogénico de daratumumab. Los estudios de rutina de genotoxicidad y carcinogenicidad generalmente no son aplicables a los medicamentos biológicos debido a que las proteínas grandes no pueden difundirse en las células y no pueden interactuar con el ADN o el material cromosómico.
Toxicología reproductiva: No se han llevado a cabo estudios en animales para evaluar los efectos potenciales de daratumumab en la reproducción o el desarrollo.
Fertilidad: No se han llevado a cabo estudios en animales para determinar los efectos potenciales en la fertilidad en pacientes masculinos o femeninos.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se han llevado a cabo estudios de interacción entre fármacos.
Las evaluaciones de farmacocinética clínica de daratumumab en combinación con lenalidomida, pomalidomida, talidomida, bortezomib y dexametasona no indicaron interacciones medicamentosas clínicamente relevantes entre daratumumab y estos medicamentos de moléculas pequeñas.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Interferencia con la prueba de antiglobulina indirecta (prueba de Coombs indirecta): Daratumumab se une a CD38 en los GRs e interfiere con la prueba de compatibilidad, incluyendo la examinación de anticuerpos y pruebas cruzadas. Los métodos de mitigación de la interferencia de daratumumab incluyen el tratamiento de los GRs reactivos con ditiotreitol (DTT) para romper la unión o la genotipificacion eritrocitaria de daratumumab. Debido a que el grupo sanguíneo Kell también es sensible al tratamiento con DTT, las unidades Kell negativas deben ser administradas hasta que se descarte o identifiquen aloanticuerpos usando GRs tratados con DTT.
Interferencia con electroforesis de proteínas séricas y pruebas de inmunofijación: Daratumumab puede ser detectado en electroforesis de proteínas en séricas (EPS) y ensayos de inmunofijación (EIF) usados para monitorear inmunoglobulinas monoclonales de enfermedad (proteína M). Esto puede llevar a resultados falsos positivos en los ensayos EPS y EIF para pacientes con proteína kappa IgG de mieloma que impacta el análisis inicial de las Respuestas Completas (RC) por los criterios del Grupo de Trabajo Internacional de Mieloma (IMWG). En pacientes con muy buena respuesta parcial (MBRP) persistente, donde se sospecha interferencia de daratumumab, considere usar un ensayo validado EIF especifico de daratumumab para distinguir daratumumab de cualquier proteína M endógena remanente en el suero del paciente, para facilitar la determinación de una RC (ver Propiedades farmacodinámicas–Estudios Clínicos).
PRECAUCIONES GENERALES:
Reacciones relacionadas con la infusión (RRIs): DARZALEX® puede causar reacciones severas relacionadas con la infusión, incluyendo reacciones anafilácticas.
Monitorear a los pacientes durante toda la infusión y el periodo posterior a la infusión.
En estudios clínicos, se reportaron RRIs en aproximadamente la mitad de todos los pacientes tratados con DARZALEX®.
La mayoría de las RRIs ocurrieron en la primera infusión y fueron Grado 1-2. Cuatro por ciento de los pacientes tuvo una RRI en más de una infusión. Han ocurrido reacciones severas, incluyendo broncoespasmo, hipoxia, disnea e hipertensión, edema laríngeo y edema pulmonar. Los signos y síntomas pueden incluir síntomas respiratorios, tales como congestión nasal, tos, irritación de la garganta, así como escalofríos, vómito y náusea. Los síntomas menos comunes fueron disnea, rinitis alérgica, pirexia, molestia en el pecho, prurito e hipotensión (ver Reacciones secundarias y adversas).
Medicar previamente a los pacientes con antihistamínicos, antipiréticos y corticosteroides para reducir el riesgo de RRIs antes del tratamiento con DARZALEX®. Interrumpir la infusión de DARZALEX® para las RRIs de cualquier severidad. Establecer manejo médico/tratamiento de soporte según sea necesario. Para los pacientes con reacciones Grado 1, 2 o 3, al reiniciar la infusión reducir la velocidad. Si ocurre una reacción anafiláctica o que amenace la vida (Grado 4), descontinuar permanentemente la administración de DARZALEX® y establecer el cuidado de emergencia apropiado (ver Dosis y vía de administración).
Para reducir el riesgo de RRIs retardadas, administrar corticosteroide oral a todos los pacientes después de todas las infusiones de DARZALEX®. Adicionalmente, considere el uso de medicamentos posteriores a la infusión (por ejemplo, corticosteroides inhalados, broncodilatadores de acción corta y prolongada) para los pacientes con un antecedente de enfermedad pulmonar obstructiva crónica para manejar las complicaciones respiratorias si estas llegaran a ocurrir (ver Dosis y vía de administración).
Neutropenia/Trombocitopenia: DARZALEX® puede aumentar la neutropenia y trombocitopenia inducidas por la terapia de base (ver Reacciones secundarias y adversas).
Monitorear los conteos de células sanguíneas completos periódicamente durante el tratamiento de acuerdo con la información para prescribir del fabricante para las terapias de base. Monitorear a los pacientes con neutropenia para signos de infección. Puede ser necesario retrasar la dosis de DARZALEX® para permitir la recuperación del conteo de células sanguíneas. No se recomienda reducir la dosis de DARZALEX®. Considerar cuidados de soporte con transfusiones o factores de crecimiento.
Interferencia con la prueba de antiglobulina indirecta (prueba de Coombs indirecta): Daratumumab se une a CD38, que se encuentra en niveles bajos en los glóbulos rojos (GRs) y puede resultar en una prueba falsa positiva de Coombs indirecta. La prueba de Coombs indirecta positiva mediada por daratumumab puede persistir por hasta 6 meses después de la última infusión de daratumumab. Debe reconocerse que daratumumab unido a los GRs puede enmascarar la detección de anticuerpos de antígenos menores en el suero del paciente. La determinación de ABO y el tipo de Rh sanguíneo de un paciente no resulta impactada.
Determinar el grupo sanguíneo y examinar a los pacientes previo a iniciar DARZALEX®.
En el caso de una transfusión planificada, notifique a los centros de transfusiones sanguíneas de esta interferencia con la prueba de antiglobulina indirecta (ver Interacciones medicamentosas y de otro género). Si se requiere una transfusión de emergencia, GRs ABO/RhD compatibles no cruzados pueden administrarse según las prácticas locales del banco de sangre.
Reactivación del virus de la hepatitis B (VHB): Se ha reportado reactivación del virus de la hepatitis B (VHB), en algunos casos fatal, en pacientes tratados con DARZALEX®. Se debe realizar un examen de detección del VHB en todos los pacientes antes de iniciar el tratamiento con DARZALEX®.
Para los pacientes con evidencia de serología positiva para el VHB, monitorear los signos clínicos y de laboratorio de la reactivación del VHB durante, y por al menos seis meses después de la finalización del tratamiento con DARZALEX®. Manejar a los pacientes de acuerdo con las guías clínicas actuales. Considere consultar a un experto en hepatitis, según lo indicado clínicamente.
En pacientes que desarrollan reactivación del VHB mientras están en tratamiento con DARZALEX®, suspenda el tratamiento con DARZALEX® y cualquier esteroide, quimioterapia concomitante e instituya el tratamiento apropiado. La reanudación del tratamiento con DARZALEX® en pacientes cuya reactivación del VHB está adecuadamente controlada debe discutirse con médicos con experiencia en el manejo del VHB.
Efectos en la habilidad para conducir y utilizar maquinaria: La influencia de DARZALEX® en la habilidad para conducir o utilizar maquinaria es nula o insignificante. Sin embargo, se ha reportado fatiga en pacientes utilizando daratumumab y esto debe considerarse al conducir o utilizar maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN: DARZALEX® debe ser administrado por un profesional de la salud, con acceso inmediato a equipo de urgencias y el apoyo médico apropiado para manejar reacciones relacionadas con la infusión (RRIs) si éstas ocurren.
Deben administrarse medicamentos previos y posteriores a la infusión (ver Medicamentos concomitantes recomendados a continuación).
Dosis-Adultos (≥ 18 años)
Dosis recomendada:
El esquema de dosificación de DARZALEX® en la Tabla 21 es para la terapia combinada con regímenes de ciclos de 4 semanas (por ejemplo, lenalidomida, pomalidomida) y para la monoterapia como se indica a continuación:
• Terapia de combinación con lenalidomida y dexametasona en dosis bajas para pacientes con mieloma múltiple que no han recibido tratamiento previo y que no son candidatos para el trasplante autólogo de células hematopoyéticas (TACHP).
• Tratamiento combinado con lenalidomida o pomalidomida y dosis bajas de dexametasona para pacientes con mieloma múltiple recurrente/refractario.
• Monoterapia para pacientes con mieloma múltiple recurrente/refractario.
La dosis recomendada de DARZALEX® de 16 mg/kg de peso corporal administrados como una infusión intravenosa de acuerdo con el siguiente esquema de dosificación (velocidades de infusión presentadas en la tabla 25):
Tabla 21: Régimen de dosificación de DARZALEX® para monoterapia o en combinación con regímenes de dosificación de ciclos de 4 semanas
|
Semanas |
Régimen |
|
Semanas 1 a 8 |
Semanalmente (8 dosis en total) |
|
Semanas 9 a 24a |
Cada dos semanas (8 dosis en total) |
|
Semana 25 hasta la progresión de la enfermedadb |
Cada cuatro semanas |
a La primera dosis del régimen de dosis cada 2 semanas se administra en la semana 9.
b La primera dosis del régimen de dosis cada 4 semanas se administra en la semana 25.
Para las instrucciones de dosificación de los medicamentos administrados con DARZALEX®, ver Estudios clínicos y la información para prescribir del fabricante.
El régimen de dosificación de DARZALEX® en la Tabla 22 es para el tratamiento combinado con bortezomib, melfalán y prednisona (regímenes de dosificación de ciclos de 6 semanas) para pacientes con mieloma múltiple que no han recibido tratamiento previo y que no son candidatos para TACHP.
La dosis recomendada es de DARZALEX® 16 mg/kg de peso corporal administrada como infusión intravenosa de acuerdo con el régimen de dosificación (velocidades de infusión presentadas en la tabla 25).
Tabla 22. Régimen de dosificación de DARZALEX® en combinación con bortezomib, melfalán y prednisona ([VMP]; régimen de dosificación de ciclos de 6 semanas)
|
Semanas |
Régimen |
|
Semanas 1 a 6 |
Semanalmente (total de 6 dosis) |
|
Semanas 7 a 54a |
Cada tres semanas (total de 16 dosis) |
|
Semana 55 en adelante hasta la progresión de la enfermedadb |
Cada cuatro semanas |
a La primera dosis del régimen de cada 3 semanas se administra en la Semana 7.
b La primera dosis del régimen de cada 4 semanas se administra en la Semana 55.
Bortezomib se administra dos veces a la semana en las Semanas 1, 2, 4 y 5 para el primer ciclo de 6 semanas, seguido de una vez a la semana en las Semanas 1, 2, 4 y 5 durante 8 ciclos más de 6 semanas. Para información sobre la dosis VMP y el régimen de dosis cuando se administra con DARZALEX® (ver Estudios clínicos).
El régimen de dosificación de DARZALEX® en la Tabla 23 es para el tratamiento combinado con bortezomib, talidomida y dexametasona (regímenes de dosificación de ciclos de 4 semanas) para el tratamiento de pacientes con mieloma múltiple que no han recibido tratamiento previo y que son candidatos para TACHP.
La dosis recomendada es de DARZALEX® 16 mg/kg de peso corporal administrada como infusión intravenosa de acuerdo con el régimen de dosificación (velocidades de infusión presentadas en la tabla 25).
Tabla 23: Régimen de dosificación para DARZALEX® en combinación con bortezomib, talidomida y dexametasona ([VTd]; régimen de dosificación de ciclos de 4 semanas)
|
Fase del tratamiento |
Semanas |
Régimen |
|
Inducción |
Semanas 1 a 8 |
Semanalmente (8 dosis en total) |
|
Semanas 9 a 16a |
Cada dos semanas (4 dosis en total) |
|
|
Detenerse para quimioterapia de dosis altas y TACHP |
||
|
Consolidación |
Semanas 1 a 8b |
Cada dos semanas (4 dosis en total) |
a La primera dosis del régimen de dosis cada 2 semanas se administra en la semana 9.
b La primera dosis del régimen de dosis cada 2 semanas se administra en la semana 1 al reiniciar el tratamiento después del TACHP.
Para las instrucciones de dosificación de los medicamentos administrados con DARZALEX®, ver Estudios clínicos y la información para prescribir del fabricante.
El esquema de dosificación de DARZALEX® en la Tabla 24 es para la terapia de combinación en el régimen de ciclos de 3 semanas (por ejemplo, bortezomib) para pacientes con mieloma múltiple recurrente/refractario.
La dosis recomendada de DARZALEX® es de 16 mg/kg de peso corporal administrada como infusión intravenosa de acuerdo con el siguiente esquema de dosificación (velocidades de infusión presentadas en la tabla 25):
Tabla 24. Régimen de dosificación para DARZALEX® con regímenes de dosificación de ciclos de 3 semanas
|
Semanas |
Régimen |
|
Semanas 1 a 9 |
Semanalmente (9 dosis en total) |
|
Semanas 10 a 24a |
Cada tres semanas (5 dosis en total) |
|
Semana 25 en adelante hasta la progresión de la enfermedadb |
Cada cuatro semanas |
a La primera dosis del régimen de dosificación cada 3 semanas se administra en la semana 10.
b La primera dosis del régimen de dosificación cada 4 semanas se administra en la semana 25.
Para las instrucciones de dosificación de los medicamentos administrados con DARZALEX®, ver Estudios clínicos y la información para prescribir del fabricante.
Dosis omitida(s): Si se omite una dosis planificada de DARZALEX®, administre la dosis tan pronto como sea posible y ajuste el esquema de dosificación consecuentemente, manteniendo el intervalo del tratamiento.
Modificaciones de dosis: No se recomienda reducir la dosis de DARZALEX®. Puede ser necesario retrasar la dosis para permitir la recuperación de los conteos de células sanguíneas en el evento de toxicidad hematológica (ver Precauciones generales). Para información relativa a los medicamentos administrados en combinación con DARZALEX®, consulte la información para prescribir del fabricante.
Medicamentos concomitantes recomendados:
Medicación previa a la infusión: Administre los siguientes medicamentos previos a la infusión para reducir el riesgo de RRIs a todos los pacientes 1-3 horas antes de cada infusión de DARZALEX®:
• Corticosteroides (de acción prolongada o acción intermedia).
Monoterapia: Metilprednisolona 100 mg, o equivalente, administrada por vía intravenosa. Después de la segunda infusión, puede reducirse la dosis de corticosteroide (metilprednisolona 60 mg por vía oral o intravenosa).
Terapia de combinación: Administrar 20 mg de dexametasona (o equivalente) antes de cada infusión de DARZALEX®. Cuando dexametasona es el corticosteroide específico del régimen de fondo, la dosis de tratamiento con dexametasona servirá como premedicación en los días de infusión de DARZALEX® (ver Estudios clínicos).
Dexametasona se administra por vía intravenosa antes de la primera infusión de DARZALEX® y puede considerarse la administración oral antes de las infusiones subsecuentes. Los corticosteroides adicionales específicos del régimen de referencia (por ejemplo, prednisona) no deben tomarse en los días de infusión de DARZALEX® cuando los pacientes han recibido dexametasona como medicamento previo.
• Antipiréticos (paracetamol/acetaminofén oral 650 a 1000 mg).
• Antihistamínicos (difenhidramina 25 a 50 mg oral o intravenosa o equivalente).
Medicación posterior a la infusión: Administrar medicación posterior a la infusión para reducir el riesgo de reacciones retardadas relacionadas con la infusión, de la siguiente manera:
Monoterapia: Administrar un corticosteroide oral (20 mg de metilprednisolona o una dosis equivalente de un corticosteroide de acción intermedia o acción prolongada de acuerdo con los estándares locales) en cada uno de los 2 días después de todas las infusiones de DARZALEX® (comenzando el día después de la infusión).
Terapia de combinación: Considerar administrar una dosis baja de metilprednisolona oral (≤ 20 mg) o su equivalente el día después de la infusión de DARZALEX®.
Sin embargo, si se administra un corticosteroide de base específico del régimen (por ejemplo, dexametasona, prednisona) al día siguiente de la infusión de DARZALEX®, pueden no ser necesarios los medicamentos adicionales después de la infusión (ver Estudios clínicos).
Adicionalmente, para los pacientes con un antecedente de enfermedad pulmonar obstructiva crónica, considerar el uso de medicamentos posteriores a la infusión incluyendo broncodilatadores de acción corta y prolongada, y corticosteroides inhalados. Después de las primeras cuatro infusiones, si el paciente no experimenta RRIs importantes, estos medicamentos inhalados posteriores a la infusión pueden ser descontinuados a discreción del médico.
Profilaxis para la reactivación del virus del herpes zóster: La profilaxis antiviral se debe considerar para la prevención de la reactivación del virus del herpes zóster.
Poblaciones especiales:
Pediátricos (17 años de edad y más jóvenes): La seguridad y la eficacia de DARZALEX® no han sido establecidas en pacientes pediátricos.
Adultos Mayores (65 años de edad en adelante): No se observaron diferencias generales en la seguridad o efectividad entre pacientes adultos mayores y los más jóvenes.
No se consideran necesarios ajustes en las dosis (ver Propiedades Farmacocinéticas).
Insuficiencia renal: No se han conducido estudios formales de daratumumab en pacientes con insuficiencia renal. Con base en los análisis farmacocinéticos (PK) de la población, no es necesario un ajuste en la dosificación para pacientes con insuficiencia renal (ver Propiedades farmacocinéticas).
Insuficiencia hepática: No se han conducido estudios formales de daratumumab en pacientes con insuficiencia hepática. Es improbable que los cambios en la función hepática tengan algún efecto en la eliminación de daratumumab ya que las moléculas IgG1 tales como daratumumab no son metabolizadas por vía hepática. Con base en un análisis PK de la población, no son necesarios ajustes en la dosificación para pacientes con insuficiencia hepática (ver Propiedades farmacocinéticas).
Administración: DARZALEX® es administrado como una infusión intravenosa después de la dilución con cloruro de sodio al 0.9%. Para las instrucciones sobre la dilución del medicamento antes de su administración, ver Instrucciones de uso y manejo y desecho.
Después de la dilución, la infusión de DARZALEX® debe ser administrada por la vía intravenosa a la velocidad de infusión inicial apropiada, como se presenta en la tabla 25 a continuación. El escalamiento en incrementos de la velocidad de infusión debe ser considerado únicamente en ausencia de reacciones a la infusión.
Para facilitar la administración, la primera dosis prescrita de 16 mg/kg en la semana 1 puede dividirse en dos días consecutivos, es decir, 8 mg/kg en el Día 1 y en el Día 2, respectivamente, ver la tabla 25 a continuación.
Tabla 25. Velocidades de infusión para la administración de DARZALEX® (16 mg/kg)
|
Volumen de dilución |
Velocidad inicial (primera hora) |
Aumentos de la velocidada |
Velocidad máxima |
|
|
Infusión Semana 1 |
||||
|
Opción 1 (infusión de dosis única) |
||||
|
Semana 1 Día 1 (16 mg/kg) |
1000 mL |
50 mL/hora |
50 mL/hora cada hora |
200 mL/hora |
|
Opción 2 (infusión de dosis dividida) |
||||
|
Semana 1 Día 1 (8 mg/kg) |
500 mL |
50 mL/hora |
50 mL/hora cada hora |
200 mL/hora |
|
Semana 1 Día 2 (8 mg/kg) |
500 mL |
50 mL/hora |
50 mL/hora cada hora |
200 mL/hora |
|
Infusión Semana 2 (16 mg/kg)b |
500 mL |
50 mL/hora |
50 mL/hora cada hora |
200 mL/hora |
|
Infusiones subsecuentes (a partir de la Semana 3, 16 mg/kg)c |
500 mL |
100 mL/hora |
50 mL/hora cada hora |
200 mL/hora |
a Considerar el escalamiento en incrementos de la velocidad de infusión sólo en la ausencia de reacciones a la infusión.
b El volumen de dilución de 500 mL para la dosis de 16 mg/kg solo debe usarse si no se presentan reacciones a la infusión la semana previa. De lo contrario, usar un volumen de dilución de 1000 mL.
c Usar una tasa inicial modificada (100 mL/hora) para las infusiones subsecuentes (es decir, a partir de la Semana 3) solamente si no se presentan reacciones a la infusión durante la infusión previa. De lo contrario, continúe usando las instrucciones indicadas en la tabla para la velocidad de infusión de la Semana 2.
Manejo de reacciones relacionadas con la infusión: Administre los medicamentos previos a la infusión para reducir el riesgo de RRIs antes del tratamiento con DARZALEX®.
Para las RRIs de cualquier grado/gravedad, interrumpa de inmediato la infusión de DARZALEX® y maneje los síntomas.
El manejo de RRIs puede requerir además de la reducción en la velocidad de infusión, o la descontinuación del tratamiento con DARZALEX® como se describe a continuación (ver Precauciones generales).
• Grado 1-2 (leves a moderadas): Una vez que se resuelven los síntomas de la reacción, reanude la infusión a no más de la mitad de la velocidad a la que ocurrió la RRI. Si el paciente no experimenta ningún otro síntoma de RRI, el escalamiento de la velocidad de infusión puede reanudarse en incrementos e intervalos según sea clínicamente apropiado hasta una velocidad máxima de 200 mL/h (tabla 25).
• Grado 3 (severas): Una vez que se resuelven los síntomas de la reacción, considere reiniciar la infusión a no más de la mitad de la velocidad a la que ocurrió la reacción. Si el paciente no experimenta síntomas adicionales, reanude el escalamiento de la velocidad de infusión en incrementos e intervalos según sea apropiado (tabla 25). Repita el procedimiento anterior en el caso de una reincidencia de síntomas Grado 3. Descontinúe DARZALEX® de manera permanente a la tercera ocurrencia de una reacción a la infusión Grado 3 o mayor.
• Grado 4 (que amenazan la vida): Descontinúe de manera permanente el tratamiento con DARZALEX®.
Instrucciones de uso, manejo y desecho:
Prepare la solución para la infusión usando una técnica aséptica como sigue:
• Calcule la dosis (mg), el volumen total (mL) de la solución de DARZALEX® requerida y el número de frascos ámpula de DARZALEX® requerido con base en el peso del paciente.
• Verifique que la solución de DARZALEX® sea de incolora a amarilla. No se use si se observan partículas opacas, decoloración u otras partículas extrañas presentes.
• Usando una técnica aséptica, retire un volumen de cloruro de sodio al 0.9% de la bolsa/contenedor de infusión que sea igual al volumen requerido de la solución de DARZALEX®.
• Retire la cantidad necesaria de la solución de DARZALEX® y diluya hasta el volumen apropiado añadiéndola a una bolsa/contenedor de infusión que contenga cloruro de sodio al 0.9% (ver Dosis y vía de administración). Las bolsas/contenedores de infusión deben estar hechos de polivinilcloruro (PVC), polipropileno (PP), polietileno (PE) o una mezcla de poliolefinas (PP+PE). Diluya bajo condiciones asépticas apropiadas. Deseche cualquier porción no usada que quede en el frasco ámpula.
• Invierta cuidadosamente la bolsa/contenedor para mezclar la solución. No agite ni congele.
• Los medicamentos parenterales deben ser inspeccionados visualmente para material particulado y decoloración antes de la administración siempre que la solución y el contenedor lo permitan. La solución diluida puede desarrollar partículas proteináceas muy pequeñas, de translúcidas a blancas, debido a que daratumumab es una proteína. No se use si se observan partículas opacas visibles, decoloración o partículas extrañas.
• Debido a que DARZALEX® no contiene conservadores, la solución diluida debe ser administrada dentro de 15 horas (incluyendo el tiempo de infusión) a temperatura ambiente 15°C-25°C (59°F-77°F) y con exposición a la luz interior.
• Si no se usa inmediatamente, la solución diluida puede ser guardada previa a la administración hasta por 24 horas en condiciones de refrigeración de 2°C-8°C (36°F-46°F) y protegida de la luz. No congelar.
• Administre la solución diluida mediante una infusión intravenosa usando un equipo de infusión ajustado con un regulador de flujo y con un filtro en línea de poliéter sulfona (PES) de baja unión a proteínas, no pirogénico, estéril (tamaño de poros de 0.22 o 0.2 micrómetros). Deben usarse equipos de administración de poliuretano (PU), polibutadieno (PBD), PVC, PP o PE.
• No infunda DARZALEX® de manera concomitante en la misma línea intravenosa con otros agentes.
• No guarde porciones sin usar de la solución de infusión para su reutilización. Cualquier producto no usado o material de desecho debe ser desechado de acuerdo a los requerimientos locales.
Incompatibilidades: Este medicamento no debe ser mezclado con otros medicamentos, excepto aquellos mencionados previamente.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Síntomas y signos: No ha habido experiencia de sobredosificación en los estudios clínicos. Se han administrado dosis de hasta 24 mg/kg por la vía intravenosa en un estudio clínico sin llegar a la dosis máxima tolerada.
Tratamiento: No existe un antídoto específico conocido para una sobredosis de DARZALEX®. En el evento de una sobredosis, el paciente debe ser monitoreado por cualquier signo o síntoma de efectos adversos y deberá instituirse de inmediato el tratamiento sintomático apropiado.
PRESENTACIONES: Caja de cartón con frasco ámpula con 100 mg en 5 mL (20 mg/mL) o 400 mg en 20 mL (20 mg/mL).
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese en refrigeración entre 2°-8° C, no se congele. Protéjase de la luz.
Después de la dilución: Debido a que las soluciones de daratumumab no contienen un conservador, a menos que el método de apertura/dilución excluya el riesgo de contaminación microbiana, el producto deberá ser usado de inmediato.
Si no se usa de inmediato, la solución puede ser almacenada en un refrigerador protegida de la luz a 2° C–8° C (36°F-46°F) hasta por 24 horas antes de su uso, seguido por 15 horas (incluyendo el tiempo de infusión) a temperatura ambiente a 15°C-25°C (59°F-77°F) y exposición a la luz interior.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños.
No se use en el embarazo y la lactancia. 
Léase instructivo anexo. Si no se administra todo el producto, deséchese el sobrante. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se administre si el cierre ha sido violado. Este medicamento debe ser administrado únicamente por médicos especialistas en oncología y con experiencia en quimioterapia antineoplásica. Dilúyase con cloruro de sodio al 0.9% al volumen indicado.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
atencionaclientes@its.jnj.com
JANSSEN-CILAG, S.A. de C.V.
Carretera Federal México-Puebla Km. 81.5,
San Mateo Capultitlán, C.P. 74160,
Huejotzingo, Puebla, México.
®Marca Registrada