
EVEREST DX - Cápsulas
Sustancia(s):
- Desloratadina, Montelukast
Presentaciones:
- 1 Caja, 15 Cápsulas, 10/5 mg/mg
- 1 Caja, 30 Cápsulas, 10/5 mg/mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada CÁPSULA contiene:
Montelukast 10 mg
Desloratadina 5 mg
Excipiente cbp 1 cápsula
INDICACIONES TERAPÉUTICAS: EVEREST-DX® (montelukast/desloratadina) está indicado como tratamiento de segunda línea para el alivio de los síntomas de la rinitis alérgica de moderada a grave y de tipo persistente, en mayores de 18 años.
FARMACOCINÉTICA Y FARMACODINAMIA: EVEREST-DX® cápsula oral es una formulación combinada que contiene desloratadina, un antagonista del receptor H1 de histamina y montelukast sódico, un antagonista de los receptores de leucotrienos.
Farmacocinética:
Absorción: Montelukast y desloratadina se absorbieron rápidamente después de la administración por vía oral de cápsulas de EVEREST-DX®. Como parte del desarrollo clínico de EVEREST-DX®, se condujo un estudio de farmacocinética comparada entre la combinación de dosis fija y los fármacos administrados de forma individual. En el estudio se incluyeron 23 sujetos sanos, adultos, de ambos sexos, quienes recibieron una dosis oral única de los productos farmacéuticos en condiciones de ayuno. Los parámetros farmacocinéticos se muestran a continuación.
Tabla 1. Parámetros farmacocinéticos de desloratadina administrada en combinación de dosis fija o monoterapia
|
Parámetro |
Combinación de dosis fija |
Monoterapia |
|
ABC0-1 (pg/mL*h) |
38837.96 ± 15891.19 |
36772.30 ± 15064.46 |
|
Cmáx. (pg/mL) |
2348.83 ± 504.06 |
2148.80 ± 667.04 |
|
Tmáx. (h) |
3.37 ± 1.11 |
3.63 ± 1.47 |
Tabla 2. Parámetros farmacocinéticos de montelukast administrado en combinación de dosis fija o monoterapia
|
Parámetro |
Combinación de dosis fija |
Monoterapia |
|
ABC0-1 (ng/mL*h) |
3301.43 ± 1479.65 |
3080.92 ± 1240.21 |
|
Cmáx. (ng/mL) |
483.15 ± 194.75 |
449.96 ± 147.30 |
|
Tmáx. (h) |
3.74 ± 1.20 |
3.89 ± 1.21 |
|
Ke (l/h) |
0.139 ± 0.056 |
0.136 ± 0.052 |
|
T½ (h) |
5.44 ± 1.36 |
5.63 ± 1.78 |
Los resultados demostraron que la biodisponibilidad comparativa entre la combinación de dosis fija de montelukast/desloratadina 10 mg/5 mg cápsula es estadísticamente equivalente (p < 0.05, Prueba t doble unilateral de Schuirmann) a la de los medicamentos administrados de forma independiente. Por tanto, fue posible asumir que no existe interacción farmacocinética entre los activos de la combinación de dosis fija montelukast/desloratadina 10 mg/5 mg cápsula.
Un estudio (Cingi et al) demostró de forma similar, que la tasa de absorción de montelukast no se alteró cuando se administró con desloratadina. Los autores de este estudio concluyen que la desloratadina no influye en la biodisponibilidad de montelukast, y se puede usar su terapia de combinación. Las concentraciones plasmáticas máximas (Cmáx.) de montelukast y desloratadina se alcanzaron en un tiempo mediano máximo (Tmáx.) de 2 horas después de la administración. Los valores medios de concentración no fueron estadísticamente diferentes según los grupos (montelukast (M, solo), montelukast/Desloratadina (MD); F = 2.99; P = .0869, P > .05) y las diferencias no fueron estadísticamente significativas según el tiempo (2 horas, 3 horas, 4 horas; F = 11.49; P ≥ .0001, P > .05). Esto demostró que la tasa de eliminación de montelukast no se alteró con administración de desloratadina.
Tabla 3. Comparación marginal del área bajo la curva (ABC) de los grupos de prueba según los intervalos de tiempo
|
Tiempo |
|||||||||||||
|
2H |
3H |
4H |
Total |
ANOVA |
|||||||||
|
Tratamiento |
n |
Media |
DS |
n |
Media |
DS |
n |
Media |
DS |
n |
Media |
DS |
F = 2.99, P .0869, ≥ 05 |
|
MD |
22 |
488.89 |
279.9 |
22 |
410.14 |
165.65 |
23 |
255.19 |
80.79 |
67 |
382.8 |
213.12 |
|
|
M |
23 |
365.62 |
194.49 |
22 |
355.79 |
132.86 |
23 |
292.34 |
105.5 |
68 |
337.65 |
150.63 |
F = 11.49, P = .0001 ≤ .05 |
|
Total |
45 |
425.88 |
245.06 |
44 |
382.96 |
150.92 |
46 |
273.77 |
94.79 |
135 |
360.06 |
185.01 |
|
ANOVA = análisis de las variancias; M = montelukast (10 mg) únicamente; MD = montelukast (10 mg)/desloratadina (5 mg); DS = desviación estándar.
La evaluación de los valores marginales del ABC de los grupos en este estudio, de acuerdo con los intervalos de tiempo, de la tabla a continuación, demuestran que la diferencia entre los grupos para el intervalo de 0 a 2 horas no fue estadísticamente significativa (t = 1.72; P = .0919, P ≥ .05). Para el intervalo de tiempo de 2 a 3 horas, la diferencia entre los grupos no fue estadísticamente significativa (t = 1.76; P = .0855, P ≥ .05). Adicionalmente para el intervalo de 3 a 4 horas, la diferencia entre grupos no es estadísticamente significativa (t =1.7; P=.8644, P≥.05).
Tabla 4. Comparaciones de los valores marginales del área bajo la curva de los grupos de prueba de acuerdo con los intervalos de tiempo
|
Tiempo |
|||||||||
|
0-2 horas |
2-3 horas |
3-4 horas |
|||||||
|
Tratamiento |
n |
Media |
DS |
n |
Media |
DS |
n |
Media |
DS |
|
MD |
22 |
488.89 |
279.39 |
22 |
449.51 |
198.72 |
23 |
329.54 |
109.88 |
|
M |
23 |
365.62 |
194.49 |
22 |
362.48 |
119.32 |
23 |
334.72 |
93.9 |
|
Prueba de t |
t = 1.72, P = .0919, ≥ .05 |
t = 1.76, P = .0855, ≥ .05 |
t = 1.7, P = 0.8644, ≥ .05 |
||||||
MD = montelukast/desloratadina (10 mg/5 mg; M = montelukast solo, SD = desviación estándar.
En la evaluación de valores acumulados, como se muestra en la siguiente tabla para el intervalo de tiempo de 0 a 2 horas, la diferencia entre los grupos no fue estadísticamente significativa (t = 1.72; P = .0919, P > .05). Tampoco hubo estadísticamente diferencias significativas para el intervalo de tiempo de 0 a 3 horas (t = 1.73; P = .0912, P > .05) o de 0 a 4 horas intervalo de tiempo (t = 1.49; P = .1450, P > .05).
Tabla 5. Comparaciones de los valores acumulados del ABC de los grupos de prueba de acuerdo con los intervalos de tiempo
|
Tiempo |
|||||||||
|
0-2 horas |
2-3 horas |
3-4 horas |
|||||||
|
Tratamiento |
n |
Media |
DS |
n |
Media |
DS |
n |
Media |
DS |
|
MD |
22 |
488.89 |
279.39 |
22 |
938.4 |
470.99 |
22 |
1270.81 |
554.97 |
|
M |
23 |
365.62 |
194.49 |
22 |
731.64 |
304.75 |
22 |
1068.13 |
319.03 |
|
Prueba de t |
t = 1.72, P = .0919, ≥.05 |
t = 1.73, P = .0912, ≥ .05 |
t = 1.49 P = 0.1450, ≥ .05 |
||||||
MD = montelukast/desloratadina (10 mg/5 mg; M = montelukast solo, SD = desviación estándar.
En un estudio a dosis única de montelukast/desloratadina comparada con cada uno de los componentes por separado, los intervalos de confianza al 90% (IC 90) estimados a partir de la transformación logarítmica de los parámetros farmacocinéticas ABC0-4 y Cmax se encuentran en rango de 80-125%. Los valores P de t1 y t2 de la prueba de t doble unilateral de·Schuirmann fueron menores a 0.05, permitiendo rechazar la hipótesis nula de no equivalencia en la biodisponibilidad. Además, la potencia de la prueba para ambas moléculas fue superior al 0.80. Se puede asumir que no hay interacción farmacocinética entre los activos de la combinación fija montelukast/Desloratadina cápsulas de 10 mg/5 mg.
Tabla 6. Intervalos de confianza (IC90%) de la transformación logarítmica de los parámetros de farmacocinética ABC0-4 y Cmáx. para montelukast
|
Parámetro |
Intervalo clásico |
Poder |
|
|
ABC0-4 |
97.184 |
118.6153 |
0.9774 |
|
ABC0-∞ |
97.3249 |
117.8686 |
0.9835 |
|
Cmáx. |
93.6579 |
121.9025 |
0.8774 |
Intervalos de confianza de T1 vs T3 sin el sujeto B09.
Tabla 7. Intervalos de confianza (IC90%) de la transformación logarítmica de los parámetros de farmacocinética ABC0-4 y Cmáx. para Desloratadina
|
Parámetro |
Intervalo clásico |
Poder |
|
|
ABC0-4 |
97.1078 |
116.7155 |
0.9883 |
|
Cmáx. |
105.5880 |
117.1460 |
0.9999 |
Intervalos de confianza de T1 vs T3 sin el sujeto B09.
Distribución:
Montelukast: En estado de ayuno, la biodisponibilidad oral media es 64%. Noventa y nueve por ciento o más, se une a las proteínas del plasma. El volumen de distribución de montelukast es pequeño; estudios en animales indican distribución mínima a través de la barrera hematoencefálica.
Desloratadina: La vida media de fase terminal es de aproximadamente 27 horas. El grado de acumulación de la desloratadina se correlaciona con su vida media y con su administración de una vez al día. La biodisponibilidad de la desloratadina es proporcional a la dosis entre 5 y 20 mg.
La desloratadina se conjuga moderadamente (83 a 87%) con las proteínas plasmáticas. No hay evidencia de acumulación clínicamente significativa del fármaco después de su administración una vez al día (a dosis de 5 a 20 mg) hasta por 14 días.
Metabolismo y excreción:
Montelukast: El metabolismo de montelukast en el hígado es extenso; estudios in vitro indican la participación de isoenzimas del citocromo P450 3A4 y 2C9. Además, estudios In vitro demuestran que montelukast es un potente inhibidor de la isoenzima hepática CYP2C8; sin embargo, datos in vivo muestran que una interacción con los sustratos 2C8 es improbable. En estado de equilibrio son indetectables las concentraciones plasmáticas de metabolitos de montelukast.
Montelukast y sus metabolitos se excretan casi de forma exclusiva a través de la bilis; menos del 0.2% del fármaco se excreta en orina. La vida media de eliminación (vida media) de montelukast es de 2.7-5-5 horas en adultos jóvenes saludables.
Desloratadina: Es extensamente metabolizada a 3-hidroxidesloratadina, un metabolito activo, el cual es posteriormente glucuronizado. La enzima responsable del metabolismo de la desloratadina aún no ha sido identificada, por lo que algunas interacciones con otros medicamentos no pueden ser completamente excluidas. Estudios in vivo con inhibidores específicos de CYP3A4 y CYP2D6 han mostrado que estas enzimas no son importantes en el metabolismo de la desloratadina. La desloratadina no inhibe CYP3A4 ni CYP2D6 y no es sustrato ni inhibidor de la glucoproteína P.
El promedio de la vida media de eliminación es 27 horas. Los valores de Cmáx. y del ABC se incrementan en forma proporcional con el incremento de la dosis oral de los 5 mg a los 20 mg. El grado de acumulación después de 14 días de dosificación es consistente con la vida media y la frecuencia de dosificación.
Después de la administración oral, la desloratadina bloquea específicamente los receptores periféricos de histamina H1 debido a que el agente no penetra al sistema nervioso central (SNC).
Farmacodinamia:
Mecanismo de acción: EVEREST-DX® proporciona los efectos como antagonista de los leucotrienos de montelukast y los efectos antihistamínicos de la desloratadina.
Montelukast: Es un antagonista potente y selectivo de leucotrienos D4 (LTD4.) en el receptor cisteinil de leucotrienos, CisLT1, presente en las vías aéreas del humano. Los leucotrienos cisteinil (LTC4, LTD4, y LTE4) son productos del metabolismo del ácido araquidónico y se liberan de varias células, incluyendo mastocitos y eosinófilos.
La unión de leucotrienos cisteinil a CisLT se ha relacionado con la fisiopatología del asma y la rinitis alérgica, incluyendo el incremento de la permeabilidad en la membrana endotelial de las vías aéreas que conduce a edema, contracción del músculo liso, y aumento de la secreción de moco espeso, viscoso. Montelukast mejora los signos y síntomas, inhibiendo las acciones fisiológicas de LTD4 en el receptor CisLT1. Clínicamente se ha demostrado que el fármaco inhibe las fases temprana y tardía de la broncoconstricción inducida por el antígeno en 75% y 57%, respectivamente. Montelukast no tiene propiedades agonistas en receptores de leucotrienos y no antagoniza las contracciones de músculo liso debidas a LTC4, acetilcolina o serotonina. También se ha vinculado a los leucotrienos en los síntomas de rinitis alérgica (por ejemplo: estornudos, prurito nasal, rinitis y congestión en la última etapa); las acciones de montelukast ayudan al control clínico de estos síntomas.
En pacientes con rinitis alérgica que recibieron Montelukast, las cuentas de eosinófilos periféricos incrementaron únicamente el 0.2%, en comparación a un 12.5% de incremento en el grupo de pacientes con placebo.
Desloratadina: Es un antagonista de la histamina no sedante, de acción prolongada, con potente actividad antagonista del receptor H1 periférico. La desloratadina ha demostrado tener actividad antialérgica, antihistamínica y antiinflamatoria. La desloratadina no penetra hacia el sistema nervioso central. A la dosis recomendada de 5 mg diarios, la incidencia de somnolencia fue similar a la del placebo. En los estudios clínicos con dosis de hasta 7.5 mg diarios, no afectó el desempeño psicomotor. Una dosis de 5 mg de desloratadina no afectó los indicadores estándar de capacidad de vuelo de pilotos incluyendo exacerbación de la somnolencia subjetiva o los procesos relacionados con el pilotaje.
Además de la actividad antihistamínica, la desloratadina ha demostrado actividad antialérgica y antiinflamatoria en numerosos estudios in vitro e in vivo (principalmente realizados en células de origen humano).
Estos estudios han demostrado que la desloratadina inhibe la amplia cascada de eventos que inician y propagan la inflamación alérgica, entre los que se incluye:
• La liberación de las citocinas proinflamatorias como: IL-4, IL-6, IL-8, IL-13.
• La liberación de quimocinas proinflamatorias importantes como RANTES.
• Producción de superóxidos aniónicos por los polimorfonucleares neutrófilos activados.
• Adherencia y quimiotaxis de eosinófilos.
• La expresión de moléculas de adherencia como la P-selectina.
• Liberación de histamina, prostaglandina PGD2 y leucotrieno LTC4 dependiente de IgE.
• La respuesta broncoconstrictora alérgica aguda y la tos alérgica en modelos animales.
Estudios clínicos:
Rinitis Alérgica Estacional (RAE): La eficacia clínica del montelukast con desloratadina en dosis fija, para el tratamiento de la rinitis alérgica estacional, se ha investigado en varios estudios diseñados de forma similar, aleatorios, doble ciego, grupo paralelo, controlado con placebo y con activo (montelukast y desloratadina). Los estudios emplearon una combinación fija de montelukast y desloratadina. Las edades de los pacientes fueron de 15 a 82 años con antecedentes de rinitis alérgica estacional.
En un estudio se informó si los pacientes tuvieron una prueba positiva cutánea para alérgeno estacional y síntomas activos de rinitis alérgica al inicio del estudio. Los síntomas principales recolectados en los estudios fueron rinorrea, congestión nasal, estornudos, comezón, ojos llorosos, enrojecimiento.
La variable primaria de desenlace fue la puntuación de los síntomas nasales, incluida la congestión nasal, estornudos, picazón y secreción. La congestión nasal también fue objetivamente medida utilizando rinometría acústica.
Las variables secundarias incluyeron la evaluación global del médico, puntaje del síntoma ocular diurno, puntaje total de síntomas y Concentración de la proteína catiónica en eosinófilos (ECP) en el fluido de lavado nasal. Los resultados de las puntuaciones, después de 6 semanas de tratamiento se pueden observarse a continuación:
Figura 1. Puntuaciones medias de la congestión nasal después de 6 semanas de tratamiento con montelukast solo, desloratadina sola, o la combinación
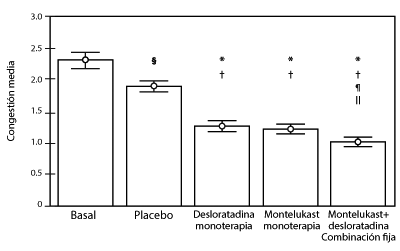
§P .01 vs basal. ¶P .05 vs desloratadina. //P .05 vs montelukast. Las barras representan el error estándar ES.
Figura 2. Puntuaciones medias de los síntomas nasales después de seis semanas de tratamiento con montelukast solo, desloratadina sola, o la combinación
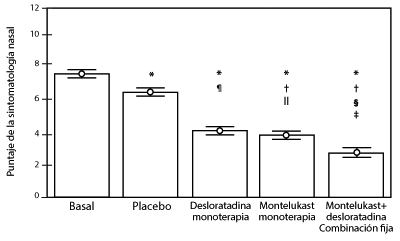
*P .001 vs basal. †P .001 vs placebo. ‡P .05 vs montelukast. §P .001 vs montelukast. ¶P .05 vs placebo. P .05 vs desloratadina. ||P .001 vs desloratadina. Las barras representan el error estándar ES.
La terapia combinada de montelukast/desloratadina dio como resultado un aumento significativo del MCA (área mínima de sección transversal por sus siglas en inglés) de las cavidades nasales comparado con la basal.
Aunque la comezón nasal, los estornudos y la rinorrea se reducen por el uso de los antagonistas del receptor H1, el bloqueo nasal es sólo parcialmente modificado por estos medicamentos. Por lo tanto, es razonable considerar la adición del receptor de leucotrienos antagonista montelukast con el propósito de reducción de la obstrucción nasal durante el tratamiento de rinitis alérgica.
Lo anterior proporciona evidencia relacionada a que la combinación de montelukast con un antihistamínico (desloratadina) ofrecen un beneficio adicional significativo en la reducción de síntomas nasales.
La seguridad de montelukast/desloratadina se investigó en varios estudios con pacientes con asma o con asma concurrente con rinitis alérgica. No se observó evidencia de un efecto adverso en las mediciones de la función pulmonar o el control del asma, y los resultados de los estudios respaldan la seguridad de la administración de montelukast/desloratadina a pacientes adultos con rinitis alérgica y asma de leve a moderada.
No hubo diferencias significativas en la efectividad del montelukast/desloratadina tabletas entre los subgrupos de pacientes definidos por género, edad o raza.
Estudios en la calidad de vida de los pacientes con Rinitis Alérgica (RA). El tratamiento óptimo de los pacientes con RA con farmacoterapia debe controlar los síntomas de la RA y además considerar el incrementar la calidad de vida de éstos.
La mejoría en la calidad de vida de los pacientes depende principalmente de la reducción de la obstrucción nasal, como el bloqueo u obstrucción nasal es un síntoma crucial en la persistencia de la rinitis alérgica, que conduce al deterioro del sueño, y somnolencia diurna con la subsecuente fatiga y reducción de la productividad.
Utilizando el RQLQ (evaluación específica de alergia sobre la calidad de vida relacionada con la salud), en pacientes con rinitis alérgica a los cuales se les administro la combinación de (montelukast/desloratadina), se demostró mejoró significativamente la calidad de vida de los pacientes comparado con cada uno de los componentes por separado.
Los resultados de los estudios sobre la calidad de vida de pacientes tratados con la combinación fija de montelukast/desloratadina informan de mejoría significativa en la calidad de vida (RQLQ) de los pacientes con rinitis alérgica, los beneficios de esta combinación fueron evidentes en la mayoría de los dominios medidos por este cuestionario y específicamente en aquellos relacionados con síntomas nasales, oculares y limitaciones en sus actividades diarias.
Figura 3. Puntuaciones de síntomas nocturnos en pacientes tratados durante seis semanas con desloratadina (grupo 1) y terapia combinada de desloratadina más montelukast (Grupo 2)
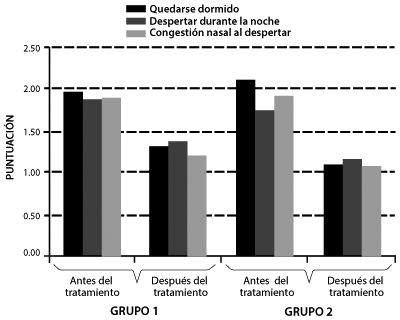
Un estudio investigó la no inferioridad de Montelukast (10 mg)/desloratadina (5 mg) para el tratamiento de la rinitis alérgica persistente en un estudio clínico controlado, prospectivo, longitudinal, aleatorizado, doble ciego, paralelo con 2 brazos de tratamiento activo, multicéntrico, en una muestra de pacientes de ambos sexos, adultos mexicanos con diagnóstico realizado al menos 1 año previo a su ingreso al estudio.
El objetivo primario comparó la eficacia de montelukast (10 mg)/desloratadina (5 mg) EVEREST- DX® cápsulas versus montelukast (10 mg)/loratadina (10 mg) encapsulada para su enmascaramiento, determinada a través del cambio de la puntuación basal global y la puntuación de la última visita del cuestionario SNOT-20.
Los objetivos secundarios incluyeron comparaciones de los resultados entre los grupos que recibieron montelukast (10 mg)/desloratadina (5 mg) y el medicamento de referencia (montelukast 10 mg)/loratadina (10 mg), en los siguientes parámetros:
- Área bajo la curva de las puntuaciones obtenidas del tiempo 0 [basal] hasta la última visita en los sujetos tratados a través de la escala SNOT-20.
- La mejoría en la sintomatología, mediante el cuestionario sobre cinco síntomas (T5SS) de los pacientes con rinitis alérgica persistente tratados.
- La puntuación en el dominio de calidad de vida del cuestionario SNOT-20 en los pacientes con rinitis alérgica persistente tratados.
Adicionalmente, en aquellos que fue necesario el uso de medicación de rescate (furoato de mometasona 0.05 g/100 mL en solución spray), se documentó el número de días que lo recibieron en los grupos de tratamiento. Los tratamientos se administraron, de acuerdo al grupo por aleatorización, de la siguiente manera:
- Medicamento de prueba: EVEREST-DX® Montelukast (10 mg)/Desloratadina (5 mg), 1 cápsula una vez al día por 6 semanas.
- Medicamento de referencia: Montelukast (10 mg)/Loratadina (10 mg) encapsulada para su enmascaramiento, 1 cápsula una vez al día por 6 semanas.
Adicionalmente se incluyó como medicamento de rescate (furoato de mometasona 0.05 g/100 mL) spray nasal en 2 disparos (bombeos) en cada fosa nasal 1 vez al día, el cual fue empleado cuando la sintomatología del padecimiento no pudo ser controlada por los medicamentos de prueba o referencia.
Se reclutaron un total de 86 pacientes de ambos sexos, mayores de 18 años con antecedente de rinitis alérgica persistente con presencia de síntomas de rinitis como: prurito nasal y faríngeo, estornudos en salva, moco nasal y síntomas conjuntivales (tales como: lagrimeo, picor y escozor ocular) por más de 4 días, durante más de cuatro semanas continuas; sintomatología de moderada a severa al momento de selección, de acuerdo a los criterios de la guía de tratamiento de la rinitis alérgica y su repercusión en el asma; calificación mínima basal de 3 en el cuestionario SNOT-20 y calificación promedio de las 2 mediciones basales ≥ 3 en el cuestionario SNOT-20 en la semana de inducción. En el análisis por protocolo se incluyeron a 74 pacientes (37 en cada grupo).
Tabla 8. Resumen de las variables demográficas y signos vitales para las visitas 1 y 4
|
Variable o dato (media ± desviación estándar) |
Visita 1 |
Visita 4 |
||
|
Referencia |
Prueba |
Referencia |
Prueba |
|
|
Género (n= masculino/femenino) |
15/29 |
17/25 |
- |
- |
|
Edad (años) |
35.5 (14.0) |
32.2 (12.9) |
- |
- |
|
Estatura (m) |
1.67 (0.08) |
1.68 (0.08) |
- |
- |
|
Peso (kg) |
73.3 (11.7) |
69.3 (10.9) |
72.4 (12.2) |
68.6 (10.6) |
|
IMC (%) |
26.2 (3.9) |
24.7 (4.1) |
25.9 (4.2) |
24.5 (4.0) |
|
Temperatura corporal (°C) |
36.1 (0.2) |
36.2 (0.3) |
36.2 (0.3) |
36.2 (0.3) |
|
Frecuencia cardiaca (latidos por minuto) |
73.4 (8.3) |
71.3 (7.4) |
72.8 (6.7) |
72.7 (8.6) |
|
Frecuencia respiratoria (respiraciones por minuto) |
18.6 (2.3) |
18.5 (1.9) |
18.5 (1.8) |
18.5 (1.8) |
|
Tensión arterial sistólica (mmHg) |
114.8 (12.2) |
110.8 (14.3) |
117.3 (12.9) |
115.3 (12.0) |
|
Tensión arterial diastólica (mmHg) |
74.7 (9.5) |
72.5 (8.3) |
75.1 (7.4) |
75.9 (6.9) |
|
-No se determinaron valores en la visita 4 por no esperarse cambios |
||||
Análisis de eficacia de las variables primarias.
Tabla 9. Estadística descriptiva del cambio global del cuestionario SNOT-20 por tratamiento
|
Media |
Mediana |
Desviación estándar |
Mínimo |
Máximo |
n |
|
|
Referencia |
3.535 |
3.725 |
1.012 |
-0.775 |
4.800 |
37 |
|
Prueba |
3.271 |
3.600 |
1.117 |
0.025 |
4.350 |
37 |
Tabla 10. Prueba de no-inferioridad (t de Student) para el cambio de puntuación global SNOT-20 Población por protocolo
(n = 74)
|
Tratamientos |
Media |
Desviación estándar |
n |
|
Referencia |
3.535 |
1.012 |
37 |
|
Prueba |
3.271 |
1.117 |
37 |
Prueba de no-inferioridad
|
IC 97.5% |
||||||
|
Diferencia de medias |
Error estándar |
t |
Grados de libertad* |
p |
límite inferior |
límite superior |
|
-0.264 |
0.248 |
2.162 |
71.3 |
0.01698 |
-0.758 |
Infinito |
* La prueba considera desigualdad de varianzas.
De los anteriores se permite concluir que el tratamiento de prueba no es inferior al de referencia.
Prueba de no-inferioridad (U de Mann-Whitney) población por protocolo:
Los resultados de la prueba de no-inferioridad no paramétrica se presentan en la siguiente tabla:
Tabla 11. Prueba de no-inferioridad (U de Mann-Whitney) para el cambio de puntuación global SNOT-20 Población por protocolo (n = 74)
|
IC 97.5% |
||||
|
Mediana de las diferencias estimada |
Estadístico·estandarizado (z) |
p |
Límite inferior |
Límite superior |
|
-0.175 |
3.276 |
0.0005 |
-0.5 |
Infinito |
* La prueba está basada en el método de Hodges-Lehmann.
Prueba de no inferioridad con la población de intención de tratar (todos los pacientes aleatorizados) resultados de la prueba en su versión paramétrica (t de Student).
Tabla 12. Prueba de no-inferioridad (t de Student) para el cambio de puntuación global SNOT-20 Población de intención de tratar (n = 86)
|
Tratamientos |
Media |
Desviación estándar |
n |
|
Referencia |
3.394 |
0.983 |
44 |
|
Prueba |
3.173 |
1.082 |
42 |
Prueba de no-inferioridad
|
Diferencia de medias |
Error estándar |
t |
Grados de libertad* |
p |
C 97.5% |
|
|
Límite inferior |
Límite superior |
|||||
|
-0.221 |
0.223 |
2.594 |
82.3 |
0.0056 |
-0.665 |
Infinito |
* La prueba considera desigualdad de varianzas.
Los resultados con la población de intención a tratar también apoyan la conclusión de no-inferioridad obtenida con la población por protocolo.
La diferencia de medias entre ambos tratamientos fue de -0.221 puntos (cercana a la obtenida con la población por protocolo de -0.264 puntos), con intervalo de confianza unilateral de confianza del 97.5% de -0.665 puntos a infinito. El intervalo no rebasa el margen de inferioridad de -0.8 puntos y la probabilidad de rebasar dicho margen es baja (0.0056) menor que el valor de significancia de 0.025%. Lo cual permite concluir que el tratamiento de prueba no es inferior, con respecto al de tratamiento de referencia.
Resultados de la prueba en su versión ni paramétrica (U de Mann-Whitney):
Tabla 13. Prueba de no-inferioridad (U de Mann-Whitney) para el cambio de puntuación global SNOT-20 Población de intención de tratar (n = 86)
|
Mediana de las diferencias estimada |
Estadístico·estandarizado (z) |
p |
IC 97.5% |
|
|
Límite inferior |
Límite superior |
|||
|
-0.150 |
3.327 |
0.0004 |
-0.5 |
Infinito |
* La prueba está basada en el método de Hodges-Lehmann.
Análisis de eficacia de las variables secundarias: Estadística descriptiva para el ABC de las puntuaciones globales del cuestionario SNOT-20 por tratamiento.
Tabla 14. Estadística descriptiva para el ABC de las puntuaciones globales del SNOT-20 por tratamiento (ABC en unidades de área) n = 74
|
Media |
Mediana |
Desviación estándar |
Mínimo |
Máximo |
n |
|
|
Referencia |
11.1 |
11.1 |
4.0 |
5.1 |
20.3 |
37 |
|
Prueba |
10.8 |
9.7 |
3.8 |
5.8 |
20.6 |
37 |
Prueba t de Student y por la prueba de Mann-Whitney y sus resultados de muestran en la siguiente tabla:
Tabla 15. Comparaciones para el ABC de las puntuaciones globales del SNOT-20, n = 74
|
Prueba t de Student* |
||||||
|
Diferencia de medias |
Error estándar |
t |
G.L. |
p |
IC95% |
|
|
-0.393 |
0.908 |
-0.4324 |
71.62 |
0.6667 |
-2.202 |
1.417 |
|
Prueba U de Mann-Whitney** |
||||
|
Diferencia de medianas |
Estadístico estandarizado |
p |
IC 95% |
|
|
-0.5 |
0.486 |
0.6266 |
-2.325 |
1.450 |
* La prueba t considera varianzas desiguales.
** La prueba U está basada en el método de Hodges-Lehmann.
No se encontró diferencia significativa para el ABC entre ambos tratamientos usando tanto la prueba paramétrica como la no-paramétrica. El hecho de que las medias y medianas del ABC de los tratamientos no difieran significativamente entres sí, es consistente con la conclusión de no-inferioridad obtenida del análisis con la variable de eficacia primaria.
Tabla 16. Indicadores del cuestionario SNOT-20
Medias de las puntuaciones por cada uno de los indicadores del cuestionario SNOT-20
|
Visita |
Basal |
2da Semana |
3ra Semana |
4ta Semana |
6tasemana |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Medicamento |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
|
Necesidad de sonarse la nariz |
4.8 |
4.7 |
3.3 |
3.3 |
2.4 |
2.3 |
1.6 |
1.9 |
1.1 |
1.1 |
|
Estornudos |
4.8 |
4.7 |
3.3 |
3.1 |
2.2 |
2.2 |
1.2 |
1.5 |
0.8 |
O.8 |
|
Secreción nasal continua |
4.7 |
4.7 |
3.1 |
3.1 |
1.9 |
2 |
1.2 |
1.5 |
0.5 |
0.7 |
|
Tos |
4.3 |
4.1 |
2.7 |
2.4 |
1.5 |
1.2 |
0.8 |
0.9 |
0.2 |
0.4 |
|
Secreción hacia la garganta |
4.3 |
4.2 |
2.8 |
2.4 |
1.6 |
1.5 |
1 |
1.1 |
0.6 |
0.6 |
|
Secreción nasal espesa |
4.1 |
4 |
2.7 |
2.3 |
1.4 |
1.4 |
1 |
0.9 |
0.5 |
0.5 |
|
Sensación de oído tapado |
3.9 |
3.9 |
2.5 |
1.8 |
1.3 |
1.2 |
0.9 |
0.8 |
0.4 |
0.5 |
|
Mareo |
3.1 |
3.2 |
1.9 |
1.6 |
0.7 |
0.9 |
1.1 |
1.1 |
0.2 |
0.3 |
|
Dolor de oído |
3.4 |
3.3 |
2.1 |
1.5 |
1.1 |
1 |
0.6 |
0.6 |
0.3 |
0.3 |
|
Dolor y presión en la cara |
4.2 |
3.7 |
2.6 |
2 |
1.5 |
1.1 |
0.8 |
1.1 |
0.5 |
0.5 |
|
Dificultad para quedar dormido |
4.4 |
4.2 |
3 |
2.7 |
1.7 |
1.8 |
1.3 |
1.3 |
0.4 |
0.7 |
|
Despierta durante la noche |
4.3 |
4.4 |
3 |
2.9 |
1.8 |
1.7 |
1.3 |
1.1 |
0.4 |
0.5 |
|
Sensación que durmió mal |
4.5 |
4.5 |
3.1 |
2.8 |
1.8 |
2 |
1.2 |
1.3 |
0.5 |
0.6 |
|
Despierta cansado |
4.5 |
4.4 |
2.8 |
3 |
1.9 |
1.9 |
1.2 |
1.4 |
0.4 |
0.8 |
|
Fatiga o cansancio |
4.4 |
4.4 |
2.8 |
2.8 |
1.6 |
1.8 |
1.1 |
1.3 |
0.5 |
0.8 |
|
Menor productividad |
4.2 |
4.2 |
2.6 |
2.4 |
1.6 |
1.5 |
0.9 |
1.1 |
0.4 |
0.8 |
|
Menor concentración |
4.2 |
4.1 |
2.4 |
2.3 |
1.4 |
1.3 |
0.7 |
0.9 |
0.4 |
0.6 |
|
Frustrado, inquieto o irritable |
3.4 |
3.3 |
1.9 |
1.5 |
0.8 |
1.1 |
0.4 |
0.7 |
0.3 |
0.4 |
|
Triste |
2.1 |
2 |
1.2 |
0.7 |
0.5 |
0.5 |
0.4 |
0.4 |
0.1 |
0.3 |
|
Avergonzado |
2.1 |
2.1 |
1.3 |
0.7 |
0.6 |
0.4 |
0.3 |
0.2 |
0.2 |
0.2 |
ML/LRT = montelukast/loratadina.
ML/DSLT = montelukast/desloratadina.
Escala: 0 = Ningún problema; 1 = muy leve; 2 = leve ;3 = moderado; 4 = severo; 5 = no puede ser peor.
Puntuaciones medias de los indicadores (1-10) SNOT-20 por tratamiento y semana (basal, 2, 3, 4 y 6 semanas).
Se observa que ambos tratamientos mitigaron los 10 síntomas evaluados por el cuestionario SNOT-20 sin que se noten diferencias apreciables entre ellos. Se observa que ambos tratamientos redujeron los problemas asociados a 10 condiciones de calidad de vida de las pacientes evaluadas por el cuestionario SNOT-20 sin que se noten diferencias apreciables entre ellos.
Tabla 17. Cuestionario SNOT-20 interpretado, de acuerdo a sus 6 niveles de severidad expresado en porcentaje
|
Visita |
Basal |
2da Semana |
3ra Semana |
4ta Semana |
6ta Semana |
|||||
|
Medicamento |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
|
Severidad |
||||||||||
|
5 |
5.9 |
8.6 |
||||||||
|
4 |
70.6 |
65.7 |
29.7 |
10.8 |
2.7 |
2.7 |
2.7 |
5.6 |
||
|
3 |
23.5 |
25.7 |
18.9 |
37.8 |
18.9 |
16.2 |
5.4 |
10.8 |
2.8 |
|
|
2 |
37.8 |
24.3 |
18.9 |
16.2 |
10.8 |
16.2 |
5.4 |
|||
|
1 |
10.8 |
27 |
37.8 |
67.6 |
35.1 |
35.1 |
10.8 |
16.7 |
||
|
0 |
2.7 |
21.6 |
45.9 |
37.8 |
81.1 |
75 |
||||
ML/LRT = montelukast/loratadina.
ML/DSLT = montelukast/desloratadina.
Escala: 0 = Ningún problema; 1 = muy leve; 2 = leve ;3 = moderado; 4 = severo; 5 = no puede ser peor.
Puntuaciones medias de los indicadores (1-10) SNOT-20 por tratamiento y semana.
Para ambos tratamientos, en condiciones basales aproximadamente el 75% de los pacientes fue clasificado en los niveles de severo o no puede ser peor. Para la sexta semana más del 90% (91.9% referencia y 91.7% prueba) de los pacientes lograron ser clasificados en los niveles muy leve o ningún problema, lo cual indica que ambos tratamientos mejoraron de manera global los síntomas y calidad de vida de los pacientes.
Tabla 18. Cambio de la puntuación global del cuestionario T5SS
|
Visita |
Basal |
3ra Semana |
6ta Semana |
|||
|
Medicamento |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
|
Congestión nasal |
2.8 |
2.8 |
1.1 |
1.3 |
0.4 |
0.5 |
|
Estornudos |
2.8 |
2.7 |
1.2 |
1.3 |
0.4 |
0.6 |
|
Rinorrea/escurrimiento nasal |
2.7 |
2.7 |
1 |
1.2 |
0.4 |
0.4 |
|
Prurito nasal |
2.6 |
2.6 |
0.8 |
1 |
0.2 |
0.4 |
|
Prurito ocular |
2.5 |
2.5 |
0.8 |
1 |
0.1 |
0.3 |
ML/LRT = montelukast/loratadina.
ML/DSLT = montelukast/desloratadina.
Puntuaciones medias T5SS de cada indicador, con respecto a las semanas en la que se aplicó el cuestionario: basal (media semanas 1 y 2), semana 3 (21 días) y semana 6 (42 días).
Figura 4. Puntuaciones globales medias T5SS por tratamiento y semana 0 = ninguna, 1 = leve, 2 = moderada, 3 = severa
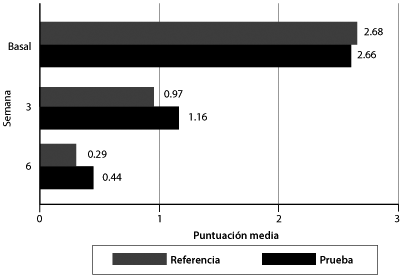
Las puntuaciones basales globales para ambos tratamientos son prácticamente iguales siendo cercanas al nivel severo. A la sexta semana, las puntuaciones de ambos tratamientos se encontraron entre los niveles ninguna (molestia) y leve, indicando mejoría de los 5 síntomas evaluados por el cuestionario T5SS.
Tabla 19. Clasificación de los pacientes por severidad T5SS (porcentajes)
|
Visita |
Basal |
3ra Semana |
6ta Semana |
|||
|
Medicamento |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
ML/LRT |
ML/DSLT |
|
Severidad |
||||||
|
3 |
81.1 |
73 |
2.7 |
2.7 |
5.6 |
|
|
2 |
18.9 |
27 |
24.3 |
29.7 |
8.1 |
2.8 |
|
1 |
40.5 |
54.1 |
10.8 |
16.7 |
||
|
0 |
32.4 |
13.5 |
81.1 |
75 |
||
ML/LRT = montelukast/loratadina.
ML/DSLT = montelukast/desloratadina.
Escala: 0 = ningún; 1 = leve; 2 = moderado; 3 = severo.
Uso de medicación de rescate: Únicamente 5 de los 86 pacientes aleatorizados necesitó usar el medicamento de rescate. En la siguiente tabla se presentan los datos de los 5 pacientes.
Tabla 20. Uso del medicamento de rescate
|
Sitio |
Paciente |
Tratamiento |
Semana 3 |
Semana 6 |
|---|---|---|---|---|
|
40 |
7 |
Prueba |
0 |
1 |
|
50 |
3 |
Referencia |
0 |
1 |
|
50 |
4 |
Prueba |
0 |
3 |
|
50 |
6 |
Prueba |
18 |
0 |
|
50 |
12 |
Prueba |
4 |
3 |
En la tercera semana, dos pacientes (ambos reclutados por el sitio 50) emplearon el medicamento de rescate. Durante la sexta semana 3 pacientes adicionales emplearon el medicamento de rescate. La duración en días del uso del medicamento de rescate varió desde 1 día a 18 días. Debido a que sólo un número pequeño de pacientes empleó el medicamento de rescate, no es posible inferir si su uso y su duración estuvo relacionado con el tratamiento asignado.
Tabla 21. Cuestionario sobre la satisfacción con el medicamento (TSQM).
Puntuaciones promedio para las cuatro dimensiones del cuestionario
|
ML/LRT |
ML/DSLT |
|
|
Efectividad |
84 |
85.5 |
|
Eventos adversos |
100 |
100 |
|
Conveniencia |
88.3 |
89.4 |
|
Global |
86.4 |
88.7 |
Para ambos tratamientos, las puntuaciones medias por dimensión fueron prácticamente iguales. Se observa que la puntuación para la dimensión de eventos adversos fue igual para ambos tratamientos y con la más alta puntuación posible (100%), indicando una excelente tolerabilidad de éstos. Las otras 3 dimensiones tuvieron puntuaciones superiores al 80% para ambos tratamientos, lo cual indica una evaluación satisfactoria por parte de los pacientes en lo que se refiere a efectividad, conveniencia de uso y satisfacción global con el medicamento.
Evaluación de seguridad: Para ambos tratamientos, 4 pacientes de los 86 pacientes aleatorizados reportaron un total de 12 eventos adversos (EAs). De los cuales 3 pacientes recibieron el tratamiento de referencia con 8 eventos adversos reportados y un paciente recibió el tratamiento de prueba con 4 eventos adversos reportados. Sólo existió un caso con elevación de aminotransferasas, que fue atribuido por el investigador al medicamento utilizado (referencia) y el evento fue cerrado por mejoría después de suspender el tratamiento.
Tabla 22. Información de eventos adversos de montelukast/desloratadina en rinitis aguda
|
Edad |
Sexo |
Descripción |
Relación |
Severidad |
Causalidad |
|
años |
M = 0/F = 1 |
Si=1/No=0 |
1 = Leve, 2 = Moderada, 3 = Severa |
1 = Cierta, 2 = Probable, 3 = Posible, 4 = Dudosa, 5 = Condicional, 6 = Inclasificable |
|
|
30 |
1 |
Gastritis aguda |
0 |
1 |
6 |
|
20 |
0 |
Pediculosis |
0 |
2 |
5 |
|
0 |
Pediculosis |
0 |
2 |
5 |
|
|
0 |
Irritación Ocular |
0 |
2 |
6 |
|
|
0 |
Veisalgia |
0 |
2 |
5 |
|
|
38 |
1 |
Cólico menstrual |
0 |
2 |
5 |
|
1 |
Somnolencia |
0 |
1 |
2 |
|
|
1 |
Incremento de aminotransferasas |
1 |
2 |
2 |
|
|
46 |
1 |
Gastritis y colitis |
0 |
2 |
6 |
|
1 |
Gastritis y colitis |
0 |
2 |
6 |
|
|
1 |
Diarrea |
0 |
2 |
6 |
|
|
1 |
Colitis |
0 |
2 |
6 |
Conclusión del estudio: De los anteriores resultados de este estudio se concluye que el medicamento de prueba EVEREST-DX® (montelukast 10 mg/desloratadina 5 mg) contra el medicamento de referencia (montelukast 10 mg/loratadina 10 mg) es seguro y eficaz para el tratamiento de la rinitis alérgica persistente y se demuestra la no inferioridad al no diferir significativamente entre las variables de ambos tratamientos; con mejoría global de los síntomas y en consecuencia una mejora en la calidad de vida de los pacientes.
CONTRAINDICACIONES: EVEREST-DX® (montelukast/desloratadina) está contraindicado en pacientes con hipersensibilidad a montelukast, a la desloratadina o a cualquier otro componente de este producto. Está contraindicado en pacientes con insuficiencia hepática grave porque pueden tener una menor depuración de desloratadina. Está contraindicado en el embarazo, lactancia y en menores de 18 años.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No existen estudios adecuados y bien controlados en mujeres embarazadas. EVEREST-DX® debe ser usado durante el embarazo sólo si es claramente necesario.
montelukast: Se han reportado de forma esporádica defectos congénitos de madres con asma que recibieron terapia, en su mayoría con múltiples fármacos, incluyendo montelukast. No se ha establecido alguna relación de causa entre montelukast y estos defectos.
Desloratadina: Como no se cuenta con datos clínicos sobre embarazos con exposición a la desloratadina, el uso sin riesgo de EVEREST-DX® durante el embarazo no ha sido establecido.
EVEREST-DX® solamente se debe usar si el beneficio potencial justifica el riesgo potencial para el feto o el recién nacido.
Lactancia: Se debe tener precaución cuando se administre EVEREST-DX® a una madre lactante. Este fármaco debe ser usado durante el embarazo solo si es claramente necesario.
Montelukast: No se sabe si el montelukast se excreta en la leche humana, por lo tanto, se debe tener precaución cuando se administre medicamentos que contienen montelukast a una mujer que está lactando.
La desloratadina se excreta en la leche materna, por lo que EVEREST-DX® no se recomienda en mujeres que estén lactando. La desloratadina solamente debe ser usada si el beneficio potencial justifica el riesgo potencial para el feto o el recién nacido.
REACCIONES SECUNDARIAS Y ADVERSAS: EVEREST-DX® (montelukast/desloratadina) fue estudiado en 397 pacientes en 6 estudios clínicos. Los estudios realizados con la combinación fija de montelukast desloratadina no mostraron una incidencia mayor adversos vs placebo o el uso de sus componentes por separado montelukast (monoterapia):
Montelukast es bien tolerado generalmente. Las reacciones adversas usualmente fueron leves, no fue necesario suspender el tratamiento. La incidencia total de estas reacciones adversas reportada con montelukast fue similar a la del placebo.
Reacciones adversas reportadas post-comercialización: Hipersensibilidad (incluyendo anafilaxis, angioedema, erupción cutánea, prurito, urticaria y, muy rara vez, infiltración eosinofílica hepática), eritema nodoso, trastornos del sueño y alucinaciones, somnolencia, vértigo, hiperactividad psicomotora (incluyendo irritabilidad. Agitación y comportamiento agresivo, inquietud y temblores), depresión, pensamientos y acciones suicidas (suicidalidad), insomnio, parestesia/hipoestesia y rara vez convulsiones; náusea, vómito, dispepsia, diarrea, incremento en ALAT y ASAT, y rara vez hepatitis colestática; artralgia, mialgia incluyendo calambres musculares, aumento en la incidencia de sangrado, contusión, palpitaciones y edema.
Desloratadina (monoterapia): En estudios clínicos en un rango de indicaciones que incluyeron rinitis alérgica y urticaria crónica idiopática a dosis recomendada de 5 mg diarios, se comunicaron efectos indeseables de Desloratadina en 3% más de los pacientes que recibieron placebo. Los efectos adversos reportados con mayor frecuencia, en comparación al placebo, fueron fatiga (1.2%), boca seca (0.8%), y cefalea (0.6%).
Desde que inició la comercialización de desloratadina, se han reportado de forma excepcional, reacciones de hipersensibilidad como anafilaxia y erupción cutánea. Además, casos de taquicardia, palpitaciones, hiperactividad psicomotora, crisis convulsivas, elevación de enzimas hepáticas, hepatitis e incremento de bilirrubinas se han reportado muy raramente.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Los estudios de carcinogenicidad se condujeron con montelukast o desloratadina, pero no con la combinación de ambos fármacos.
Montelukast: Estudios en animales han reportado que montelukast no es carcinogénico, genotóxico o mutagénico y no afectó la fertilidad ni la conducta reproductora, sin embargo, los resultados de estudios realizados en animales no siempre son predictivos de la respuesta humana.
Desloratadina: No se observaron efectos de la desloratadina sobre la fertilidad de la rata a una exposición 34 veces mayor que la exposición en el ser humano resultante de la administración del agente a la dosis clínica recomendada. La desloratadina no presenta riesgo carcinogénico en el hombre, conforme a los datos disponibles en los estudios originales para loratadina. La desloratadina no mostró efectos mutagénicos en los estudios de mutagénesis in vitro e in vivo. La desloratadina no fue teratógeno en ratas o conejos, a exposiciones 228 y 864 veces mayores, respectivamente, que la exposición en los humanos a la dosis clínica recomendada.
La desloratadina es el metabolito activo primario de la loratadina. Los estudios preclínicos conducidos con desloratadina y loratadina demostraron que no hubo diferencias significativas en el perfil toxicológico de ambas a niveles de exposición comparativos con desloratadina. Los datos preclínicos con desloratadina revelaron que no existe riesgo especial en humanos en estudios convencionales de seguridad farmacológica, toxicidad a dosis repetidas, toxicidad genética, y toxicidad en la reproducción. La falta de potencial carcinogénico fue demostrada en estudios conducidos con loratadina.
La desloratadina solamente se debe usar si el beneficio potencial justifica el riesgo potencial para el feto o el recién nacido.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se realizaron estudios específicos de interacción farmacológica con EVEREST-DX®.
Interacciones de montelukast: Montelukast se puede administrar con otros tratamientos usados comúnmente en la prevención y el tratamiento crónico del asma y rinitis alérgica; a la dosis clínica. En los estudios con montelukast no tuvo efectos clínicamente importantes sobre la farmacocinética de los siguientes medicamentos: Teofilina, prednisona, prednisolona, anticonceptivos orales (etinilestradiol/noretindrona-35/1), terfenadina, digoxina y warfarina.
Es importante señalar que montelukast en estudios clínicos se ha empleado con diversos medicamentos de prescripción común, sin reportes de interacciones clínicas adversas. Dentro de éstos se incluyen hormonas tiroideas, hipnóticos sedantes, agentes antiinflamatorios no esteroides, benzodiacepinas y descongestionantes.
El ABC de concentración plasmática-tiempo de montelukast disminuyó 40% aproximadamente durante la coadministración de fenobarbital, pero no se recomienda ningún ajuste de la dosificación de montelukast.
Estudios in vitro han demostrado que montelukast es un inhibidor del CYP 2C8, sin embargo, los datos de un estudio clínico de interacción fármaco-fármaco, que involucró a montelukast y rosiglitazona (un sustrato probado como representativo de fármacos metabolizados principalmente por CYP2C8, demostró que montelukast no inhibe al CYP2C8 in vivo.
Por lo tanto, no se espera que montelukast altere el metabolismo de fármacos metabolizados por esta enzima (por ejemplo, paclitaxel, rosiglitazona, repaglinida, entre otras).
Interacciones de Desloratadina: Desloratadina puede administrarse junto con los alimentos. No administrar junto con azitromicina, cimetidina, eritromicina, fluoxetina, ketoconazol, ya que aumenta la concentración plasmática de desloratadina.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No hubo diferencias significativas entre los grupos tratados con la combinación de montelukast/desloratadina y con placebo en la aparición de reacciones adversas de laboratorio relacionadas con el fármaco en los estudios clínicos en pacientes adultos.
Interacciones en las pruebas de laboratorio: Los antihistamínicos (desloratadina) pueden disminuir o inclusive prevenir las reacciones de pruebas cutáneas de hipersensibilidad, por lo que EVEREST-DX® debe ser suspendido al menos 48 horas antes de estas pruebas.
Con el uso de desloratadina, se han reportado de forma excepcional elevación de enzimas hepáticas y muy raramente el incremento de bilirrubinas.
PRECAUCIONES GENERALES: Montelukast/desloratadina no debe usarse de manera concomitante con otros productos que contengan los mismos ingredientes activos, montelukast o desloratadina (antihistamínicos).
Pacientes con asma y rinitis alérgica: Si se decide incluir a EVEREST-DX® como parte de la terapia; debe considerarse que Montelukast no es efectivo para el tratamiento agudo de asma, incluyendo estatus asmaticus y broncoespasmo agudo. Por tanto, debe advertirse a los pacientes sobre tener disponible medicación de rescate apropiada (por ejemplo, agonistas beta inhalados). Además, montelukast/desloratadina no debe usarse como monoterapia para el manejo de broncoespasmo inducido por ejercicio. En caso de requerirse, disminuir gradualmente bajo supervisión médica la dosis del corticosteroide inhalado concomitante, no se deben sustituir súbitamente con montelukast/desloratadina los corticosteroides inhalados por vía oral.
La reducción de la dosis de corticosteroides sistémicos en pacientes que reciben agentes antiasmáticos, incluyendo a los antagonistas de receptores de leucotrienos ha sido seguida en raros casos por la ocurrencia de uno o más de lo siguiente: Eosinofilia, erupción cutánea vasculítica, empeoramiento de los síntomas pulmonares, complicaciones cardiacas, y/o neuropatía diagnosticada algunas veces como síndrome de Churg-Strauss, una vasculitis eosinofílica sistémica.
Se recomienda cuidado y la adecuada supervisión clínica cuando se considere la reducción de corticosteroides sistémicos en pacientes que estén recibiendo medicamentos que contengan montelukast.
Empleo de montelukast en pacientes de edad avanzada: No se reportan diferencias relacionadas con la edad en los perfiles de seguridad o eficacia. La desloratadina no requiere de ajuste de dosis en pacientes mayores de 65 años.
En pacientes con asma que han recibido montelukast (monoterapia) se han reportado de eventos neuropsiquiátricos tales como agitación, irritación, insomnio, anormalidades del sueño, alucinaciones, depresión, desorientación, irritabilidad. Lo anterior se ha informado durante la reducción de la terapia corticoesteroide por vía oral. Debe indicarse a los pacientes y/o cuidadores que notifiquen a su médico si se presentan alteraciones de este tipo.
La desloratadina debe administrarse con precaución en pacientes con antecedentes personales o familiares de crisis convulsivas y, principalmente en niños pequeños, que son más susceptibles de desarrollar nuevas crisis cuando están en tratamiento con desloratadina. Los profesionales sanitarios pueden considerar la suspensión de desloratadina en pacientes que experimenten una crisis durante el tratamiento.
Con el uso de desloratadina no se han observado efectos sobre la capacidad para guiar vehículos y usar maquinarias.
Uso pediátrico: La seguridad y la eficacia de EVEREST-DX® no han sido evaluadas en pacientes menores de 18 años.
Se han reportado eventos adversos en sistema nervioso central que pueden ser significativos con el uso de montelukast en población pediátrica, por lo que lo que los productos con este principio activo no deben ser utilizados en esta población. Estos eventos no se han presentado con frecuencia en población adulta.
Este medicamento contiene lactosa, por lo que pacientes con problemas hereditarios de intolerancia a la galactosa, deficiencia de lactasa, Lapp o malabsorción de glucosa-galactosa, no deben tomar este medicamento.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Adultos (mayores de 18 años): La dosis recomendada de EVEREST-DX® es una cápsula (montelukast 10 mg/desloratadina 5 mg) una vez al día.
El tiempo de administración puede ser individualizado en los pacientes con rinitis alérgica para ajustarse a las necesidades del paciente; se puede administrar de día o noche.
En pacientes con uso de corticoides inhalados; EVEREST-DX® puede incorporarse al régimen de terapia. En caso de indicarse disminución de la dosis del corticosteroide inhalado; este debe reducirse de forma gradual y bajo supervisión médica. Asimismo en algunos pacientes EVEREST-DX® no debe ser substituido por corticosteroides inhalados de forma abrupta.
EVEREST-DX® no debe usarse de manera concomitante con otros productos que contengan los mismos ingredientes activos montelukast, desloratadina o inclusive antihistamínicos (como loratadina).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: En caso de sobredosis con EVEREST-DX® deben instituirse las medidas generales, de apoyo y sintomáticas. Estas deben mantenerse por el tiempo que fuera necesario.
Montelukast: No hay información específica sobre el tratamiento de la sobredosificación con montelukast.
En estudios con pacientes con diagnóstico de asma crónica, se ha administrado montelukast a pacientes adultos a dosis de hasta 200 mg diarios durante 22 semanas y, en estudios de corta duración hasta 900 mg diarios durante una semana aproximadamente, sin reacciones adversas de importancia clínica.
Casos de sobredosificación aguda se han reportado en adultos y niños en dosis de 1000 mg o·más; los eventos adversos de estas sobredosificaciones fueron relativamente benignos e incluyeron dolor de cabeza, sequedad de boca, somnolencia o hiperactividad, vómito y dolor abdominal.
Los hallazgos clínicos y de laboratorio observados fueron consistentes con el perfil de seguridad en adultos. En la mayoría de los casos de sobredosificación no se han reportado efectos adversos.
Los efectos adversos que ocurrieron con más frecuencia fueron consistentes con el perfil de seguridad de montelukast e incluyeron dolor abdominal, somnolencia, dolor de cabeza, vómito e hiperactividad psicomotora.
Desloratadina: Síntomas por ingestión de altas dosis en forma aguda, se puede producir nerviosismo, palpitaciones, convulsiones y molestias gastrointestinales, como diarrea y sequedad de la boca. Si las condiciones son adecuadas, se debiese practicar lavado gástrico para extraer la sustancia activa que no se haya absorbido. Se recomienda el tratamiento sintomático y coadyuvante. Basado en estudios clínicos en adolescentes y adultos con múltiples dosis, en los cuales se administró 45 mg de desloratadina (9 veces la dosis clínica recomendada), se determinó la ausencia de efectos colaterales relevantes. La desloratadina no se elimina por hemodiálisis. No se conoce si EVEREST-DX® puede eliminarse a través de hemodiálisis o diálisis peritoneal.
PRESENTACIONES: Caja con 15 y 30 cápsulas de 10 mg/5 mg e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese la caja bien cerrada. Consérvese a no más de 30 °C.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. No se deje al alcance de los niños. Su venta requiere receta médica. No use durante el embarazo o lactancia. No deberá usar en menores de 18 años. Este medicamento contiene lactosa, que puede causar reacciones de hipersensibilidad.
Reporte las sospechas de reacción adversa a los correos:
farmacovigilancia@cofepris.gob.mx y
farmacovigilancia@liomont.com.mx
LABORATORIOS LIOMONT, S.A. de C.V.
Adolfo López Mateos No.68, Col. Cuajimalpa, C.P. 05000, Cuajimalpa de Morelos, Ciudad de México, México.
Reg. Núm. 157M2022 SSA IV
Versión: Julio-2022
®Marca registrada