
FASLODEX - Solución inyectable
Sustancia(s):
- Fulvestrant
Presentaciones:
- 1 Jeringa(s) precargada(s) , 5 ml
- 2 Jeringa(s) precargada(s) , 5 ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada jeringa contiene:
Fulvestrant 250 mg
Vehículo, c.b.p. 5 mL.
INDICACIONES TERAPÉUTICAS: FASLODEX® está indicado para el tratamiento del cáncer de mama localmente avanzado o metastásico en mujeres postmenopáusicas de cualquier edad que hayan recibido antes tratamiento endocrino (anti estrógenos o tratamiento con inhibidores de la aromatasa), sin considerar si su estado menopáusico fue natural o se indujo en forma artificial.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas: La farmacología y el modo de acción establecido en los estudios de fulvestrant es el primer agente en una nueva clase de anti-estrógenos que regula el receptor de estrógeno (RE), y que por tanto puede ser descrito como bajo-regulador. Fulvestrant ejerce estos efectos farmacológicos mediante la unión con alta afinidad a los receptores de estrógenos alfa (REa), y tiene un novedoso modo de acción que induce una pérdida rápida de la proteína ERa de las células de cáncer de mama.
Fulvestrant es un potente inhibidor reversible del crecimiento de las células de cáncer de mama sensibles al estrógeno in vitro y tiene mayor potencia y eficacia que tamoxifeno. Fulvestrant inhibe el crecimiento de xenoinjertos de cáncer de mama sensibles a estrógenos en ratones inmunosuprimidos, es más efectivo que tamoxifeno para prevenir el establecimiento del tumor de xenoinjertos de células cancerosas mamarias humanas y suprime el crecimiento de tumores mamarios durante un tiempo dos veces más prolongado que con tamoxifeno. Fulvestrant inhibe el crecimiento de las células cancerosas de mama resistentes a tamoxifeno in vitro y de tumores mamarios resistentes a tamoxifeno in vivo.
Efectos en el tejido canceroso de mama in vivo: Los estudios clínicos en mujeres postmenopáusicas con cáncer de mama primario mostraron que fulvestrant causa una desregulación significativa de la expresión de RE en los tumores RE-positivos de una manera dosis dependiente. También se observó una disminución significativa en la expresión (marcador de acción de los estrógenos) de receptores para progesterona (RP) consistente con los resultados preclínicos, que demuestran que fulvestrant carece de actividad agonistas estrogénicos intrínsecos. Estos cambios en la expresión del RE y RP fueron acompañados por reducciones en la expresión del Ki67, un marcador de proliferación celular tumoral, que también se relacionó con la dosis de fulvestrant 500 mg teniendo un efecto significativamente mayor que la dosis de 250 mg.
Efectos en el cáncer de mama avanzado: Un estudio clínico de fase III (Estudio D6997C00002; CONFIRM) fue completado en 736 mujeres postmenopáusicas con cáncer de mama avanzado que habían presentado recurrencia de la enfermedad durante o después del tratamiento endocrino adyuvante, o progresión después del tratamiento endocrino para la enfermedad avanzada.
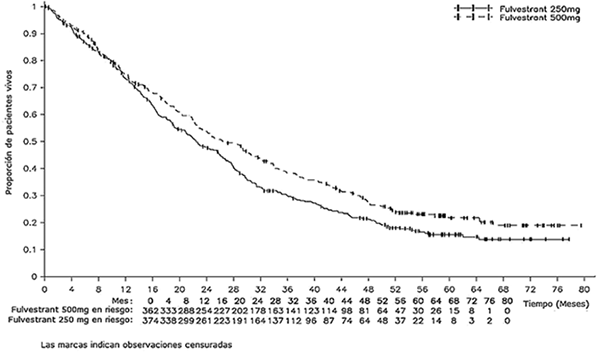
El estudio incluyó a 423 pacientes con recurrencia o progresión durante el tratamiento antiestrogénico (grupo AE) y 313 pacientes con recurrencia o progresión durante el tratamiento con un inhibidor de la aromatasa (subgrupo AI). El estudio comparó la eficacia y seguridad de FASLODEX® 500 mg con FASLODEX® 250 mg. La supervivencia sin progresión (SSP) fue el criterio de valoración primario, los criterios de valoración secundarios de eficacia incluyeron tasas de respuesta objetiva (ORR, por sus siglas en inglés); tasa de beneficio clínico (CBR, por sus siglas en inglés) y supervivencia global (SG). La SSP para FASLODEX® 500 mg se mostró significativamente más larga que la de FASLODEX® 250 mg. El análisis SG final a 75% de madurez mostró que FASLODEX® 500 mg está asociado con un aumento de 4.1 meses de la SG mediana y con una reducción de 19% del riesgo de muerte comparado con FASLODEX® 250 mg (HR = 0.81; 95% IC 0.69-0.96; p = 0.016 [valor nominal p, ya que no se hizo ajuste por multiplicidad]). Se resumen los resultados de eficacia en la tabla a continuación.
Tabla 1. Resumen de los resultados del criterio de valoración primario (SSP) y de los criterios de valoración secundarios clave del estudio CONFIRM
|
Variable |
Tipo de estimación; comparación de tratamientos |
FASLODEX® 500 mg |
FASLODEX® 250 mg |
Comparación entre grupos (FASLODEX® 500 mg/FASLODEX® 250) |
||
|
Tasa de riesgo |
95% IC |
Valor p |
||||
|
SSP |
Mediana K-M en meses; tasa de riesgo |
|||||
|
Todas las pacientes |
6.5 |
5.5 |
0.80 |
0.68, 0.94 |
0.006 |
|
|
Subgrupo AE (n = 423) |
8.6 |
5.8 |
0.76 |
0.62, 0.94 |
0.013 |
|
|
Subgrupo AI (n = 313)a |
5.4 |
4.1 |
0.85 |
0.67, 1.08 |
0.195 |
|
|
SG actualizadab |
Mediana K-M en meses; tasa de riesgo |
|||||
|
Todas las pacientes |
26.4 |
22.3 |
0.81 |
0.69, 0.96 |
0.016c |
|
|
Subgrupo AE (n = 423) |
30.6 |
23.9 |
0.79 |
0.63, 0.99 |
0.038c |
|
|
Subgrupo AI (n = 313)a |
24.4 |
20.8 |
0.86 |
0.67, 1.11 |
0.241c |
|
|
Variable |
Tipo de estimación; comparación de tratamientos |
FASLODEX® 500 mg |
FASLODEX® 250 mg |
Comparación entre grupos (FASLODEX® 500 mg/FASLODEX® 250) |
||
|
Posible radio |
95% IC |
|||||
|
ORRd |
% de pacientes con OR; posible radio |
|||||
|
Todas las pacientes |
13.8 |
14.6 |
0.94 |
0.57, 1.55 |
||
|
Subgrupo AE (n = 296) |
18.1 |
19.1 |
0.93 |
0.52, 1.68 |
||
|
Subgrupo AI (n = 205)a |
7.3 |
8.3 |
0.87 |
0.30, 2.44 |
||
|
CBRe |
% de pacientes con CB; posible radio |
|||||
|
Todas las pacientes |
45.6 |
39.6 |
1.28 |
0.95, 71 |
||
|
Subgrupo AE (n = 423) |
52.4 |
45.1 |
1.34 |
0.92, 1.97 |
||
|
Subgrupo AI (n = 313)a |
36.2 |
32.3 |
1.19 |
0.74, 1.90 |
||
ª FASLODEX® está indicado en pacientes con enfermedad recidivante o progresiva con tratamiento antiestrogénico. Los resultados del grupo AI no son concluyentes.
b Se presentan las SG de los análisis actualizados y de supervivencia final a 75% de madurez.
c Valor nominal de p sin ajustar por multiplicidad entre el análisis inicial global de supervivencia a 50% de madurez y el análisis final de supervivencia a 75% madurez (duración mínima de seguimiento de 50 meses).
d Se evaluó la ORR en pacientes que estaban evaluables para respuesta en la línea de base (es decir, aquéllas con enfermedad medible en la línea de base: 240 pacientes en el grupo de FASLODEX® 500 mg y 261 pacientes en el grupo de FASLODEX® 250 mg).
e Pacientes con una respuesta objetiva mejor de la respuesta total, respuesta parcial o enfermedad estable ³ 24 semanas.
SSP: Supervivencia sin progresión (el tiempo entre la aleatorización y la progresión más temprana o muerte por cualquier causa). Seguimiento de duración mínima de 18 meses).
ORR: Tasa de respuesta objetiva.
OR: Respuesta objetiva.
CBR: Tasa de beneficio clínico.
CB: Beneficio clínico.
SG: Supervivencia global.
K-M: Kaplan-Meier.
IC: Intervalo de confianza.
AI: Inhibidor de la aromatasa.
AE: Antiestrogénico.
Figura 1. Muestra una grafica Kaplan-Meier de los datos de supervivencia general del estudio CONFIRM
Los estudios clinicos de fase III (estudios 9238IL/0020 y 9238IL/0021) compararon la eficacia y seguridad de FASLODEX® 250 mg con un inhibidor de la aromatasa de tercera generación, anastrozol.
Los dos estudios clínicos de fase III fueron completados en un total de 851 mujeres postmenopáusicas con cáncer de mama avanzado que habían presentado recurrencia de la enfermedad durante o después del tratamiento endocrino adyuvante, o progresión después del tratamiento endocrino para la enfermedad avanzada. En el estudio 9238IL/0021 el TTP (tiempo hasta la progresión) para la comparación de FASLODEX® 250 mg contra anastrozol fue la siguiente; índice de riesgo (IC 95.14%) = 0.92 (0.74 a 1.14), p = 0.43. En el estudio 9238IL/0020 el TTP para la comparación de FASLODEX® 250 mg contra anastrozol fue la siguiente; índice de riesgo (IC 95.14%) = 0.98 (0.80 a 1.21), p = 0.84.
En general, FASLODEX® 250 mg fue por lo menos tan efectivo como anastrozol en términos de respuesta objetiva, beneficio clínico, tiempo hasta la progresión, tiempo hasta la falla del tratamiento y calidad de vida.
FASLODEX® 250 mg mostró una respuesta duradera en ambos estudios. En Norteamérica (estudio 9238IL70021), las pacientes en estudio tuvieron una mediana de duración de la respuesta de 19.3 meses para FASLODEX® 250 mg y de 10.5 meses para anastrozol. En el resto de los estudios mundiales (estudio 9238IL/0020), la mediana de duración de la respuesta fue de 14.3 y 14.0 para FASLODEX® 250 mg y anastrozol, respectivamente.
Efectos en el endometrio postmenopáusico: Los datos preclínicos para FASLODEX® sugieren que no tendrá efecto estimulante en el endometrio postmenopáusico. Una prueba en voluntarias postmenopáusicas sanas mostró que, en comparación con placebo, el tratamiento previo con 250 mg de FASLODEX® disminuía mucho la estimulación del endometrio postmenopáusico en voluntarias tratadas con 20 µg al día de etinilestradiol. Esto demuestra un potente efecto antiestrogénico en el endometrio postmenopáusico.
El tratamiento neoadyuvante para un máximo de 16 semanas en pacientes con cáncer de mama tratadas con fulvestrant 500 mg o 250 mg no produjeron cambios clínicamente significativos en el grosor endometrial, lo que indica una falta de efecto agonista. No hay evidencia de efectos negativos de endometrio en las pacientes estudiados con cáncer de mama.
Efectos sobre el hueso: El tratamiento neoadyuvante para un máximo de 16 semanas en pacientes con cáncer de mama tratadas ya sea con fulvestrant 500 mg o 250 mg no produjeron cambios clínicamente significativos en los marcadores séricos de hueso metabolizados. No hay evidencia de efectos negativos de endometrio en las pacientes estudiados con cáncer de mama.
Propiedades farmacocinéticas: Después de la administración intravenosa o intramuscular, fulvestrant se elimina con rapidez a una velocidad próxima al flujo sanguíneo hepático (nominalmente, 10.5 mL de plasma/min/kg). Sin embargo, la inyección intramuscular de acción prolongada de FASLODEX® mantiene concentraciones plasmáticas de fulvestrant dentro de un rango estrecho (hasta 3 veces) durante un periodo de por lo menos 28 días después de la inyección. La administración de FASLODEX® 500 mg alcanza los niveles de exposición en o cerca del estado de equilibrio dentro de un mes de dosificación (promedio [CV]):AUC 475 (33.4%) ng.días/mL, Cmáx. 25.1 (35.3%) ng/mL, Cmín. 16.3 (25.9%) ng/mL, respectivamente.
Los resultados de los estudios con dosis única de fulvestrant predicen la farmacocinética de las dosis múltiples.
No se detectó diferencia alguna en el perfil farmacocinético de fulvestrant en relación con la edad (rango 33 a 89 años).
No se detecto diferencia en el perfil farmacocinético del fulvestrant en relación con los grupos étnicos.
La administración de 250 mg de fulvestrant una vez al mes (28 días ± 3 días) produce acumulación limitada, y se aproxima a niveles de estado estable después de 3 a 6 dosis, con un aumento aproximado de hasta 2 veces en la exposición (promedio ± SD: AUC 328 ± 48 ng.días/mL, Cmáx. 15.8 ± 2.4 ng/mL, Cmín. 7.4 ± 1.7 ng/mL, respectivamente).
Absorción: Fulvestrant no se administra por vía oral.
Distribución: Fulvestrant se somete a una distribución amplia y rápida, el volumen de distribución aparente en el estado estable fue grande (aproximadamente 3 a 5 L/kg), lo que sugiere que la distribución del compuesto es sobre todo extravascular. Fulvestrant se une a proteínas plasmáticas en un porcentaje alto (99%) en concentraciones mucho mayores a las probables en el uso clínico. Las fracciones VLDL, LDL, y HDL de lipoproteínas parecen ser los principales elementos de unión. No pudo identificarse la función, si la hay, de la globulina transportadora de hormonas sexuales. No se realizaron estudios sobre las interacciones competitivas de enlace entre fármacos, ya que la mayoría de las interacciones publicadas de este tipo implican el enlace con albúmina y glucoproteínas ácidas a-1.
Metabolismo: Se determinó la biotransformación y disposición de fulvestrant en humanos después de la administración intramuscular e intravenosa de fulvestrant marcado con 14C. Parece que el metabolismo del fulvestrant incluye combinaciones de varias vías posibles de biotransformación análogas a las de los esteroides endógenos; incluyendo oxidación, hidroxilación aromática y conjugación con ácido glucurónico y sulfato en las posiciones 2-, 3- y 17- del núcleo esteroide, así como oxidación de la cadena lateral sulfóxido. El metabolismo de fulvestrant en humanos produce un perfil de metabolitos similar al que se observa en otras especies. Los metabolitos identificados son menos activos o tienen una actividad similar a fulvestrant en modelos antiestrogénicos. Los estudios con preparaciones de hígado humano y enzimas humanas recombinantes indican que CYP 3A4 es la única isoenzima P-450 implicada en la oxidación de fulvestrant; sin embargo, in vivo parece que ninguna vía distinta a P-450 es predominante.
Excreción: Fulvestrant se elimina con rapidez por la vía hepatobiliar; la velocidad total de la excreción depende de la vía de administración. La excreción se produjo por las heces, y la eliminación renal del material relacionado con el fármaco fue insignificante (menos de 1%).
Poblaciones especiales – insuficiencia hepática: La farmacocinética del fulvestrant ha sido evaluada en un estudio clínico a dosis única realizado en sujetos categoría Child-Pugh A y B con insuficiencia o cirrosis hepática, utilizando una dosis alta de una menor duración de la formulación de una inyección intramuscular. Hubo una reducción de 1.3 a 2 veces en promedio en el aclaramiento en pacientes categoría Child-Pugh A y B con insuficiencia hepática respectivamente en comparación con sujetos sanos. Que condujo a un incremento similar en la AUC. Sujetos con categoría Child-Pugh C no fueron evaluados.
Las concentraciones medias plasmáticas en el estado estable del modelo intramuscular del fulvestrant en sujetos con insuficiencia hepática categoría Child-Pugh A y B caen dentro del rango superior de las concentraciones esperadas para pacientes con función hepática normal a los que se les administro la formulación intramuscular. Dado el perfil de seguridad conocido del fulvestrant, no se considera necesario un ajuste de dosis.
CONTRAINDICACIONES: FASLODEX® está contraindicado en pacientes con hipersensibilidad conocida a la sustancia farmacológica o a cualquiera de los excipientes.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: Como se esperaba con un potente antiestrogénico, los estudios en animales mostraron toxicidad en órganos reproductivos (véase Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Fulvestrant se encuentra en la leche de ratas en niveles mucho más altos que los del plasma de la rata. Se desconoce el riesgo potencial para los humanos. Por lo tanto, debe evitarse el uso de FASLODEX® en mujeres embarazadas o durante la lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS:
FASLODEX® 500 mg: Las categorías de la siguiente frecuencia de reacciones adversas a medicamentos (RAM´s) se calculó en base al grupo de tratamiento con FASLODEX® 500 mg en los análisis de seguridad agrupados de CONFIRM (estudio D6997C00002), FINDER 1 (estudio D6997C00004), FINDER 2 (estudio D6997C00006) y NEWEST (estudio D6997C00003) que compararon FASLODEX® 500 mg con FASLODEX® 250 mg. Las frecuencias presentadas en la siguiente tabla se basaron en todos los eventos reportados, independientemente de la evaluación de la causalidad por parte del investigador.
Tabla 2. Resumen de las RAM´s observadas en los estudios clínicos de 500 mg
|
Frecuencia |
SOC |
RAM |
|
Muy común (³ 10%) |
Trastornos generales y condiciones del lugar |
Reacciones del sitio de inyección. |
|
Trastornos hepatobiliares |
Elevación de las enzimas hepáticas (ALT, AST, ALP). |
|
|
Trastornos gastrointestinales |
Náuseas. |
|
|
Común (³ 1- < 10%) |
Trastornos vasculares |
Bochornos. |
|
Trastornos del sistema nervioso |
Dolor de cabeza. |
|
|
Trastornos hepatobiliares |
Bilirrubina elevadaa |
|
|
Trastornos gastrointestinales |
Vómito, diarrea. |
|
|
Trastornos de metabolismo y nutrición |
Anorexia. |
|
|
Trastornos de piel y tejido subcutáneo |
Salpullido. |
|
|
Infecciones e infestaciones |
Infecciones del tracto urinario. |
|
|
Trastornos del sistema inmune |
Reacciones de hipersensibilidad. |
|
|
Poco común (³ 0.1% y <1%) |
Trastornos hepatobiliares |
Insuficiencia hepáticab, hepatitisb, gamma-GT elevada. |
|
Sangre y sistema linfático |
Disminución de la cuenta plaquetaria. |
a Basado sobre cualquier cambio grado CTC desde línea base.
b El evento no se observó en estudios clínicos importantes (CONFIRM, FINDER 1, FINDER 2, NEWEST). Se ha calculado la frecuencia con un límite superior del intervalo de confianza de 95% para la estimación de punto. Esto se calcula como 3/560 (donde 560 es el número de pacientes en los principales estudios clínicos) lo cual equivale a una categoría de frecuencias de "poco común".
Sobre la base de los datos, no hay evidencia de una relación causal entre FASLODEX® y eventos raros o poco frecuentes en los estudios clínicos de FASLODEX®.
FASLODEX® 250 mg: Las categorías de la siguiente frecuencia de reacciones adversas a medicamentos (RAM´s) se calculo en base al grupo de tratamiento con FASLODEX® 250 mg en los análisis agrupados de seguridad de los estudios 9238IL/0020, 9238IL/0021, 9238IL/0025, D6997C00002 (CONFIRM), D6997C00004 (FINDER 1), D6997C00006 (FINDER 2), y D6997C00003 (NEWEST).
Tabla 3. Resumen de las RAM´s observadas en los estudios clínicos para FASLODEX® 250 mg
|
Frecuencia |
SOC |
RAM |
|
Muy común (³ 10%) |
Trastornos generales y condiciones del lugar |
Reacciones del sitio de inyección. Astenia. |
|
Trastornos hepatobiliares |
Elevación de las enzimas hepáticas (ALT, AST, ALP). |
|
|
Trastornos gastrointestinales |
Náuseas. |
|
|
Trastornos del sistema nervioso |
Dolor de cabeza. |
|
|
Común ³ 1- < 10%) |
Trastornos vasculares |
Bochornos. |
|
Trastornos gastrointestinales |
Vómito, diarrea. |
|
|
Trastornos de metabolismo y nutrición |
Anorexia. |
|
|
Trastornos de piel y tejido subcutáneo |
Salpullido. |
|
|
Infecciones e infestaciones |
Infecciones del tracto urinario. |
|
|
Trastornos del sistema inmune |
Reacciones de hipersensibilidad. |
|
|
Trastornos hepatobiliares |
Bilirrubina elevadab |
|
|
Poco común (³ 0.1% y < 1%) |
Trastornos hepatobiliares |
Insuficiencia hepáticac, hepatitisc, gamma-GT elevada |
|
Sangre y sistema linfático |
Disminución de la cuenta plaquetaria |
a En 4 de 7 estudios incluidos en el conjunto de análisis, pacientes en el brazo de FASLODEX® 250 mg que recibieron 2 inyecciones. En los otros 3 estudios (Estudios 9238IL/0020, 9238IL/0025 y NEWEST), los pacientes del brazo de FASLODEX® 250 mg que recibieron una inyección única y la frecuencia de reacciones en el lugar de la inyección en cada uno de estos 3 estudios fue 7.3% (16/219), 13.5 (42/310) y 5.0% (5/101), respectivamente.
b Basado sobre cualquier cambio grado CTC desde línea base.
c El evento no se observó en estudios clínicos importantes (9238IL/0020, 9238IL/0021, 9238IL/0025, CONFIRM, FINDER 1, FINDER 2, NEWEST). Se ha calculado la frecuencia con un límite superior del intervalo de confianza de 95% para la estimación de punto. Esto se calcula como 3/1,263 (donde 1,263 es el número de pacientes en los principales estudios clínicos) lo cual equivale a una categoría de frecuencias de "poco común".
Sobre la base de los datos, no hay evidencia de una relación causal entre FASLODEX® y eventos raros o poco frecuentes en los estudios clínicos de FASLODEX®.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Toxicidad aguda: La toxicidad aguda de fulvestrant es baja. En los roedores, la dosis letal media fue mayor a 70 mg/kg después de la administración intramuscular (más de 400 veces la dosis clínica); mayor de 50 mg/kg después de la administración intravenosa y mayor de 2,000 mg/kg después de la administración oral.
Toxicidad crónica: Fulvestrant fue bien tolerado en todas las especies animales en las que se probó. En estudios de toxicidad con dosis múltiple intramuscular en ratas y perros, la actividad antiestrogénica de fulvestrant causó la mayoría de los efectos observados, sobre todo en el sistema reproductor femenino, pero también en otros órganos sensibles a las hormonas en ambos sexos. No hubo evidencia de otros efectos tóxicos sistémicos en ratas que recibieron dosis de hasta 10 mg/rata/15 días por 6 meses, ni en perros con dosis de hasta 40 mg/kg/28 días durante 12 meses.
En estudios con perros después de la administración oral e intravenosa, se observaron efectos en el sistema cardiovascular (elevaciones ligeras en el segmento ST del ECG [oral] y paro sinusal en un perro [intravenosa]), pero esto ocurrió en animales expuestos a fulvestrant a niveles mucho mayores que los registrados en pacientes (Cmáx. > 15 veces) y por lo tanto, se consideran sin importancia para la seguridad de los humanos con la dosis clínica.
Mutagenicidad: Fulvestrant no mostró potencial genotóxico.
Toxicología reproductiva: Fulvestrant mostró efectos en la reproducción y el desarrollo embrionario/fetal consistente con su actividad antiestrogénica con dosis similares a la dosis clínica. En las ratas, fulvestrant causó reducción reversible de la fertilidad femenina y la sobrevivencia embrionaria con dosis de 0.01 mg/kg/día o más, además de distocia y mayor incidencia de anormalidades fetales, incluida flexión del tarso. Las conejas que recibieron fulvestrant en dosis ³ 1 mg/kg/día no pudieron mantener el embarazo y con dosis de hasta 0.25 mg/kg/día se observaron aumentos en el peso placentario y pérdida posterior a la implantación, pero sin efectos en el desarrollo fetal.
Carcinogenicidad: Un estudio de dos años sobre oncogenicidad en ratas (administración intramuscular) mostró aumento en la incidencia de tumores benignos de células de la granulosa ovárica en las hembras con la dosis alta, 10 mg/rata/15 días. En un estudio de dos años de oncogenicidad en ratones, la administración oral fue asociada con un aumento en la incidencia de tumores del estroma del cordón sexual (benigno y maligno) en el ovario a dosis de 150 y 500 mg/kg/día. El nivel sin efecto observado (NOEL, por sus siglas en inglés) de estos hallazgos fue de 10 mg/rata/30 días en ratas y 20 mg/kg/día en ratones, respectivamente. La inducción de estos tumores es consistente con las alteraciones en la retroalimentación endocrina, relacionadas con la farmacología en los niveles de gonadotropina, causados por el efecto antiestrogénico en los animales con ciclos hormonales. Por lo tanto, no se considera que este hallazgo tenga relevancia clínica para el uso de fulvestrant en mujeres postmenopáusicas con cáncer de mama avanzado.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Fulvestrant no inhibe en forma significativa ninguna de las isoenzimas principales de citocromo P-450 (CYP) in vitro; los resultados de una prueba farmacocinética clínica incluyeron la administración concomitante de fulvestrant con midazolam también sugiere que la dosis terapéutica de fulvestrant no tiene efectos inhibidores en CYP3A4. Adicionalmente, aunque fulvestrant puede metabolizarse por CYP3A4 in vitro, un estudio clínico con rifampicina mostró que no había cambio en la depuración de fulvestrant como resultado de la inducción de CYP3A4. Los resultados de un estudio clínico con ketoconazol, un potente inhibidor de CYP3A4, también indicaron que no hay cambios de importancia clínica en la depuración de fulvestrant. No es necesario ajustar la dosis en pacientes que al mismo tiempo reciben inductores o inhibidores de CYP3A4.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No se han reportado.
PRECAUCIONES GENERALES: Fulvestrant se metaboliza sobre todo en el hígado. Se debe tener precaución con FASLODEX® en pacientes con insuficiencia hepática, ya que el metabolismo se puede ver reducido (véase Dosis y vía de administración y Farmacocinética y farmacodinamia).
Debe tenerse precaución antes de tratar a pacientes con depuración de creatinina menor de 30 mL/min (véase Farmacocinética y farmacodinamia).
Debe tenerse cuidado cuando se trate a pacientes con diátesis hemorrágica o trombocitopenia, así como en pacientes que usan anticoagulantes debido a la ruta de administración.
Efectos en la habilidad para conducir u operar maquinaria: Es improbable que FASLODEX® afecte la capacidad de los pacientes para conducir u operar maquinaria. Sin embargo, ha habido informes de astenia durante el tratamiento con FASLODEX® y los pacientes que presenten este síntoma deben tener precaución cuando conducen u operan maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Mujeres adultas (incluidas pacientes de edad avanzada): La dosis recomendada es de 500 mg a ser administrada por vía intramuscular con dos inyecciones de 5 mL aplicadas en el glúteo a intervalos de 1 mes con una dosis adicional de 500 mg administrada dos semanas después de la dosis inicial. Se recomienda administrar la inyección lentamente (1-2 minutos/inyección).
La dosis de 250 mg por vía intramuscular aplicada en el glúteo a intervalos de 1 mes como inyección única de 5 mL. Se recomienda administrar la inyección lentamente.
Niños: No se recomienda su empleo en niños o adolescentes, ya que no se ha establecido su seguridad y efectividad en este grupo de edad.
Pacientes con insuficiencia renal: No se recomiendan ajustes en la dosis para pacientes con depuración de creatinina mayor de 30 mL/min. No se ha hecho una evaluación adicional de la seguridad y eficacia en pacientes con depuración de creatinina menor de 30 mL/min (véase Precauciones generales).
Pacientes con insuficiencia hepática: No se recomiendan ajustes en la dosis para pacientes con categoría Child-Pugh A y B con daño hepático. El uso de FASLODEX® no ha sido evaluado en paciente Child-Pugh C y daño hepático (véase Precauciones generales y Farmacocinética y farmacodinamia).
Pacientes de edad avanzada: No se requieren ajustes en la dosis para los pacientes geriátricos.
Interacciones que requieren ajuste de dosis: No hay interacciones farmacológicas conocidas que requieran un ajuste en la dosis.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No hay experiencia de sobredosis en humanos. Los estudios en animales sugieren que no hay efectos distintos a los relacionados en forma directa o indirecta con la actividad antiestrogénica cuando se administraron dosis más altas de fulvestrant. Si ocurre una sobredosis, debe emplearse tratamiento sintomático.
PRESENTACIONES: Caja de cartón con 1 o 2 jeringas prellenadas con 250 mg/5 mL e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese entre 2 y 8°C. No se congele. Consérvese en su empaque original. Protéjase de la luz.
LEYENDAS DE PROTECCIÓN:
No se deje al alcance de los niños.
Su venta requiere receta médica.
No se use en el embarazo ni en la lactancia.
Este medicamento deberá ser administrado
únicamente por médicos especialistas en oncología
con experiencia en quimioterapia antineoplásica.
Literatura exclusiva para médicos.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx

ASTRAZENECA, S. A. de C. V.
Súper Avenida Lomas Verdes No. 67
Fracc. Lomas Verdes
C.P. 53120, Naucalpan de Juárez, México
Reg. Núm. 197M2005, SSA IV