
FLANAX 660
NAPROXENO
Tabletas de liberación prolongada
1 Caja, 2 Tabletas, 660 mg
1 Caja, 4 Tabletas, 660 mg
1 Caja, 8 Tabletas, 660 mg
1 Caja, 12 Tabletas, 660 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Naproxeno sódico 660 mg
Equivalente a 603 mg de naproxeno Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
Antiinflamatorio no esteroideo con acción analgésica, antiinflamatoria y antipirética, indicado para el tratamiento de procesos que no requieran más de 10 días de tratamiento.
Infecciones de las vías respiratorias: Dolor e inflamación asociados con el resfriado común, como el dolor de cabeza y de garganta, así como para el alivio de la fiebre. Asociado al tratamiento de malestares o síntomas de enfermedades respiratorias como sinusitis, dolor asociado a amigdalitis, faringitis, faringoamigdalitis, rinofaringitis, laringitis, laringotraqueítis, bronquitis, bronconeumonía y neumonía; sólo o en conjunto con el antibiótico o antiviral indicado por el médico, que no requieran más de 10 días de tratamiento.
Traumatología: Dolor muscular y dolor de cuerpo moderado a severo. Dolor e inflamación de articulaciones, ligamentos, músculos y tendones por contracturas, torceduras, desgarres, esguinces o luxaciones. Dolor e inflamación ocasionados por sobreesfuerzo físico, ejercicio o vicios posturales. Dolor e inflamación por golpes, contusiones y traumatismos menores en tejidos blandos, o posteriores a manipulaciones ortopédicas. Dolor de espalda, cuello, columna, lumbago. Inflamación y rigidez muscular y/o articular, por los cuales no se requieran más de 10 días de tratamiento.
Usos ginecológicos: En dismenorrea primaria y secundaria con síntomas asociados: cólico, dolor de espalda y/o cintura, dolor de cabeza (cefalea), dolor muscular moderado a severo; los cuales no requieran más de 10 días de tratamiento. Como analgésico y antiinflamatorio en el postparto en mujeres que no vayan a amamantar. Después de la aplicación de un dispositivo intrauterino (DIU).
Reumatología: Para el alivio de dolor e inflamación en artritis y procesos reumáticos agudos que no requieran de más de 10 días de tratamiento; incluyendo la artrosis, fibromialgia o cualquier enfermedad de etiología reumatológica.
Usos odontológicos: Para el alivio de dolor moderado a severo e inflamación de muelas y después de extracciones dentales; que no requieran más de 10 días de tratamiento.
FARMACOCINÉTICA Y FARMACODINAMIA:
Grupo farmacoterapéutico de naproxeno: Sistema músculo-esquelético, productos antiinflamatorios y antirreumáticos no esteroideos, derivado del ácido propiónico.
Código ATC: MO1A E02.
Farmacodinamia: El naproxeno pertenece al grupo de fármacos antiinflamatorios no esteroideos, los cuales ejercen funciones analgésicas, antipiréticas y antiinflamatorias a través de la inhibición reversible de la síntesis de prostaglandinas. El naproxeno es un inhibidor no selectivo de COX, funciona inhibiendo tanto la enzima COX-1 como la enzima COX-2. Inhibe la formación de la tromboxano-sintasa dependiente de COX-1, A2 (TXA2), la cual reduce la agregación plaquetaria, y la prostaciclina dependiente de COX-2 (PGI2), que es un importante mediador de la vasodilatación. El naproxeno proporciona alivio del dolor, disminuye la fiebre y reduce la respuesta inflamatoria.
Farmacocinética:
Absorción: El naproxeno se disuelve rápidamente en el medio gástrico y se absorbe rápida y completamente desde el tracto gastrointestinal. Se pueden obtener niveles plasmáticos significativos del naproxeno y un inicio de alivio del dolor dentro de los 20 minutos a partir de la ingesta, y los niveles máximos (Cmáx) se logran en aproximadamente 1 hora (tmáx).
El naproxeno de liberación prolongada de 660 mg es una tableta de dos capas, con una capa de liberación inmediata y una capa de liberación prolongada. La capa de liberación inmediata comienza a disolverse inmediatamente, mientras que la capa de liberación prolongada se disuelve lentamente en el tracto gastrointestinal. Los niveles máximos (Cmáx) se alcanzan en aproximadamente 45-60 minutos (tmáx) en condiciones de ayuno. El naproxeno sódico de LP está diseñado para liberar 264 mg inmediatamente y 396 mg en forma de liberación prolongada. La cantidad de naproxeno sódico en el porcentaje de liberación inmediata del producto es menor que la dosis inicial aprobada de naproxeno sódico 440 mg de LI.
Distribución: Después de la absorción, más del 99% está unido a la albúmina sérica. El volumen de distribución es de aproximadamente 0.1 L/kg y la vida media de eliminación (t½) es de aproximadamente 14 horas.
Metabolismo y excreción: El naproxeno, después del metabolismo hepático, se excreta principalmente (≥ 95%) a través de los riñones. Los datos farmacocinéticos muestran linealidad en la dosis recomendada. Los pacientes con insuficiencia hepática grave pueden tener mayores niveles de naproxeno libre. En insuficiencia renal severa, la eliminación se deteriora, pero no se ha observado una acumulación significativa en la dosis recomendada.
Bayer ha desarrollado una forma farmacéutica de naproxeno sódico de liberación prolongada para el alivio efectivo del dolor hasta por 24 horas con dosificación una vez al día. Para esta presentación, Bayer desarrolló tres formas farmacéuticas de comprimidos de naproxeno sódico de Liberación prolongada LP de 660 mg (prototipos 1, 2 y 3) y realizó un estudio farmacocinético piloto (estudio [12656]) para determinar el perfil farmacocinético de una dosis única de estas tres formas farmacéuticas de comprimidos de ER en relación con el comprimido de naproxeno sódico de liberación inmediata LI comercial establecido (220 mg) dosificado cada 8 horas. Los prototipos 1, 2 y 3 se formularon con 20%, 30% y 40% de excipiente, respectivamente, que controlan la velocidad de liberación de naproxeno sódico del porcentaje de liberación prolongada de la forma farmacéutica propuesta. El objetivo primario del estudio farmacocinético piloto fue elegir el prototipo óptimo a desarrollar.
El estudio [12656] fue un estudio cruzado de cuatro vías, de cuatro secuencias, aleatorizado, abierto. A cada paciente se le administraron secuencias de tratamiento de cuatro fármacos diferentes con un periodo de reposo farmacológico de 7 días entre cada tratamiento.
Los tratamientos fueron:
• Una sola dosis del comprimido de 660 mg de prototipo LP 1.
• Una sola dosis de 660 mg del comprimido de LP prototipo 2.
• Una sola dosis del comprimido de 660 mg de LP prototipo 3
• Una dosis del comprimido de 220 mg de naproxeno sódico LI cada 8 horas. Los muestreos de sangre para la evaluación farmacocinética se obtuvieron antes de la dosis y 48 horas después de la dosis.
Cada uno de los tres prototipos mostró un perfil farmacocinético que coincide con el diseño de la forma farmacéutica, con un rápido aumento inicial en concentraciones plasmáticas que posteriormente se mantuvieron durante 24 horas, indicando el componente liberación prolongada y liberación retardada de las formas farmacéuticas.
En comparación con el comprimido de naproxeno sódico de LI de 220 mg, los tres prototipos mostraron una exposición total similar según lo que se midió por los valores de área bajo la curva, con un intervalo de confianza (IC) de 90% que está dentro del rango de 80-125%. Inicialmente, la exposición fue mayor durante el primer intervalo de dosificación de 8 horas para los tres prototipos que para el comprimido de naproxeno sódico de LI. Esto refleja la contribución del componente de IR de los prototipos (264 mg; 40% de la forma farmacéutica de 660 mg) en comparación con la dosis de 220 mg del comprimido de naproxeno sódico de LI. Durante el segundo intervalo de dosificación de 8 horas, la exposición fue similar de los prototipos y el comprimido de naproxeno sódico de LI. Por último, para el último intervalo de dosificación de 8 horas, las concentraciones plasmáticas de los tres prototipos fueron menores que después de la administración de la tercera dosis de 220 mg con el comprimido de naproxeno sódico de LI. Por lo tanto, un estudio clínico se llevó a cabo para caracterizar la eficacia analgésica, con énfasis en demostrar la eficacia en las últimas ocho horas del periodo de dosificación de 24 horas.
Las concentraciones plasmáticas máximas (Cmáx) fueron más altas después de la administración de una sola dosis de cada uno de los tres prototipos en comparación con las concentraciones plasmáticas máximas correspondientes para el comprimido de naproxeno sódico de LI. Para los prototipos 1 y 2, el IC de 90% resultante estaba fuera de los límites de 80-125%, aunque solo de forma marginal para el prototipo 2 (IC de 90%: 113.35-127.80). Para el prototipo 3, el IC de 90% para Cmáx estaba dentro de los límites de 80-125%, así como para el área bajo la curva; sin embargo, este prototipo se consideró el menos deseable en términos de características de fabricación.
Las concentraciones plasmáticas máximas para el comprimido de naproxeno sódico de LI generalmente ocurrieron después de la tercera dosis; por lo tanto, el valor de la mediana del tmáx fue de 17 horas, mientras que el componente de liberación inmediata de los tres prototipos de LP garantiza un tmáx rápido, con valores promedio de 0.75-2 horas. Las semividas terminales calculadas del naproxeno después de la administración de los tres prototipos y comprimido de naproxeno sódico de LI fueron comparables (13-15 horas).
Con base en los resultados del estudio farmacocinético piloto, el prototipo 2 con excipiente al 30% se seleccionó para la evaluación adicional en el programa de desarrollo clínico para la forma farmacéutica del comprimido de naproxeno sódico de LP.
Se realizaron tres estudios de biodisponibilidad relativa. Las características clave de los estudios se presentan en la siguiente tabla.
Tabla 1. Características de los estudios de biodisponibilidad
|
ID del estudio |
Objetivo primario |
Características del estudio |
|
[13106] |
Determinar el perfil farmacocinético de un comprimido de naproxeno sódico de LP de 660 mg en relación con un comprimido de naproxeno sódico comercial de 220 mg establecido después de la administración de un solo día y en el estado estable después de la administración de múltiples dosis durante 4 días de ayuno |
Estudio aleatorizado, abierto, cruzado doble, una sola dosis y múltiples dosis, en un solo centro en 28 pacientes sanos |
|
[13965] |
Determinar el perfil farmacocinético de un comprimido de naproxeno sódico (660 mg) de LP en relación con un comprimido de naproxeno sódico comercial de 220 mg establecido después de la administración de un solo día y la administración de múltiples dosis durante 4 días tras ingerir alimentos |
Estudio aleatorizado, abierto, de dos tratamientos, de dos secuencias, cruzado, en un solo centro en 32 pacientes sanos |
|
[13080] |
Determinar la biodisponibilidad de una sola dosis oral de un comprimido de naproxeno sódico de ER administrado en ayuno y tras ingerir alimentos |
Estudio aleatorizado, abierto, de dos tratamientos, de dos secuencias, cruzado, en un solo centro en 32 pacientes sanos |
Los estudios [13106] y [13965] mostraron exposición general comparable para el producto de LP en comparación con el producto de LI para la dosis diaria total equivalente (660 mg) según lo que se midió por los valores de área bajo la curva. Bajo condiciones de ayuno (estudio [13106]), la concentración plasmática máxima (Cmáx) fue menor para la forma farmacéutica de LP que para el comprimido de LI después de la dosificación de un solo día o varios días. Esto era de esperarse, ya que el naproxeno sódico de LI se administró como una dosis inicial de 440 mg en comparación con la dosis de 264 mg disponible del componente de LI de la forma farmacéutica propuesta de LI. Tras ingerir alimentos (estudio [13965]), Cmáx fue similar entre las formas farmacéuticas de LP y LI.
Figura 1. Concentraciones plasmáticas de naproxeno de la media aritmética contra tiempo en estado estable para los productos de LP y LI después de dosis múltiples en ayuno (estudio [13106])
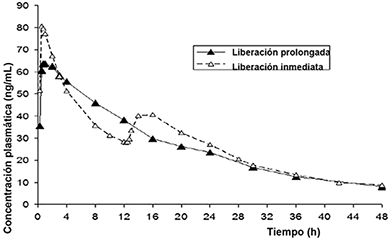
En general, dos estudios (estudios [13080] y [13965]) evaluaron el efecto de los alimentos en el perfil farmacocinético de la forma farmacéutica de naproxeno sódico de LP. El estudio [13080] evaluó el perfil farmacocinético de una sola dosis de la forma farmacéutica de naproxeno sódico de LP tras ingerir alimentos y en ayuno. El estudio [13965] fue un estudio de biodisponibilidad relativa que compara los perfiles farmacocinéticos de la forma farmacéutica de naproxeno sódico de LP y el comprimido de naproxeno sódico de LI de 220 mg tras ingerir alimentos después de una dosificación de un solo día y de varios días. Ambos estudios mostraron que los alimentos no tuvieron ningún efecto en el grado de absorción con base en los valores comparables del área bajo la curva, pero retrasaron la velocidad de absorción, como se muestra por un valor Tmáx mayor.
En general, tras ingerir alimentos, la administración de la forma farmacéutica de LP demostró un perfil farmacocinético con dos picos, aunque este fenómeno no se observó en ninguno de los perfiles en ayuno. Además, hubo una amplia gama de los valores de Tmáx individuales después de la administración de una sola dosis tras ingerir alimentos, el segundo pico ocurre tarde en el intervalo de dosificación (hasta 24 horas en algunos pacientes) y en algunos casos que representan la concentración plasmática más alta para ese paciente. El retraso significativo en el segundo pico es probablemente reflejo de un retraso en el vaciado gástrico en presencia de una comida, como indicaron los resultados de la simulación in vitro de TIM-1; y el retraso subsecuente en el transporte de la fórmula de LP en el tracto gastrointestinal (GI) inferior.
Después de la administración de varios días durante cuatro días, la variabilidad en los valores de Tmáx fue menos con perfiles de farmacocinética menores de la forma farmacéutica de LP con un segundo pico que representa la mayor concentración plasmática y sin perfiles farmacocinéticos que tienen concentración plasmática máxima de hasta 24 horas posteriores a la dosis. Existían dudas sobre que el inicio de la eficacia analgésica de la forma farmacéutica de LP pueda no ser lo suficientemente rápido como para el control de condiciones de dolor agudo durante las primeras horas del intervalo de dosificación de 24 horas. En consecuencia, se llevaron a cabo dos ensayos clínicos en el modelo de dolor dental (estudio [13130], estudio [15142]) para caracterizar la eficacia analgésica. Los datos demostraron que el naproxeno sódico de LP de 660 mg ofreció alivio del dolor a partir de los 30 minutos posteriores a la dosificación y se prolongaron hasta el periodo de dosificación de 24 horas. Esto coincide con los resultados del naproxeno sódico de LI de 220 mg que muestran un inicio de la eficacia analgésica dentro de 30 a 40 minutos posteriores a la dosis.
El estudio puente farmacocinético de naproxeno sódico (estudio [14566]) comparó el perfil farmacocinético del comprimido de naproxeno sódico de LP de 660 mg en relación con dos comprimidos de Naproxeno sódico 500 mg (equivalentes a naproxeno sódico 1100 mg) después de la administración de una sola dosis en ayuno. La concentración plasmática media del naproxeno después del tratamiento con naproxeno sódico excedió rápidamente la concentración plasmática media después del tratamiento con comprimido de naproxeno sódico de LP y seguía siendo más alta para la duración del tiempo de muestreo de 36 horas. Tal como se esperaba, debido a una dosis más alta de aproximadamente 67% de naproxeno sódico para naproxeno sódico que para el comprimido de naproxeno sódico de LP (1100 mg y 660 mg, respectivamente), la exposición total media (medida por el área bajo la curva) y la exposición máxima (medida por el Cmáx) fueron mayores para naproxeno sódico en comparación con las del comprimido de naproxeno sódico de LP. La semivida terminal media (t1/2) y tiempo de permanencia medio (MRT) del naproxeno fueron mayores que los del comprimido de naproxeno sódico de LP. El Tmáx promedio de Naproxeno sódico fue 4.00 horas, más de las 2.00 horas que se observaron para los comprimidos de naproxeno sódico de LP. Los resultados de este estudio apoyan la conclusión que la dosis habitual de naproxeno sódico (1100 mg una vez al día) excede la exposición de la dosis propuesta del comprimido de naproxeno sódico de LP (660 mg una vez al día).
Bayer estableció la eficacia del comprimido de naproxeno sódico de LP de 660 mg a dos brazos: administrado por vía oral una vez al día y en dos dosis para evaluar los ensayos de eficacia del dolor dental doble ciego, aleatorizados, paralelos, controlados con placebo, de una sola dosis (estudio [13130], estudios [15142]).
En la siguiente tabla se presenta una descripción de los estudios 13130 y 15142.
Tabla 2. Descripción de los estudios de dolor dental utilizando el comprimido de naproxeno sódico de LP de 660 mg obtenido de la evaluación del prototipo final
|
ID del estudio |
Diseño |
Tratamientos |
N |
Duración |
Sexo H/M Edad promedio (intervalo) |
Diagnóstico |
Variable de evaluación primaria |
|
[13130] |
Doble ciego, aleatorizado, controlado con placebo |
Naproxeno sódico de LP de 660 mg (todos los días) Placebo |
153 159 |
1 día |
N = 312 H = 117; M = 195 20.6 (16-39) |
Pacientes sanos programados para someterse a la extirpación quirúrgica de uno o dos terceros molares retenidos, uno de los cuales debe haber sido al menos una retención ósea mandibular parcial, y que tenían dolor postoperatorio moderado a intenso durante 4 horas posteriores a la cirugía. |
Diferencia resumida de la intensidad del dolor para 0 a 24 horas y 16 a 24 horas |
|
[15142] |
Doble ciego, aleatorizado, controlado con placebo |
Naproxeno sódico de LP de 660 mg (todos los días) Naproxeno sódico de LP de 220 mg (QD) Placebo |
120 120 60 |
1 día |
N = 300 H = 118; M = 182 23.8 (18-43) |
Pacientes sanos programados para someterse a la extirpación quirúrgica de hasta dos terceros molares retenidos, uno de los cuales debe haber sido una retención ósea parcial o mandibular completa, y que tenían dolor postoperatorio moderado a intenso durante 4 horas posteriores a la cirugía. |
Diferencia resumida de la intensidad del dolor para 0 a 24 horas |
En el estudio [13130], a un total de 153 pacientes se les administró una sola dosis de comprimido de naproxeno sódico de LP de 660 mg y 159 pacientes se trataron con placebo. Los dos grupos de tratamiento eran comparables con respecto a la demografía y características iniciales. La intensidad del dolor postquirúrgico también fue similar en los dos grupos. Las puntuaciones de intensidad de dolor promedio inicial fueron 2.4 en ambos grupos, y las puntuaciones de la escala análoga visual (VAS) iniciales fueron 72.3 (tratamiento con naproxeno sódico de LP) y 72.6 (tratamiento con placebo).
Los resultados de los análisis de eficacia demostraron otras diferencias significativas entre el grupo de naproxeno sódico de LP y el grupo de placebo en el efecto de la intensidad del dolor. Los pacientes en el grupo de naproxeno sódico de LP tuvieron una reducción mayor consistente y significativa (p < 0.001) en la intensidad del dolor desde el inicio, empezando de 0.5 horas hasta 24 horas posteriores a la dosis en comparación con los pacientes en el grupo de placebo.
En el estudio 15142, el objetivo fue establecer la no inferioridad del comprimido de naproxeno sódico de LP de 660 mg administrado todos los días en comparación con comprimidos de naproxeno sódico 220 mg LI tres veces al día durante 24 horas con respecto a la clinimetría establecida.
Un total de 300 pacientes (118 hombres, 182 mujeres) se asignaron aleatoriamente a una dosis oral única de comprimidos de naproxeno sódico de LP de 660 mg (n = 120), naproxeno sódico de LI de 220 mg cada 8 horas (n = 120) o placebo (n = 60).
El grupo de naproxeno sódico de LP tuvo valores estadística y significativamente mayores en comparación con el grupo de placebo (p < 0.05), lo que indica alivio del dolor sostenido durante las 24 horas.
El naproxeno sódico de LP de 660 mg y el naproxeno sódico de LI de 220 mg tres veces al día fueron estadística y significativamente mejores que el placebo, y el naproxeno sódico de LP de 660 mg todos los días y naproxeno sódico de LI de 220 mg tres veces al día fueron similares.
El estudio demostró que el naproxeno sódico de LP de 660 mg todos los días y el naproxeno sódico 220 mg LI tres veces al día fueron estadística y significativamente mejores que el placebo (p ≤ 0.029) en todos los intervalos de tiempo a partir de 30 minutos posteriores a la dosificación. El naproxeno sódico de LP de 660 mg todos los días y naproxeno sódico de LI de 220 mg tres veces al día fueron similares. Esto demuestra que el inicio del efecto analgésico es similar para el producto de LP en comparación con el producto de LI.
Para el alivio del dolor general categórico, el naproxeno sódico de LP de 660 mg todos los días y el naproxeno sódico de LI de 220 mg tres veces al día fueron estadística y significativamente mejores que el placebo (p < 0.001) en todos los intervalos de tiempo en los que fue medido y después de 30 minutos.
En conclusión, los datos sobre la eficacia de ambos estudios demostraron que el naproxeno sódico de LP es un analgésico eficaz que ofrece alivio del dolor a partir de 30 minutos posteriores a la dosis y se mantiene durante 24 horas. Por otra parte, la eficacia analgésica del naproxeno sódico de LP todos los días no era inferior a la del naproxeno sódico de LI de 220 mg comercial tres veces al día en el dolor dental postquirúrgico inmediatamente después de la dosis y durante todo el periodo de 24 horas.
Antecedentes de estudios publicados con formulación 660mg: No existen datos publicados de la forma farmacéutica de naproxeno sódico de LP de 660 mg. Sin embargo, los perfiles de seguridad y eficacia del naproxeno sódico de LI pueden proporcionar una perspectiva y apoyan la seguridad favorable y la tolerabilidad del naproxeno sódico de LP de 660 mg todos los días, ya que la exposición general del naproxeno sódico con el naproxeno sódico de LP y el naproxeno sódico de LI son similares como se demostró en tres estudios farmacocinéticos (estudios [12656], [13106] y [13965]). Además, la no inferioridad clínica del naproxeno sódico de LP de 660 mg con el naproxeno sódico de LI se ha establecido en un estudio de dolor dental postquirúrgico (estudio [15142]).
Reporte en seguridad: Los eventos adversos más comúnmente reportados fueron los trastornos gastrointestinales y los relacionados con el sistema nervioso con una relación plausible con el fármaco de estudio a partir de los dos ensayos controlados de fase 3. Los eventos adversos se informaron con mayor frecuencia por los pacientes en los grupos de placebo (26.9%, 59/219) y naproxeno sódico de LI de 220 mg tres veces al día (28.3%, 34/120), que en el grupo de naproxeno sódico de LP de 660 mg todos los días (13.6%, 37/273). Este hallazgo de perfiles de eventos adversos comparables entre el placebo y el naproxeno sódico de LP coincide con el naproxeno sódico de LI donde los perfiles de eventos adversos son comparables entre el placebo y el naproxeno sódico de LI.
El potencial de mal uso del producto de naproxeno sódico de LP de 660 mg se examinó durante el estudio del uso real que se llevó a cabo en un entorno realista. Los resultados de este estudio apoyan el uso seguro y adecuado del producto de naproxeno sódico de LP de 660 mg. Los cinco eventos adversos informados con mayor frecuencia en este estudio fueron: dispepsia (2.0%), cefalea (2.0%), náuseas (1.6%), dolor abdominal superior (1.4%) y diarrea (1.4%).
Los resultados de una amplia evaluación de seguridad posterior a la comercialización del naproxeno de la base de datos de la OMS y una revisión de la bibliografía médica apoyan la seguridad del naproxeno sódico de LI como analgésico y antipirético. No hubo resultados que sugieran que más advertencias o precauciones deban aplicarse al naproxeno sódico. Con respecto a la sobredosis aguda, el riesgo para la forma farmacéutica de naproxeno sódico de LP de 660 mg es probable que sea comparable o menor que el naproxeno sódico de LI, ya que las características de liberación atenuarán la concentración plasmática máxima asociada con la ingestión en comparación con la misma dosis de productos de LI.
No se espera que el comprimido de naproxeno sódico de LP de 660 mg muestre un perfil de seguridad diferente del producto de naproxeno sódico de LI de 220 mg aprobado con una dosis diaria total de 660 mg idéntica. Esta expectativa se basa en los hallazgos del programa de desarrollo clínico del naproxeno sódico de LP de 660 mg que no revela ningún hallazgo nuevo de seguridad y demostró perfiles de exposición, seguridad y tolerabilidad comparables entre el naproxeno sódico de LP de 660 mg y el naproxeno sódico de LI.
CONTRAINDICACIONES:
• Hipersensibilidad conocida al naproxeno sódico, o cualquier otro ingrediente del medicamento.
• Historia de asma, urticaria o reacciones de tipo alérgico después de tomar ácido acetilsalicílico u otros medicamentos antiinflamatorios no esteroideos (AINE).
• Historia de hemorragia digestiva o perforación relacionados con la terapia previa con AINE.
• Antecedentes de, o úlcera péptica activa recurrente o hemorragia (dos o más episodios diferentes de ulceración o sangrado demostrado).
• Insuficiencia cardiaca severa.
• Insuficiencia renal severa.
• Insuficiencia hepática severa.
• Embarazo y lactancia.
• Menores de 12 años.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: Como con cualquier otro medicamento de este tipo, el naproxeno sódico produce retrasos en el parto en animales y también afecta el sistema cardiovascular fetal humano (cierre del conducto arterioso).
Por lo tanto, el naproxeno sódico no debería ser administrado salvo una valoración médica previa.
El uso de naproxeno sódico en el embarazo requiere un balance precavido de los posibles beneficios contra los riesgos potenciales a la madre y al feto, especialmente durante el primer y tercer trimestre.
Lactancia: El naproxeno sódico se ha encontrado en la leche materna, por lo tanto, no deberá administrarse naproxeno sódico en mujeres que están amamantando.
REACCIONES SECUNDARIAS Y ADVERSAS:
Trastornos cardiacos/trastornos vasculares: Se ha reportado edema, hipertensión e insuficiencia cardiaca en relación con el tratamiento con AINE.
Datos epidemiológicos y de estudios clínicos indican que el uso de coxibs y de algunos AINE (particularmente en altas dosis y en tratamientos a largo plazo) puede estar asociado con un pequeño aumento del riesgo de eventos trombóticos arteriales (por ejemplo, infarto al miocardio y accidente cerebrovascular).
Trastornos gastrointestinales: Los eventos adversos observados con mayor frecuencia son de naturaleza gastrointestinal. Pueden ocurrir úlceras pépticas, perforación o sangrado GI, algunas veces fatales, particularmente en personas mayores. Se han reportado náuseas, vómito, diarrea, flatulencias, estreñimiento, dispepsia, dolor abdominal, melena, hematemesis, estomatitis ulcerativa, exacerbación de colitis y enfermedad de Crohn después de la administración. Se ha observado gastritis con menor frecuencia.
Trastornos de la piel y del tejido subcutáneo: Reacciones ampollosas, incluyendo el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica (muy raro).
El naproxeno sódico causa tiempos de sangrado modestamente elevados dependientes de la dosis y transitorios. Sin embargo, estos valores no exceden con frecuencia el límite superior del rango de referencia.
Se han observado las siguientes reacciones adversas al naproxeno o el naproxeno sódico, incluidas aquéllas que se presentan con dosis de prescripción.
Tabla de efectos no deseados
|
Sistema/clase de órgano (MedDRA) |
Frecuencia |
Efectos |
|
Trastornos del sistema inmune |
Muy raro < 0.01% y reportes aislados |
Reacciones anafilácticas/anafilactoides incluyendo shock con resultado fatal. |
|
Trastornos de la sangre y del sistema linfático |
Muy raro < 0.01% y reportes aislados |
Trastornos hematopoyéticos (leucopenia, trombocitopenia, agranulocitosis, anemia aplásica, eosinofilia, anemia hemolítica). |
|
Trastornos psiquiátricos |
Muy raro < 0.01% y reportes aislados |
Trastornos psiquiátricos, depresión, anormalidades del sueño, incapacidad para concentrarse. |
|
Trastornos del sistema nervioso |
Común = 1% -< 10% |
Vértigo, cefalea, mareo. |
|
Poco común = 0.1% -< 1% |
Adormecimiento, insomnio, somnolencia. |
|
|
Muy raro < 0.01% y reportes aislados |
Meningitis aséptica, disfunción cognitiva, convulsiones. |
|
|
Trastornos oculares |
Muy raro < 0.01% y reportes aislados |
Trastorno visual, opacidad córnea, papilitis, neuritis óptica retrobulbar, papiledema. |
|
Trastornos del oído y laberinto |
Poco común = 0.1% -< 1% |
Vértigo. |
|
Muy raro < 0.01% y reportes aislados |
Disfunción auditiva, tinnitus, trastornos auditivos. |
|
|
Trastornos cardiacos |
Muy raro < 0.01% y reportes aislados |
Insuficiencia cardiaca congestiva, hipertensión, edema pulmonar, palpitaciones. |
|
Trastornos vasculares |
Muy raro < 0.01% y reportes aislados |
Vasculitis. |
|
Trastornos respiratorios, torácicos y mediastinales |
Muy raro < 0.01% y reportes aislados |
Disnea, asma, neumonitis eosinofílica. |
|
Trastornos gastrointestinales |
Común = 1% -< 10% |
Dispepsia, náuseas, acidez, dolor abdominal. |
|
Poco común = 0.1% -< 1% |
Diarrea, estreñimiento, vómito. |
|
|
Raro = 0.01% -< 0.1% |
Úlceras pépticas con o sin sangrado o perforación, sangrado gastrointestinal, hematemesis, melena. |
|
|
Muy raro < 0.01% y reportes aislados |
Pancreatitis, colitis, úlceras aftosas, estomatitis, esofagitis, ulceraciones intestinales. |
|
|
Trastornos hepatobiliares |
Muy raro < 0.01% y reportes aislados |
Hepatitis (incluyendo casos fatales), ictericia. |
|
Poco común = 0.1% -< 1% |
Exantema (sarpullido), prurito, urticaria. |
|
|
Raro = 0.01% -< 0.1% |
Edema angioneurótico. |
|
|
Trastornos de la piel y tejido subcutáneo |
Muy raro < 0.01% y reportes aislados |
Alopecia (normalmente reversible), fotosensibilidad, porfiria, eritema multiforme exudativo, reacciones ampollosas incluyendo el síndrome de Stevens-Johnson y necrólisis epidérmica tóxica, eritema nodoso, erupción fija por fármacos, liquen plano, reacción pustular, erupciones cutáneas, lupus sistémico eritematoso, reacciones de fotosensibilidad incluyendo porfiria cutánea tardía (“pseudoporfiria”) o epidermólisis ampollosa, equimosis, púrpura, sudoración. |
|
Trastornos renales y urinarios |
Raro = 0.01% -< 0.1% |
Insuficiencia renal. |
|
Muy raro < 0.01% y reportes aislados |
Nefritis intersticial, necrosis papilar renal, síndrome nefrótico, insuficiencia renal, enfermedad renal, hematuria, proteinuria. |
|
|
Congénito |
Muy raro < 0.01% y reportes aislados |
Cierre del conducto arterioso. |
|
Trastornos del sistema reproductivo y pecho |
Muy raro < 0.01% y reportes aislados |
Femenino: Infertilidad. |
|
Trastornos generales |
Raro = 0.01% -< 0.1% |
Edema periférico, particularmente en pacientes con hipertensión o insuficiencia renal, pirexia (incluyendo escalofríos y fiebre). |
|
Muy raro < 0.01% y reportes aislados |
Edema, sed, malestar. |
|
|
Investigaciones |
Muy raro < 0.01% y reportes aislados |
Creatinina sérica elevada, prueba de función hepática anormal, hipercalcemia. |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Hasta el momento no existen reportes que relacionen el uso de naproxeno sódico en humanos con efectos carcinogénicos, mutagénicos o teratogénicos.
Existe cierta evidencia acerca de que los fármacos que inhiben la síntesis de ciclooxigenasa/prostaglandina pueden ocasionar deterioro de la fertilidad femenina mediante un efecto en la ovulación. Esto es reversible al retiro del tratamiento.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Ciclosporina: Las concentraciones de ciclosporina pueden aumentar, elevando el riesgo de nefrotoxicidad.
Litio: Los niveles de litio pueden aumentar, lo que podría inducir náuseas, polidipsia, poliuria, temblores, confusión.
Metotrexato utilizado en dosis de 15 mg/semana o más: La concentración elevada de metotrexato eleva el riesgo de toxicidad a esta sustancia.
Fármacos antiinflamatorios no esteroideos (AINE) incluido el ácido acetilsalicílico: Riesgo elevado de úlceras y sangrado gastrointestinal.
Dosis bajas de ácido acetilsalicílico: El naproxeno sódico puede atenuar la inhibición plaquetaria irreversible inducida por el ácido acetilsalicílico. Datos farmacodinámicos clínicos sugieren que el uso simultáneo (mismo día) con naproxeno sódico por más de un día consecutivo, inhibe el efecto del ácido acetilsalicílico (dosis bajas) en la actividad de las plaquetas y esta inhibición puede persistir durante varios días después de haber suspendido la terapia con naproxeno sódico. La relevancia clínica de esta interacción no se conoce. El tratamiento con naproxeno sódico en pacientes con mayor riesgo cardiovascular puede limitar la protección cardiovascular del ácido acetilsalicílico.
Anticoagulantes: Los AINE pueden mejorar los efectos de los anticoagulantes, como la warfarina. Los anticoagulantes y otros fármacos que influyen en la hemostasia aumentan el riesgo de sangrado y requieren de un monitoreo cuidadoso.
Agentes antiplaquetarios e inhibidores selectivos de la recaptación de serotonina (SSRI, por sus siglas en inglés): Riesgo elevado de sangrado gastrointestinal.
Corticosteroides: Riesgo elevado de sangrado o ulceración gastrointestinal.
Diuréticos y fármacos antihipertensivos incluidos los inhibidores de la ECA: Se puede reducir la eficacia diurética y antihipertensiva, particularmente en pacientes con nefropatía pre-existente.
Durante el uso a corto plazo de naproxeno sódico, las interacciones de importancia clínica no parecen ser relevantes para los siguientes medicamentos:
• Antiácidos.
• Agentes antidiabéticos.
• Hidantoínas.
• Probenecid.
• Zidovudina.
Interacciones fármaco-alimento: La absorción del naproxeno sódico puede retrasarse con los alimentos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
El naproxeno sódico puede interferir con los análisis de orina de esteroides 17-cetógenos y con el ácido 5-hidroxindolacético (5 HIAA).
PRECAUCIONES GENERALES:
El dolor de origen gastrointestinal no es una indicación para usar naproxeno sódico.
No deberá utilizarse naproxeno sódico en combinación con inhibidores específicos de la COX-2 y otros AINE.
Los efectos no deseados pueden minimizarse con el uso de la dosis efectiva más baja durante la duración más corta necesaria para controlar los síntomas.
Perforación, ulceración y sangrado gastrointestinal: Se ha reportado perforación, ulceración y sangrado GI, que pueden ser fatales, con todos los AINE (incluido el naproxeno) en cualquier momento durante el tratamiento, con o sin síntomas de advertencia o un historial previo de eventos GI graves.
El riesgo de perforación, ulceración o sangrado GI es mayor con dosis elevadas de AINE en los pacientes con un antecedente de úlcera, particularmente si se complica con hemorragia o perforación, y en las personas mayores. Estos pacientes deben comenzar el tratamiento con la dosis más baja disponible. Se debe considerar una terapia de combinación con agentes gastroprotectores (por ejemplo, misoprostol o inhibidores de la bomba de protones) para estos pacientes, y también para los pacientes que requieran una dosis baja concomitante de ácido acetilsalicílico, o de otros fármacos que tengan la posibilidad de incrementar los riesgos gastrointestinales.
Los pacientes con un antecedente de toxicidad gastrointestinal, particularmente las personas mayores, deben reportar cualquier síntoma abdominal inusual (especialmente sangrado GI) en particular en las etapas iniciales del tratamiento. Se aconseja precaución en pacientes que reciben medicamentos concomitantes que puedan incrementar el riesgo de ulceración o sangrado, tales como los corticosteroides orales, los anticoagulantes como la warfarina, los inhibidores selectivos de la recaptación de serotonina o agentes antiplaquetarios tales como el ácido acetilsalicílico.
Cuando ocurre ulceración o sangrado GI en pacientes que reciben naproxeno sódico, se debe interrumpir el tratamiento.
Los AINE, como el naproxeno deben administrarse con precaución en pacientes con un antecedente de enfermedad gastrointestinal (colitis ulcerativa, enfermedad de Crohn) ya que su condición puede exacerbarse. Debido al perfil de seguridad que tienen los antiinflamatorios AINE se recomienda que en pacientes con condiciones gastrointestinales consulten a su médico antes de su uso.
Retención de líquidos/sodio en condiciones cardiovasculares y edema periférico: Se requiere precaución antes de iniciar el tratamiento en pacientes con antecedentes de hipertensión y/o insuficiencia cardiaca, se deberá evaluar la relación riesgo-beneficio, ya que se ha reportado retención de líquidos, hipertensión y edema en relación con la terapia con AINE.
Efectos cardiovasculares y cerebrovasculares: Datos epidemiológicos y de estudios clínicos indican que el uso de coxibs y de algunos AINE (particularmente en altas dosis y en tratamientos a largo plazo) puede estar asociado con un pequeño aumento del riesgo de eventos trombóticos arteriales (por ejemplo, infarto al miocardio o accidente cerebrovascular). Aunque los datos indican que el uso de naproxeno (1000 mg al día) puede estar asociado con un menor riesgo, no puede excluirse. No hay datos suficientes sobre los efectos de las dosis bajas (naproxeno sódico 220 mg-660 mg diarios o naproxeno 200 mg-600 mg diarios) para sacar conclusiones firmes sobre los posibles riesgos trombóticos.
El naproxeno sódico puede atenuar el efecto antiplaquetario del ácido acetilsalicílico. Verificar si el paciente está en un régimen de ácido acetilsalicílico y planea tomar naproxeno sódico.
Reacciones en la piel: Se han reportado con muy poca frecuencia en relación con el uso de AINE reacciones cutáneas graves, algunas de ellas fatales, incluyendo la dermatitis exfoliativa, el síndrome de Stevens-Johnson, y la necrólisis epidérmica tóxica.
Los pacientes parecen tener un mayor riesgo de estas reacciones en las etapas tempranas de la terapia. Se debe descontinuar el uso de naproxeno sódico al momento de la primera aparición de erupciones en la piel, lesiones de las mucosas, o cualquier otro signo de hipersensibilidad.
Reacciones anafilácticas (anafilactoides): Pueden ocurrir reacciones de hipersensibilidad, incluyendo reacciones anafilácticas (anafilactoides), en pacientes con y sin antecedentes de hipersensibilidad con exposición al ácido acetilsalicílico a otros fármacos antiinflamatorios no esteroideos o a productos que contienen naproxeno sódico. Estas reacciones también pueden ocurrir en individuos con antecedentes de angioedema, reactividad broncoespasmódica (por ejemplo, asma), rinitis, pólipos nasales, enfermedad alérgica, enfermedad respiratoria crónica o sensibilidad al ácido acetilsalicílico. Esto también aplica para pacientes que exhiben reacciones alérgicas (por ejemplo, reacciones cutáneas, comezón o urticaria) al naproxeno sódico o a otros AINE. Las reacciones anafilactoides, como la anafilaxia, pueden tener un resultado fatal.
Efectos hepáticos: Se han reportado reacciones hepáticas graves, incluidas la ictericia y la hepatitis (algunos casos de hepatitis han sido fatales), con el naproxeno sódico al igual que con otros fármacos antiinflamatorios no esteroideos. Se ha reportado reacción cruzada.
Precauciones para pacientes mayores: Las personas mayores tienen una frecuencia elevada de reacciones adversas a los AINE, especialmente perforación y sangrado gastrointestinal, mismos que pueden ser fatales. Mujeres embarazadas o que planean embarazarse:
Precauciones relacionadas a la fertilidad: Hay cierta evidencia que los medicamentos que inhiben la síntesis de ciclooxigenasa/prostaglandinas pueden causar alteraciones en la fertilidad femenina por un efecto en la ovulación. Esto es reversible con el retiro del tratamiento.
Pacientes con historia médica: Los sujetos con los siguientes historiales médicos adicionales deben estar bajo una supervisión adecuada y cuidadosa cuando toman naproxeno sódico:
• Quienes están tomando cualquier otro analgésico.
• Quienes están tomando esteroides.
• Con trastornos de coagulación o quienes toman fármacos que influyan en la hemostasia.
• En terapia diurética intensiva.
• Con insuficiencia renal, insuficiencia hepática o cardiaca.
• Con diabetes mellitus tipo 2 sin tratamiento y control médico de larga evolución y que presenten un daño renal preestablecido.
• Con hipertensión arterial sistémica sin tratamiento y control médico de larga evolución y que presenten un daño renal preestablecido.
No se ha realizado ningún estudio sobre el efecto en la capacidad de manejar y utilizar maquinaria. Sin embargo, se han observado efectos no deseados tales como somnolencia, mareo, vértigo e insomnio con el uso de naproxeno sódico. Se aconseja a los pacientes que observen cómo reaccionan antes de manejar o de operar maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN:
La vía de administración de FLANAX® 660 es oral. Cada dosis debe tomarse con abundante líquido y se toma preferentemente antes de los alimentos. Si se toma con alimentos, el efecto puede tardar más.
La dosis máxima diaria (24 horas) de equivalente a naproxeno base no debe exceder los 800 mg (equivalente a 880 mg de naproxeno sódico).
Dosis:
Adultos y adolescentes de 12 años en adelante: Tomar una tableta cada 24 horas mientras duran los síntomas.
El naproxeno sódico no debe tomarse por más de 10 días, salvo valoración médica. Si el dolor o la fiebre persisten o si los síntomas cambian, se debe valorar nuevamente al paciente.
Niños: Niños menores de 12 años no deben tomar este producto salvo valoración médica.
Pacientes geriátricos: Los adultos mayores son más propensos a eventos adversos, por lo que deberá evaluarse el riesgo beneficio.
Pacientes con insuficiencia cardiaca, hepática y/o renal severas: Deberá evaluarse el riesgo beneficio, mediante la evaluación médica.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Una sobredosificación significativa puede caracterizarse por vértigo, somnolencia, mareo, dolor epigástrico, malestar abdominal, acidez estomacal, indigestión, náuseas y vómito, alteraciones transitorias en la función hepática, hipoprotrombinemia, disfunción renal, acidosis metabólica, apnea, o desorientación. Debido a que el naproxeno sódico puede absorberse rápidamente, deben anticiparse niveles sanguíneos altos y tempranos. Pocos pacientes han experimentado convulsiones, pero no está claro si éstas estuvieron relacionadas con el naproxeno sódico o no. Se han descrito algunos casos con disfunción renal aguda reversible. No se sabe qué dosis del fármaco sería potencialmente mortal.
Si un paciente ingiere una gran cantidad de naproxeno sódico, puede hacerse un lavado gástrico y emplearse medidas de apoyo habituales como la administración de carbón activado. La hemodiálisis no disminuye la concentración plasmática del naproxeno sódico debido al alto grado de su unión proteica. No existe ningún antídoto específico.
PRESENTACIONES:
Caja de cartón con 2, 4, 8 o 12 tabletas de 660 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese la caja bien cerrada a no más de 30 °C.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se administre durante el embarazo y la lactancia. No se administre a niños menores de 12 años. No tomar por más de 10 días.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
BAYER DE MÉXICO, S.A. de C.V.
Carretera. México-Toluca Km. 52.5, C.P. 52000,
Lerma, México, México.
Reg. Núm. 036M2022 SSA IV


