HALAVEN - Solución
Sustancia(s):
- Mesilato De Eribulina
Presentaciones:
- 1 Caja , 1 Frasco(s) ámpula , 2 ml , 0.5 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada frasco ámpula contiene:
|
Mesilato de eribulina |
1 mg |
|
Vehículo cbp 2 mL. |
|
INDICACIONES TERAPÉUTICAS: Tercera línea en el tratamiento para cáncer de mama localmente avanzado o metastásico, posterior al tratamiento con al menos dos regímenes de quimioterapia que incluyan una antraciclina y un taxano.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética:
Distribución: La farmacocinética de eribulina es lineal, con una vida media de eliminación de aproximadamente 40 horas, un volumen de distribución medio de 43 L/m2 a 114 L/m2 y una depuración media de 1.16 L/hr/m2 a 2.42 L/hr/m2 en un rango de dosis de entre 0.25 mg/m2 y 4.0 mg/m2. La unión de la eribulina a las proteínas plasmáticas humanas en concentraciones de 100 ng/mL a 1,000 ng/mL varía de 49% a 65%. La exposición a eribulina después de la administración de dosis múltiples es comparable a la administración de una dosis única. Con la administración semanal no se observa acumulación de eribulina.
Biotransformación: Eribulina sin cambios fue la especie de mayor circulación en el plasma después de la administración de 14C-eribulina a los pacientes. Las concentraciones de metabolito representaron < 0.6% del compuesto madre, confirmando que no hubo metabolitos humanos mayores de eribulina.
Eliminación: Eribulina se elimina sin cambios principalmente en las heces. Después de la administración de 14C-eribulina a los pacientes, aproximadamente 82% de la dosis se eliminó en las heces y 9% en orina.
Eribulina sin cambios representó la mayor parte de la radiactividad total en heces y orina.
Insuficiencia hepática: La farmacocinética de eribulina fue evaluada en un estudio Fase 1 en pacientes con insuficiencia hepática leve (Child-Pugh A) y moderada (Child-Pugh B). En comparación con los pacientes que tienen función hepática normal, la exposición a eribulina aumentó 1.75 veces y 2.79 veces en pacientes con insuficiencia hepática leve y moderada, respectivamente.
Farmacodinamia:
Grupo farmacoterapéutico: Agentes antineoplásicos, Código del Sistema de Clasificación Anatómica Terapéutica y Química (ATC): L01XX41.
Eribulina es un inhibidor no taxano de la dinámica microtubular perteneciente a la clase de agentes antineoplásicos de la halicondrina. Es un análogo sintético estructuralmente simplificado de la halicondrina B, un producto natural aislado de la esponja marina Halichondria okadai.
Eribulina inhibe la fase de crecimiento de los microtúbulos sin afectar la fase de acortamiento y fija la tubulina a los agregados no productivos. Eribulina ejerce sus efectos a través de un mecanismo antimitótico basado en la tubulina que provoca un bloqueo del ciclo celular G2/M, la alteración de las células mitóticas y finalmente, la muerte de las células apópticas después de un bloqueo mitótico prolongado.
Experiencia clínica: La eficacia de HALAVEN® en cáncer de mama está soportada por dos estudios Fase 2, de grupo único realizado en 403 pacientes y el estudio comparativo aleatorizado Fase 3 realizado en 762 pacientes. Los pacientes del estudio pivotal Fase 3 EMBRACE tenían cáncer de mama localmente recurrente o metastásico y habían recibido previamente al menos dos y máximo cinco ciclos de quimioterapia, incluyendo una antraciclina y un taxano (a menos que estuviera contraindicado).
Los pacientes debían haber progresado dentro de los 6 meses posteriores a su último ciclo de quimioterapia. Los pacientes fueron aleatorizados 2:1 para recibir HALAVEN® (1.4 mg/m2 los días 1 y 8 en un ciclo de 21 días administrados por vía intravenosa en 2 a 5 minutos) o el tratamiento elegido por el médico (TPC, por sus siglas en inglés), definido como quimioterapia de un solo fármaco, tratamiento hormonal o terapia biológica aprobada para el tratamiento de cáncer, o tratamiento paliativo o radioterapia, que refleje la práctica local. El grupo del TPC consistió en 97% de quimioterapia (26% vinorelbina, 18% gemcitabina, 18% capecitabina, 16% taxano, 9% antraciclina, 10% de otros tipos de quimioterapia) y 3% de terapia hormonal.
En el grupo de eribulina, el 17.7% de los pacientes recibió G-CSF como profilaxis secundaria.
En el estudio 305, se observó una mejora estadísticamente significativa en la supervivencia total en los pacientes aleatorizados en el grupo de HALAVEN®, en comparación con el grupo control (Tabla 1). Se realizó una actualización de análisis de supervivencia no planeado cuando se había observado el 77% de los eventos (Figura 1) fue consistente con el análisis primario. En pacientes aleatorizados con HALAVEN® (la población con intención de ser tratada [ITT]), la tasa de respuesta objetiva según los criterios de RECIST fue de 11% (95% IC: 8.6%, 14.3%) y la duración de la respuesta media fue de 4.2 meses (95% IC: 3.8, 5.0 meses).
Tabla 1. Comparación de la supervivencia total en el grupo de mesilato de eribulina y de control
|
Supervivencia total |
Mesilato de eribulina (n = 508) |
Grupo Control (n = 254) |
|
Análisis primario de supervivencia |
||
|
Número de muertes |
274 |
148 |
|
Mediana, meses (95% CI) |
13.1 (11.8, 14.3) |
10.6 (9.3, 12.5) |
|
Proporción de riesgo (95% CI)a |
0.81 (0.66, 0.99) |
|
|
Valor P b |
0.041 |
|
|
Análisis de supervivencia actualizado |
||
|
Número de muertes |
386 |
203 |
|
Mediana, meses (95% CI) |
13.2 (12.1, 14.4) |
10.6 (9.2, 12.0) |
CI = Intervalo de confianza; HER2 = receptor 2 de factor de crecimiento epidémico humano.
a Basado en el modelo de daños proporcionales Cox estratificado por región geográfica, estado HER2 y terapia anterior capecitabina.
b Basado en una prueba de rango logarítmico estratificado por región geográfica, estado HER2 y terapia anterior capecitabina.
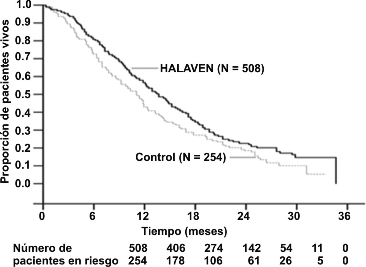
Figura 1. Análisis de supervivencia total actualizada.
CONTRAINDICACIONES: Hipersensibilidad al fármaco o a los componentes de la fórmula.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: Las mujeres en edad fértil deben ser advertidas para evitar quedar embarazadas mientras ellas o sus parejas, estén recibiendo HALAVEN® y deben utilizar un método anticonceptivo efectivo durante y hasta por 3 meses, después del tratamiento.
Embarazo: No existe información sobre el uso de HALAVEN® en mujeres embarazadas. El mesilato de eribulina es embriotóxico, fetotóxico y teratogénico en ratas. HALAVEN® no deberá utilizarse durante el embarazo, a menos que sea claramente necesario y después de considerar cuidadosamente las necesidades de la madre y el riesgo para el feto.
Lactancia: No existe información sobre la excreción de mesilato de eribulina o sus metabolitos en la leche materna humana o de animales. Sin embargo, no se puede excluir el riesgo para los recién nacidos o infantes por lo que no debe utilizarse HALAVEN® durante la lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS: La tabla siguiente muestra las tasas de incidencia de eventos adversos de tratamiento emergente contra reacciones adversas de tratamiento emergente observadas en 827 pacientes con cáncer de mama que recibieron la dosis recomendada en Estudios Fase 2 y 3 de cáncer de mama. La información entre paréntesis demuestra la tasa de incidencia de eventos adversos en 503 pacientes tratados con HALAVEN® en el estudio pivotal de cáncer de mama sólo para el estudio 305. Todos los eventos adversos ocurrieron a una velocidad mayor o igual a 5% se incluyen en la Tabla. Los eventos adicionales que ocurrieron a una velocidad menor a 5% están incluidos, basados en una evaluación de la farmacología conocida de eribulina y toma en consideración la ocurrencia de línea base del evento en pacientes con cáncer de mama en ausencia de tratamiento con medicamentos.
Tabla 2. Eventos adversos de Tratamiento Emergente (%) y Reacciones Adversas (%) reportadas para HALAVEN® en los estudios 201, 211 & 305
|
Clase Órgano Sistema MedDRA Términos Preferidos |
Todos los grados n = 827 (n = 503) |
Grados 3 y 4 n = 827 (n = 503) |
||
|
Eventos Adversos |
Reacciones Adversas |
Eventos Adversos |
Reacciones Adversas |
|
|
Trastornos Sanguíneos y del Sistema Linfáticoa |
||||
|
Neutropenia |
55.3% (51.7%) |
54.5% |
48.9% (45.2%) |
48.3% |
|
Leucopenia |
22.5% (23.3%) |
22.1% |
14.1% (13.9%) |
14.0% |
|
Anemia |
22.6% (61.2%) |
20.3% |
2% (2%) |
1.4% |
|
Neutropenia febril |
4.7% (4.6%) |
4.7%d |
4.7%d (4.4%)d |
4.6%d |
|
Linfopenia |
2.3% (2.4%) |
2.3% |
0.8% (1.2%) |
0.8% |
|
Trombocitopenia |
2.9% (2.6%) |
2.8% |
0.9% (0.8%) |
0.9% |
|
Trastornos del Sistema Nervioso |
||||
|
Neuropatía Periféricab |
34.9% (34.6%) |
32% |
7.6% (8.2%) |
6.9% |
|
Dolor de Cabeza |
20.4% (19.3%) |
11% |
0.7% (0.4%) |
0.1% |
|
Disgeusia |
9.9% (7.8%) |
9.6% |
0 |
0 |
|
Mareo |
8.8% (7.6%) |
4.5%c |
0.4% (0.6%) |
0.1%c |
|
Trastornos Psiquiátricos |
||||
|
Ansiedad |
6.7% (5.4%) |
0.7% |
0.6 (0.6%) |
0 |
|
Depresión |
5.3% (5.0%) |
1.2% |
0.7 (0.6%) |
0 |
|
Insomnio |
8.8% (7.6%) |
2.5% |
0.1% (0%) |
0 |
|
Trastornos del Ojo |
||||
|
Aumento de lagrimeo |
8.2% (7.2%) |
7.3% |
0 |
0 |
|
Trastornos Generales y Condiciones en el Lugar de Administración |
||||
|
Astenia/Fatiga |
59.9% (53.5%) |
52.8% |
10.4% (8.8%) |
8.4% |
|
Inflamación de la Mucosa |
10.5% (8.3%) |
9.8%c |
1.6% (1.2%) |
1.3%c |
|
Pirexia |
23.9% (20.9%) |
16.6% |
0.6% (0.2%) |
0.2%c |
|
Edema Periférico |
10.9% (9.1%) |
5.0%c |
0.5% (0.4%) |
0 |
|
Dolor |
7.4% (4.8%) |
2.7% |
1.5% (0.4%) |
0 |
|
Trastornos Gastrointestinales |
||||
|
Constipación |
27.7% (24.7%) |
16.3%c |
1% (0.6%) |
0.4%c |
|
Diarrea |
20.0% (18.3%) |
15.0%c |
0.7%c (0%) |
0.5%c |
|
Náusea |
39.8% (34.6%) |
35.1%c |
1.9%c (1.2%) |
1.1%c |
|
Vómito |
20.6% (18.1%) |
14.5%c |
1.3% (1%) |
0.5%c |
|
Estomatitis |
9.1% (7.8%) |
8.3%c |
0.7% (0.4%) |
0.7%c |
|
Boca seca |
7.0% (5.6%) |
5.7% |
0 |
0 |
|
Dispepsia |
8.7% (8.3%) |
5.3% |
0.2% |
0.2% |
|
Dolor Abdominal |
9.8% (7.8%) |
9.4%c,h |
1.6% (0.6%) |
0.6%c |
|
Eventos Hepatobiliares |
||||
|
Incremento de Aspartato Aminotransferasa |
4.0% (4.2%) |
2.3%c |
1.2% (1.2%)c |
0.6%c |
|
Incremento de Alanina Aminotransferasa |
4.4% (5.2%) |
3.0%c |
1.3% (1.6%)c |
1.1%c |
|
Incremento Gama Glutamil transferasa |
0.2% (0.2%) |
0.1% |
0.2% (0.2%) |
0.1% |
|
Hiperbilirubinemia |
1.3% (1.8%) |
0.4% |
0.2% (0.4%) |
0 |
|
Trastornos Músculo-esqueléticos y del Tejido Conectivo |
||||
|
Artralgia/Mialgia |
23.2% (21.9%) |
12.7%c |
1.2% (0.4%) |
0.4%c |
|
Dolor de Espalda |
15.2% (15.5%) |
1.9% |
1.8% (0.8%) |
0 |
|
Dolor de Huesos |
10.6% (11.9%) |
1.7%c |
1.8% (1.8%) |
0.1%c |
|
Dolor de Extremidades |
11.7% (11.3%) |
4.7%c |
1.1% (1%) |
0 |
|
Espasmo Muscular |
6.5% (7.2%) |
4.1%c |
0.2% (0) |
0.2%c |
|
Debilidad Muscular |
5.4% (5.4%) |
2.5% |
0.8 (0.6%) |
0.4% |
|
Investigaciones |
||||
|
Disminución de Peso |
16.6% (21.3%) |
9.7c |
0.5% (0.6%) |
0.1%c |
|
Trastorno del Metabolismo y Nutrición |
||||
|
Disminución del Apetito |
24.9% (22.5%) |
18.5%c |
0.7% (0.4%) |
0.4%c |
|
Hipocalemia |
8.0% (7.2%) |
3.5% |
2.5% (2.6%) |
0.7% |
|
Hipomagnesemia |
4.4% (4.4%) |
2.8% |
0.2% (0.4%) |
0.1% |
|
Deshidratación |
3.3% (2.4%) |
1.8%c |
0.6% (0.6%) |
0.2%c |
|
Trastornos Respiratorios, Torácicos y Mediastínicos |
||||
|
Tos |
16.4% (14.3%) |
4.8%i |
0.5% (0) |
0 |
|
Disnea |
17.4% (15.7%) |
5.7%c, d, j |
4.1%d (3.6%) |
0.5%c, d |
|
Trastorno de la Piel y del Tejido Subcutáneo |
||||
|
Alopecia |
50.4% (44.5%) |
49.7% |
N/A |
N/A |
|
Erupción |
5.9% (6.2%) |
4.1% |
0.1% (0%) |
0.1% |
|
Prurito |
4.2% (4.4%) |
2.8%c, k |
0.1% (0.2%) |
0.1%c |
|
Infecciones e Infestaciones |
42.7% (42.1%) |
15.1% |
5.8% (4.4%) |
2.7% |
|
Sepsis |
0.4% (0.2%) |
0.1% |
0.4%d (0.2%)d |
0.1% |
|
Neumonía |
1.3% (1.2%) |
0.5% |
0.9% (0.8%) |
0.2% |
|
Infección de Tracto Respiratorio Superior |
5.6 (5.2%) |
1.3% |
0.1% (0.2%) |
0.1% |
|
Infección de Tracto Urinario |
10.8% (9.7%) |
5.0%c, l |
0.6% (0.8%) |
0.2%c |
Nota: La tasa de incidencia (%) de eventos adversos reportados para mesilato de eribulina en el estudio de cáncer de mama pivote 305 es n = 503.
a. Basado en datos de laboratorio.
b. Incluye neuropatía periférica, neuropatía, neuropatía motora periférica, polineuropatía, neuropatía sensorial periférica, neuropatía sensitivo-motora periférica, polineuropatía desmielinizante y parestesia.
c. No hubo eventos Grado 4.
d. Hubo eventos Grado 5: disnea 0.2%, neutropenia febril 0.1%, sepsis 0.1%.
e. No aplica (Sistema de clasificación no especificado > Grado 2 para alopecia).
f Incluye todo tipo de infecciones.
g Reacciones adversas como base del investigador en la evaluación del evento como posible o probable relacionadas con el fármaco estudiado.
h incluye términos combinados "dolor abdominal" + "dolor abdominal superior".
i Incluye términos combinados "tos" + "tos productiva" +.
j Incluye términos combinados "disnea" + "de esfuerzo disnea".
k Incluye términos combinados "prurito" + "prurito generalizado".
l Incluye términos combinados "infección de tracto urinario" + "cistitis" + "Infección de tracto urinario por E. Coli".
La Tabla 3 muestra las tasas de incidencia de eventos adversos de tratamiento emergente contra las reacciones adversas de tratamiento emergente observados en 1503 pacientes con cáncer de mama que recibieron la dosis recomendada de cada cinco Fase 2 y dos Estudios Fase 3 con cáncer de mama. Todos los eventos adversos que ocurren a una velocidad mayor o igual al 5% se incluyen en la Tabla. Los eventos adicionales que se producen a una velocidad menor al 5% están incluidos basados en una evaluación de la farmacología conocida de eribulina y toma en consideración la ocurrencia de línea base del evento en pacientes con cáncer de mama en ausencia de tratamiento con medicamentos.
Tabla 3. Eventos adversos de Tratamiento Emergente (%) y Reacciones Adversas (%) reportadas para mesilato de eribulina en los estudios 201, 209, 211, 221, 224, 301 & 305
|
Clase Órgano Sistema MedDRA |
Todos los grados n = 1503 |
Grados 3 y 4 n = 1503 |
||
|
Términos Preferidos |
Eventos Adversos |
Reacciones Adversas |
Eventos Adversos |
Reacciones Adversas |
|
Trastornos Sanguíneos y del Sistema Linfáticoa |
||||
|
Neutropenia |
57.0% |
56.4% |
49.7% |
49.0% |
|
Leucopenia |
29.3% |
28.9% |
17.3% |
17.1% |
|
Anemia |
20.6% |
18.3% |
2.0% |
1.5% |
|
Neutropenia Febril |
4.7% |
4.6%d |
4.5%d |
4.5%d |
|
Linfopenia |
4.9% |
4.9% |
1.4% |
1.4% |
|
Trombocitopenia |
4.3% |
4.0% |
0.7% |
0.7% |
|
Trastornos del Sistema Nervioso |
||||
|
Neuropatía Periféricab |
35.6% |
31.1% |
7.6% |
6.9% |
|
Dolor de Cabeza |
17.2% |
8.8% |
0.8% |
0.2% |
|
Disgeusia |
8.8% |
8.5% |
0 |
0 |
|
Mareo |
7.9% |
3.4% |
0.5%c |
0.1%c |
|
Trastornos del Ojo |
||||
|
Aumento de lagrimeo |
6.0% |
5.3% |
0.1%c |
0.1%c |
|
Trastornos Generales y Condiciones en el Lugar de Administración |
||||
|
Astenia/Fatiga |
47.9% |
41.2% |
7.8% |
6.2% |
|
Inflamación de la Mucosa |
8.3% |
7.6% |
1.1% |
1.0% |
|
Pirexia |
20.4% |
13.6% |
0.6% |
0.3%c |
|
Edema Periférico |
8.6% |
3.4% |
0.3%c |
0 |
|
Dolor |
5.2% |
1.6% |
0.9% |
0 |
|
Trastornos Gastrointestinales |
||||
|
Constipación |
19.6% |
11.2% |
0.6%c |
0.3% |
|
Diarrea |
17.9% |
13.1% |
0.8%c |
0.6%c |
|
Náusea |
33.8% |
29.5% |
1.1%c |
0.6%c |
|
Vómito |
17.6% |
12.2% |
0.9% |
0.3%c |
|
Estomatitis |
9.3% |
8.8% |
0.8%c |
0.8%c |
|
Boca seca |
5.3% |
4.3% |
0 |
0 |
|
Dispepsia |
5.9% |
3.7% |
0.2%c |
0.1%c |
|
Dolor Abdominal |
8.0% |
3.9% |
1.1% |
0.4% |
|
Eventos Hepatobiliares |
||||
|
Incremento de Aspartato aminotransferasa |
7.4% |
5.3% |
1.5%c |
1.0%c |
|
Incremento de Alanina aminotransferasa |
7.6% |
5.7% |
2.1%c |
1.7%c |
|
Incremento Gama glutamil transferasa |
1.8% |
1.2% |
0.9%c |
0.5%c |
|
Hiperbilirubinemia |
1.5% |
0.7% |
0.3%c |
0.1%c |
|
Trastornos Músculo-esqueléticos y del Tejido Conectivo |
||||
|
Artralgia/Mialgia |
19.4% |
9.6% |
1.1% |
0.3%c |
|
Dolor de Espalda |
13.0% |
2.0% |
1.5% |
0.1%c |
|
Dolor de Huesos |
9.6% |
1.6% |
1.7% |
0.1%c |
|
Dolor de Extremidades |
10.0% |
3.7% |
0.7%c |
0 |
|
Espasmo Muscular |
5.1% |
3.0% |
0.1%c |
0.1%c |
|
Investigaciones |
||||
|
Disminución de Peso |
11.3% |
6.6% |
0.3%c |
0.1%c |
|
Trastorno del Metabolismo y Nutrición |
||||
|
Disminución del Apetito |
21.9% |
17.0% |
0.7%c |
0.4%c |
|
Hipocalemia |
6.1% |
2.9% |
1.7% |
0.6% |
|
Hipomagnesemia |
2.9% |
1.7% |
0.2% |
0.1% |
|
Deshidratación |
2.8% |
1.6% |
0.5%c |
0.3%c |
|
Trastornos Respiratorios, Torácicos y Mediastínicos |
||||
|
Tos |
13.6% |
3.5% |
0.6%c |
0.1%c |
|
Disnea |
13.9% |
3.7% |
3.1%d |
0.7% |
|
Trastorno de la Piel y del Tejido Subcutáneo |
||||
|
Alopecia |
44.6% |
43.6% |
N/Ae |
N/Ae |
|
Erupción |
5.1% |
3.3% |
0% |
0% |
|
Prurito |
3.9% |
2.0% |
0.1%c |
0.1%c |
|
Infecciones e Infestaciones |
37.7% |
11.7% |
4.8% |
2.1% |
|
Sepsis/Sepsis neutropénica |
0.6% |
0.4% |
0.6%d |
0.4%d |
|
Neumonía |
1.2% |
0.3% |
0.8%c |
0.2%c |
|
Infección de Tracto Urinario |
8.0% |
2.3% |
0.5% |
0.1%c |
Nota: La tasa de incidencia (%) de eventos adversos reportados para mesilato de eribulina en el estudio de cáncer de mama pivote 305 es n = 503.
a Basado en datos de laboratorio.
b Incluye todos los términos en el más amplio Sistema Estándar MedDRA de consulta.
c No hubo eventos Grado 4.
d Hubo eventos Grado 5: disnea 0.6%, neutropenia febril 0.1 %, sepsis 0.2 %, infestaciones e infecciones 0.1%.
e No aplica (Sistema de clasificación no especificado > Grado 2 para alopecia).
f Incluye todo tipo de infecciones.
Reacciones Adversas Post-comercialización: Debido a que estas reacciones son reportadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera fiable la frecuencia o establecer una relación causal con la exposición al medicamento.
Trastornos del Sistema Inmunológico: Hipersensibilidad al medicamento.
Trastornos Hepatobiliares: Hepatitis.
Trastornos Gastrointestinales: Pancreatitis.
Respiratorios, Torácicos y Mediastínicos: Enfermedad pulmonar intersticial.
Poblaciones especiales:
Población de edad avanzada: El perfil de seguridad de HALAVEN® en pacientes de edad avanzada (> 65 años de edad) fue similar al de los pacientes ≤ 65 años de edad. No se recomienda hacer ajustes a la dosis con base en la edad de pacientes de edad avanzada.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Fertilidad: Se ha observado toxicidad testicular en ratas y perros.
No se realizó ningún estudio de fertilidad con eribulina, pero en base a los hallazgos preclínicos en estudios de dosis repetidas que muestran toxicidad testicular en ratas y perros la fertilidad masculina puede verse comprometida por el tratamiento con eribulina.
Eribulina no fue mutagénica in vitro en un estudio de mutación reversa bacteriana (prueba Ames). Eribulina fue positiva en la prueba de mutagénesis de linfoma en ratones y fue clastogénica en el estudio de micronúcleos de rata in vivo.
No se han realizado estudios de carcinogenicidad con eribulina.
Un estudio de desarrollo embriofetal en ratas confirmó la toxicidad desarrollada y el potencial teratogénico de mesilato de eribulina. Ratas embarazadas fueron tratadas con 0.01, 0.03, 0.1 y 0.15 mg/kg en los días de gestación 8, 10 y 12. Se observó incremento en el número de resorciones y reducción de peso fetal, relacionados con la dosis, a ≥ 0.1 mg/kg e incremento en la incidencia de malformaciones (ausencia de mandíbula, lengua, estómago y bazo) registradas con dosis de 0.15 mg/kg.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se espera que haya interacciones medicamentosas con los inhibidores de CYP3A4, inductores de CYP3A4 o inhibidores de P-glicoproteína (P-gp). No hay efecto en la exposición a eribulina (área bajo la curva [AUC]) y la concentración máxima (Cmax) cuando eribulina fue administrada con o sin ketoconazol, un potente inhibidor de CYP3A4 o cuando es administrado con rifampicina un potente inductor de CYP34.
Eribulina no inhibe a las enzimas CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 o CYP3A4 ni induce a las enzimas CYP1A2, CYP2C9, CYP2C19 o CYP3A4 a concentraciones clínicamente relevantes.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Ninguna.
PRECAUCIONES GENERALES:
Hematología: La mielosupresión depende de la dosis y se manifiesta principalmente como neutropenia. Se presentó neutropenia febril en pacientes tratados con mesilato de eribulina.
Se debe realizar un monitoreo del conteo sanguíneo completo antes de cada dosis en todos los pacientes que reciban HALAVEN®.
Los pacientes con neutropenia febril, neutropenia severa o trombocitopenia deben ser tratados de acuerdo con las recomendaciones.
Los pacientes con alanina aminotransferasa (ALT) o aspartato aminotransferasa (AST) > 3 x ULN (límite superior al normal) tuvieron una mayor incidencia de neutropenia Grado 4 y neutropenia febril. Aunque los datos son limitados, los pacientes con bilirrubina > 1.5 x ULN también tienen una mayor incidencia de neutropenia Grado 4 y neutropenia febril.
Neuropatía periférica: Monitorear de cerca a los pacientes para detectar signos de neuropatía motora periférica y sensorial. La neuropatía periférica debe tratarse retrasando y ajustando la dosis de acuerdo con las recomendaciones.
Efectos sobre la capacidad para conducir y utilizar máquinas: Mesilato de eribulina puede causar efectos secundarios, como cansancio y mareos que pueden provocar una influencia leve a moderada sobre la capacidad para conducir o utilizar máquinas. Los pacientes deben ser advertidos de no conducir ni operar máquinas si se sienten cansados o mareados.
Prolongación del intervalo QT: En un estudio ECG abierto no controlado en 26 pacientes, se observó prolongación QT en el día 8, independiente de la concentración de eribulina, con prolongación QT no observada en el día 1. Monitoreo de ECG es recomendado si la terapia es iniciada en pacientes con insuficiencia cardiaca congestiva, bradiarritmias, medicamentos conocidos para prolongar el intervalo QT, incluyendo Clase Ia y III antiarrítmicos, y alteraciones electrolíticas. Hipopotasemia o hipomagnesemia correcta antes de iniciar HALAVEN® y monitorear estos electrólitos periódicamente durante la terapia. Evitar HALAVEN® en pacientes con síndrome de QT largo congénito.
DOSIS Y VÍA DE ADMINISTRACIÓN: HALAVEN® debe ser administrado únicamente bajo supervisión de un médico calificado con experiencia en el uso de medicamentos citotóxicos.
La dosis recomendada de HALAVEN® en solución lista para utilizarse es de 1.4 mg/m2, debe ser administrada por vía intravenosa durante 2 a 5 minutos los días 1 y 8 de cada ciclo de 21 días.
Retrasos en la dosis durante la terapia:
No administrar HALAVEN® el día 1 o el día 8 por cualquiera de las siguientes razones:
• Cuenta absoluta de neutrófilos (ANC) < 1 x 109/L.
• Plaquetas < 75 x 109/L.
• Toxicidades no hematológicas Grado 3 o 4.
Ajustes en la dosis durante la terapia: Los pacientes deben ser evaluados clínicamente durante el tratamiento mediante un examen físico y pruebas de laboratorio, incluyendo conteo de sangre completo. Si se observa la presencia de toxicidades grado 3/4, el tratamiento tendrá que retrasarse para permitir la recuperación. Los pacientes únicamente deberán ser tratados si el conteo absoluto de neutrófilos (ANC, por sus siglas en inglés) sea ≥ 1 x 109/L y las plaquetas son ≥ 75 x 109/L y cuando se haya recuperado completamente de la toxicidad grado 2 o menor de un ciclo previo.
Las recomendaciones para la reducción de la dosis para el retratamiento se presentan en la siguiente tabla. Si las toxicidades vuelven a aparecer, se debe hacer una reducción adicional de la dosis.
Tabla 4. Recomendaciones para la reducción de la dosis.
|
Reacción adversa |
Dosis recomendada |
|
Hematológica |
|
|
Neutropenia grado 4 que dura más de 7 días |
1.1 mg/m2 |
|
Neutropenia grado 3 o 4 complicada por fiebre o infección |
|
|
Trombocitopenia grado 4 |
|
|
Trombocitopenia grado 3 complicada por hemorragia o que requiere transfusión de sangre o plaquetas |
|
|
No hematológicas |
|
|
Cualquier grado 3 o 4 en el ciclo previo |
|
|
Recurrencia de reacción adversa |
|
|
Cualquier grado 3 o 4 independiente de la reducción a 1.1 mg/m2 |
0.7 mg/m2 |
|
Cualquier grado 3 o 4 independiente de la reducción a 0.7 mg/m2 |
Considerar descontinuación |
Pacientes con insuficiencia hepática: La dosis recomendada de HALAVEN® en pacientes con insuficiencia hepática leve (Child-Pugh A) es de 1.1 mg/m2 administrada por vía intravenosa durante 2 a 5 minutos los días 1 y 8 de un ciclo de 21 días.
La dosis recomendada de HALAVEN® en pacientes con insuficiencia hepática moderada (Child Pugh B) es de 0.7 mg/m2 administrada por vía intravenosa durante 2 a 5 minutos los días 1 y 8 de un ciclo de 21 días.
Pacientes con insuficiencia renal: En pacientes con insuficiencia renal moderada o severa se debe considerar la reducción de la dosis inicial.
Pacientes pediátricos: La seguridad y la efectividad de HALAVEN® en pacientes pediátricos menores a 18 años de edad no han sido establecidas.
Pacientes de edad avanzada: En pacientes de más de 65 años de edad no se recomiendan los ajustes específicos en la dosis.
Vía de administración: Intravenosa.
Método de administración: La dosis puede ser diluida en hasta 100 ml de solución para inyección de cloruro de sodio 9 mg/mL (0.9%).
Precauciones especiales para la eliminación y otros manejos: Mesilato de eribulina es un medicamento anticancerígeno citotóxico y, al igual otros compuestos tóxicos, deben tomarse precauciones durante su manipulación. Se recomienda el uso de guantes, lentes de seguridad y ropa de protección. Si la piel se pone en contacto con la solución, deberá lavarse inmediata y perfectamente con agua y jabón. Si se pone en contacto con las membranas mucosas, se deben enjuagar perfectamente con agua. Mesilato de eribulina debe ser preparado y administrado únicamente por personal capacitado en el manejo de agentes citotóxicos. Las mujeres que estén embarazadas no deben manipular HALAVEN®.
Para ver las instrucciones sobre la dilución del producto antes de su administración.
Utilizando una técnica aséptica, HALAVEN® puede ser diluido hasta 100 mL con una solución para inyección de cloruro de sodio 9 mg/mL (0.9%).
Cualquier producto sin usar o material de desecho debe ser dispuesto de acuerdo los requerimientos locales.
Incompatibilidades: Mesilato de eribulina solución no debe ser diluido en solución para infusión de glucosa al 5%.
HALAVEN® no debe ser mezclado con otros medicamentos.
Vida útil en uso: Desde el punto de vista microbiológico, HALAVEN® debe ser utilizado inmediatamente. El producto no está diseñado para ser almacenado después de abrirse o después de diluirse, a menos que se realice bajo condiciones asépticas controladas y validadas. Si no se utiliza inmediatamente, los tiempos y las condiciones de almacenamiento una vez abierto son responsabilidad del usuario.
Dependiendo de los controles microbiológicos apropiados, si no se usa inmediatamente la solución no diluida en una jeringa, HALAVEN® no debe almacenarse más de 4 horas a 25°C con luz ambiental o 24 horas a 2-8°C.
Dependiendo de los controles microbiológicos apropiados, las soluciones diluidas de HALAVEN® (0.02 mg/mL a 0.2 mg/mL en cloruro de sodio 9 mg/mL [0.9%]) solución inyectable no se debe almacenar más de 24 horas entre 2 y 8°C.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Se han reportado casos de sobredosis de mesilato de eribulina con aproximadamente 4 veces la dosis recomendada que provocó neutropenia grado 3 que duró siete días y una reacción de hipersensibilidad grado 3 que duró un día.
No existe ningún antídoto conocido para la sobredosis de HALAVEN®.
PRESENTACIÓN: Caja con 1 frasco ámpula de 2 mL (0.5 mg/mL).
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 25°C. No congelar o refrigerar.
LEYENDAS DE PROTECCIÓN:
No se use durante el embarazo o la lactancia. No se deje al alcance de los niños. Su venta requiere receta médica. Literatura exclusiva para el médico.
Eisai Inc.
6611 Tributary Street
Baltimore Maryland (MD) 21224, EUA
Reg. Núm. 237M2014, SSA IV

