IMBRUVICA - Cápsulas
Sustancia(s):
- Ibrutinib
Presentaciones:
- 1 Caja, 1 Frasco(s), 90 Cápsulas, 140 Miligramos
- 1 Caja, 1 Frasco(s), 120 Cápsulas, 140 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada CÁPSULA contiene:
Ibrutinib 140 mg
Excipiente cbp
INDICACIONES TERAPÉUTICAS:
Linfoma de células del manto (LCM): IMBRUVICA® está indicado para el tratamiento de pacientes adultos con LCM que han recibido por lo menos un tratamiento previo, el tratamiento deberá continuar hasta pérdida de respuesta o intolerancia al medicamento.
Leucemia linfocítica crónica/linfoma de linfocitos pequeños (LLC/LLP): IMBRUVICA® está indicado para el tratamiento de pacientes con LLC/LLP, y para el tratamiento de paciente con Leucemia Linfocítica Crónica (LLC) con deleción 17p.
Macroglobulinemia de Waldenström (MW): IMBRUVICA® está indicado para el tratamiento de pacientes con Macroglobulinemia de Waldenström (MW).
Linfoma de la zona marginal (LZM): IMBRUVICA® está indicado para el tratamiento de pacientes con Linfoma de la zona marginal (LZM) que requieren tratamiento sistémico y que han recibido al menos un tratamiento previo basado en anti-CD20.
Enfermedad de injerto contra huésped crónica (EICHc): IMBRUVICA® está indicado para el tratamiento de pacientes adultos con enfermedad de injerto contra huésped crónica (EICHc) posterior a la falla de una o más líneas de terapia sistémica.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacocinéticas:
Absorción: Ibrutinib es absorbido rápidamente después de la administración oral con una mediana de Tmax de 1 a 2 horas. La biodisponibilidad absoluta en condiciones de ayuno (n=8) fue de 2.9% (IC 90%=2.1-3.9) y se duplicó cuando se combina con una comida. La farmacocinética de ibrutinib no difiere de manera significativa en pacientes con diferentes neoplasias de células B. La exposición a ibrutinib aumenta con dosis de hasta 840 mg. El ABC (área bajo la curva) en estado estacionario en pacientes con 560 mg es (media ± desviación estándar) 953 ± 705 ng*h/mL y en pacientes con 420 mg con LLC/LLP es 732 ± 521 ng*h/mL (680 ± 517 ng*h/mL en el subgrupo de pacientes R/R) y con EICHc es 1159 ± 583 ng*h/mL. La administración de ibrutinib en condiciones de ayuno resultó en aproximadamente el 60% de la exposición (ABCfinal) en comparación ya sea con 30 minutos antes, 30 minutos después (condición con alimentos) o 2 horas después de un desayuno alto en grasas.
Distribución: La unión reversible de ibrutinib a las proteínas plasmáticas humanas, in vitro, fue de 97.3% sin dependencia de la concentración en el intervalo de 50 a 1000 ng/mL. El volumen de distribución (Vd) es 683 L y el volumen de distribución aparente en estado estacionario (Vd,ss/F) es aproximadamente de 10000 L.
Metabolismo: Ibrutinib es metabolizado primariamente por el citocromo P450, CYP3A4/5 para producir un metabolito prominente dihidrodiol con una actividad inhibitoria hacia BTK aproximadamente 15 veces menor que aquella de ibrutinib. La exposición sistémica en estado estacionario al metabolito dihidrodiol es comparable a la del fármaco padre.
Estudios in vitro indicaron que la participación de CYP2D6 en el metabolismo oxidativo de ibrutinib es <2%. Adicionalmente, como parte del estudio balance masa humana, los sujetos genotipificados como metabolizadores lentos de CYP2D6, mostraron un perfil farmacocinético similar a los metabolizadores rápidos. Por lo tanto, no son necesarias precauciones en pacientes con diferentes genotipos de CYP2D6.
Resultados de estudios en microsomas hepáticos y hepatocitos no indicaron ninguna participación de CYP1A, CYP2B6, CYP2C8, CYP2C9 o CYP2C19 en el metabolismo de ibrutinib.
Eliminación: La depuración intravenosa fue de 62 y 76 L/h en condiciones de ayuno y alimentación, respectivamente. En línea con el alto efecto de primer paso, la depuración oral aparente es de aproximadamente 2000 y 1000 L/h en condiciones de ayuno y alimentación, respectivamente. La vida media de ibrutinib es de 4 a 6 horas.
Después de una administración oral única de [14C]-Ibrutinib radiomarcado en sujetos sanos, aproximadamente 90% de la radioactividad se excretó dentro de 168 horas, con la mayoría (80%) excretada en las heces y menos del 10% se presentó por la orina. Ibrutinib inalterado representó aproximadamente el 1% de la excreción del producto radiomarcado en heces y nada en la orina, con el resto de la dosis siendo metabolitos.
Poblaciones especiales:
Adultos mayores (65 años de edad y mayores): La farmacocinética de la población indicó que en pacientes de edad avanzada (67 a 81 años), se predice una exposición 14% mayor de ibrutinib. No se justifica el ajuste de la dosis según la edad.
Pacientes pediátricos (18 años de edad y menores): No se realizaron estudios de farmacocinética con IMBRUVICA® en pacientes menores de 18 años de edad.
Género: Los datos de farmacocinética de la población indicaron que el género no influye significativamente la depuración de ibrutinib de la circulación.
Insuficiencia renal: Ibrutinib tiene depuración renal mínima; la excreción urinaria de los metabolitos es <10% de la dosis. No se han realizado estudios clínicos específicos en sujetos con insuficiencia renal. No es necesario ajustar la dosis en pacientes con insuficiencia renal leve o moderada (depuración de creatinina mayor a 30 mL/min). No existen datos en pacientes con insuficiencia renal severa o en pacientes en diálisis.
Insuficiencia hepática: Ibrutinib se metaboliza en el hígado. Se llevó a cabo un estudio de insuficiencia hepática en sujetos no oncológicos que recibieron una dosis única de 140 mg de IMBRUVICA® en condiciones de ayuno, el ABCfinal de ibrutinib aumentó 2.7-, 8.2- y 9.8- veces, en sujetos con insuficiencia hepática leve (n=6; Child Pugh clase A), moderada (n=10; Child Pugh clase B) y severa (n=8; Child Pugh clase C), respectivamente. La fracción libre de ibrutinib también aumentó con el grado de insuficiencia, con 3.0, 3,8 y 4,8% en sujetos con insuficiencia hepática leve, moderada y severa, respectivamente, en comparación con 3.3% en el plasma de los controles sanos en este estudio. El aumento correspondiente en la exposición ibrutinib no unida del área bajo la curva (ABCno unida,final), se estima que sea 4.1, 9.8 y 13 veces mayor en sujetos con insuficiencia hepática leve, moderada y severa, respectivamente.
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Agentes antineoplásicos, inhibidores de la proteína quinasa, código ATC: L01XE27.
Mecanismo de acción: Ibrutinib es un potente inhibidor de molécula pequeña de la tirosina quinasa de Bruton (BTK). Ibrutinib forma un enlace covalente con un residuo de cisteína (Cys-481) en el sitio activo de la BTK, llevando a la inhibición sostenida de la actividad enzimática de la BTK. La BTK, un miembro de la familia de las quinasas Tec, es una importante molécula de señalización de las vías del receptor del antígeno de las células B (BCR) y del receptor de citosinas. La vía de los BCR está involucrada en la patogénesis de diversas neoplasias de células B, incluyendo LCM, Linfoma Difuso de Células B Grandes (LDCBG), Linfoma Folicular y LLC de células B. El papel fundamental de la BTK en la señalización a través de los receptores de superficie de las células B, resulta en la activación de las vías necesarias para el tráfico, la quimiotaxis y la adhesión de las células B. Los estudios preclínicos han mostrado que ibrutinib inhibe la proliferación y la sobrevida de neoplasias de células B, in vivo, así como la migración celular y la adhesión al sustrato in vitro.
Linfocitosis: Al inicio del tratamiento, se ha observado un aumento reversible en el recuento de linfocitos (es decir, aumento ≥50% del valor basal y un recuento absoluto ≥5000/mcL), a menudo asociado con la reducción de la linfadenopatía, en la mayoría de los pacientes (66%) con LLC/LLP tratados con IMBRUVICA® como un agente simple. Este efecto también se ha observado en algunos pacientes (35%) con LCM tratados con IMBRUVICA®. Esta linfocitosis observada es un efecto farmacodinámico y no debe ser considerada una enfermedad progresiva en ausencia de otros hallazgos clínicos. En ambos tipos de enfermedades, la linfocitosis típicamente ocurre durante el primer mes de la terapia con IMBRUVICA® y por lo general se resuelve dentro de una media de 8 semanas en pacientes con LCM y 14 semanas en los pacientes con LLC/LLP (rango, 0.1 a 104 semanas).
Un gran aumento en el número de linfocitos circulantes (por ejemplo, > 400000/mcL) se ha observado en algunos pacientes.
No se observó linfocitosis en pacientes con MW tratados con IMBRUVICA®.
Cuando se administró IMBRUVICA® con quimioinmunoterapia, la linfocitosis fue infrecuente (7% con IMBRUVICA® + BR frente a 6% con placebo + BR).
Agregación plaquetaria in vitro: En un estudio in vitro, ibrutinib demostró inhibición de la agregación plaquetaria inducida por colágeno en muestras de los sujetos de cohortes ya sea con insuficiencia renal, aquellos con warfarina o sujetos sanos. La magnitud de la inhibición de la agregación plaquetaria inducida por colágeno en la cohorte de sujetos con aspirina fue menos pronunciada ya que la agregación plaquetaria inducida por colágeno ya se había reducido sin ibrutinib. Ibrutinib no mostró una inhibición significativa de la agregación plaquetaria para los 4 agonistas adenosina difosfato (ADP), ácido araquidónico, ristocetina y péptido activador del receptor de trombina 6 (TRAP-6, por sus siglas en inglés) a través de cualquiera de estas cohortes de sujetos o sujetos sanos.
Efecto sobre el intervalo QT/QTc y la electrofisiología cardiaca: El efecto de ibrutinib sobre el intervalo QTc se evaluó en 20 sujetos masculinos y femeninos sanos en un estudio, aleatorizado, doble ciego de QT completo con placebo y controles positivos. A una dosis supra-terapéutica de 1680 mg, ibrutinib no prolongó el intervalo QTc en ningún grado clínicamente relevante. El límite superior más grande del IC del 90% de dos lados para las diferencias de medias ajustadas en la basal entre ibrutinib y placebo fueron inferiores a 10 ms. En este mismo estudio, se observó un acortamiento dependiente de la concentración en el intervalo QTc (-5.3 ms [IC del 90%: -9.4, -1.1] a una Cmax de 719 ng/mL después de la dosis supra-terapéutica de 1680 mg) que no fue considerado clínicamente significativo.
Estudios clínicos:
Linfoma de células del manto (LCM): La seguridad y la eficacia de IMBRUVICA® en pacientes con LCM quienes recibieron por lo menos un tratamiento previo fueron evaluadas en un estudio de Fase 2 abierto, multicéntrico (PCYC-1104-CA) de 111 pacientes. La mediana de la edad fue 68 años (rango, 40 a 84 años), el 77% eran hombres y el 92% eran caucásicos. La mediana de tiempo desde el diagnóstico fue de 42 meses y la mediana del número de tratamientos previos fue 3 (rango, 1 a 5 tratamientos), incluyendo 35% con dosis altas de quimioterapia previa, 43% con bortezomib previo, 24% con lenalidomida previa y 11% con trasplante previo de células madre. En la basal, 39% de los pacientes tenía enfermedad voluminosa (≥5 cm), 49% tenían una puntuación de riesgo alto por el Índice de Pronóstico Internacional de LCM Simplificado (MIPI) y 72% tenían enfermedad avanzada (implicación extranodal y/o de médula ósea) en el tamizaje.
IMBRUVICA® fue administrado por la vía oral en dosis de 560 mg una vez al día, hasta la progresión de la enfermedad o hasta la toxicidad inaceptable. La respuesta tumoral fue evaluada de acuerdo con los criterios revisados del Grupo de Trabajo Internacional (IWG) para linfoma no-Hodgkin (LNH). El criterio de valoración primario en este estudio fue la tasa global de respuesta (ORR) evaluada por investigador. Las respuestas a IMBRUVICA® se muestran en la Tabla 1.
Tabla 1: Tasa de respuesta global (ORR) y duración de la respuesta (DOR), basadas en la evaluación del investigador, en pacientes con linfoma de células del manto
|
Total N=111 |
|
|
ORR (%) |
67.6 |
|
IC 95% (%) |
(58.0, 76.1) |
|
CR (%) |
20.7 |
|
PR (%) |
46.8 |
|
Mediana de DOR (CR+PR) (meses) |
17.5 (15.8, NR) |
|
Mediana de Tiempo a la Respuesta Inicial, meses (rango) |
1.9 (1.4-13.7) |
|
Mediana de Tiempo a CR, meses (intervalo) |
5.5 (1.7, 11.5) |
IC = intervalo de confianza; CR = respuesta completa; PR = respuesta parcial; NR = no alcanzado.
Los datos de eficacia fueron evaluados posteriormente por un Comité de Revisión Independiente (IRC), demostraron una ORR de 69% con una tasa de CR de 21% y una tasa de PR de 48%. La mediana de DOR estimada por el IRC fue de 19.6 meses.
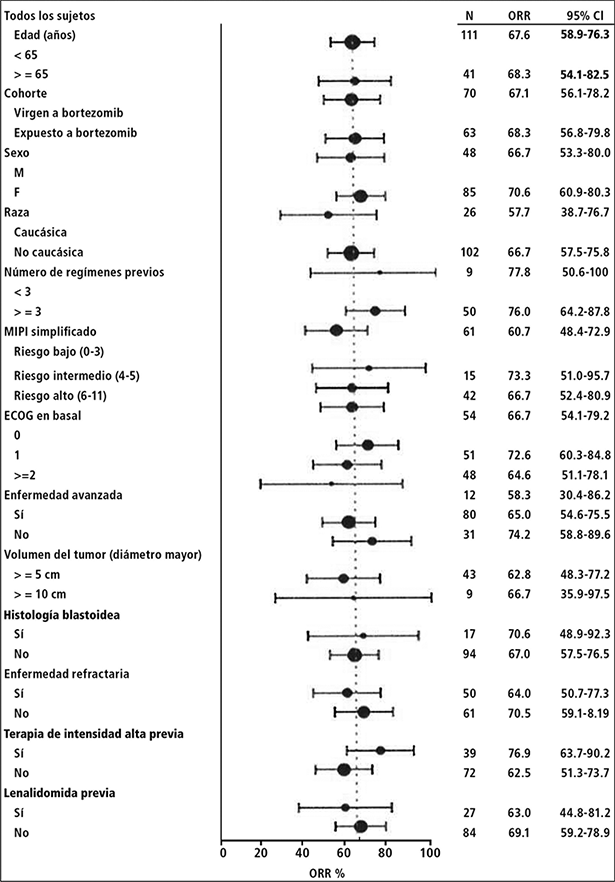
La respuesta global a IMBRUVICA® fue independiente del tratamiento previo que incluyó bortezomib y lenalidomida, del riesgo/pronóstico subyacente, enfermedad voluminosa, del género o de la edad (Figura 1).
Figura 1: Análisis de subgrupos de la tasa de respuesta general por evaluación del investigador (Estudio PCYC-1104-CA; 560 mg)
La seguridad y eficacia de IMBRUVICA® se demostraron en un estudio aleatorizado Fase 3, abierto, multicéntrico, que incluyó 280 pacientes con LCM que recibieron al menos una terapia previa (estudio MCL3001). Los pacientes fueron aleatorizados 1:1 para recibir ya sea IMBRUVICA® por vía oral a 560 mg una vez al día en un ciclo de 21 días o temsirolimus por vía intravenosa a 175 mg en los días 1, 8, 15 del primer ciclo, seguido por 75 mg en los días 1, 8, 15 de cada ciclo subsecuente de 21 días. El tratamiento en ambos brazos continuó hasta la progresión de la enfermedad o toxicidad inaceptable. La mediana de edad fue de 68 años (rango, 34-88), 74% eran hombres y 87% eran de raza blanca. La mediana del tiempo desde el diagnóstico fue de 43 meses, y la mediana del número de tratamientos previos fue de 2 (rango: 1 a 9 tratamientos), incluyendo 51% con dosis altas de quimioterapia previa, 18% con bortezomib previo, 5% con lenalidomida previa, y 24% con trasplante de células madre previo. En la basal, el 53% de los pacientes tenía enfermedad voluminosa (≥ 5 cm), 21% tenía puntuación de alto riesgo por MIPI simplificado, 60% tenía enfermedad extraganglionar y 54% tenía implicación de la médula ósea en el tamizaje.
La supervivencia libre de progresión (SLP), evaluada por el IRC de acuerdo con los criterios revisados del IWG para el linfoma no Hodgkin (LNH) mostró una reducción estadísticamente significativa del 57% en el riesgo de muerte o progresión para los pacientes en el brazo de IMBRUVICA®. Los resultados de eficacia para el estudio MCL3001 se muestran en la Tabla 2 y la curva de Kaplan Meier para la SLP en la Figura 2.
Tabla 2: Resultados de Eficacia en el estudio MCL3001
|
Criterio de valoración |
IMBRUVICA® N=139 |
Temsirolimus N=141 |
|
Supervivencia libre de progresióna |
||
|
Número de eventos (%) |
73 (52.5) |
111 (78.7) |
|
Mediana en meses de la supervivencia libre de progresión (IC 95%) |
14.6 (10.4, NE) |
6.2 (4.2, 7.9) |
|
HR (IC 95%) |
0.43 (0.32, 0.58) |
|
|
Tasa de respuesta global (CR+PR) |
71.9% |
40.4% |
|
Valor p |
p<0.0001 |
|
NE = no estimable; HR = índice de riesgo; IC = intervalo de confianza.
a IRC evaluado.
Una proporción menor de pacientes tratados con IMBRUVICA® experimentó un empeoramiento clínicamente significativo de los síntomas de linfoma en comparación con temsirolimus (27% contra 52%) y el tiempo hasta el empeoramiento de los síntomas ocurrió más lentamente con IMBRUVICA® frente a temsirolimus (HR 0.27, p <0.0001).
Figura 2: Curva de Kaplan-Meier de supervivencia libre de progresión (población con intención de tratar) en el Estudio MCL 3001

Leucemia linfocítica crónica/Linfoma de linfocitos pequeños (LLC/LLP): La seguridad y la eficacia de IMBRUVICA® en pacientes con LLC/LLP se demostró en un estudio no controlado y dos estudios controlados, aleatorizados.
Agente único:
Pacientes con LLC/LLP sin tratamiento previo: Se realizó un estudio aleatorizado, multicéntrico, abierto, fase 3 (PCYC-1115-CA) de IMBRUVICA® contra clorambucilo en pacientes con LLC/LLP sin tratamiento previo que tenían 65 años de edad o más. Los pacientes (n = 269) fueron aleatorizados 1:1 para recibir ya sea IMBRUVICA® 420 mg al día hasta la progresión de la enfermedad o toxicidad inaceptable, o clorambucilo a una dosis inicial de 0.5 mg/kg en los Días 1 y 15 de cada ciclo de 28 días por un máximo de 12 ciclos, con una tolerancia de incrementos de dosis intra pacientes de hasta 0.8 mg/kg con base a la tolerabilidad. Después de progresión confirmada de la enfermedad, los pacientes con clorambucilo pudieron cambiarse a ibrutinib.
La mediana de edad fue de 73 años (rango, 65 a 90 años), 63% eran hombres y 91% eran caucásicos. El noventa y uno por ciento de los pacientes tenía una escala de actividad ECOG basal de 0 o 1 y 9% tenía una escala de actividad ECOG de 2. El estudio incluyó 269 pacientes con LLC o LLP. En la basal, 45% tenía una etapa clínica avanzada (Etapa de Rai III o IV), 35% de los pacientes tenía al menos un tumor ≥ 5 cm, 39% tenía anemia basal, 23% tenía trombocitopenia basal, 65% tenía niveles elevados de microglobulina β2 >3500 mcg/L, 47% tenían CrCL de <60 mL/min y 20% de los pacientes presentaron del11q.
La supervivencia libre de progresión (SLP), evaluada por el IRC de acuerdo con los criterios del Taller Internacional sobre Leucemia Linfocítica Crónica (IWCLL) indicó una reducción estadísticamente significativa del 84% en el riesgo de muerte o de progresión en el brazo de IMBRUVICA®. Con una mediana de seguimiento de 18 meses, la mediana de SLP no se alcanzó en el brazo de ibrutinib y fue de 19 meses en el brazo de clorambucilo. Se observó una mejoría significativa en la ORR en el brazo de ibrutinib (82%) frente al brazo de clorambucilo (35%). El análisis de la supervivencia global (OS) también demostró una reducción de 84% estadísticamente significativa en el riesgo de muerte para los pacientes en el brazo de IMBRUVICA®. Los resultados de eficacia del estudio PCYC-1115-CA se muestran en la Tabla 3 y las curvas de Kaplan-Meier para la SLP y la OS se muestran en las Figuras 3 y 4, respectivamente.
Hubo una mejoría sostenida estadísticamente significativa de plaquetas o hemoglobina en la población ITT (intención de tratar) a favor de ibrutinib contra clorambucilo. En pacientes con citopenias basales, la mejoría hematológica sostenida fue: plaquetas 77% contra 43%; hemoglobina 84% contra 45% para ibrutinib y clorambucilo respectivamente.
Tabla 3: Resultados de eficacia del estudio PCYC-1115-CA
|
Criterio de valoración |
IMBRUVICA® N=136 |
Clorambucilo N=133 |
|
Supervivencia Libre de Progresióna |
||
|
Número de eventos (%) |
15 (11.0) |
64 (48.1) |
|
Mediana (IC 95%), meses |
No alcanzado |
18.9 (14.1, 22.0) |
|
HR (IC 95%) |
0.161 (0.091, 0.283) |
|
|
Tasa de Respuesta Globala (CR +PR) |
82.4% |
35.3% |
|
Valor de p |
<0.0001 |
|
|
Supervivencia globalb |
||
|
Número de muertes (%) |
3 (2.2) |
17 (12.8) |
|
HR (IC 95%) |
0.163 (0.048, 0.558) |
|
IC = intervalo de confianza, HR = índice de riesgo; CR = respuesta completa; PR = respuesta parcial.
a IRC evaluado.
b Mediana OS no alcanzada en ambos grupos.
p <0.005 para la OS.
Figura 3: Curva Kaplan-Meier de supervivencia libre de progresión (Población con intención de tratar) en el estudio PCYC-1115-CA
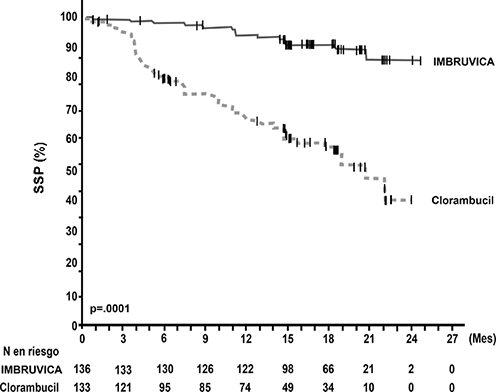
Figura 4: Curva Kaplan-Meier de supervivencia global (Población con intención de tratar) en el estudio PCYC-1115-CA

Pacientes con LLC/LLP quienes recibieron por lo menos un tratamiento previo: Se realizó un estudio abierto, multicéntrico (PCYC-1102-CA) en 51 pacientes que tenían LLC/LLP quienes recibieron 420 mg diario. IMBRUVICA® se administró hasta la progresión de la enfermedad o toxicidad inaceptable. La mediana de edad fue de 68 (rango, de 37 a 82 años), la mediana del tiempo desde el diagnóstico fue de 80 meses y mediana del número de tratamientos previos fue de 4 (rango, 1 a 12 tratamientos), incluyendo 92%, con un análogo de nucleósido previo, 98% con rituximab previo, 86%, con un alquilante previo, 39% con bendamustina previa y 20% con ofatumumab previo. En la basal, el 39.2% de los pacientes tenían Etapa de Rai IV, 45% tenían enfermedad voluminosa (≥ 5 cm), 35% tenían del17p, el 31% tenían del11q.
La ORR fue evaluada por el investigador de acuerdo con los criterios del Taller Internacional en LLC de 2008 (IWCLL). A la mediana de duración del seguimiento de 16 meses, las respuestas para IMBRUVICA® de los 51 pacientes se muestran en la Tabla 4.
Tabla 4: Tasa de respuesta global en pacientes con leucemia linfocítica crónica tratados con 420 mg de IMBRUVICA® (Estudio PCYC-1102-CA) (N=51)
|
ORR (CR+PR) (IC 95%) (%) |
78.4 (64.7, 88.7) |
|
CR (%) |
3.9 |
|
PR (%) |
74.5 |
|
ORR incluyendo la respuesta parcial con Linfocitosis (PRL) (%) |
92.2 |
|
Mediana de DOR (CR+PR) |
NR1 |
|
Mediana del Tiempo a la Respuesta Inicial, meses (rango) |
1.8 (1.4, 12.2) |
IC = intervalo de confianza; CR = respuesta completa; PR = respuesta parcial.
1 92.5 de respondedores fueron censurados (es decir, libres de progresión y vivos) con una mediana de seguimiento de hasta 16.4 meses.
NR: No alcanzado.
Los datos de eficacia fueron evaluados adicionalmente usando los criterios IWCLL por un IRC, demostrando un ORR de 65% (IC 95%: 50%, 78%), de todas las respuestas parciales. El DOR varió de 4 a 24+ meses. No se alcanzó la mediana de DOR.
Se realizó un estudio aleatorizado, multicéntrico, abierto, Fase 3 de IMBRUVICA® contra ofatumumab (PCYC-1112-CA) en pacientes con LLC/LLP. Los pacientes (n=391) fueron aleatorizados 1:1 para recibir ya sea IMBRUVICA® 420 mg diarios hasta progresión de la enfermedad o toxicidad inaceptable, u ofatumumab por hasta 12 dosis (300/2000 mg). Cincuenta y siete pacientes aleatorizados a ofatumumab cruzaron después de la progresión para recibir IMBRUVICA®. La mediana de edad fue de 67 años (rango, 30 a 88 años), 68% eran hombres y 90% eran de raza blanca. Todos los pacientes tenían un estado de desempeño ECOG basal de 0 o 1. La mediana del tiempo desde el diagnóstico fue de 91 meses y la mediana del número de tratamientos previos fue de 2 (rango, 1 a 13 tratamientos). En la basal, 58% de los pacientes tenían al menos un tumor ≥ 5 cm. Treinta y dos por ciento de los pacientes tenían del17p y 31% tenían del11q.
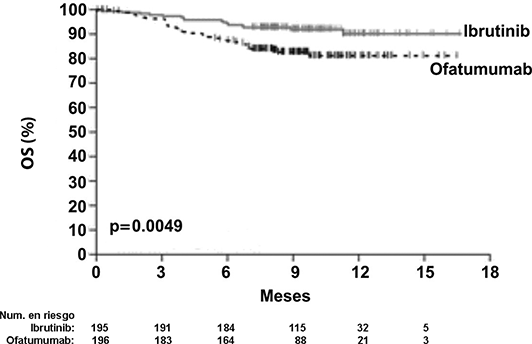
La supervivencia libre de progresión (SLP), evaluada por un IRC de acuerdo con los criterios IWCLL indicó un 78% de reducción estadísticamente significativa en el riesgo de muerte o progresión para los pacientes en el brazo de IMBRUVICA®. El análisis de la supervivencia global (OS) demostró una reducción del 57% estadísticamente significativa en el riesgo de muerte para los pacientes en el brazo de IMBRUVICA®. Los resultados de eficacia del estudio PCYC-1112-CA se muestran en la Tabla 5.
Tabla 1: Resultados de eficacia en pacientes con leucemia linfocítica crónica/linfoma de linfocitos pequeños (Estudio PCYC-1112-CA)
|
Criterio de valoración |
IMBRUVICA N=195 |
Ofatumumab N=196 |
|
Supervivencia libre de progresión |
||
|
Mediana de supervivencia libre de progresión, meses |
No alcanzada |
8.1 |
|
HR (IC 95%) |
0.215 (0.146; 0.317) |
|
|
Supervivencia generala |
||
|
HR (IC 95%) |
0.434 (0.238; 0.789)b |
|
|
HR (IC 95%) |
0.387 (0.216; 0.695)c |
|
|
Tasa de respuesta globald,e (%) |
42.6 |
4.1 |
|
Tasa de respuesta global incluyendo respuesta parcial con linfocitosis (PRL)d (%) |
62.6 |
4.1 |
HR = índice de riesgo; IC = intervalo de confianza; PR = respuesta parcial.
a Mediana de OS no alcanzó para ambos brazos.
b Los pacientes aleatorizados a ofatumumab que progresaron fueron censurados al iniciar ibrutinib, si aplica.
c Análisis de sensibilidad en el que los pacientes cruzados del brazo ofatumumab no fueron censurados en la fecha de la primera dosis de IMBRUVICA®.
d Por IRC Repetir tomografías computarizadas si se requieren para confirmar la respuesta.
e Todos los PRs lograron. p <0,0001 para la ORR.
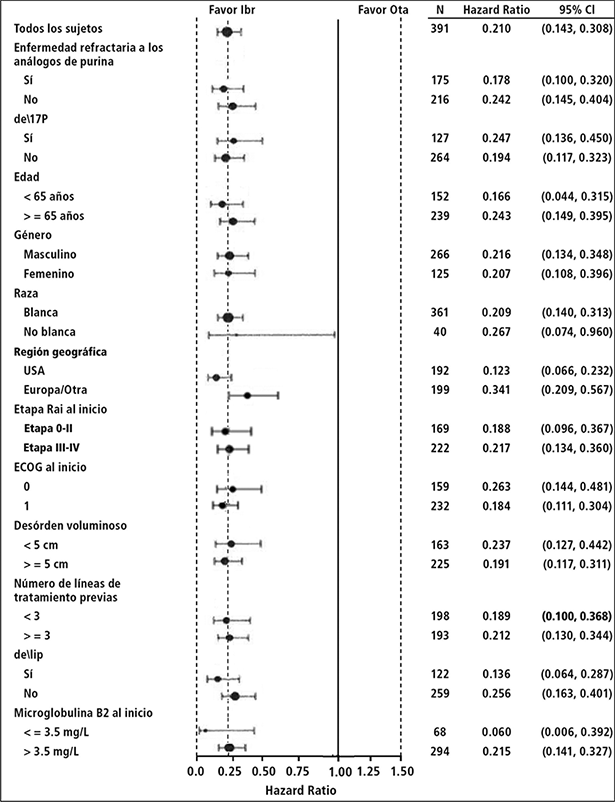
La eficacia fue similar entre todos los subgrupos examinados, incluso en pacientes con y sin deleción 17p, un factor de estratificación pre-especificado (Figura 5).
Figura 5: Análisis de subgrupos de supervivencia libre de progresión por IRC (Estudio PCYC-1112; 420 mg)
Las curvas Kaplan-Meier para SLP y OS se muestran en las Figuras 6 y 7, respectivamente.
Figura 6: Curva Kaplan-Meier de supervivencia libre de progresión (Población con intención de tratar) en el estudio PCYC-1112-CA
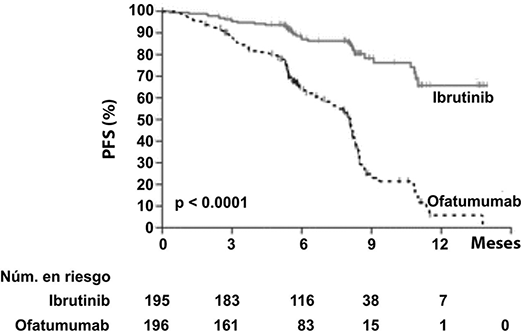
Figura 7: Curva Kaplan-Meier de supervivencia global (Población con intención de tratar) en el estudio PCYC-1112-CA
LLC/LLP con deleción 17p: El estudio PCYC-1112-CA incluyó a 127 pacientes con LLC/LLP con deleción 17p. La mediana de la edad fue de 67 años (rango, 30 a 84 años), 62% eran hombres y 88% eran caucásicos. Todos los pacientes tenían un estado de desempeño ECOG basal de 0 o 1. La SLP y ORR se evaluaron mediante IRC. En la Tabla 6 se muestran los resultados de la eficacia para la LLC/LLP con deleción 17p.
Tabla 6: Resultados de eficacia en pacientes con LLC con deleción del 17p
|
Criterio de valoración |
IMBRUVICA N=63 |
Ofatumumab N=64 |
|
Supervivencia libre de progresión |
||
|
Mediana de supervivencia libre de progresión, meses |
No alcanzada |
5.8 |
|
HR (IC 95%) |
0.25 (0.14; 0.45) |
|
|
Tasa de respuesta global |
47.6% |
4.7% |
|
Tasa de respuesta global incluyendo PRL |
66.7% |
4.7% |
a IRC evaluada. Se alcanzaron todas las respuestas parciales; ninguno de los pacientes alcanzó una respuesta completa.
HR = índice de riesgo; IC = intervalo de confianza; PRL = respuesta parcial con linfocitosis.
Terapia de combinación: La seguridad y eficacia de IMBRUVICA® en pacientes tratados previamente para LLC/LLP fueron evaluados además en un estudio aleatorizado, multicéntrico, doble ciego, fase 3 de IMBRUVICA® en combinación con BR (bendamustina y rituximab) contra placebo + BR (Estudio CLL3001). Los pacientes (n=578) fueron aleatorizados 1:1 para recibir ya sea IMBRUVICA® 420 mg o placebo en combinación con BR hasta la progresión de la enfermedad o toxicidad inaceptable. Todos los pacientes recibieron BR por un máximo de seis ciclos de 28 días. Bendamustina se dosificó a 70 mg/m2 infundido IV durante 30 minutos en el ciclo 1, los días 2 y 3, y en los ciclos 2-6, los días 1 y 2 hasta por 6 ciclos. Rituximab se administró en una dosis de 375 mg/m2 en el primer ciclo, el día 1, y de 500 mg/m2 en los ciclos de 2 a 6, el día 1. Noventa pacientes aleatorizados a placebo + BR se cruzaron para recibir IMBRUVICA® después de la progresión confirmada por el IRC. La mediana de edad fue de 64 años (rango, 31 a 86 años), 66% eran hombres y 91% eran de raza blanca. Todos los pacientes tenían un estado de desempeño ECOG basal de 0 o 1. La mediana del tiempo desde el diagnóstico fue de 5.9 años y la mediana del número de tratamientos previos fue de 2 (rango, 1 a 11 tratamientos). En la basal, 56% de los pacientes tenía al menos un tumor >5 cm, 26% se presentó con del11q.
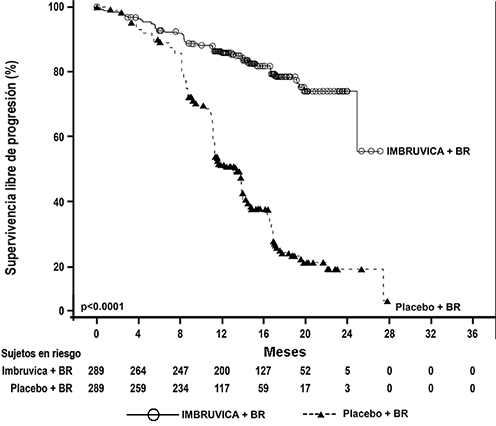
La supervivencia libre de progresión (SLP), evaluada por el IRC de acuerdo con criterios IWCLL indicó una reducción estadísticamente significativa del 80% en el riesgo de muerte o progresión. Los resultados de eficacia del estudio CLL3001 se muestran en la Tabla 7 y las curvas de Kaplan-Meier para la SLP se muestran en la Figura 8.
Tabla 7: Resultados de eficacia del Estudio CLL3001
|
Criterio de valoración |
IMBRUVICA + BR N=289 |
Placebo + BR N=289 |
|
Supervivencia libre progresióna |
||
|
Número de eventos (%) |
56 (19.4) |
183 (63.3) |
|
Mediana (IC 95%), meses |
No alcanzado |
13.3 (11.3, 13.9) |
|
HR (IC 95%) |
0.20 (0.15, 0.28) |
|
|
Tasa de respuesta globalb (%) |
82.7% |
67.8% |
|
CR/CRib |
10.4 |
2.8 |
|
Supervivencia global |
||
|
HR (IC 95%) |
0.628 (0.385, 1.024) |
|
|
Enfermedad residual mínima-estado negativod (%) |
12.8 |
4.8 |
IC = intervalo de confianza; HR = índice de riesgo; CR = respuesta completa; CRi = respuesta completa con recuperación medular incompleta.
a IRC evaluado.
b IRC evaluado; ORR (CR, CRi, respuesta ganglionar parcial, respuesta parcial).
c La mediana de OS no alcanza para los dos brazos.
d MRD se evaluó en pacientes con sospecha de respuesta completa; 120 pacientes para IMBRUVICA®, 57 pacientes de placebo tuvieron muestras obtenidas MRD.
Figura 8: Curva Kaplan-Meier de supervivencia libre de progresión (población ITT) en el Estudio CLL3001
Macroglobulinemia de Waldenström (MW): La seguridad y eficacia de IMBRUVICA® en MW (linfoma linfoplasmocítico excretor de IgM) se evaluaron en un estudio abierto, multicéntrico, de un solo brazo de 63 pacientes previamente tratados. La mediana de edad fue de 63 años (rango, 44 a 86 años), 76% eran hombres y 95% eran caucásicos. Todos los pacientes tenían un estado de desempeño ECOG basal de 0 o 1. La mediana del tiempo desde el diagnóstico fue de 74 meses, y la mediana del número de tratamientos previos fue 2 (rango, 1 a 11 tratamientos). En la basal, el valor de la mediana de IgM en suero fue de 3,5 g/dL (rango, 0.7-8.4 g/dL), y 60% de los pacientes eran anémicos (hemoglobina ≤11 g/dL).
IMBRUVICA® se administró por vía oral a 420 mg una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable. El criterio de valoración primario de este estudio fue la ORR evaluada el investigador. La ORR y la DOR fueron evaluadas utilizando criterios adoptados del Tercer Taller Internacional de Macroglobulinemia de Waldenström. Las respuestas a IMBRUVICA® se muestran en la Tabla 8.
Tabla 8: La tasa global de respuesta (ORR) y la duración de la respuesta (DOR), basado en la evaluación del investigador en pacientes con MW
|
Total (N=63) |
|
|
ORR (%) |
87.3 |
|
IC 95% (%) |
(76.5, 94.4) |
|
VGPR (%) |
14.3 |
|
PR (%) |
55.6 |
|
MR (%) |
17.5 |
|
Mediana duración de la respuesta en meses (rango) |
NR (0.03+, 18.8+) |
IC = intervalo de confianza; NR = no alcanzado; MR = respuesta menor; PR = respuesta parcial; VGPR = muy buena respuesta parcial; ORR = MR+PR+VGPR.
La mediana del tiempo de respuesta fue de 1.0 mes (rango, 0.7 13.4 meses). Los resultados de eficacia también fueron evaluados por un IRC demostrando una ORR de 82.5%, con una tasa de VGPR 11% y una tasa de PR 51%.
Linfoma de la zona marginal: La seguridad y eficacia de IMBRUVICA® en LZM fueron evaluados en un estudio abierto, multicéntrico, de un solo brazo, fase 2 (PCYC-1121) de pacientes que recibieron al menos una terapia previa. El análisis de eficacia incluyó a 60 pacientes con 3 subtipos de LZM: tejido linfoide asociado a mucosas (TLAM; n=30), nodal (n=17), y esplénico (n=13). La mediana de la edad fue de 66 años (rango, 30 a 92 años), 57% eran mujeres, y 85% eran caucásicos. Noventa y dos por ciento de pacientes tenía un estado de desempeño ECOG basal de 0 o 1 y 8% tenía un estado de desempeño ECOG basal de 2. La mediana del tiempo desde el diagnóstico fue de 3.7 años y la mediana del número de tratamientos previos fue de 2 (rango, 1 a 9 tratamientos).
IMBRUVICA® se administró oralmente a 560 mg una vez al día hasta la progresión de la enfermedad o toxicidad inaceptable. El criterio de valoración primario en este estudio fue la ORR por evaluación del IRC de acuerdo a los criterios revisados de IWG para NHL. Las respuestas a IMBRUVICA® se muestran en la Tabla 9.
Tabla 9: Tasa de respuesta global (ORR) y duración de respuesta (DOR) basado en evaluación IRC en pacientes con LZM
|
Total (N=60) |
|
|
ORR (CR +PR) (%) |
48.3 |
|
IC 95% (%) |
(35.3, 61.7) |
|
Respuesta completa (CR) (%) |
3.3 |
|
Respuesta parcial (PR) (%) |
45.0 |
|
Mediana de DOR, meses (rango) |
NR (16.7, NR) |
IC = intervalo de confianza; NR = no alcanzado.
Media de seguimiento de hasta 19.4 meses.
La mediana del tiempo a la respuesta inicial fue de 4.5 meses (rango, 2.3 a 16.4 meses).
La eficacia fue consistente entre los 3 subtipos de MZL.
Enfermedad de injerto contra huésped crónica (EICHc): La seguridad y eficacia de IMBRUVICA® en EICHc fueron evaluadas en un estudio abierto, multicéntrico, de un brazo, de 42 pacientes con EICHc después de falla de la terapia de corticoesteroides de primera línea y que requirieron terapia adicional. La mediana de edad fue de 56 años (rango, 19 a 74 años), 52% eran hombres, y 93% caucásicos. Las malignidades subyacentes más comunes que llevaron a trasplante fueron leucemia linfocítica aguda, leucemia mieloide aguda, y LLC. La mediana de tiempo desde el diagnóstico fue de 14 meses y la mediana del número de tratamientos previos de EICHc fue de 2 (rango, 1 a 3 tratamientos). La mayoría de los pacientes (88%) tuvo al menos 2 órganos involucrados al diagnóstico, siendo los órganos más comúnmente involucrados boca (86%), piel (81%), y tracto gastrointestinal (33%). Los pacientes incluidos en este estudio presentaron >25% de área de superficie corporal (BSA), “erupción eritematosa” definida por los criterios NIH (puntuación cutánea NIH de 2 y 3) o puntuación bucal >4 definida por los criterios NIH. La mediana de dosis diaria de esteroides por peso corporal en la medición basal fue de 0.3 mg/kg/día.
IMBRUVICA® fue administrada oralmente a 420 mg una vez al día hasta la progresión de la enfermedad, toxicidad inaceptable o recurrencia de malignidad subyacente. El criterio principal en este estudio fue la mejor ORR de acuerdo a evaluación de investigador usando los Criterios de Respuesta del Panel de Consenso de Institutos Nacionales de Salud 2005 con modificación. Las respuestas se vieron a través de los órganos involucrados en EICHc (piel, boca, tracto gastrointestinal, e hígado). Los resultados de eficacia se muestran en la Tabla 10.
Tabla 10: Mejor tasa de respuesta global (ORR) y tasa de respuesta sostenida basadas en la evaluación del investigador en pacientes con EICHc
|
Total (N=42) |
|
|
ORR (%) |
66.7 |
|
IC 95% (%) |
(50.5, 80.4) |
|
Respuesta completa (CR) (%) |
21.4 |
|
Respuesta parcial (PR) (%) |
45.2 |
|
Tasa de respuesta sostenida* (%) |
71.4 |
IC = intervalo de confianza.
* La tasa de respuesta sostenida se define como la proporción de pacientes que alcanzaron una CR o PR (N=28) que fue sostenida por al menos 20 semanas.
CONTRAINDICACIONES: IMBRUVICA® está contraindicado en pacientes quienes tienen hipersensibilidad conocida (por ejemplo, reacciones anafilácticas y anafilactoides) a ibrutinib o a los excipientes en su formulación.
Embarazo, lactancia y pacientes menores de 18 años de edad.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No existen estudios adecuados y bien controlados de IMBRUVICA® en mujeres embarazadas. Con base en hallazgos en animales, IMBRUVICA® puede causar daño fetal cuando se administra a mujeres embarazadas.
IMBRUVICA® no se debe utilizar durante el embarazo. Las mujeres en edad fértil deben utilizar medidas anticonceptivas altamente efectivas mientras toman IMBRUVICA®. Aquellas que usan métodos hormonales de control de la natalidad deben agregar un método de barrera. Las mujeres deben evitar quedar embarazadas mientras están tomando IMBRUVICA® y por hasta mes luego de finalizado el tratamiento. Si este fármaco se utiliza durante el embarazo o si la paciente se embaraza mientras toma este fármaco, se debe notificar a la paciente sobre el riesgo potencial al feto. Se desconoce el periodo de tiempo después del tratamiento con IMBRUVICA® en el que sea seguro embarazarse.
Los hombres deben ser advertidos de no ser padre o donar esperma mientras reciben IMBRUVICA®, y durante los 3 meses siguientes a la finalización del tratamiento (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Ibrutinib fue estudiado por efectos sobre el desarrollo embrio-fetal en ratas preñadas que recibieron dosis orales de 10, 40 y 80 mg/kg/día. Ibrutinib a una dosis de 80 mg/kg/día (aproximadamente 14 veces el ABC de ibrutinib y 9.5 veces el ABC del metabolito dihidrodiol comparado con pacientes a una dosis de 560 mg diarios) se asoció con aumento de pérdidas post-implante y aumento de las malformaciones viscerales (corazón y principales vasos). Ibrutinib en dosis ≥ 40 mg/kg/día (≥ aproximadamente 5.6 veces el ABC de ibrutinib y 4.0 veces el ABC del metabolito dihidrodiol comparado con pacientes a una dosis de 560 mg diarios) se asoció con disminución de los pesos fetales.
Ibrutinib también se administró por vía oral a conejas preñadas durante el periodo de organogénesis a dosis orales de 5, 15 y 45 mg/kg/día. Ibrutinib a una dosis de 15 mg/kg/día o mayor se asoció con malformaciones esqueléticas (esternebras fusionadas) e ibrutinib a una dosis de 45 mg/kg/día se asoció con un aumento de la pérdida post-implantación. Ibrutinib causó malformaciones en conejos a una dosis de 15 mg/kg/día (aproximadamente 2.0 veces la exposición (ABC) en pacientes con LCM o LZM que recibieron 560 mg de ibrutinib al día y 2.8 veces la exposición en pacientes con LLC o MW quienes recibían dosis de 420 mg ibrutinib por día).
Lactancia: Se desconoce si ibrutinib o sus metabolitos se excretan en leche materna humana. Debido a que varios fármacos se excretan por esta vía y debido al potencial para reacciones adversas serias en lactantes por IMBRUVICA®, se debe descontinuar la lactancia durante el tratamiento con IMBRUVICA®.
REACCIONES SECUNDARIAS Y ADVERSAS: En esta sección, se presentan las reacciones adversas (RA). Las reacciones adversas son aquellos eventos adversos que se pudieron asociar razonablemente con el uso de ibrutinib basado en una evaluación integral de la información de eventos adversos disponible. No se puede establecer una relación causal confiable con el uso de ibrutinib en casos individuales. Además, debido a que los estudios clínicos se realizan bajo condiciones muy variables, la tasa de eventos adversos observada en los estudios clínicos de un medicamento no puede ser comparada directamente con las tasas de estudios clínicos de otros medicamentos y pueden no reflejar las tasas observadas en la práctica clínica.
Reacciones adversas de los estudios integrados en pacientes con neoplasias malignas de células B: Los datos descritos a continuación reflejan la exposición a IMBRUVICA® en tres estudios fase 2 (PCYC-1102-CA, PCYC-1104-CA y PCYC-1118E) y cuatro estudios fase 3 (PCYC-1112-CA, PCYC-1115-CA, CLL3001 y MCL3001), que incluyeron 981 pacientes con neoplasias malignas de células B. Los pacientes recibieron IMBRUVICA® hasta la progresión de la enfermedad o toxicidad inaceptable.
Las reacciones adversas que ocurrieron con mayor frecuencia en los pacientes tratados con IMBRUVICA® para neoplasias malignas de células B (≥20%) fueron diarrea, hemorragia (por ejemplo, hematoma), neutropenia, dolor músculo esquelético, náuseas, exantema y pirexia.
Las reacciones adversas de grado 3/4 más comunes (≥5%) fueron: neutropenia, neumonía, trombocitopenia y neutropenia febril.
Tabla 11: Reacciones adversas reportadas en pacientes tratados con IMBRUVICA® para neoplasias malignas de células B (N=981)
|
Clase de sistema/órgano |
Reacción adversa |
Todos los grados (%) |
Grado 3 o 4 (%) |
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
19 |
1 |
|
Neumonía*† |
16 |
10 |
|
|
Sinusitis* |
11 |
1 |
|
|
Infección cutánea* |
10 |
3 |
|
|
Infección del tracto urinario |
9 |
2 |
|
|
Sepsis*† |
4 |
3 |
|
|
Neoplasias benignas y malignas (incluyendo quistes y pólipos) |
Cáncer de piel no-melanoma * |
6 |
1 |
|
Carcinoma células basales |
3 |
<1 |
|
|
Carcinoma células escamosas |
2 |
<1 |
|
|
Trastornos de la sangre y sistema linfático |
Neutropenia |
30 |
26 |
|
Trombocitopenia |
20 |
10 |
|
|
Neutropenia febril |
5 |
5 |
|
|
Leucocitosis |
2 |
1 |
|
|
Linfocitosis |
2 |
1 |
|
|
Síndrome de Leucostasis |
<1 |
<1 |
|
|
Trastornos del metabolismo y la nutrición |
Hiperuricemia |
7 |
2 |
|
Síndrome de lisis tumoral |
1 |
1 |
|
|
Trastorno del sistema nervioso |
Cefalea |
13 |
1 |
|
Mareos |
9 |
0 |
|
|
Trastornos oculares |
Visión borrosa |
7 |
0 |
|
Trastornos cardiacos |
Fibrilación auricular |
6 |
3 |
|
Trastornos vasculares |
Hemorragia* |
30 |
1 |
|
Hematoma * |
22 |
<1 |
|
|
Epistaxis |
8 |
<1 |
|
|
Petequias |
7 |
0 |
|
|
Hematoma Subdural† |
1 |
1 |
|
|
Hipertensión* |
10 |
4 |
|
|
Trastornos gastrointestinales |
Diarrea |
41 |
3 |
|
Náusea |
27 |
1 |
|
|
Constipación |
16 |
<1 |
|
|
Vómito |
14 |
<1 |
|
|
Estomatitis* |
13 |
1 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Erupción* |
22 |
2 |
|
Eritema |
2 |
0 |
|
|
Urticaria |
1 |
<1 |
|
|
Angioedema |
<1 |
<1 |
|
|
Trastornos músculo esqueléticos y del tejido conectivo |
Dolor músculo esquelético* |
28 |
3 |
|
Espasmos musculares |
14 |
<1 |
|
|
Artralgia |
12 |
1 |
|
|
Trastornos generales y condiciones del sitio de administración |
Pirexia |
20 |
2 |
|
Edema periférico |
14 |
1 |
Descontinuación y reducción de la dosis debido a RAs: De los 981 pacientes tratados con IMBRUVICA® con neoplasias malignas de células B, 5% descontinuó el tratamiento debido a reacciones adversas. Las reacciones adversas más frecuentes que condujeron a la descontinuación del tratamiento incluyeron neumonía, fibrilación auricular y hemorragia.
Las reacciones adversas que condujeron a la reducción de la dosis ocurrieron en el 5% de los pacientes.
Leucostasis: Se han observado casos aislados de leucostasis (ver Precauciones generales).
Adultos mayores: De los 981 pacientes tratados con IMBRUVICA®, 62% tenían 65 años de edad o más. La neumonía Grado 3 o mayor ocurrió más frecuentemente, (≥5%), entre los pacientes de edad avanzada tratados con IMBRUVICA® (13% de los pacientes de edad ≥65 frente al 7% de los pacientes <65 años).
Cáncer de piel no-melanoma: Basado en un análisis integrado de los estudios aleatorizados, controlados, fase 3 (PCYC-1112-CA, PCYC-1115-CA, CLL3001 y MCL3001), la incidencia de cáncer de piel no-melanoma fue del 6% en pacientes tratados con IMBRUVICA® y 3% en pacientes tratados con el comparador.
Linfoma de células del manto: Los datos descritos a continuación reflejan la exposición a IMBRUVICA® en un estudio clínico fase 2 (PCYC-1104-CA) y un estudio aleatorizado fase 3 (MCL3001) en pacientes con LCM (n=250).
Las reacciones adversas que ocurrieron con mayor frecuencia (≥20%) fueron diarrea, hemorragia (por ejemplo: hematoma), fatiga, dolor músculo esquelético, náuseas, infección del tracto respiratorio superior, tos y erupción.
Las reacciones adversas de Grado 3/4 más comunes (≥5%) fueron: neutropenia, trombocitopenia, neumonía y anemia.
Descontinuación y reducción de la dosis debido a RAs: De los 250 pacientes tratados con IMBRUVICA® para LCM, siete (3%) descontinuaron el tratamiento debido a reacciones adversas. Las reacciones adversas más frecuentes que llevaron a la descontinuación del tratamiento fueron: hemorragia, neumonía y trombocitopenia. Las reacciones adversas que llevaron a la reducción de la dosis ocurrieron en 6% de los pacientes.
Las reacciones adversas del Estudio 1104 se describen a continuación en la Tabla 12 para reflejar la exposición a IMBRUVICA® en pacientes con LCM que recibieron al menos una terapia previa con una mediana de duración del tratamiento de 8.3 meses.
Tabla 12: Reacciones adversas reportadas en ≥10% de pacientes con Linfoma de Células del Manto tratados con 560 mg de IMBRUVICA®-Estudio PCYC-1104-CA (n=111)
|
Clase de sistema/órgano |
Reacción Adversa |
Todos los Grados (%) |
Grado 3/4 (%) |
|
Infecciones e infestaciones |
Neumonía |
12 |
5 |
|
Infección del tracto urinario |
14 |
3 |
|
|
Sinusitis |
14 |
1 |
|
|
Infección del tracto respiratorio superior |
26 |
0 |
|
|
Trastornos de la sangre y del sistema linfático |
Neutropenia |
19 |
17 |
|
Trombocitopenia |
21 |
12 |
|
|
Anemia |
15 |
10 |
|
|
Trastornos del metabolismo y la nutrición |
Deshidratación |
14 |
4 |
|
Hiperuricemia |
17 |
5 |
|
|
Disminución del apetito |
23 |
2 |
|
|
Trastorno del sistema nervioso |
Mareos |
14 |
0 |
|
Cefalea |
12 |
0 |
|
|
Trastornos respiratorios, torácicos y mediastinales |
Disnea |
28 |
4 |
|
Epistaxis |
11 |
0 |
|
|
Tos |
18 |
0 |
|
|
Trastornos gastrointestinales |
Diarrea |
53 |
5 |
|
Dolor abdominal |
18 |
5 |
|
|
Vómito |
23 |
0 |
|
|
Estomatitis |
13 |
1 |
|
|
Estreñimiento |
28 |
0 |
|
|
Náusea |
32 |
1 |
|
|
Dispepsia |
11 |
0 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Erupción |
16 |
2 |
|
Trastornos músculo esqueléticos y del tejido conectivo |
Espasmos musculares |
14 |
0 |
|
Mialgia |
14 |
0 |
|
|
Artralgia |
14 |
0 |
|
|
Dolor de espalda |
14 |
1 |
|
|
Dolor en extremidades |
12 |
0 |
|
|
Trastornos generales y condiciones del sitio de administración |
Pirexia |
19 |
1 |
|
Fatiga |
43 |
5 |
|
|
Astenia |
12 |
3 |
|
|
Edema periférico |
30 |
2 |
|
|
Lesión, envenenamiento y complicaciones de procedimientos |
Contusión |
18 |
0 |
Reacciones adversas severas: En el estudio fase 2, se reportaron reacciones adversas severas en 60% de los pacientes (frecuencias emergentes al tratamiento). Las reacciones adversas serias que ocurrieron en más del 2% de los pacientes fueron fibrilación auricular (6%), neumonía (5%), infección del tracto urinario (4%), dolor abdominal (3%), hematoma subdural (3%), neutropenia febril (3%), falla renal aguda (3%), edema periférico (3%) y pirexia (3%).
Las reacciones adversas del estudio MCL3001 se describen a continuación en la Tabla 13 que refleja la exposición a IMBRUVICA® en pacientes con LCM que recibieron al menos una terapia previa, tratados con una mediana de duración del tratamiento de 14.4 meses.
Tabla 13: Reacciones adversas reportadas en pacientes con LCM tratados con 560 mg de IMBRUVICA®-Estudio MCL3001 (n=139)
|
Clase de sistema/órgano |
Clase de sistema/órgano |
IMBRUVICA® (n=139) |
Temsirolimus (n=139) |
||
|
Todos los grados (%) |
Grado 3 o 4 (%) |
Todos los grados (%) |
Grado 3 o 4 (%) |
||
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
19 |
2 |
12 |
1 |
|
Neumonía* |
14 |
10 |
19 |
12 |
|
|
Trastornos oculares |
Conjuntivitis |
12 |
0 |
5 |
0 |
|
Trastornos cardiacos |
Fibrilación auricular |
4 |
4 |
2 |
1 |
|
Trastornos gastrointestinales |
Dolor abdominal |
8 |
4 |
8 |
1 |
|
Trastornos músculo esqueléticos y del tejido conectivo |
Espasmos musculares |
19 |
0 |
3 |
0 |
* Incluye múltiples términos de reacciones adversas.
Leucemia linfocítica crónica/Linfoma de linfocitos pequeños: Los datos descritos a continuación reflejan la exposición a IMBRUVICA® en un estudio clínico de un solo brazo, abierto (Estudio PCYC-1102-CA) y tres estudios clínicos aleatorizados (Estudio PCYC-1115-CA, Estudio PCYC-1112-CA y Estudio CLL3001) en pacientes con LLC/LLP (n=668).
Las reacciones adversas más frecuentes que ocurrieron en los estudios PCYC-1102-CA, PCYC-1112-CA, PCYC-1115-CA y CLL3001 (≥20%) fueron diarrea, neutropenia, dolor músculo esquelético, náuseas, hemorragia (por ejemplo: hematoma), erupción, pirexia y trombocitopenia.
Las reacciones adversas Grado 3/4 más comunes (≥5%) fueron: neutropenia, neumonía, trombocitopenia y neutropenia febril.
Descontinuación y reducción de la dosis debido a RAs: Seis por ciento de los pacientes que recibieron IMBRUVICA® en los estudios PCYC-1102-CA, PCYC-1112-CA, PCYC-1115-CA y CLL3001 descontinuaron el tratamiento debido a reacciones adversas. Éstos incluyeron neumonía, fibrilación auricular, hemorragia, neutropenia, erupción y sepsis. Las reacciones adversas que condujeron a una reducción de la dosis ocurrieron en 5% de los pacientes aproximadamente.
Pacientes con LLC/LLP no tratados previamente: Las reacciones adversas se describen a continuación en la Tabla 14, reflejan la exposición a IMBRUVICA® con una mediana de duración de 17.4 meses, que es aproximadamente 2.5 veces la mediana de exposición a clorambucilo de 7.1 meses en el Estudio PCYC-1115-CA.
Tabla 14: Reacciones adversas reportadas en pacientes no tratados previamente con LLC/LLP tratado con 420 mg IMBRUVICA®-Estudio PCYC-1115-CAA.
|
Clase de Sistema/Órgano |
IMBRUVICA® (N=135) |
Clorambucilo (N=132) |
||
|
Reacción adversa |
Todos los Grados (%) |
Grado 3 o 4 (%) |
Todos los Grados (%) |
Grado 3 o 4 (%) |
|
Infecciones e infestaciones |
||||
|
Infección cutánea* |
15 |
2 |
3 |
1 |
|
Neumonía* |
14 |
8 |
7 |
4 |
|
Neoplasias benignas, malignas e inespecíficas (incluyendo quistes y pólipos) |
||||
|
Carcinoma de células basales |
9 |
1 |
2 |
0 |
|
Trastornos del metabolismo y la nutrición |
||||
|
Hiponatremia |
7 |
3 |
1 |
0 |
|
Trastornos oculares |
||||
|
Ojo seco |
17 |
0 |
5 |
0 |
|
Aumento del lagrimeo |
13 |
0 |
6 |
0 |
|
Visión borrosa |
13 |
0 |
8 |
0 |
|
Agudeza visual disminuida |
11 |
0 |
2 |
0 |
|
Trastornos cardiacos |
||||
|
Fibrilación auricular |
6 |
1 |
1 |
0 |
|
Trastornos vasculares |
||||
|
Hipertensión* |
14 |
4 |
1 |
0 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||
|
Tos |
22 |
0 |
15 |
0 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
42 |
4 |
17 |
0 |
|
Estomatitis* |
14 |
1 |
4 |
1 |
|
Dispepsia |
11 |
0 |
2 |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción* |
21 |
4 |
12 |
2 |
|
Hematoma* |
19 |
0 |
7 |
0 |
|
Trastornos del tejido músculo esquelético y conectivo |
||||
|
Dolor músculo esquelético* |
36 |
4 |
20 |
0 |
|
Artralgia |
16 |
1 |
7 |
1 |
|
Espasmos musculares |
11 |
0 |
5 |
0 |
|
Trastornos generales y condiciones del sito de administración |
||||
|
Edema periférico |
19 |
1 |
9 |
0 |
a Sujetos con múltiples eventos para una reacción adversa dada se cuentan una sola vez por cada término de reacción adversa.
* Incluye varios términos de reacciones adversas.
Pacientes con LLC/LLP que recibieron al menos una terapia previa:
Agente único: En la tabla 15 se muestran las reacciones adversas a la exposición de IMBRUVICA® con una mediana de duración de 8.6 meses y una exposición a ofatumumab con una mediana de duración de 5.3 meses en el estudio PCYC-1112-CA.
Tabla 15: Reacciones adversas reportadas en pacientes con LLC/LLP tratados con IMBRUVICA® como agente único en el estudio PCYC-1112-CAa
|
Clase de Sistema/Órgano |
IMBRUVICA® (N=195) |
Ofatumumab (N=191) |
||
|
Reacción adversa |
Todos los Grados (%) |
Grado 3 o 4 (%) |
Todos los Grados (%) |
Grado 3 o 4 (%) |
|
Infecciones e infestaciones |
||||
|
Infección del tracto respiratorio superior |
16 |
1 |
10 |
2 |
|
Neumonía* |
15 |
10 |
13 |
9 |
|
Sinusitis* |
11 |
1 |
6 |
0 |
|
Infección del tracto urinario |
10 |
4 |
5 |
1 |
|
Infección cutánea* |
7 |
2 |
3 |
1 |
|
Sepsis* |
4 |
2 |
4 |
3 |
|
Trastornos de la sangre y del sistema linfático |
||||
|
Anemia |
23 |
5 |
17 |
8 |
|
Neutropenia |
22 |
16 |
15 |
14 |
|
Trombocitopenia |
17 |
6 |
12 |
4 |
|
Linfocitosis |
4 |
2 |
3 |
1 |
|
Leucocitosis |
4 |
3 |
1 |
0 |
|
Neutropenia Febril |
2 |
2 |
3 |
3 |
|
Trastornos del sistema nervioso |
||||
|
Cefalea |
14 |
1 |
6 |
0 |
|
Mareos |
11 |
0 |
5 |
0 |
|
Trastornos oculares |
||||
|
Visión borrosa |
10 |
0 |
3 |
0 |
|
Trastornos cardiacos |
||||
|
Fibrilación Auricular |
5 |
3 |
1 |
0 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||
|
Epistaxis |
9 |
0 |
3 |
1 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
48 |
4 |
18 |
2 |
|
Náusea |
26 |
2 |
18 |
0 |
|
Estomatitis* |
17 |
1 |
6 |
1 |
|
Estreñimiento |
15 |
0 |
9 |
0 |
|
Vómito |
14 |
0 |
6 |
1 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción* |
24 |
3 |
13 |
0 |
|
Hematoma* |
21 |
0 |
4 |
0 |
|
Petequias |
14 |
0 |
1 |
0 |
|
Trastornos músculo esqueléticos y del tejido conectivo |
||||
|
Dolor músculo esquelético* |
28 |
2 |
18 |
1 |
|
Artralgia |
17 |
1 |
7 |
0 |
|
Trastornos generales y condiciones del sitio de administración |
||||
|
Pirexia |
24 |
2 |
15 |
1 |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos |
||||
|
Hematoma subdural |
1 |
0 |
0 |
0 |
a Ocurriendo con ≥10% de incidencia y 5% mayor en el brazo de IMBRUVICA® en comparación con el brazo de ofatumumab o reacciones adversas graves con ≥2% de incidencia y 2% mayor en el brazo de IMBRUVICA® en comparación con el brazo de ofatumumab o biológicamente plausible.
* Incluye varios términos sobre reacciones adversas.
Los pacientes con múltiples eventos, para una reacción adversa dada se cuentan una sola vez por cada término de reacción adversa.
Los eventos se clasifican por órganos y sistemas y por frecuencia decreciente de término reacción adversa en el brazo IMBRUVICA®.
Terapia de combinación: Las reacciones adversas descritas a continuación en la Tabla 16 reflejan la exposición a IMBRUVICA® + BR con una mediana de duración de 14.7 meses y la exposición a placebo + BR con una mediana de 12.8 meses en el Estudio CLL3001.
Tabla 16: Reacciones adversas reportadas en pacientes con LLC/LLP tratados con IMBRUVICA® en combinación con la BR en el Estudio CLL3001
|
Clase de Sistema/Órgano |
IMBRUVICA® + BR (N=287) |
Placebo + BR (N=287) |
||
|
Reacción adversa |
Todos los grados % |
Grado 3 o 4 % |
Todos los grados % |
Grado 3 o 4 % |
|
Trastornos de la sangre y del sistema linfático |
||||
|
Trombocitopenia |
31 |
15 |
24 |
15 |
|
Trastornos cardiacos |
||||
|
Fibrilación Auricular |
7 |
3 |
2 |
1 |
|
Trastornos vasculares |
||||
|
Hipertensión* |
10 |
5 |
5 |
2 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
36 |
2 |
23 |
1 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción* |
24 |
3 |
18 |
1 |
|
Hematomas* |
18 |
<1 |
6 |
0 |
|
Trastornos músculo esqueléticos y del tejido conectivo |
||||
|
Dolor músculo esquelético* |
29 |
2 |
20 |
0 |
|
Espasmos musculares |
12 |
<1 |
5 |
0 |
a Ocurrieron con una incidencia al menos 5% más alto para las RAs o 2% más alto para las RASs.
Los eventos están ordenados por clase de órgano y sistema y por frecuencia decreciente del término de reacción adversa en el brazo de IMBRUVICA®.
* Incluye múltiples términos de reacciones adversas
<1 se utiliza para frecuencias por encima de 0 y por debajo de 0.5%.
Macroglobulinemia de Waldenström (MW): Los datos descritos a continuación reflejan la exposición a IMBRUVICA® en un estudio clínico abierto que incluyó a 63 pacientes con MW tratados previamente.
Las reacciones adversas más frecuentes que ocurrieron en el estudio MW (≥20%) fueron neutropenia, trombocitopenia, diarrea, erupción, náuseas, espasmos musculares y fatiga.
Descontinuación y reducción de la dosis debido a RAs: Seis por ciento de los pacientes que recibieron IMBRUVICA® en el estudio de MW descontinuaron el tratamiento debido a reacciones adversas. Las reacciones adversas que condujeron a la reducción de la dosis ocurrieron en 11% de los pacientes.
Las reacciones adversas descritas a continuación en la Tabla 17, reflejan la exposición a IMBRUVICA® con una mediana de duración de 11.7 meses en el estudio MW.
Tabla 17: Reacciones adversas reportadas en ≥10% de los pacientes con MW tratados con 420 mg de IMBRUVICA® Estudio 1118E (N = 63)
|
Clase de Sistema/Órgano |
Reacción Adversa |
Todos los grados (%) |
Grados 3-4 (%) |
|
Infecciones e infestaciones |
Sinusitis |
19 |
0 |
|
Infección del tracto respiratorio superior |
19 |
0 |
|
|
Neumonía* |
14 |
6 |
|
|
Infección de la piel* |
14 |
2 |
|
|
Neoplasias benignas, malignas e inespecíficas (incluyendo quistes y pólipos) |
Cáncer de piel* |
11 |
0 |
|
Trastornos de la sangre y del sistema linfático |
Neutropenia |
25 |
17 |
|
Trombocitopenia |
17 |
13 |
|
|
Anemia |
16 |
3 |
|
|
Trastornos del sistema nervioso |
Mareos |
14 |
0 |
|
Cefalea |
13 |
0 |
|
|
Trastornos respiratorios, torácicos y mediastinales |
Epistaxis |
19 |
0 |
|
Tos |
13 |
0 |
|
|
Trastornos gastrointestinales |
Diarrea |
37 |
0 |
|
Náusea |
21 |
0 |
|
|
Estomatitis* |
16 |
0 |
|
|
Reflujo gastroesofágico |
13 |
0 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Erupción* |
22 |
0 |
|
Hematomas* |
16 |
0 |
|
|
Prurito |
11 |
0 |
|
|
Trastornos músculo esqueléticos y del tejido conectivo |
Espasmos musculares |
21 |
0 |
|
Artralgia |
13 |
0 |
|
|
Trastornos generales y condiciones del sito de administración |
Fatiga |
21 |
0 |
* Incluye múltiples términos de reacciones adversas
Linfoma de la zona marginal: Los datos descritos a continuación reflejan exposición a IMBRUVICA® en un estudio clínico abierto que incluyó 63 pacientes con LZM que recibieron al menos una terapia previa.
Las reacciones adversas que ocurrieron con mayor frecuencia en el estudio LZM (≥20%) fueron fatiga, diarrea, hematomas, dolor músculo esquelético, anemia, hemorragia, erupción, náusea, trombocitopenia, artralgia, edema periférico, tos, disnea e infección de tracto respiratorio superior.
Descontinuación y reducción de la dosis debido a RAs: Trece por ciento de los pacientes que recibieron IMBRUVICA® en el ensayo LZM descontinuó el tratamiento debido a reacciones adversas. Las reacciones adversas que llevaron a reducción de la dosis ocurrieron en aproximadamente 10% de los pacientes.
Las reacciones adversas descritas a continuación en la Tabla 18 reflejan la exposición a IMBRUVICA® con una mediana de duración de 11.6 meses en el estudio LZM.
Tabla 18: Reacciones adversas reportadas en ≥10% de los pacientes con LZM tratados con 560 mg de IMBRUVICA® Estudio 1121 (N=63)
|
Clase de Sistema/Órgano |
Reacción Adversa |
Todos los grados (%) |
Grados 3-4 (%) |
|
Infecciones e infestaciones |
Infección del tracto respiratorio superior |
21 |
0 |
|
Sinusitis* |
19 |
0 |
|
|
Bronquitis |
11 |
0 |
|
|
Neumonía* |
11 |
10 |
|
|
Trastornos de la sangre y del sistema linfático |
Anemia |
33 |
14 |
|
Trombocitopenia* |
25 |
2 |
|
|
Neutropenia* |
8 |
8 |
|
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito |
16 |
2 |
|
Hiperuricemia |
16 |
0 |
|
|
Hipoalbuminemia |
14 |
0 |
|
|
Hipocalemia |
13 |
0 |
|
|
Trastornos psiquiátricos |
Ansiedad |
16 |
2 |
|
Trastornos del sistema nervioso |
Mareos |
19 |
0 |
|
Cefalea |
13 |
0 |
|
|
Trastornos vasculares |
Hemorragia* |
30 |
0 |
|
Hipertensión* |
14 |
5 |
|
|
Trastornos respiratorios, torácicos y mediastinales |
Tos |
22 |
2 |
|
Disnea |
21 |
2 |
|
|
Trastornos gastrointestinales |
Diarrea |
43 |
5 |
|
Náusea |
25 |
0 |
|
|
Dispepsia |
19 |
0 |
|
|
Estomatitis* |
17 |
2 |
|
|
Dolor abdominal |
16 |
2 |
|
|
Estreñimiento |
14 |
0 |
|
|
Dolor abdominal superior |
13 |
0 |
|
|
Vómito |
11 |
2 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Hematomas* |
41 |
0 |
|
Erupción * |
29 |
5 |
|
|
Prurito |
14 |
0 |
|
|
Trastornos músculo esqueléticos y del tejido conectivo |
Dolor músculo esquelético |
40 |
3 |
|
Artralgia |
24 |
2 |
|
|
Espasmos musculares |
19 |
3 |
|
|
Trastornos generales y condiciones del sito de administración |
Fatiga |
44 |
6 |
|
Edema periférico |
24 |
2 |
|
|
Pirexia |
17 |
2 |
* Incluye múltiples términos de reacciones adversas
Enfermedad de injerto contra huésped crónica: Los datos descritos a continuación reflejan la exposición a IMBRUVICA® en un estudio clínico abierto que incluyó 42 pacientes con EICHc después de la falla de la terapia de corticoesteroides de primera línea y que requirió terapia adicional.
Las reacciones adversas que ocurrieron de forma más común en el estudio EICHc (≥20%) fueron fatiga, hematomas, diarrea, estomatitis, espasmos musculares, náusea, hemorragia y neumonía. Ocurrió fibrilación auricular en un paciente (2%), que fue Grado 3.
Descontinuación y reducción de dosis debido a RAs: Veinticuatro por ciento de los pacientes que recibieron IMBRUVICA® en el estudio EICHc descontinuó el tratamiento debido a reacciones adversas. Las reacciones adversas que llevaron a reducción de la dosis ocurrieron en 26% de los pacientes.
Las reacciones adversas descritas a continuación en la Tabla 19 reflejan la exposición a IMBRUVICA® con una mediana de duración de 4.4 meses en el estudio EICHc.
Tabla 19: Reacciones adversas reportadas por ≥10% de los pacientes con EICHc tratados con 420 mg de IMBRUVICA®-Estudio 1129 (N=42)
|
Clase de Sistema/Órgano |
Reacción Adversa |
Todos los grados (%) |
Grados 3-4 (%) |
|
Infecciones e infestaciones |
Neumonía* |
21 |
10 |
|
Infección del tracto respiratorio superior |
19 |
0 |
|
|
Sepsis* |
10 |
10 |
|
|
Trastornos del metabolismo y de la nutrición |
Hipocalemia |
12 |
7 |
|
Trastornos del sistema nervioso |
Cefalea |
17 |
5 |
|
Trastornos vasculares |
Hemorragia* |
26 |
0 |
|
Trastornos respiratorios, torácicos y mediastinales |
Tos |
14 |
0 |
|
Disnea |
12 |
2 |
|
|
Trastornos gastrointestinales |
Diarrea |
36 |
10 |
|
Estomatitis* |
29 |
2 |
|
|
Náusea |
26 |
0 |
|
|
Estreñimiento |
12 |
0 |
|
|
Trastornos de la piel y del tejido subcutáneo |
Hematomas* |
41 |
0 |
|
Erupción * |
12 |
0 |
|
|
Trastornos músculo esqueléticos y del tejido conectivo |
Espasmos musculares |
29 |
2 |
|
Dolor músculo esquelético* |
14 |
5 |
|
|
Trastornos generales y condiciones del sito de administración |
Fatiga |
57 |
12 |
|
Pirexia |
17 |
5 |
|
|
Edema periférico |
12 |
0 |
|
|
Lesiones, intoxicaciones y complicaciones de procedimientos |
Caídas |
17 |
0 |
* Incluye múltiples términos de reacciones adversas.
Datos post-comercialización: Además de las reacciones adversas reportadas durante los estudios clínicos y que se enlistan anteriormente, se han reportado las siguientes reacciones adversas durante la experiencia post- comercialización (Tabla 20). Debido a que estas reacciones fueron reportadas de manera voluntaria por una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco. En la tabla se proporcionan las frecuencias de acuerdo a la siguiente convención:
|
Muy común |
≥1/10 (≥10%) |
|
Común |
≥1/100 y <1/10 (≥1% y <10%) |
|
Poco común |
≥1/1000 y <1/100 (≥0.1% y <1%) |
|
Raro |
≥1/10000 y <1/1000 (≥0.01% y <0.1%) |
|
Muy raro |
<1/10000, incluyendo reportes aislados (<0.01%) |
|
Se desconoce |
No se puede estimar a partir de los datos disponibles. |
En la Tabla 20, se presentan las reacciones adversas por categoría de frecuencia con base en la incidencia en los estudios clínicos de estudios epidemiológicos, cuando éstos se conocen.
Tabla 20: Reacciones adversas identificadas durante la experiencia post-comercialización con IMBRUVICA®
|
Clase de Sistema/Órgano Reacción Adversa |
Categoría de frecuencia estimada a partir de los estudios clínicos con IMBRUVICA® |
|
Trastornos del sistema inmunológico |
|
|
Enfermedad pulmonar intersticial*† |
Común |
|
Trastornos del metabolismo y de la nutrición |
|
|
Síndrome de lisis tumoral |
Poco común |
|
Trastornos hepatobiliares |
|
|
Falla hepática* |
Se desconoce |
|
Trastornos de la piel y de tejidos subcutáneos |
|
|
Angioedema |
Poco común |
|
Eritema |
Se desconoce |
|
Onicoclasia |
Común |
|
Síndrome de Stevens-Johnson |
Se desconoce |
|
Urticaria |
Poco común |
* Incluye múltiples términos de las reacciones adversas.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Los siguientes efectos adversos fueron vistos en estudios de hasta 13 semanas de duración en ratas y perros. Se encontró que ibrutinib induce efectos gastrointestinales (heces blandas/diarrea y/o inflamación) en ratas a dosis equivalentes a las humanas (HEDs) ≥ 16 mg/kg/día y en perros a HEDs de ≥ 32 mg/kg/día. Los efectos en el tejido linfoide (depleción linfoide) también fueron inducidos a HEDs de ≥ 28 mg/kg/día en ratas y ≥ 32 mg/kg/día en perros. En ratas se observó atrofia moderada de células acinares pancreáticas a HEDs de ≥ 6 mg/kg/día. Se observó una leve disminución ósea trabecular y cortical en ratas administradas a HEDs de ≥ 16 mg/kg/día por 13 semanas. Todos los hallazgos notables en ratas y perros se revirtieron completa o parcialmente después de periodos de recuperación de 6 a 13 semanas.
Carcinogenicidad y mutagenicidad: No se han conducido estudios de carcinogenicidad con ibrutinib.
Ibrutinib no tiene propiedades genotóxicas cuando se probó en bacterias, células de mamíferos o en ratones.
Fertilidad: No se observaron efectos sobre la fertilidad y la capacidad reproductiva en ratas macho o hembra hasta la dosis máxima ensayada, 100 mg/kg/día (dosis equivalente en humanos [HED] de 16 mg/kg/día).
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Ibrutinib es primariamente metabolizado por la enzima 3A4 del citocromo P450 (CYP3A4).
Agentes que pueden aumentar las concentraciones plasmáticas de ibrutinib: El uso concomitante de IMBRUVICA® y fármacos que inhiben fuerte o moderadamente CYP3A puede aumentar la exposición a ibrutinib y los inhibidores de CYP3A deben evitarse.
Inhibidores fuertes de CYP3A: La co-administración de ketoconazol, un inhibidor fuerte de CYP3A, en 18 sujetos sanos, aumentó la exposición (Cmáx y ABC0-última) de ibrutinib en 29 y 24 veces, respectivamente. La exposición máxima observada de ibrutinib (ABC) fue ≤ 2 veces en 37 pacientes tratados con inhibidores de CYP3A leves y/o moderados en comparación con la exposición a ibrutinib en 76 pacientes no tratados concomitantemente con inhibidores de CYP3A. Los datos clínicos de seguridad en 66 pacientes tratados con inhibidores moderados (n=47) o fuertes (n=19) de CYP3A no presentaron aumentos significativos de las toxicidades.
Los inhibidores fuertes de CYP3A (por ejemplo, ketoconazol, indinavir, nelfinavir, ritonavir, saquinavir, claritromicina, telitromicina, itraconazol, nefazodona, cobicistat y posaconazol) deben ser evitados. Si el beneficio es superior al riesgo y un inhibidor de CYP3A fuerte debe ser utilizado, reducir la dosis de IMBRUVICA® a 140 mg o detener temporalmente el tratamiento con IMBRUVICA® (durante 7 días o menos).
Inhibidores moderados y leves de CYP3A: En pacientes con neoplasias malignas de células B, la co-administración de los inhibidores de CYP3A eritromicina y voriconazol, incrementaron la Cmáx por 3.4 veces y 6.7 veces e incrementaron el ABC por 3.0 veces y 5.7 veces, respectivamente. Si se indica un inhibidor moderado de CYP3A (por ejemplo, fluconazol, voriconazol, eritromicina, amprenavir, aprepitant, atazanavir, ciprofloxacino, crizotinib, diltiazem, fosamprenavir, imatinib, verapamilo, amiodarona, dronedarona), reducir la dosis de IMBRUVICA® a 140 mg para la duración de uso del inhibidor. No es necesario ajustar la dosis en combinación con inhibidores leves. Monitorear de cerca al paciente para toxicidad y seguir la guía de modificación de la dosis según sea necesario. Evitar el consumo de toronja y de naranja de Sevilla durante el tratamiento con IMBRUVICA® ya que contienen inhibidores moderados de CYP3A. La administración de ibrutinib con alimentos puede aumentar su concentración (ver Propiedades farmacocinéticas).
Agentes que pueden disminuir las concentraciones plasmáticas de ibrutinib: La administración de IMBRUVICA® con inductores fuertes de CYP3A puede disminuir las concentraciones plasmáticas de ibrutinib hasta 90%.
Evitar el uso concomitante de inductores potentes de CYP3A (por ejemplo, carbamazepina, rifampicina, fenitoína y hierba de San Juan). Considere agentes alternativos con menor inducción de CYP3A.
Fármacos cuyas concentraciones plasmáticas pueden ser alteradas por ibrutinib: Los estudios in vitro indicaron que ibrutinib es un inhibidor reversible débil hacia CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4/5 y no muestra una inhibición de CYP450 dependiente del tiempo. El metabolito dihidrodiol de ibrutinib es un inhibidor débil hacia CYP2B6, CYP2C8, CYP2C9 y CYP2D6. Tanto ibrutinib como el metabolito dihidrodiol son a lo mucho, inductores débiles de las isoenzimas del CYP450 in vitro. Por lo tanto, es poco probable que IMBRUVICA® presente alguna interacción medicamentosa clínicamente relevante con medicamentos que pueden ser metabolizados por las enzimas del CYP450.
Los estudios in vitro indicaron que ibrutinib no es un sustrato de P-gp ni de otros transportadores mayores, salvo de OCT2. El metabolito dihidrodiol y otros metabolitos son sustratos de la P-gp. Ibrutinib es un inhibidor leve de P-gp y de la proteína de resistencia en el cáncer de mama (BCRP). No se espera que ibrutinib tenga interacciones fármaco-fármaco sistémicas con sustratos de P-gp. Sin embargo, no se puede excluir que ibrutinib pudiera inhibir la P-gp intestinal y BCRP después de una dosis terapéutica. No existen datos clínicos disponibles. Para minimizar el potencial de una interacción en el tracto gastrointestinal (GI), deben administrarse sustratos de P-gp o BCRP con índice terapéutico estrecho como digoxina o metotrexato al menos 6 horas antes o después de IMBRUVICA®. Ibrutinib también puede inhibir la BCRP sistemáticamente e incrementar la exposición de los fármacos que presentan un flujo hepático mediado por la BCRP, como rosuvastatina.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Los principales cambios observados en las pruebas de laboratorio durante el tratamiento de ibrutinib están asociados con parámetros hematológicos. En el estudio aleatorizado, controlado PCYC-1112-CA, la incidencia de las toxicidades hematológicas de laboratorio fue similar en el brazo de ibrutinib y ofatumumab para disminuciones de Grado 3/4 en neutrófilos (ibrutinib: 23.1%, ofatumumab: 26.2%), hemoglobina (0%, 0%), y plaquetas (51%, 9.9%) utilizando la escala de calificación IWCLL. La incidencia de Grado 3/4 de toxicidad en los parámetros de química clínica fue baja y similar en el brazo ibrutinib (0% - 3.1%) y el brazo de ofatumumab (0%-5.8%).
PRECAUCIONES GENERALES:
Eventos relacionados con sangrado: Ha habido reportes de eventos hemorrágicos en pacientes tratados con IMBRUVICA®, tanto con y sin trombocitopenia. Éstos incluyen eventos hemorrágicos menores tales como contusión, epistaxis y petequias; y eventos hemorrágicos mayores, algunos fatales, incluyendo sangrado gastrointestinal, hemorragia intracraneal, y hematuria.
Los pacientes fueron excluidos de la participación en los estudios fase 2 y 3 de IMBRUVICA® si requerían warfarina u otros antagonistas de la vitamina K. Warfarina u otros antagonistas de la vitamina K no deben ser administrados concomitantemente con IMBRUVICA®. Se debe evitar el uso de suplementos tales como aceite de pescado y preparaciones de vitamina E.
En un estudio in vitro de función plaquetaria, se observaron efectos inhibitorios de ibrutinib en la agregación plaquetaria inducida por colágeno (ver Propiedades farmacocinéticas). El uso de IMBRUVICA® en pacientes que requieren otros anticoagulantes o medicamentos que inhiban la función plaquetaria puede incrementar el riesgo de sangrado.
IMBRUVICA® se debe suspender por lo menos 3 a 7 días antes y después de una cirugía, dependiendo del tipo de cirugía y el riesgo de sangrado.
No se ha estudiado a pacientes con diátesis hemorrágica congénita.
Leucostasis: Se reportaron casos aislados de leucostasis en pacientes tratados con IMBRUVICA®. Un alto número de linfocitos en circulación (> 400,000/mcL) puede conferir un mayor riesgo. Considerar suspender temporalmente IMBRUVICA®. Los pacientes deben ser monitoreados de cerca. Administrar cuidados de soporte incluyendo hidratación y/o citorreducción como se indique.
Infecciones: Se observaron infecciones (incluyendo sepsis, infecciones bacterianas, virales o micóticas) en pacientes tratados con IMBRUVICA®. Algunas de estas infecciones han sido asociadas con hospitalización y muerte. Considerar la profilaxis de acuerdo con el estándar de atención en pacientes que están en mayor riesgo de infecciones oportunistas. Aunque no se ha establecido la causalidad, se han presentado casos de leucoencefalopatía multifocal progresiva (LMP) en pacientes tratados con IMBRUVICA®. Los pacientes deben ser monitoreados por síntomas (fiebre, escalofríos, debilidad, confusión) y se debe instituir la terapia adecuada como se indique.
Citopenias: Se reportaron citopenias grado 3 o grado 4 (neutropenia, trombocitopenia y anemia) emergentes al tratamiento en pacientes tratados con IMBRUVICA®. Monitorear el recuento sanguíneo completo mensualmente.
Enfermedad pulmonar intersticial (EPI): Se han reportado casos de EPI en pacientes tratados con IMBRUVICA®. Monitorear a los pacientes por síntomas pulmonares indicativos de EPI. Si se desarrollan síntomas, interrumpir IMBRUVICA® y maneje adecuadamente la EPI. Si los síntomas persisten, considerar los riesgos y beneficios del tratamiento con IMBRUVICA® y seguir las guías de modificación de la dosis.
Fibrilación Auricular: Se reportaron fibrilación auricular y flutter auricular en pacientes tratados con IMBRUVICA®, particularmente en pacientes con factores de riesgo cardiaco, hipertensión, infecciones agudas, y antecedente de fibrilación auricular. Monitorear clínicamente a los pacientes de manera periódica para fibrilación auricular. Los pacientes que desarrollen síntomas de arritmia (por ejemplo, palpitaciones, aturdimiento) o nueva aparición de disnea deben ser evaluados clínicamente y si se indica, realizar un ECG. Para las fibrilaciones auriculares que persisten, considerar riesgos y beneficios del tratamiento con IMBRUVICA® y seguir las guías de modificación de dosis.
Síndrome de lisis tumoral: Se ha reportado síndrome de lisis tumoral durante la terapia con IMBRUVICA®. Los pacientes con riesgo de síndrome de lisis tumoral son aquellos con una alta carga tumoral previa al tratamiento. Se debe monitorear de cerca a los pacientes y tomar las precauciones apropiadas.
Cáncer de piel no-melanoma: Han ocurrido cánceres de piel no-melanoma en pacientes tratados con IMBRUVICA®. Monitorear a los pacientes por la aparición de cáncer de piel no-melanoma.
Efectos sobre la habilidad para manejar o utilizar maquinarias: Se han reportado fatiga, mareo y astenia en algunos pacientes tomando IMBRUVICA® y debería considerarse cuando se evalúa la habilidad de los pacientes para manejar u operar maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis: IMBRUVICA® debe administrarse por vía oral una vez al día con un vaso con agua, aproximadamente a la misma hora todos los días. Las cápsulas se deben tragar completas con agua y no se deben abrir, romper o masticar. IMBRUVICA® no se debe tomar con jugo de toronja. La administración de IMBRUVICA® deberá continuarse hasta la progresión de la enfermedad o hasta que no sea tolerado por el paciente.
Linfoma de células del manto y Linfoma de la zona marginal: La dosis recomendada de IMBRUVICA® para LCM o LZM es de 560 mg una vez al día hasta la progresión de la enfermedad o hasta que no sea tolerado por el paciente.
Leucemia linfocítica crónica/Linfoma de linfocitos pequeños (LLC/LLP) y Macroglobulinemia de Waldenström (MW): La dosis recomendada de IMBRUVICA® para LLC/LLP o MW es de 420 mg una vez al día, hasta la progresión de la enfermedad o hasta que no sea tolerado por el paciente.
La dosis recomendada de IMBRUVICA® para la LLC/LLP cuando se utiliza en combinación con bendamustina y rituximab (BR) (administrada cada 28 días por hasta 6 ciclos) es de 420 mg por vía oral una vez al día hasta la progresión de la enfermedad o hasta que no sea tolerado por el paciente.
Para obtener información adicional relativa a BR, consulte la información para prescribir correspondiente de BR.
Enfermedad de injerto contra huésped crónica (EICHc): La dosis recomendada de IMBRUVICA® para EICHc es de 420 mg una vez al día hasta la progresión de la EICHc, recurrencia de una malignidad subyacente, o hasta que ya no sea tolerada por el paciente. Cuando un paciente ya no requiere terapia para el tratamiento de EICHc, IMBRUVICA® se debe descontinuar considerando la evaluación médica del paciente de forma individual.
Guías de modificación de la dosis: Se requieren modificaciones de la dosis para el uso concomitante de inhibidores moderados y fuertes de CYP3A ya que éstos pueden aumentar la exposición de ibrutinib (ver Interacciones medicamentosas y de otro género).
La terapia con IMBRUVICA® debe suspenderse para cualquier nuevo inicio o empeoramiento de toxicidades no hematológicas Grado ≥3, neutropenia Grado 3 o mayor con infección o fiebre o toxicidades hematológicas Grado 4.
Una vez que los síntomas de la toxicidad se han resuelto a Grado 1 o al nivel basal (recuperación), la terapia con IMBRUVICA® puede ser reiniciada a la dosis inicial. Si la toxicidad vuelve a ocurrir, reducir la dosis en 140 mg por día. Se puede considerar una segunda reducción de la dosis en 140 mg según sea necesario. Si estas toxicidades persisten o recurren después de dos reducciones de la dosis, descontinuar IMBRUVICA®.
Las modificaciones recomendadas de la dosis para estas toxicidades se describen a continuación:
|
Ocurrencia de la toxicidad |
Modificación de la dosis para LCM/LZM después de la recuperación |
Modificación de la dosis para LLC/LLP/MW/EICHc después de la recuperación |
|
Primera |
Reiniciar en 560 mg diarios |
Reiniciar en 420 mg diarios |
|
Segunda |
Reiniciar en 420 mg diarios |
Reiniciar en 280 mg diarios |
|
Tercera |
Reiniciar en 280 mg diarios |
Reiniciar en 140 mg diarios |
|
Cuarta |
Descontinuar IMBRUVICA® |
|
Dosis omitida: Si no se toma una dosis de IMBRUVICA® a la hora programada, se puede tomar tan pronto como sea posible en el mismo día con un regreso al programa normal al siguiente día. El paciente no deberá tomar cápsulas adicionales para compensar la dosis omitida.
Poblaciones especiales:
Pediátricos (18 años de edad y menores): Aún no se ha evaluado la seguridad y eficacia de IMBRUVICA® en niños.
Insuficiencia renal: Ibrutinib tiene depuración renal mínima. No se han conducido estudios clínicos específicos en pacientes con insuficiencia renal. Se trataron pacientes con insuficiencia renal leve o moderada en los estudios clínicos de IMBRUVICA®. No se necesita ajuste de dosis para pacientes con insuficiencia renal leve o moderada (depuración de creatinina mayor a 30 mL/min). Se debe mantener la hidratación y se deben monitorear periódicamente los niveles de creatinina sérica. No existen datos en pacientes con insuficiencia renal severa o en pacientes en diálisis (ver Propiedades farmacocinéticas).
Insuficiencia hepática: Ibrutinib se metaboliza en el hígado. En un estudio de insuficiencia hepática, los datos mostraron un incremento en la exposición a ibrutinib (ver Propiedades farmacocinéticas). Para pacientes con insuficiencia hepática leve (Child-Pugh clase A), la dosis recomendada es de 280 mg diarios. Para pacientes con insuficiencia hepática moderada (Child-Pugh clase B) la dosis recomendada es de 140 mg diarios. Monitorear a los pacientes para signos de toxicidad por IMBRUVICA® y seguir las guías de modificación de dosis cuando se requiera. No se recomienda administrar IMBRUVICA® a pacientes con insuficiencia hepática severa (Child-Pugh clase C).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Síntomas y signos: Existen datos limitados sobre los efectos de la sobredosis de IMBRUVICA®. No se alcanzó la Dosis Máxima Tolerada en el estudio fase I en el que los pacientes recibieron hasta 12.5 mg/kg/día (1400 mg/día). En un estudio separado, un sujeto sano que recibió una dosis de 1680 mg experimentó incrementos reversibles Grado 4 de las enzimas hepáticas [aspartato aminotransferasa (AST) y alanina aminotransferasa (ALT)]. No existe un antídoto específico para IMBRUVICA®. Los pacientes que ingirieron una dosis mayor que la dosis recomendada debieran ser vigilados estrechamente y recibir tratamiento de soporte apropiado.
PRESENTACIONES: Caja de cartón con frasco con 90 o 120 cápsulas de 140 mg e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 30°C.
Consérvese el frasco bien cerrado.
LEYENDAS DE PROTECCIÓN:
No se deje al alcance de los niños. Su venta requiere receta médica. No se use durante el embarazo, la lactancia y en menores de 18 años de edad. Literatura exclusiva para médicos. Prohibida la venta fraccionada del producto. Este medicamento puede producir mareo y afectar el estado de alerta, por lo que no deberá conducir vehículos automotores ni maquinaria pesada durante su uso. Este medicamento deberá ser recetado únicamente por médicos especialistas con experiencia en quimioterapia.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
atencionaclientes@its.jnj.com
JANSSEN-CILAG, S.A. de C.V.
Carretera Federal México-Puebla Km. 81.5,
San Mateo Capultitlán, C.P. 74160,
Huejotzingo, Puebla, México.
Registro Núm. 003M2015, SSA IV
®Marca Registrada