KEYTRUDA®
PEMBROLIZUMAB
Solución
1 Caja, 1 Frasco(s) ámpula, 100/4 mg/ml
III. FORMA FARMACÉUTICA Y FORMULACIÓN
Forma Farmacéutica: Solución.
Fórmula:
El frasco ámpula contiene:
Pembrolizumab 100 mg
Vehículo cs 4 mL
Anticuerpo monoclonal humanizado IgG4 de origen ADN recombinante expresado en células de ovario de hámster chino (CHO).
XIX. NÚMERO DE REGISTRO DEL MEDICAMENTO ANTE LA SECRETARÍA
Reg. No. 277M2016 SSA IV
Número Tracer: MK3475-MEX-2019-020702
No. de Entrada: 193300415U0004
Número de Tracer cambios COFEPRIS: 3475-MEX-2019-021305
CCDS-MK3475-IV-092018 & S-CCDS-MK3475-IV-102018
IV. INDICACIONES TERAPÉUTICAS
Melanoma
KEYTRUDA (pembrolizumab) está indicado para el tratamiento de pacientes con melanoma no resecable o metastásico.
Cáncer de Pulmón de Células No Pequeñas
KEYTRUDA, como monoterapia, está indicado para la primera línea de tratamiento de pacientes con cáncer de pulmón de células no pequeñas (NSCLC, por sus siglas en inglés) metastásico cuyos tumores expresen PD-L1 con puntuación de proporción de expresión tumoral (TPS, por sus siglas en inglés) ≥50% determinado por una prueba validada, sin aberraciones genómicas tumorales EGFR y ALK.
KEYTRUDA, en combinación con quimioterapia con pemetrexed y platino, está indicado para la primera línea de tratamiento de pacientes con NSCLC no escamoso, metastásico, sin aberraciones tumorales genómicas EGFR o ALK.
KEYTRUDA, en combinación con carboplatino y paclitaxel, o nab-paclitaxel, está indicado para el tratamiento de primera línea de pacientes con NSCLC escamoso metastásico.
KEYTRUDA, como monoterapia, está indicado para el tratamiento de pacientes con NSCLC avanzado, cuyos tumores expresen PD-L1 con un TPS ≥1%, determinado por una prueba validada y que han recibido quimioterapia que contenga platino. Los pacientes con aberraciones genómicas tumorales EGFR o ALK deben haber recibido terapia previa para estas aberraciones antes de recibir KEYTRUDA.
Carcinoma Urotelial
KEYTRUDA está indicado para el tratamiento de pacientes con carcinoma urotelial localmente avanzado o metastásico, que han recibido quimioterapia que contiene platino.
V. FARMACOCINÉTICA Y FARMACODINAMIA
Clase Terapéutica
KEYTRUDA (pembrolizumab) es un agente antineoplásico de la clase de los anticuerpos monoclonales.
Mecanismo de Acción
PD-1 es un receptor de control inmunológico que limita la actividad de los linfocitos T en los tejidos periféricos. La vía del PD-1 es un punto de control inmunológico que puede estar comprometido por las células tumorales para inhibir la vigilancia inmunológica de las células T activas. KEYTRUDA es un anticuerpo de alta afinidad frente al PD-1, que ejerce un doble bloqueo del ligando de la vía PD-1, incluyendo PD-L1 y PD-L2, en células presentadoras de antígeno o tumorales. Al inhibir al receptor PD-1 para unirse a sus ligandos, KEYTRUDA reactiva los linfocitos T citotóxicos específicos para tumor en el microambiente tumoral y reactiva la inmunidad antitumoral.
Farmacodinamia
En la sangre periférica de pacientes que recibieron 2 mg/kg de KEYTRUDA cada 3 semanas o 10 mg/kg cada 2 o 3 semanas, se observó un aumento en el porcentaje de linfocitos T CD4+ y CD8+ activados (es decir, HLA-DR+) después del tratamiento en todas las dosis y esquemas sin un aumento en el número de linfocitos T circulantes.
Farmacocinética
Se estudió la farmacocinética de pembrolizumab en 2,993 pacientes con distintos tipos de cáncer que recibieron dosis en el rango de 1 a 10 mg/kg cada 2 semanas, de 2 a 10 mg/kg cada 3 semanas, o 200 mg cada 3 semanas. No hay diferencias clínicamente significativas en la farmacocinética de pembrolizumab a través de las indicaciones.
Absorción
KEYTRUDA se administra por vía IV y por lo tanto está biodisponible completamente y de inmediato.
Distribución
De manera consistente con una distribución extravascular limitada, el volumen de distribución de pembrolizumab en estado de equilibrio farmacocinético es pequeño (6.0 L; coeficiente de variación [CV]: 20%). Como se espera de un anticuerpo, pembrolizumab no se une a las proteínas plasmáticas de una manera específica.
Metabolismo
Pembrolizumab se cataboliza a través de vías no específicas; el metabolismo no contribuye a su depuración.
Eliminación
La depuración de pembrolizumab (CV%) es aproximadamente 23% menor [media geométrica, 195 mL/día (40%)] después de alcanzar el cambio máximo en el estado de equilibrio farmacocinético, comparado con la primera dosis (252 mL/día [CV%: 37%]); esta disminución en la eliminación con el tiempo no se considera como clínicamente importante. El valor de la media geométrica (CV%) para la vida media (t½) terminal es de 22 días (32%).
Las concentraciones de pembrolizumab en estado de equilibrio se alcanzaron a las 16 semanas después de dosificación repetida con un esquema de cada 3 semanas y la acumulación sistémica fue de 2.1 veces. La concentración máxima (Cmáx), concentración mínima (Cmín) y el área bajo la curva de concentración plasmática versus la curva de tiempo (AUCss) en el estado de equilibrio de pembrolizumab incrementó de manera proporcional a la dosis en el rango de dosis de 2 a 10 mg/kg cada 3 semanas.
Poblaciones Especiales
En análisis farmacocinéticos de la población se evaluaron los efectos de varias covariables sobre la farmacocinética de pembrolizumab. Los siguientes factores no tuvieron un efecto clínicamente importante en la depuración de pembrolizumab: edad (rango 15-94 años), sexo, raza, insuficiencia renal leve o moderada, insuficiencia hepática leve y carga tumoral. La relación entre el peso corporal y la depuración apoya el uso, ya sea de la dosis fija o la dosis basada en el peso corporal, para proporcionar un control a la exposición adecuado y similar.
Insuficiencia Renal
Mediante análisis farmacocinéticos en población se evaluó el efecto de la insuficiencia renal sobre la depuración de pembrolizumab, en pacientes con insuficiencia renal leve (TFG <90 y ≥60 mL/min/1.73 m2) o moderada (TFG <60 y ≥30 mL/min/1.73 m2), comparado con pacientes con función renal normal (TFG ≥90 mL/min/1.73 m2). No se encontraron diferencias clínicamente importantes en la depuración de pembrolizumab entre los pacientes con insuficiencia renal leve o moderada y en pacientes con función renal normal. No se ha estudiado KEYTRUDA en pacientes con insuficiencia renal grave (TFG <30 y ≥15 mL/min/1.73 m2) [ver XIII. Dosis y Vía de Administración].
Insuficiencia Hepática
Mediante análisis farmacocinéticos de población se evaluó el efecto de la insuficiencia hepática sobre la depuración de pembrolizumab, en pacientes con insuficiencia hepática leve (bilirrubina total [TB] 1.0 a 1.5 x ULN (límite superior normal, por sus siglas en inglés), o AST >ULN como se define utilizando los criterios del Instituto Nacional del Cáncer (EE. UU.) de insuficiencia hepática, comparado con pacientes con función hepática normal (TB y AST ≤ULN). No se encontraron diferencias clínicamente importantes en la depuración de pembrolizumab entre los pacientes con insuficiencia hepática leve y en pacientes con función hepática normal. No se ha estudiado KEYTRUDA en pacientes con insuficiencia hepática moderada (TB >1.5 a 3 x ULN y cualquier valor de AST) o grave (TB >3 x ULN y cualquier valor de AST) [ver XIII. Dosis y Vía de Administración].
ESTUDIOS CLÍNICOS
Eficacia y seguridad clínicas
Melanoma
KEYNOTE-006: Ensayo clínico controlado en pacientes con melanoma que no habían recibido tratamiento con Ipilimumab
Se investigaron la eficacia y seguridad de KEYTRUDA en KEYNOTE-006, un estudio Fase III multicéntrico, controlado, para el tratamiento de melanoma no resecable o metastásico en pacientes que no habían recibido ipilimumab y que previamente no habían recibido tratamiento sistémico o habían recibido solamente uno. Se aleatorizó a los pacientes (1:1:1) para recibir KEYTRUDA a una dosis de 10 mg/kg cada 2 (n=279) o 3 semanas (n=277) o ipilimumab (n=278). La aleatorización fue estratificada por línea de tratamiento, estatus ECOG y expresión de PD-L1. El estudio excluyó a pacientes con enfermedad autoinmune o los que estaban recibiendo inmunosupresión; hipersensibilidad grave previa a otros anticuerpos monoclonales; e infección por VIH, hepatitis B o hepatitis C. No se requirió que los pacientes con melanoma con mutación BRAF V600E hubieran recibido previamente terapia con inhibidores de BRAF.
Los pacientes fueron tratados con KEYTRUDA hasta la progresión de la enfermedad o la presencia de toxicidad inaceptable. Se permitió que los pacientes estables clínicamente con evidencia inicial de progresión de la enfermedad permanecieran en tratamiento, hasta la confirmación de la progresión. La evaluación del estatus del tumor se realizó a las 12 semanas, posteriormente cada 6 semanas hasta la semana 48, seguidos cada 12 semanas de ahí en adelante.
De los 834 pacientes en KEYNOTE-006, 60% fueron varones, 44% tenían ≥65 años (la mediana de la edad fue de 62 años [rango 18-89]) y el 98% eran blancos. El 66% no tenían tratamientos sistémicos previos y por lo tanto recibieron el tratamiento de estudio como primera línea, mientras que el 34% tuvieron un tratamiento previo y por lo tanto recibieron el tratamiento de estudio como segunda línea. Treinta y uno por ciento tenían un ECOG de 1 y 69% un ECOG de 0. El 80% de los pacientes eran positivos a PD-L1 (con expresión de PD-L1 en membrana en ≥1% del tumor y de las células inmunes asociadas evaluados de manera prospectiva por un ensayo de inmunohistoquímica con el anticuerpo 22C3 anti-PD-L1) y 18% eran PD-L1 negativos. El 65% de los pacientes estaba en etapa M1c, 32% tenía LDH elevada y 9% tenían metástasis cerebrales. Se reportaron mutaciones BRAF en 302 (36%) de los pacientes. Entre los pacientes con tumores con mutación BRAF, 139 (46%) fueron tratados previamente con un inhibidor de BRAF. Las características basales estaban equilibradas entre los brazos de tratamiento.
Las mediciones primarias de eficacia fueron supervivencia global (OS, por las siglas en inglés para overall survival) y supervivencia libre de progresión (PFS, por progression free survival) (evaluado por Revisión Integrada de Radiología y Oncología [IRO], utilizando Criterios de Evaluación de Respuesta en Tumores Sólidos [RECIST 1.1]). Las mediciones secundarias de eficacia fueron tasa de respuesta global (ORR, overall response rate) y duración de la respuesta. La tabla 1 resume las mediciones clave de eficacia.
Tabla 1. Respuesta a 10 mg/kg de KEYTRUDA cada 2 o 3 semanas en pacientes con melanoma avanzado que no habían recibido KEYTRUDA, en KEYNOTE-006
|
Desenlace |
KEYTRUDA 10 mg/kg cada 3 semanas n=277 |
KEYTRUDA 10 mg/kg cada 2 semanas n=279 |
Ipilimumab n=278 |
|
OS* |
|||
|
Número (%) de pacientes con evento |
92 (33%) |
85 (30%) |
112 (40%) |
|
Hazard Ratio† (95% CI) |
0.69 (0.52, 0.90) |
0.63 (0.47, 0.83) |
--- |
|
Valor de p‡ |
0.00358 |
0.00052 |
--- |
|
Mediana en meses (95% CI) |
No alcanzado (NA, NA) |
No alcanzado (NA, NA) |
No alcanzado (13, NA) |
|
PFS§ por IRO¶ |
|||
|
Número (%) de pacientes con evento |
157 (57%) |
157 (56%) |
188 (68%) |
|
Hazard Ratio† (95% CI) |
0.58 (0.47, 0.72) |
0.58 (0.46, 0.72) |
--- |
|
Valor de p‡ |
<0.00001 |
<0.00001 |
--- |
|
Mediana en meses (95% CI) |
4.1 (2.9, 6.9) |
5.5 (3.4, 6.9) |
2.8 (2.8, 2.9) |
|
Mejor respuesta§ global por IRO¶ |
|||
|
ORR % (95% CI) |
33% (27, 39) |
34% (28, 40) |
12% (8, 16) |
|
Respuesta completa % |
6% |
5% |
1% |
|
Respuesta parcial % |
27% |
29% |
10% |
|
Duración de la respuesta# por IRO¶ |
|||
|
Mediana en meses (rango) |
No alcanzado (2.0+, 22.8+) |
No alcanzado (1.8+, 22.8) |
No alcanzado (1.1+, 23.8+) |
|
% en curso a 12 mesesþ |
79% |
75% |
79% |
* Basado en el segundo análisis interino.
† Hazard Ratio (KEYTRUDA comparado con ipilimumab) basado en el modelo de riesgo proporcional estratificado de Cox.
‡ Basado en prueba estratificada de rango logarítmico (Log Rank).
§ Basado en el primer análisis interino.
¶ IRO = Revisión por radiólogo independiente más revisión por oncólogo, utilizando RECIST 1.1.
# Basado en pacientes con la mejor respuesta global como la respuesta completa o parcial confirmada a partir del análisis final.
þ Basado en los estimados de Kaplan Meier.
NA = no disponible.
El análisis final se llevó a cabo después de que todos los pacientes tuvieron al menos 21 meses de seguimiento. El análisis de OS final se llevó a cabo después de 383 eventos-paciente (119 para KEYTRUDA de 10 mg/kg cada 3 semanas, 122 de KEYTRUDA de 10 mg/kg cada 2 semanas y 142 para ipilimumab). Las HRs de OS vs. Ipilimumab fueron 0.68 (95% CI: 0.53, 0.86; p<0.001) para pacientes tratados con KEYTRUDA 10 mg/kg cada 3 semanas y 0.68 (95% CI: 0.53, 0.87; p<0.001) para pacientes tratados con KEYTRUDA 10 mg/kg cada 2 semanas. La tasa de OS a los 18 meses y 24 meses fueron 62% y 55% respectivamente para KEYTRUDA 10 mg/kg cada 3 semanas; 60% y 55% respectivamente para KEYTRUDA 10 mg/kg cada 2 semanas; y 47% y 43% respectivamente para ipilimumab.
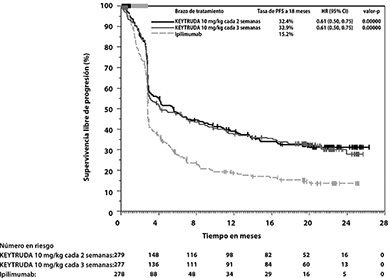
En el análisis final, un análisis de PFS a largo plazo se llevó a cabo con base en 566 eventos-paciente (183 para KEYTRUDA 10 mg/kg cada 3 semanas, 181 para KEYTRUDA 10 mg/kg cada 2 semanas y 202 para ipilimumab). Las HRs de PFS vs. ipilimumab fueron 0.61 (95% CI: 0.50, 0.75) para pacientes tratados con KEYTRUDA 10 mg/kg cada 3 semanas y 0.61 (95% CI: 0.50, 0.75) para pacientes tratados con KEYTRUDA 10 mg/kg cada 2 semanas. (Ver Figuras 1 y 2). El porcentaje de respondedores con una respuesta en curso a 18 meses fue 68% para KEYTRUDA 10 mg/kg cada 3 semanas; 71% para KEYTRUDA 10 mg/kg cada 2 semanas y 70% para ipilimumab.
Figura 1. Gráfica de Kaplan-Meier para supervivencia global por brazo de tratamiento en KEYNOTE-006 (población por intención a tratar)
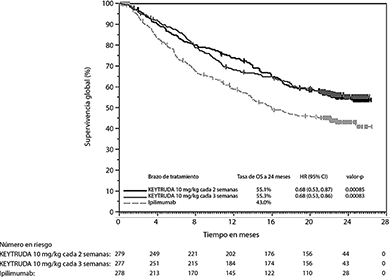
Figura 2. Gráfica de Kaplan-Meier para supervivencia libre de progresión (basada en IRO) por brazo de tratamiento en KEYNOTE-006 (población por intención a tratar)
Análisis de sub-poblaciones por mutación BRAF
Se realizó un análisis de subgrupos como parte del análisis final de los pacientes en KEYNOTE-006 que tenían BRAF tipo nativo, mutante BRAF sin tratamiento BRAF previo y mutante BRAF con tratamiento BRAF previo. Las hazard ratios (HRs) de PFS (KEYTRUDA agrupados [10 mg/kg cada 2 o 3 semanas] vs ipilimumab) fueron 0.61 (95% CI: 0.49, 0.76) para BRAF tipo nativo, 0.52 (95% CI: 0.35, 0.78) para mutante BRAF sin tratamiento previo y 0.76 (95% CI: 0.51, 1.14) para mutante BRAF con tratamiento previo. Las HRs para OS para los pacientes de KEYTRUDA agrupados vs ipilimumab fueron 0.68 (95% CI: 0.52, 0.88) para BRAF de tipo nativo, 0.70 (95% CI: 0.40, 1.22) para mutante BRAF sin tratamiento BRAF previo y 0.66 (95% CI: 0.41, 1.04) para mutante BRAF con tratamiento BRAF previo. Las ORR para pacientes de KEYTRUDA agrupados vs ipilimumab fueron 38% vs 14% para BRAF tipo nativo, 41% vs 15% para mutante BRAF sin tratamiento BRAF previo y 24% vs 10% para mutante BRAF con tratamiento BRAF previo.
Análisis de sub-poblaciones por estatus PD-L1
Se realizó un análisis de subgrupos como parte del análisis final de KEYNOTE-006 en pacientes que eran PD-L1 positivos vs PD-L1 negativos. Las HRs para PFS (pacientes de KEYTRUDA agrupados [10 mg/kg cada 2 o 3 semanas] vs ipilimumab) fueron 0.53 (95% CI: 0.44, 0.65) para pacientes PD-L1 positivos y de 0.87 (95% CI: 0.58, 1.30) para pacientes PD-L1 negativos. Las HRs para OS para pacientes de KEYTRUDA agrupados vs ipilimumab fueron 0.63 (95% CI: 0.50, 0.80) para pacientes PD-L1 positivos y de 0.76 (95% CI: 0.48, 1.19) para pacientes PD-L1 negativos.
KEYNOTE-002: Ensayo clínico controlado en pacientes con melanoma tratados previamente con ipilimumab
Se investigaron la eficacia y seguridad de KEYTRUDA en KEYNOTE-002, un estudio multicéntrico, controlado, para el tratamiento de melanoma no resecable o metastásico en pacientes tratados previamente con ipilimumab y si la mutación BRAF V600 era positiva, un inhibidor de BRAF o MEK. Se aleatorizó a los pacientes (1:1:1) para recibir KEYTRUDA a una dosis de 2 (n=180) o 10 mg/kg (n=181) cada 3 semanas o quimioterapia (n=179; incluyendo dacarbazina, temozolomida, carboplatino, paclitaxel, o carboplatino+paclitaxel). El estudio excluyó a pacientes con enfermedad autoinmune o los que estaban recibiendo tratamiento inmunosupresor, antecedentes de reacciones adversas inmunomediadas graves o que amenazaron la vida, por el tratamiento con ipilimumab, definido como cualquier toxicidad Grado 4 o Grado 3 que requiriera tratamiento con corticosteroides (mayor de 10 mg/día de prednisona o dosis equivalente) durante más de 12 semanas; hipersensibilidad grave previa a otros anticuerpos monoclonales; antecedentes de neumonitis o enfermedad pulmonar intersticial; infección por VIH, hepatitis B o hepatitis C.
Los pacientes fueron tratados con KEYTRUDA hasta la progresión de la enfermedad o toxicidad inaceptable. Se permitió que los pacientes clínicamente estables con evidencia inicial de progresión de la enfermedad permanecieran en tratamiento hasta que se confirmara la progresión de la enfermedad. La evaluación del estatus del tumor se realizó a las 12 semanas, luego cada 6 semanas hasta la semana 48, seguido por cada 12 semanas de ahí en adelante. Los pacientes con quimioterapia que experimentaron progresión de la enfermedad verificada de manera independiente después de la primera evaluación calendarizada de la enfermedad tuvieron acceso al cruzamiento y a recibir 2 mg/kg o 10 mg/kg de KEYTRUDA cada 3 semanas de manera doble ciego.
De los 540 pacientes en KEYNOTE-002, 61% eran varones, 43% tenían ≥65 años (la mediana de la edad fue de 62 años [rango 15-89] y 98% eran blancos. El 82% de los pacientes tenían etapa M1c, 73% tenían al menos dos y 32% al menos tres tratamientos sistémicos previos para melanoma avanzado. Cuarenta y cinco por ciento tenía un ECOG de 1, 40% tenían LDH elevada y 23% tenían un tumor con mutación BRAF. Las características basales estaban equilibradas entre los brazos de tratamiento.
Las medidas primarias de eficacia fueron PFS (evaluado por revisión de IRO utilizando RECIST 1.1) y OS. Las mediciones secundarias de eficacia fueron PFS (evaluada por el Investigador utilizando RECIST 1.1), ORR y duración de la respuesta. La Tabla 2 resume las mediciones clave de eficacia en pacientes tratados previamente con ipilimumab y la gráfica de Kaplan-Meier para PFS se muestra en la Figura 3. No hubo una diferencia estadísticamente significativa entre KEYTRUDA y quimioterapia en el análisis final de OS, que no fue ajustada por los efectos de confusión potenciales del cruzamiento. De los pacientes aleatorizados al brazo de quimioterapia, el 55% fueron cruzados y recibieron subsecuentemente tratamiento con KEYTRUDA.
Tabla 2. Respuesta a 2 mg/kg o 10 mg/kg de KEYTRUDA cada 3 semanas en pacientes con melanoma no resecable o metastásico en KEYNOTE-002
|
Desenlace |
KEYTRUDA 2 mg/kg cada 3 semanas n=180 |
KEYTRUDA 10 mg/kg cada 3 semanas n=181 |
Quimioterapia n=179 |
|
OS* |
|||
|
Número (%) de pacientes con evento |
123 (68%) |
117 (65%) |
128 (72%) |
|
Hazard Ratio† (95% CI) |
0.86 (0.67, 1.10) |
0.74 (0.57, 0.96) |
--- |
|
Valor de p‡ |
0.117 |
0.011è |
--- |
|
Mediana en meses (95% CI) |
13.4 (11.0, 16.4) |
14.7 (11.3, 19.5) |
11.0 (8.9, 13.8) |
|
PFS§ por IRO¶ |
|||
|
Número (%) de pacientes con evento |
129 (72%) |
126 (70%) |
155 (87%) |
|
Hazard Ratio† (95% CI) |
0.57 (0.45, 0.73) |
0.50 (0.39, 0.64) |
--- |
|
Valor de p‡ |
<0.0001 |
<0.0001 |
--- |
|
Mediana en meses (95% CI) |
2.9 (2.8, 3.8) |
2.9 (2.8, 4.7) |
2.7 (2.5, 2.8) |
|
Promedio en meses (95% CI)# |
5.4 (4.7, 6.0) |
5.8 (5.1, 6.4) |
3.6 (3.2, 4.1) |
|
PFS§ por INVþ |
|||
|
Número (%) de pacientes con evento |
122 (68%) |
112 (62%) |
157 (88%) |
|
Hazard Ratio† (95% CI) |
0.49 (0.38, 0.62) |
0.41 (0.32, 0.52) |
--- |
|
Valor de p‡ |
<0.0001 |
<0.0001 |
--- |
|
Mediana en meses (95% CI) |
3.7 (2.9, 5.4) |
5.4 (3.8, 6.8) |
2.6 (2.4, 2.8) |
|
Promedio en meses (95% CI)# |
5.8 (5.2, 6.4) |
6.5 (5.8, 7.1) |
3.7 (3.2, 4.1) |
|
Mejor respuesta§ global por IRO¶ |
|||
|
ORR % (95% CI) |
21% (15, 28) |
25% (19, 32) |
4% (2, 9) |
|
Respuesta completa % |
2% |
3% |
0% |
|
Respuesta parcial % |
19% |
23% |
4% |
|
Duraciónβ de la respuesta IRO¶ |
|||
|
Mediana en meses (rango) |
22.8 (1.4+, 25.3+) |
No alcanzado (1.1+, 28.3+) |
6.8 (2.8+, 11.3) |
|
% en curso a 12 mesesà |
73% |
79% |
No alcanzadoð |
* Basado en el análisis final.
† Hazard Ratio (KEYTRUDA comparado con quimioterapia) basada en el modelo de riesgo proporcional estratificado de Cox.
‡ Basado en prueba estratificada de rango logarítmico.
§ Basado en el segundo análisis interino.
¶ IRO = Revisión por radiólogo independiente más revisión de oncólogo, utilizando RECIST 1.1.
# Tiempo de supervivencia libre de progresión restringido, con base en un seguimiento de 12 meses.
þ INV = evaluación por el investigador utilizando RECIST 1.1.
β Basado en pacientes con mejor respuesta global, según respuesta completa o parcial confirmada a partir del análisis final.
à Basado en los estimados de Kaplan-Meier.
è No estadísticamente significativo después del ajuste para multiplicidad.
ð El seguimiento máximo para los pacientes en curso en el brazo de quimioterapia es 11.3 meses; los pacientes continúan para seguimiento.
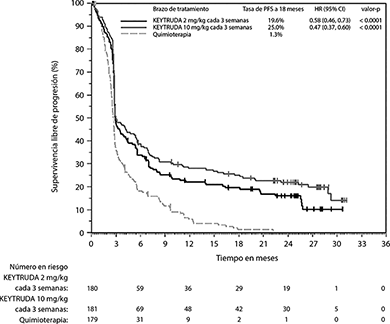
En el análisis final, un análisis de PFS a largo plazo se llevó a cabo basado en 466 eventos de PFS (150 para KEYTRUDA 2 mg/kg cada 3 semanas; 144 para KEYTRUDA 10 mg/kg cada 3 semanas y 172 para quimioterapia). Las HRs de PFS vs. quimioterapia fueron 0.58 (95% CI: 0.46, 0.73) para los pacientes tratados con KEYTRUDA 2 mg/kg cada 3 semanas y 0.47 (95% CI: 0.37, 0.60) para pacientes tratados con KEYTRUDA 10 mg/kg cada 3 semanas (Figura 3).
Figura 3. Gráfica de Kaplan-Meier para supervivencia libre de progresión (basada en IRO) por brazo de tratamiento en KEYNOTE-002 (población por intención a tratar)
KEYNOTE-001: Estudio abierto en pacientes con melanoma
La seguridad y eficacia de KEYTRUDA también se investigaron en un estudio no controlado, abierto, para el tratamiento de melanoma no resecable o metastásico. La eficacia fue evaluada para 276 pacientes de dos cohortes definidas de KEYNOTE-001, una que incluyó pacientes previamente tratados con ipilimumab (y si eran positivos a la mutación BRAF V600), un inhibidor de BRAF o MEK) y otra que incluyó pacientes sin tratamiento previo con ipilimumab. Los pacientes fueron aleatorizados para recibir KEYTRUDA a una dosis de 2 mg/kg cada 3 semanas o 10 mg/kg cada 3 semanas. Los pacientes fueron tratados con KEYTRUDA hasta la progresión de la enfermedad o toxicidad inaceptable. Se permitió que los pacientes clínicamente estables con evidencia inicial de progresión de la enfermedad, permanecieran con el tratamiento, hasta que se confirmara la progresión de la enfermedad. Los criterios de exclusión fueron similares a los de KEYNOTE-002.
De los 89 pacientes que recibieron 2 mg/kg de KEYTRUDA tratados previamente con ipilimumab, 53% eran varones, 33% tenían ≥65 años de edad y la mediana de la edad fue de 59 años (rango 18-88). Todos, excepto dos pacientes eran blancos. El 84% de los pacientes tenía etapa M1c y 8% tenían antecedentes de metástasis cerebrales. Setenta y ocho por ciento de los pacientes tenía al menos dos y 35% tenía tres o más tratamientos sistémicos previos para melanoma avanzado. Se reportaron mutaciones BRAF en el 13% de la población del estudio.
De los 51 pacientes que recibieron 2 mg/kg de KEYTRUDA que no habían recibido tratamiento con ipilimumab, 63% eran varones, 35% tenían ≥65 años de edad y la mediana de edad era de 60 años (rango 35-80). Todos, excepto un paciente eran blancos. El 63% de los pacientes tenían etapa M1c y 2% antecedentes de metástasis cerebrales. El 45% no tenía tratamientos previos para melanoma avanzado. Se reportaron mutaciones BRAF en el 39% de la población del estudio.
La medición primaria de eficacia fue ORR, evaluada por revisión independiente, utilizando respuestas confirmadas y RECIST 1.1. Las mediciones secundarias de eficacia fueron tasa de control de la enfermedad (DCR; incluyendo respuesta completa, respuesta parcial y enfermedad estable), duración de la respuesta, PFS y OS. La respuesta del tumor se evaluó a intervalos de 12 semanas. La Tabla 3 resume las mediciones clave de eficacia en los pacientes, tratados previamente o sin tratamiento previo con ipilimumab, que recibieron KEYTRUDA a una dosis de 2 mg/kg en un tiempo de seguimiento mínimo de 30 meses para todos los pacientes.
Tabla 3. Respuesta a 2 mg/kg de KEYTRUDA cada 3 semanas en pacientes con melanoma no resecable o metastásico en KEYNOTE-001
|
Desenlace |
KEYTRUDA 2 mg/kg cada 3 semanas en pacientes tratados previamente con ipilimumab n=89 |
KEYTRUDA 2 mg/kg cada 3 semanas en pacientes sin tratamiento previo con ipilimumab n=51 |
|
Mejor Respuesta* Global por IRO† |
||
|
ORR %, (95% CI) |
26% (17, 36) |
35% (22, 50) |
|
Tasa de Control de la Enfermedad %‡ |
48% |
49% |
|
Respuesta completa |
7% |
12% |
|
Respuesta parcial |
19% |
24% |
|
Enfermedad estable |
20% |
14% |
|
Duración de la Respuesta§ |
||
|
Mediana en meses (rango) |
30.5 (2.8+, 30.6+) |
27.4 (1.6+, 31.8+) |
|
% en curso a 24 meses¶ |
75% |
71%# |
|
PFS |
||
|
Mediana en meses (95% CI) |
4.9 (2.8, 8.3) |
4.7 (2.8, 13.8) |
|
Tasa de PFS a 12 meses |
34% |
38% |
|
OS |
||
|
Mediana en meses (95% CI) |
18.9 (11, no disponible) |
28.0 (14, no disponible) |
|
Tasa de OS a 24 meses |
44% |
56% |
* Incluye pacientes sin enfermedad medible a nivel basal por radiólogo independiente.
† IRO = Revisión por radiólogo independiente más revisión por oncólogo utilizando RECIST 1.1.
‡ Basado en la mejor respuesta de enfermedad estable o mejor.
§ Basado en pacientes con una respuesta confirmada por un revisor independiente, iniciando desde la fecha en que la respuesta se registró por vez primera; n=23 para pacientes tratados previamente con ipilimumab; n=18 para pacientes sin tratamiento previo con ipilimumab.
¶ Basado en la estimación de Kaplan-Meier.
Los resultados para pacientes tratados previamente con ipilimumab (n=84) y sin tratamiento previo con ipilimumab (n=52) que recibieron 10 mg/kg de KEYTRUDA cada 3 semanas fueron similares a los observados en los pacientes que recibieron 2 mg/kg de KEYTRUDA cada 3 semanas.
Cáncer de Pulmón de Células No Pequeñas
KEYNOTE-024: Ensayo clínico controlado en pacientes con NSCLC que no habían recibido tratamiento previo
La eficacia de KEYTRUDA se investigó en KEYNOTE-024, un ensayo clínico multicéntrico, aleatorizado, controlado. Los criterios clave de elección fueron NSCLC metastásico, puntuación de proporción de expresión tumoral (TPS) de 50% o mayor por una prueba inmunohistoquímica usando el kit PD-L1 IHC 22C3 pharmaDx, y sin tratamiento sistémico previo para NSCLC metastásico. Fueron inelegibles los pacientes con aberraciones tumorales genómicas EGFR y ALK; enfermedad autoinmune que requiera terapia sistémica dentro de los 2 años de tratamiento; una condición médica que requiera inmunosupresión; o quien había recibido más de 30 Gy de radiación torácica dentro de las 26 semanas previas. Los pacientes se aleatorizaron (1:1) para recibir 200 mg de KEYTRUDA cada 3 semanas (n=154) o la quimioterapia con platino de elección del investigador (n=151; incluyendo pemetrexed+carboplatino, pemetrexed+cisplatino, gemcitabina+cisplatino, gemcitabina+carboplatino o paclitaxel+carboplatino. Los pacientes con cáncer no escamoso podrían recibir mantenimiento con pemetrexed). Los pacientes se trataron con KEYTRUDA hasta una toxicidad inaceptable o progresión de la enfermedad. El tratamiento podría continuar más allá de la progresión de la enfermedad si el paciente fue clínicamente estable y se consideró por el investigador que se deriva un beneficio clínico. Los pacientes sin progresión de la enfermedad se podrían tratar por hasta 24 meses. El tratamiento con KEYTRUDA se podría reiniciar para la subsecuente progresión de la enfermedad y administrar hasta 1 año adicional. La evaluación del estatus del tumor se llevó a cabo cada 9 semanas. Los pacientes en quimioterapia que experimentaron progresión de la enfermedad verificada de manera independiente fueron capaces del cruce y recibir KEYTRUDA.
Entre los 305 pacientes en KEYNOTE-024, las características basales fueron; mediana de la edad 65 años (54% de 65 años o más); 61% varones; 82% blancos y 15% asiáticos; y 35% y 65% con un estatus ECOG de 0 y 1, respectivamente. Las características de la enfermedad fueron escamoso (18%) y no escamoso (82%); M1 (99%); y metástasis cerebral (9%).
La medición primaria de los resultados de eficacia fue PFS como se evaluó por la revisión central, independiente y cegada (BICR) usando RECIST 1.1 Las mediciones secundarias de los resultados de eficacia fueron OS y ORR (como se evaluó por la BICR usando RECIST 1.1). La Tabla 4 resume las mediciones de eficacia claves para la población ITT completa.
Tabla 4: Resultados de Eficacia en KEYNOTE-024
|
Desenlace |
KEYTRUDA 200 mg cada 3 semanas |
Quimioterapia |
|
n=154 |
n=151 |
|
|
PFS* |
||
|
Número (%) de pacientes con evento |
73 (47%) |
116 (77%) |
|
Hazard Ratio† (95% CI) |
0.50 (0.37, 0.68) |
|
|
Valor de p‡ |
<0.001 |
|
|
Mediana en meses (95% CI) |
10.3 (6.7, NA) |
6.0 (4.2, 6.2) |
|
OS |
||
|
Número (%) de pacientes con evento |
44 (29%) |
64 (42%) |
|
Hazard Ratio† (95% CI) |
0.60 (0.41, 0.89) |
|
|
Valor de p‡ |
0.005 |
|
|
Mediana en meses (95% CI) |
No alcanzado (NA, NA) |
No alcanzado (9.4, NA) |
|
Proporción de la respuesta objetiva* |
||
|
ORR % (95% CI) |
45% (37, 53) |
28% (21, 36) |
|
Respuesta completa % |
4% |
1% |
|
Respuesta parcial % |
41% |
27% |
|
Duración de la respuesta§ |
||
|
Mediana en meses (rango) |
No alcanzado (1.9+, 14.5+) |
6.3 (2.1+, 12.6+) |
|
% con duración ≥ 6 meses |
88%¶ |
59%# |
* Evaluado por BICR utilizando RECIST 1.1.
† Hazard Ratio (KEYTRUDA comparado con quimioterapia) basada en el modelo de riesgo proporcional estratificado de Cox.
‡ Basado en prueba estratificada de rango logarítmico.
§ Basado en pacientes con mejor respuesta global, según respuesta completa o parcial confirmada.
¶ Basada en los estimados de Kaplan-Meier; incluye 43 pacientes con respuestas de 6 meses o por más tiempo.
# Basada en los estimados de Kaplan-Meier; incluye 16 pacientes con respuestas de 6 meses o por más tiempo.
NA = no disponible.
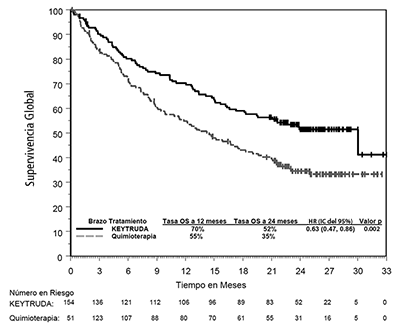
El análisis final de OS fue realizado a una mediana de seguimiento de 25 meses después de 169 eventos en pacientes (73 para KEYTRUDA y 96 para quimioterapia). La OS mediana fue de 30.0 meses (IC del 95%: 18.3, NA) para KEYTRUDA y 14.2 meses (IC del 95%: 9.8, 19.0) para quimioterapia. El HR de la OS fue de 0.63 (IC del 95%: 0.47, 0.86; p=0.002). Ver la Figura 5.
Figura 4: Gráfica de Kaplan-Meier para supervivencia libre de progresión por brazo de tratamiento en KEYNOTE-024 (población por intención a tratar)
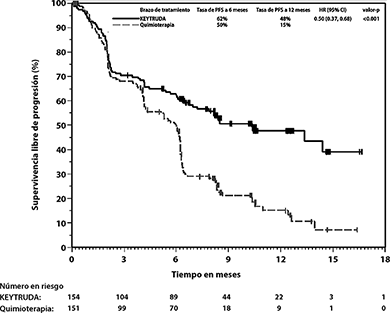
Figura 5: Gráfica de Kaplan-Meier para supervivencia global por brazo de tratamiento en KEYNOTE-024 (población por intención a tratar)
La mejoría en el beneficio como se evaluó por PFS, OS, ORR y la duración de la respuesta para KEYTRUDA como se comparó con la quimioterapia en la población estudiada se asoció con mejoras en la calidad de vida relacionadas con la salud (HRQoL). El cambio a partir de los valores basales a la semana 15 mostró una mejora significativa en el estatus global de salud/puntaje QoL del Cuestionario C30 de Calidad de Vida de la Organización Europea para la Investigación y el Tratamiento de Cáncer (EORTC QLC) C30 para los pacientes que reciben KEYTRUDA comparado con la quimioterapia (diferencia en las medias LS = 7.82; 95% CI: 2.85, 12.79; p=0.002 de dos colas). El tiempo de deterioro en el desenlace combinado en el EORTC QLQ-LC 13 de tos, disnea y dolor de pecho se prolongó para los pacientes que reciben KEYTRUDA comparado con la quimioterapia (HR = 0.66; 95% CI: 0.44, 0.97; p=0.029 de dos colas), en donde el deterioro se define como un punto 10 confirmado o una disminución de la puntuación mayor a partir del valor inicial en cualquiera de estos tres síntomas.
KEYNOTE-189: Estudio controlado de terapia de combinación en pacientes con NSCLC no escamoso sin exposición previa a tratamiento
Se investigó la eficacia de KEYTRUDA en combinación con quimioterapia con pemetrexed y platino en un estudio multicéntrico, aleatorizado, controlado con activo y doble ciego, KEYNOTE-189. Los criterios clave de elegibilidad fueron NSCLC metastásico no escamoso, ningún tratamiento sistémico previo para NSCLC metastásico y sin aberraciones tumorales genómicas EGFR o ALK. Fueron inelegibles pacientes con enfermedades autoinmunes que requirieron terapia sistémica durante 2 años de tratamiento, pacientes con alguna condición médica que requirieron inmunosupresión; o quienes habían recibido más de 30 Gy de radiación torácica dentro de las 26 semanas previas. Los pacientes fueron aleatorizados (2:1) para recibir uno de los siguientes regímenes:
• 200 mg de KEYTRUDA con 500 mg/m2 de pemetrexed y la elección del investigador entre 75 mg/m2 de cisplatino o AUC de carboplatino de 5 mg/mL/min por vía intravenosa cada 3 semanas durante 4 ciclos, seguido por 200 mg de KEYTRUDA y 500 mg/m2 de pemetrexed por vía intravenosa cada 3 semanas.
• Placebo con 500 mg/m2 de pemetrexed y la elección del investigador entre 75 mg/m2 de cisplatino o AUC de carboplatino de 5 mg/mL/min por vía intravenosa cada 3 semanas durante 4 ciclos seguido por placebo y 500 mg/m2 de pemetrexed por vía intravenosa cada 3 semanas.
El tratamiento con KEYTRUDA continuó hasta la progresión de la enfermedad definida por RECIST 1.1, según lo determinaba el investigador, toxicidad inaceptable o un máximo de 24 meses. Se permitió la administración de KEYTRUDA más allá de la progresión de la enfermedad definida por RECIST por BICR o más allá de la descontinuación de pemetrexed si el paciente estaba clínicamente estable y estaba obteniendo beneficio clínico, determinado por el investigador. Para pacientes que completaron 24 meses de tratamiento o tuvieron una respuesta completa, el tratamiento con KEYTRUDA podía re-iniciarse por progresión de la enfermedad y administrarse durante 1 año adicional. La evaluación del estatus tumoral se realizó a la Semana 6 y la Semana 12, seguido por cada 9 semanas de ahí en adelante. A los pacientes que recibieron placebo más quimioterapia quienes experimentaron progresión de la enfermedad verificada de manera independiente, se les ofreció KEYTRUDA como monoterapia.
Entre los 616 pacientes en KEYNOTE-189 (410 pacientes en el brazo de combinación de KEYTRUDA y 206 en el brazo de placebo más quimioterapia), las características basales fueron: mediana de la edad de 64 años (49% tenían 65 años de edad o mayores); 59% varones; 94% blancos y 3% asiáticos; 43% y 56% con estatus funcional ECOG de 0 o 1 respectivamente; 31% con TPS de PD-L1 <1%; y 18% con metástasis cerebrales tratadas o no tratadas a nivel basal. Un total de 67 pacientes en el brazo de placebo más quimioterapia cruzaron para recibir monoterapia con KEYTRUDA en el momento de la progresión de la enfermedad y 18 pacientes adicionales recibieron un inhibidor de puntos de control inmunitario como tratamiento subsecuente.
Las mediciones primarias de las variables de eficacia fueron OS y PFS (evaluadas por BICR utilizando RECIST 1.1). Las mediciones secundarias de las variables de eficacia fueron ORR y duración de la respuesta, evaluado por BICR utilizando RECIST 1.1. La mediana de tiempo de seguimiento fue de 10.5 meses (rango: 0.2 – 20.4 meses). La tabla 5 resume las mediciones clave de eficacia.
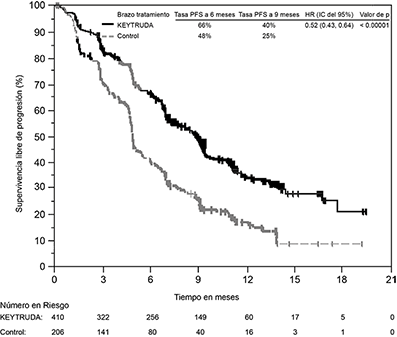
Tabla 5: Respuesta a KEYTRUDA, quimioterapia con pemetrexed y platino, en pacientes con NSCLC no escamoso en KEYNOTE-189
|
Variables de Desenlace |
KEYTRUDA + Quimioterapia con Pemetrexed + Platino n=410 |
Placebo + Quimioterapia con Pemetrexed + Platino n=206 |
|
OS |
||
|
Número (%) de pacientes con evento |
127 (31%) |
108 (52%) |
|
Hazard Ratio* (IC del 95%) |
0.49 (0.38, 0.64) |
|
|
Valor de p† |
<0.00001 |
|
|
Mediana en meses (IC del 95%) |
No alcanzado (NA, NA) |
11.3 (8.7, 15.1) |
|
PFS |
||
|
Número (%) de pacientes con evento |
244 (60%) |
166 (81%) |
|
Hazard Ratio** (IC del 95%) |
0.52 (0.43, 0.64) |
|
|
Valor de p† |
<0.00001 |
|
|
Mediana en meses (IC del 95%) |
8.8 (7.6, 9.2) |
4.9 (4.7, 5.5) |
|
Tasa de Respuesta Objetiva |
||
|
ORR‡ % (IC del 95%) |
48% (43, 53) |
19% (14, 25) |
|
Respuesta completa % |
0.5% |
0.5% |
|
Respuesta parcial % |
47% |
18% |
|
Valor de p§ |
<0.0001 |
|
|
Duración de la respuesta |
||
|
Mediana en meses (rango) |
11.2 (1.1+, 18.0+) |
7.8 (2.1+, 16.4+) |
|
% con duración ≥6 meses¶ |
81% |
63% |
|
% con duración ≥9 meses¶ |
60% |
44% |
* Con base en el modelo de riesgo proporcional estratificado de Cox.
† Con base en la prueba de rango logarítmico estratificado.
‡ Con base en pacientes con una mejor respuesta global confirmada como respuesta completa o respuesta parcial.
§ Con base en el método de Miettinen y Nurminen estratificado por estatus de PDL1, quimioterapia con platino y estatus de fumador.
¶ Con base en la estimación de Kaplan-Meier.
NA = no disponible.
Figura 6: Curva de Kaplan-Meier de supervivencia global por brazo de tratamiento en KEYNOTE-189 (población por intención a tratar)
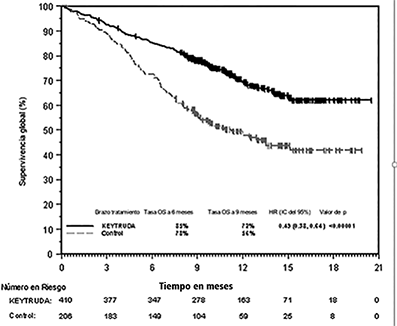
Figura 7: Curva de Kaplan-Meier para supervivencia libre de progresión por brazo de tratamiento en KEYNOTE-189 (población por intención a tratar)
Se evaluaron los desenlaces reportados por el paciente utilizando EORTC QLQ-C30 y EORTC QLQ-LC13. Los análisis exploratorios de pacientes que recibieron el tratamiento de combinación con pembrolizumab mostraron Estado Global de Salud EORTC QLQ-C30/QoL estable a la Semana 12 y la Semana 21 versus declinaciones en los pacientes que recibieron placebo más quimioterapia. Hubo una tendencia hacia una prolongación en el tiempo hasta el deterioro en el punto final de EORTC QLQ-LC13/QLQ-C30 de tos, disnea o dolor torácico observada para pacientes que recibieron tratamiento de combinación con pembrolizumab.
KEYNOTE-407: Estudio controlado de tratamiento de combinación en pacientes con NSCLC escamoso sin exposición previa a tratamiento
Se investigó la eficacia de KEYTRUDA en combinación con carboplatino y, ya sea paclitaxel o nab-paclitaxel, en el estudio KEYNOTE-407, un estudio aleatorizado, doble ciego, multicéntrico, controlado con placebo. Los criterios clave de elegibilidad para este estudio fueron NSCLC escamoso, metastásico, independientemente del estatus de expresión tumoral de PD-L1, y sin tratamiento sistémico previo para enfermedad metastásica. Fueron inelegibles pacientes con enfermedad autoinmune que requiriera tratamiento sistémico dentro de los 2 años de tratamiento; una condición médica que requiriera inmunosupresión; o quienes habían recibido más de 30 Gy de radiación torácica dentro de las 26 semanas anteriores. La aleatorización se estratificó por expresión tumoral de PD-L1 (TPS <1% [negativa] vs. TPS ≥1%), elección del investigador de paclitaxel o nab-paclitaxel, y por región geográfica (Este de Asia vs. no-Este de Asia). Los pacientes fueron aleatorizados (1:1) a uno de los siguientes tratamientos; todos los medicamentos del estudio fueron administrados vía infusión intravenosa:
• 200 mg de KEYTRUDA y AUC de carboplatino de 6 mg/mL/min el Día 1 de cada ciclo de 21 días durante 4 ciclos, y 200 mg/m2 de paclitaxel el Día 1 de cada ciclo de 21 días durante 4 ciclos o 100 mg/m2 de nab-paclitaxel los Días 1, 8 y 15 de cada ciclo de 21 días durante 4 ciclos, seguido por 200 mg de KEYTRUDA cada 3 semanas. KEYTRUDA fue administrado antes de la quimioterapia el Día 1.
• Placebo y AUC de carboplatino de 6 mg/mL/min el Día 1 de cada ciclo de 21 días durante 4 ciclos y 200 mg/m2 de paclitaxel el Día 1 de cada ciclo de 21 días durante 4 ciclos o 100 mg/m2 de nab-paclitaxel los Días 1, 8 y 15 de cada ciclo de 21 días durante 4 ciclos, seguido por placebo cada 3 semanas.
El tratamiento con KEYTRUDA o placebo continuó hasta que se determinó la progresión de la enfermedad definida por RECIST 1.1, por revisión central independiente ciega (BICR, por las siglas en inglés para Blinded Independent Central Review), toxicidad inaceptable, o un máximo de 24 meses. Se permitió la administración de KEYTRUDA más allá de la progresión de la enfermedad definida por RECIST si el paciente estaba clínicamente estable y estaba obteniendo beneficio clínico, según lo determinado por el investigador. Podía reiniciarse el tratamiento con KEYTRUDA por progresión de la enfermedad subsecuente y administrarse hasta por 1 año adicional.
A los pacientes en el brazo placebo se les ofreció KEYTRUDA como agente único en el momento de la progresión de la enfermedad.
Se realizó evaluación del estatus tumoral cada 6 semanas hasta la Semana 18, cada 9 semanas hasta la Semana 45 y cada 12 semanas en adelante. La mediciones principales de eficacia fueron supervivencia libre de progresión y tasa de respuesta objetiva (ORR, por sus siglas en inglés, Objective Response Rate), evaluado por BICR utilizando RECIST 1.1 y supervivencia en global. Una medición adicional de eficacia fue la duración de la respuesta, según lo evaluado por BICR utilizando RECIST 1.1.
Se aleatorizó un total de 559 pacientes: 278 pacientes al brazo de KEYTRUDA y 281 al brazo placebo. Las características de la población de estudio fueron: mediana de la edad de 65 años (rango: 29 a 88); 55% de 65 años de edad o mayores; 81% varones; 77% blancos; estatus de desempeño ECOG de 0 (29%) y de 1 (71%); y 8% con metástasis cerebrales tratadas a nivel basal. Treinta y cinco por ciento tenían una TPS de expresión tumoral de PD-L1 <1% [negativa]; 19% eran de la región del Este de Asia; y 60% recibieron paclitaxel.
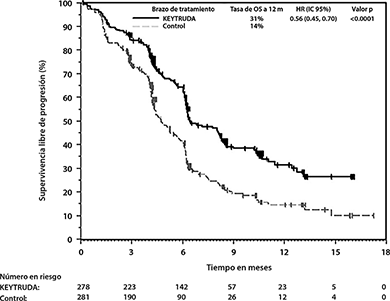
En KEYNOTE-407, hubo una mejoría estadísticamente significativa en OS, PFS y ORR en pacientes aleatorizados a KEYTRUDA en combinación con carboplatino y ya sea paclitaxel o nab-paclitaxel en comparación con pacientes aleatorizados a placebo con carboplatino y ya sea paclitaxel o nab-paclitaxel (ver Tabla 6 y Figuras 8 y 9).
Tabla 6: Resultados de Eficacia en KEYNOTE-407
|
Variables de Desenlace |
KEYTRUDA Carboplatino Paclitaxel/Nab-paclitaxel n=278 |
Placebo Carboplatino Paclitaxel/Nab-paclitaxel n=281 |
|
OS |
||
|
Número de eventos (%) |
85 (31%) |
120 (43%) |
|
Mediana en meses (IC del 95%) |
15.9 (13.2, NA) |
11.3 (9.5, 14.8) |
|
Hazard ratio* (95% IC) |
0.64 (0.49, 0.85) |
|
|
Valor de p (rango logarítmico estratificado) |
0.0008 |
|
|
PFS |
||
|
Número de eventos (%) |
152 (55%) |
197 (70%) |
|
Mediana en meses (IC del 95%) |
6.4 (6.2, 8.3) |
4.8 (4.3, 5.7) |
|
Hazard ratio* (IC del 95%) |
0.56 (0.45, 0.70) |
|
|
Valor de p (rango logarítmico estratificado) |
<0.0001 |
|
|
Tasa de Respuesta en Global |
||
|
Tasa de respuesta en global† |
58% |
38% |
|
(IC del 95%) |
(52, 64) |
(33, 44) |
|
Duración de la Respuesta |
||
|
Mediana de duración de la respuesta en meses (rango) |
7.7 (1.1+, 14.7+) |
4.8 (1.3+, 15.8+) |
|
% con duración ≥ 6 meses‡ |
62% |
40% |
* Con base en el modelo de riesgo proporcional estratificado de Cox.
† En el análisis intermedio inicial (n=101 para tratamiento de combinación con KEYTRUDA, n=102 para placebo), se observó una diferencia estadísticamente. significativa; la ORR fue de 58% [IC del 95% (48, 68)] y 35% [IC del 95% (26, 45)] para placebo, p=0.0004.
‡ Con base en estimación de Kaplan-Meier.
NA = no disponible.
Figura 8: Curva de Kaplan-Meier para Supervivencia Global en KEYNOTE-407
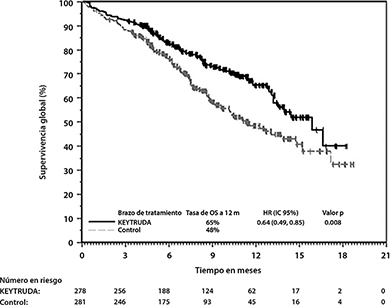
Figura 9: Curva de Kaplan-Meier para Supervivencia Libre de Progresión en KEYNOTE-407
KEYNOTE-010: Ensayo clínico controlado en pacientes con NSCLC tratados previamente con quimioterapia
Se investigó la eficacia de KEYTRUDA en KEYNOTE-010, un estudio multicéntrico, aleatorizado, controlado. Los criterios de elegibilidad fueron NSCLC avanzado que había progresado después de quimioterapia con platino, y si era adecuado, terapia dirigida para mutaciones ALK o EGFR y expresión PD-L1 de TPS 1% o mayor por una versión de ensayo clínico del kit PD-L1 IHC 22C3 pharmDx™. Fueron inelegibles pacientes con enfermedad autoinmune; una condición médica que requería inmunosupresión; o que habían recibido más de 30 Gy de radiación torácica dentro de las 26 semanas previas. Se aleatorizó a los pacientes (1:1:1) para recibir 2 mg/kg (n=344) o 10 mg/kg (n=346) de KEYTRUDA cada 3 semanas o 75 mg/m2 de docetaxel cada 3 semanas (n=343). Los pacientes fueron tratados con KEYTRUDA hasta la progresión de la enfermedad o toxicidad inaceptable. Se realizó una evaluación del estado del tumor cada 9 semanas.
Entre los 1,033 pacientes en KEYNOTE-010, las características basales fueron: mediana de la edad 63 años (42% edad de 65 o más); 61% varones; 72% blancos y 21% asiáticos, y 34% y 66% con un estatus ECOG 0 y 1, respectivamente. Las características de la enfermedad fueron escamoso (21%) y no escamoso (70%); M1 (91%); metástasis cerebrales (15%); y la incidencia de aberraciones genómicas fue EGFR (8%) o ALK (1%). La terapia previa incluyó régimen doble con platino (100%); los pacientes recibieron uno (69%) o dos o más (29%) tratamientos previos.
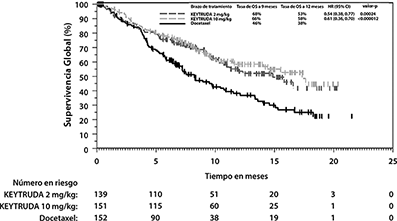
Las mediciones primarias de eficacia fueron OS y PFS, evaluados por un comité revisor independiente, utilizando RECIST1.1. Las mediciones secundarias de eficacia fueron ORR y duración de la respuesta. La Tabla 7 resume las mediciones clave de eficacia para la población ITT completa (TPS ≥1%) y para el sub-grupo de pacientes con TPS≥50%. Las gráficas de Kaplan-Meier para OS (TPS ≥1% y TPS ≥50%) se muestran en las Figuras 10 y 11.
Tabla 7. Respuesta a 2 mg/kg o 10 mg/kg de KEYTRUDA cada 3 Semanas en Pacientes con NSCLC
Tratados Previamente en KEYNOTE-010
|
Desenlace |
KEYTRUDA 2 mg/kg cada 3 semanas |
KEYTRUDA 10 mg/kg cada 3 semanas |
Docetaxel 75 mg/m2 cada 3 semanas |
|
TPS ≥1% |
|||
|
Número de pacientes |
344 |
346 |
343 |
|
OS |
|||
|
Número (%) de pacientes con evento |
172 (50%) |
156 (45%) |
193 (56%) |
|
Hazard Ratio* (95% CI) |
0.71 (0.58, 0.88) |
0.61 (0.49, 0.75) |
--- |
|
Valor de p† |
<0.001 |
<0.001 |
--- |
|
Mediana en meses (95% CI) |
10.4 (9.4, 11.9) |
12.7 (10.0, 17.3) |
8.5 (7.5, 9.8) |
|
PFS‡ |
|||
|
Número (%) de pacientes con evento |
266 (77%) |
255 (74%) |
257 (75%) |
|
Hazard Ratio* (95% CI) |
0.88 (0.73, 1.04) |
0.79 (0.66, 0.94) |
--- |
|
Valor de p† |
0.068 |
0.005 |
--- |
|
Mediana en meses (95% CI) |
3.9 (3.1, 4.1) |
4.0 (2.6, 4.3) |
4.0 (3.1, 4.2) |
|
Tasa global de respuesta‡ |
|||
|
ORR %§ (95% CI) |
18% (14, 23) |
18% (15, 23) |
9% (7, 13) |
|
Duración de la respuesta‡,¶,# |
|||
|
Mediana en meses (rango) |
No alcanzado (0.7+, 20.1+) |
No alcanzado (2.1+, 17.8+) |
6.2 (1.4+, 8.8+) |
|
% en curso |
73% |
72% |
34% |
|
TPS ≥50% |
|||
|
Número de pacientes |
139 |
151 |
152 |
|
OS |
|||
|
Número (%) de pacientes con evento |
58 (42%) |
60 (40%) |
86 (57%) |
|
Hazard Ratio* (95% CI) |
0.54 (0.38, 0.77) |
0.50 (0.36, 0.70) |
--- |
|
Valor de p† |
<0.001 |
<0.001 |
--- |
|
Mediana en meses (95% CI) |
14.9 (10.4, NA) |
17.3 (11.8, NA) |
8.2 (6.4, 10.7) |
|
PFS‡ |
|||
|
Número (%) de pacientes con evento |
89 (64%) |
97 (64%) |
118 (78%) |
|
Hazard Ratio* (95% CI) |
0.58 (0.43, 0.77) |
0.59 (0.45, 0.78) |
--- |
|
Valor de p† |
<0.001 |
<0.001 |
--- |
|
Mediana en meses (95% CI) |
5.2 (4.0, 6.5) |
5.2 (4.1, 8.1) |
4.1 (3.6, 4.3) |
|
Tasa de respuesta global‡ |
|||
|
ORR %§ (95% CI) |
30% (23, 39) |
29% (22, 37) |
8% (4, 13) |
|
Duración de la respuesta‡,¶,Þ |
|||
|
Mediana en meses (rango) |
No alcanzado (0.7+, 16.8+) |
No alcanzado (2.1+, 17.8+) |
8.1 (2.1+, 8.8+) |
|
% en curso |
76% |
75% |
33% |
* Hazard Ratio (KEYTRUDA comparado con docetaxel) basado en el modelo de riesgo proporcional estratificado de Cox.
† Basado en la prueba estratificada de rango logarítmico.
‡ Evaluado por BICR usando RECIST 1.1.
§ Todas las respuestas fueron respuestas parciales.
¶ Basado en pacientes con una mejor respuesta global, confirmado por respuesta completa o respuesta parcial.
# Incluye 30, 31, y 2 pacientes con respuestas en curso de 6 meses o más en los grupos de 2 mg/kg de KEYTRUDA, 10 mg/kg de KEYTRUDA y docetaxel, respectivamente.
Þ Incluye 22, 24, y 1 paciente con respuestas en curso de 6 meses o más en los grupos de 2 mg/kg de KEYTRUDA, 10 mg/kg de KEYTRUDA y docetaxel, respectivamente.
Figura 10: Gráfica de Kaplan-Meier para Supervivencia Global por Grupo de Tratamiento en KEYNOTE-010
(TPS ≥1%, población por intención a tratar)
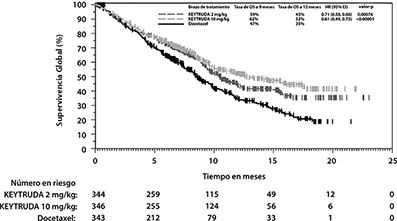
Figura 11. Gráfica de Kaplan-Meier para Supervivencia Global por Brazo de Tratamiento en KEYNOTE-010
(TPS ≥50%, población por intención a tratar)
Los resultados de eficacia fueron similares para los brazos de 2 mg/kg y 10 mg/kg de KEYTRUDA. Los resultados de eficacia para OS fueron consistentes independientemente de la edad del espécimen del tumor (nuevo versus previamente conservado).
KEYNOTE-001: Estudio abierto en pacientes con NSCLC tratados previamente con quimioterapia
La eficacia de KEYTRUDA también se investigó en un estudio multicéntrico, abierto, aleatorizado, con cohorte de dosis comparativas de KEYNOTE-001. Los pacientes tenían NSCLC avanzado que era PD-L1 positivo, con progresión de la enfermedad después de tratamiento con quimioterapia con platino. Los pacientes con aberraciones tumorales genómicas EGFR o ALK tuvieron progresión de la enfermedad con tratamiento aprobado para estas aberraciones, antes de recibir KEYTRUDA. El estudio excluyó a pacientes con enfermedad autoinmune; una condición médica que requirió inmunosupresión; o que habían recibido más de 30 Gy de radiación torácica dentro de las 26 semanas previas. Se aleatorizó a los pacientes para recibir 10 mg/kg de KEYTRUDA cada 2 (n=69) o 3 (n=87) semanas hasta la progresión de la enfermedad o toxicidad inaceptable. Se realizó evaluación del estatus tumoral cada 9 semanas. Las principales mediciones de eficacia fueron ORR (de acuerdo con RECIST 1.1, evaluado por un revisor central independiente ciego) y la duración de la respuesta.
La prevalencia de pacientes con expresión de PD-L1 con una TPS mayor de o igual a 50% entre los pacientes escrutinizados con NSCLC, como se corroboró retrospectivamente por el kit de diagnóstico PDL-1 IHC 22C3 pharmDx™ fue 26%. Entre los pacientes aleatorizados con muestras de tumor evaluables para expresión de PD-L1, 61 tenían una TPS mayor de o igual a 50%. Las características basales para esta población incluyeron: mediana de edad de 60 años (34% con 65 años o más); 61% varones, 79% blancos y 34% y 64% con un estatus ECOG de 0 y 1, respectivamente. Las características de la enfermedad fueron escamoso y no escamoso (21% y 75%, respectivamente); M1 (98%); metástasis cerebrales (11%) y uno (25%), dos (31%) o tres o más (44%) tratamientos previos. El estatus de mutaciones entre los pacientes fue EGFR (10%), ALK (0%) o Kras (16%).
Los resultados de eficacia para pacientes con NSCLC tratados con 10 mg/kg cada 2 o 3 semanas en KEYNOTE-001 se resumen en la Tabla 8.
Tabla 8. Respuesta a 10 mg/kg de KEYTRUDA cada 2 o 3 Semanas en Pacientes con NSCLC Tratados Previamente, con TPS de expresión de PD-L1 ≥50% (n=61)
|
Desenlace |
|
|
Mejor Respuesta Global* |
|
|
ORR %, (95% CI) |
43% (30, 56) |
|
Respuesta completa |
2% |
|
Respuesta parcial |
41% |
|
Duración de la Respuesta† |
|
|
Mediana en meses (rango) |
No alcanzado (2.1+, 13.4+) |
|
% en curso |
65%‡ |
|
Tiempo de Respuesta† |
|
|
Mediana en meses (rango) |
2.1 (1.4, 6.2) |
|
PFS§ |
|
|
Mediana en meses (95% CI) |
6.3 (2.1, 10.7) |
|
Tasa de PFS a 6 meses |
53% |
|
OS§ |
|
|
Tasa de OS a 12 meses |
60% |
* Basado en todos los pacientes tratados (n=61), con evaluación por revisión independiente y RECIST 1.1.
† Basado en pacientes (n=26) con una respuesta confirmada por revisión independiente.
‡ Incluye 17 pacientes con respuestas en curso de 6 meses o más.
§ Basado en todos los pacientes tratados (n=61).
Se observaron resultados de ORR similares en otro grupo de pacientes (n=25) con TPS mayor de o igual a 50%, que recibieron KEYTRUDA a una dosis de 2 mg/kg cada 3 semanas en KEYNOTE-001.
Carcinoma Urotelial
KEYNOTE-045: Estudio controlado en pacientes con carcinoma urotelial tratados previamente con quimioterapia que contenía platino
La eficacia de KEYTRUDA se evaluó en KEYNOTE-045, un estudio multicéntrico, aleatorizado (1:1), con control activo, en pacientes con carcinoma urotelial localmente avanzado o metastásico, con progresión de la enfermedad durante o después de quimioterapia que contenía platino. El estudio excluyó a pacientes con enfermedad autoinmune o una condición médica que requería inmunosupresión.
Los pacientes fueron aleatorizados para recibir ya sea 200 mg de KEYTRUDA cada 3 semanas (n=270) o la elección del investigador de cualquiera de los siguientes regímenes de quimioterapia, todos administrados intravenosamente cada 3 semanas (n=272): paclitaxel 175 mg/m2 (n=84), docetaxel 75 mg/m2 (n=84), o vinflunina 320 mg/m2 (n=87). Los pacientes recibieron KEYTRUDA hasta la aparición de toxicidad inaceptable o progresión de la enfermedad. Se permitió que los pacientes clínicamente estables con evidencia inicial de progresión de la enfermedad permanecieran bajo tratamiento hasta que se confirmara la progresión de la enfermedad. Los pacientes sin progresión de la enfermedad fueron tratados hasta por 24 meses. El tratamiento con pembrolizumab podía reiniciarse por progresión subsecuente de la enfermedad y administrarse hasta por 1 año adicional. La evaluación del estatus del tumor se realizó a las 9 semanas después de la aleatorización, después cada 6 semanas a lo largo del primer año, seguido por cada 12 semanas de ahí en adelante. Las mediciones principales de eficacia fueron OS y PFS, evaluadas por BICR por RECIST v1.1. Las mediciones adicionales de eficacia fueron ORR, evaluada por BICR por RECIST v1.1 y duración de la respuesta.
Entre los 542 pacientes aleatorizados, las características de la población de estudio fueron: mediana de la edad de 66 años (rango: 26 a 88), 58% de 65 años o más; 74% varones; 72% blancos y 23% asiáticos; 57% con escala de estado funcional ECOG de 1 o mayor; 96% fue M1 y 4% fue M0. El 87% de los pacientes tenía metástasis viscerales, incluyendo 34% con metástasis hepáticas. El 86% tuvo un tumor primario en el tracto inferior y 14% tuvieron un tumor primario en el tracto superior. El 15% de los pacientes tuvo progresión de la enfermedad después de quimioterapia previa neoadyuvante o adyuvante que contenía platino, como la línea más reciente de terapia. El 21% había recibido 2 o más regímenes sistémicos previos en los pacientes con metástasis. El 76% de los pacientes recibió cisplatino previo, 23% tuvo carboplatino previo y el 1% fue tratado con otros regímenes basados en platino.
En un análisis intermedio pre-especificado, la mediana de tiempo de seguimiento para 270 pacientes tratados con KEYTRUDA fue de 10.3 meses. El estudio demostró mejorías estadísticamente significativas en OS y ORR para los pacientes aleatorizados a KEYTRUDA, comparado con quimioterapia (Tabla 9 y Figura 12). No hubo una diferencia estadísticamente significativa entre KEYTRUDA y quimioterapia con respecto a PFS. Los resultados de eficacia se resumen en la Tabla 9.
Tabla 9: Resultados de Eficacia en Pacientes con Carcinoma Urotelial Tratado Previamente con Quimioterapia
|
Criterio de Valoración |
KEYTRUDA 200 mg cada 3 semanas n=270 |
Quimioterapia n=272 |
|
OS |
||
|
Número (%) de pacientes con evento |
155 (57%) |
179 (66%) |
|
Hazard Ratio* (IC del 95%) |
0.73 (0.59, 0.91) |
|
|
Valor de p† |
0.002 |
|
|
Mediana en meses (IC del 95%) |
10.3 (8.0, 11.8) |
7.4 (6.1, 8.3) |
|
PFS‡ |
||
|
Número (%) de pacientes con evento |
218 (81%) |
219 (81%) |
|
Hazard Ratio* (IC del 95%) |
0.98 (0.81, 1.19) |
|
|
Valor de p† |
0.416 |
|
|
Mediana en meses (IC del 95%) |
2.1 (2.0, 2.2) |
3.3 (2.3, 3.5) |
|
Tasa de Respuesta Objetiva‡ |
||
|
ORR % (IC del 95%) |
21% (16, 27) |
11% (8, 16) |
|
Respuesta Completa |
7% |
3% |
|
Respuesta Parcial |
14% |
8% |
|
Valor de p§, |
0.001 |
|
|
Duración de la respuesta‡,¶ |
||
|
Mediana en meses (rango) |
No alcanzado (1.6+, 15.6+) |
4.3 (1.4+, 15.4+) |
|
Número (%#) de pacientes con duración ≥6 meses |
41 (78%) |
7 (40%) |
|
Número (%#) de pacientes con duración ≥12 meses |
14 (68%) |
3 (35%) |
* Hazard Ratio (KEYTRUDA comparado con quimioterapia) con base en el modelo de riesgo proporcional estratificado de Cox.
† Con base en la prueba de rango logarítmico estratificado (Log Rank).
‡ Evaluado por BICR usando RECIST 1.1.
§ Con base en el método de Miettinen y Nurminen.
¶ Con base en los pacientes con una mejor respuesta global, confirmada como respuesta completa o parcial.
# Con base en estimación de Kaplan-Meier.
El análisis final de OS se realizó 13.6 meses después del análisis intermedio con 419 pacientes-eventos (200 para KEYTRUDA y 219 para quimioterapia). La mediana de la OS fue de 10.1 meses (IC del 95%: 8.0, 12.3) para KEYTRUDA y 7.3 meses (IC del 95%: 6.1, 8.1) para quimioterapia. La HR para la OS fue de 0.70 (IC del 95%: 0.57, 0.85; p<0.001). Ver la Figura 12. En el análisis final no hubo una diferencia estadísticamente significativa entre KEYTRUDA y quimioterapia con respecto a la PFS.
En el análisis final, entre los 57 pacientes con respuesta que recibieron KEYTRUDA versus 30 pacientes con respuesta que recibieron quimioterapia, no se alcanzó la mediana de duración de la respuesta (rango 1.6+ a 30.0+ meses) en pacientes que recibieron KEYTRUDA versus 4.4 meses (rango 1.4+ a 29.9+ meses) en pacientes que recibieron quimioterapia. En los pacientes que recibieron KEYTRUDA, 84% tuvieron respuestas de 6 meses o más largas y 68% tuvieron respuestas de 12 meses o más largas (con base en estimación de Kaplan-Meier) versus 47% que tuvieron respuestas de 6 meses o más largas y 35% que tuvieron respuestas de 12 meses o más largas (con base en estimación de Kaplan-Meier) en pacientes que recibieron quimioterapia. Las tasas de respuesta completa y parcial fueron 9% y 12%, respectivamente, en pacientes que recibieron KEYTRUDA versus 3% y 8%, respectivamente en pacientes que recibieron quimioterapia.
Figura 12: Curva de Kaplan-Meier para Supervivencia Global por Brazo de Tratamiento en KEYNOTE-045 (población por intención a tratar)
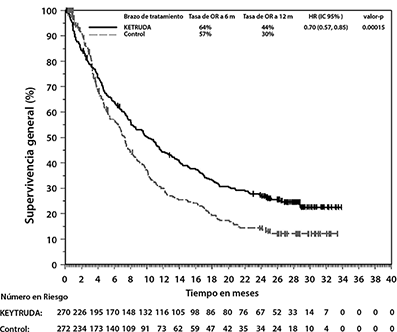
Los desenlaces reportados por los pacientes (PROs, por las siglas en inglés para Patient-Reported Outcomes) fueron evaluados utilizando el EORTC QLQ-C30. Se observó un tiempo prolongado hasta el deterioro en la puntuación del estado general de salud/QoL del EORTC QLQ-C30, para pacientes tratados con pembrolizumab, en comparación con la elección de quimioterapia hecha por el investigador (HR 0.70; IC del 95% 0.55-0.90). Con más de 15 semanas de seguimiento, pacientes tratados con pembrolizumab tuvieron puntuaciones estables en el estado general de salud/QoL, mientras que aquellos tratados con la elección de quimioterapia hecha por el investigador tuvieron una declinación en las puntuaciones de estado general de salud/QoL. Estos resultados deben interpretarse en el contexto del diseño abierto del estudio y por lo tanto, ser considerados con cautela.
Inmunogenicidad
En estudios clínicos en pacientes tratados con pembrolizumab a una dosis de 2 mg/kg cada tres semanas, 200 mg cada tres semanas, o 10 mg/kg cada dos o tres semanas, 36 (1.8%) de 2,034 pacientes evaluables dieron positivo para anticuerpos contra pembrolizumab, de los cuales, 9 pacientes (0.4%) tuvieron anticuerpos neutralizantes contra pembrolizumab. No hubo evidencia de alteración en el perfil farmacocinético o de seguridad con el desarrollo de anticuerpos neutralizantes o fijadores anti-pembrolizumab.
VI. CONTRAINDICACIONES
KEYTRUDA está contraindicado en la hipersensibilidad severa a cualquiera de los componentes de la fórmula.
VII. PRECAUCIONES GENERALES
Reacciones adversas inmunomediadas
En pacientes que recibieron KEYTRUDA han ocurrido reacciones adversas inmunomediadas, incluyendo casos graves y fatales. Las reacciones adversas inmunomediadas pueden ocurrir después de la descontinuación del tratamiento. En estudios clínicos, la mayoría de las reacciones adversas inmunomediadas fueron reversibles y manejadas con interrupciones de KEYTRUDA, administración de corticosteroides y/o cuidados de soporte. Pueden ocurrir de manera simultánea reacciones adversas inmunomediadas que afecten a más de un sistema corporal.
Para reacciones adversas inmunomediadas sospechadas, asegurar una evaluación adecuada para confirmar la etiología o excluir otras causas. Con base en la gravedad de la reacción adversa, suspenda KEYTRUDA y administre corticosteroides (ver a continuación). Luego de la mejoría a Grado 1 o menos, inicie la disminución gradual del corticosteroide y continúe su disminución gradual durante al menos 1 mes. Con base en información limitada a partir de estudios clínicos en pacientes cuyas reacciones adversas inmunomediadas no se pudieron controlar con el uso de corticosteroides, se puede considerar la administración de otros inmunosupresores sistémicos. Reinicie la administración de KEYTRUDA si la reacción adversa continúa en Grado 1 o menos después de la descontinuación gradual del corticosteroide. Si ocurre otro episodio de alguna reacción adversa grave, descontinúe KEYTRUDA de forma permanente [ver XIII. Dosis y Vía de Administración y IX. Reacciones Secundarias y Adversas].
Neumonitis inmunomediada
Se ha reportado neumonitis (incluyendo casos fatales) en pacientes que reciben KEYTRUDA [ver IX. Reacciones Secundarias y Adversas]. Haga monitoreo a los pacientes buscando signos y síntomas de neumonitis. Si se sospecha neumonitis, evalúe con imágenes radiográficas y excluya otras causas. Administre corticosteroides para eventos Grado 2 o mayor (dosis inicial de prednisona o equivalente de 1-2 mg/kg/día, seguido de una disminución gradual), suspenda la administración de KEYTRUDA por neumonitis moderada (Grado 2) y descontinúe de forma permanente KEYTRUDA por neumonitis grave (Grado 3), que amenace la vida (Grado 4) o moderada recurrente (Grado 2) [ver XIII. Dosis y Vía de Administración y Reacciones adversas inmunomediadas, arriba].
Colitis inmunomediada
Se ha reportado colitis en pacientes que reciben KEYTRUDA [ver IX. Reacciones Secundarias y Adversas]. Haga monitoreo a los pacientes buscando signos y síntomas de colitis y excluya otras causas. Administre corticosteroides para eventos de Grado 2 o mayor (dosis inicial de prednisona o equivalente de 1-2 mg/kg/día, seguido de una disminución gradual), suspenda la administración de KEYTRUDA por colitis moderada (Grado 2) o grave (Grado 3) y descontinúe de forma permanente KEYTRUDA por colitis que amenace la vida (Grado 4) [ver XIII. Dosis y Vía de Administración y Reacciones adversas inmunomediadas, arriba].
Hepatitis inmunomediada
Se ha reportado hepatitis en pacientes que reciben KEYTRUDA [ver IX. Reacciones Secundarias y Adversas]. Haga monitoreo a los pacientes buscando cambios en el funcionamiento hepático (al inicio del tratamiento, periódicamente durante el tratamiento y según esté indicado con base en la evaluación clínica) y síntomas de hepatitis, y excluya otras causas. Administre corticosteroides (dosis inicial de 0.5-1 mg/kg/día [para eventos de Grado 2] de prednisona o equivalente, a dosis de 1-2 mg/kg/día [para Grado 3 o eventos mayores], seguido de una disminución gradual) y, con base en la gravedad de la elevación de las enzimas hepáticas, suspenda o descontinúe KEYTRUDA [ver XIII. Dosis y Vía de Administración y Reacciones adversas inmunomediadas, arriba].
Nefritis inmunomediada
Se ha reportado nefritis en pacientes que reciben KEYTRUDA [ver IX. Reacciones Secundarias y Adversas]. Haga monitoreo a los pacientes buscando cambios en la función renal y excluya otras causas. Administre corticosteroides para eventos de Grado 2 o mayor (dosis inicial de prednisona o equivalente de 1-2 mg/kg/día, seguido de una disminución gradual), suspenda la administración de KEYTRUDA por nefritis moderada (Grado 2) y descontinúe de forma permanente KEYTRUDA por nefritis grave (Grado 3) o que amenace la vida (Grado 4) [ver XIII. Dosis y Vía de Administración y Reacciones adversas inmunomediadas, arriba].
Endocrinopatías inmunomediadas
Se ha reportado hipofisitis en pacientes que reciben KEYTRUDA [ver IX. Reacciones Secundarias y Adversas]. Haga monitoreo a los pacientes para detectar signos y síntomas de hipofisitis (incluyendo hipopituitarismo e insuficiencia suprarrenal secundaria) y excluya otras causas. Administre corticosteroides para tratar la insuficiencia suprarrenal secundaria y otro reemplazo hormonal como esté indicado clínicamente, suspenda la administración de KEYTRUDA por hipofisitis moderada (Grado 2), suspenda o descontinúe KEYTRUDA por hipofisitis grave (Grado 3) o que amenace la vida (Grado 4) [ver XIII. Dosis y Vía de Administración y Reacciones adversas inmunomediadas, arriba].
Se ha reportado diabetes mellitus tipo 1, incluyendo cetoacidosis diabética en pacientes que reciben KEYTRUDA [ver IX. Reacciones Secundarias y Adversas]. Haga monitoreo a los pacientes buscando hiperglucemia u otros signos y síntomas de diabetes. Administre insulina para la diabetes tipo 1 y suspenda la administración de KEYTRUDA en casos de hiperglucemia grave, hasta que se logre el control metabólico.
Se han reportado trastornos tiroideos, incluyendo hipertiroidismo, hipotiroidismo y tiroiditis, en pacientes que reciben KEYTRUDA y pueden ocurrir en cualquier momento durante el tratamiento, por lo tanto, haga monitoreo a los pacientes buscando cambios en la función tiroidea (al inicio del tratamiento, periódicamente durante el tratamiento y según esté indicado con base en la evaluación clínica) y signos y síntomas clínicos de trastornos tiroideos. El hipotiroidismo se puede manejar con terapia de reemplazo sin la descontinuación del tratamiento y sin corticosteroides. El hipertiroidismo se puede manejar sintomáticamente. Suspenda o descontinúe la administración de KEYTRUDA por hipertiroidismo grave (Grado 3) o que amenace la vida (Grado 4) [ver XIII. Dosis y Vía de Administración, IX. Reacciones Secundarias y Adversas, Reacciones adversas inmunomediadas, arriba].
Para pacientes con endocrinopatía grave (Grado 3) o que amenaza la vida (Grado 4) que mejoran a Grado 2 o inferior y se controlan con reemplazo hormonal, se puede considerar la continuación de KEYTRUDA.
Reacciones cutáneas graves
Se han reportado reacciones cutáneas graves inmunomediadas en pacientes tratados con KEYTRUDA. Haga monitoreo a los pacientes para identificar posibles reacciones cutáneas graves y para excluir otras causas. Con base en la severidad de la reacción adversa, suspenda o discontinúe permanentemente KEYTRUDA y administre corticosteroides (Ver XIII Dosis y Vía de Administración).
Se han reportado casos de Síndrome de Stevens-Johnson (SJS, por sus siglas en inglés) y de necrólisis epidérmica tóxica (TEN, por sus siglas en inglés) en pacientes tratados con KEYTRUDA, algunos de estos casos tuvieron consecuencias fatales. Suspenda KEYTRUDA en caso de signos y síntomas de SJS o TEN y refiera al paciente a que reciba atención especializada para evaluación clínica y tratamiento. Descontinúe permanentemente KEYTRUDA en caso de que el SJS o TEN sea confirmado (Ver XIII Dosis y Vía de Administración).
Otras reacciones adversas inmunomediadas
En menos del 1% de los pacientes tratados con KEYTRUDA en KEYNOTE-001, KEYNOTE-002, KEYNOTE-006, y KEYNOTE-010, se reportaron las siguientes reacciones adversas clínicamente significativas: uveítis, miositis, síndrome de Guillain-Barré, pancreatitis, encefalitis, sarcoidosis y síndrome miasténico/miastenia gravis (incluyendo exacerbaciones). En otros estudios clínicos con KEYTRUDA y también en el uso post-comercialización se ha reportado miocarditis.
En estudios clínicos o en el uso post-comercialización, se han reportado casos de estas reacciones adversas inmunomediadas, algunas de las cuales fueron graves.
Reacciones adversas relacionadas con trasplantes
En el uso post-comercialización se ha reportado rechazo de trasplante de órgano sólido en pacientes tratados con KEYTRUDA. El tratamiento con KEYTRUDA puede aumentar el riesgo de rechazo en pacientes receptores de trasplante de órgano sólido. Considere el beneficio del tratamiento con KEYTRUDA contra el riesgo de un posible rechazo de órgano en estos pacientes.
Se ha reportado enfermedad de injerto contra huésped aguda (GVHD, por las siglas en inglés para Graft-Versus-Host-Disease), incluyendo GVHD fatal, después del tratamiento con KEYTRUDA, en pacientes con antecedentes de trasplante alogénico de células madre hematopoyéticas (HSCT, por las siglas para Hematopoietic Stem Cell Transplant). Pacientes que experimentaron GVHD después de su trasplante pueden estar en mayor riesgo de GVHD después del tratamiento con KEYTRUDA. Considere el beneficio del tratamiento con KEYTRUDA contra el riesgo de una posible GVHD en pacientes con antecedentes de HSCT alogénico.
Incremento de la mortalidad en pacientes con mieloma múltiple cuando se añade KEYTRUDA a un análogo de talidomida y dexametasona
En dos estudios clínicos aleatorizados en pacientes con mieloma múltiple, la adición de KEYTRUDA a un análogo de talidomida más dexametasona, uso para el cual no están indicados los anticuerpos bloqueadores de PD-1 o PD-L1, resultó en un incremento en la mortalidad. No se recomienda el tratamiento de pacientes con mieloma múltiple con un anticuerpo bloqueador de PD-1 o PD-L1 en combinación con un análogo de talidomida más dexametasona, fuera del ambiente de estudios clínicos controlados.
Reacciones relacionadas con la infusión
Se han reportado reacciones graves a la infusión, incluyendo hipersensibilidad y anafilaxis, en 6 (0.2%) de 2,799 pacientes que recibieron KEYTRUDA en KEYNOTE-001, KEYNOTE-002, KEYNOTE-006 y KEYNOTE-010. Para reacciones graves a la infusión, suspenda la misma y descontinúe permanentemente KEYTRUDA [ver XIII. Dosis y Vía de Administración]. Los pacientes con reacción a la infusión leve o moderada pueden continuar recibiendo KEYTRUDA bajo estrecha vigilancia; se puede considerar pre-medicación con antipiréticos y antihistamínicos.
VIII. RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA
Embarazo
Resumen de riesgos
Con base a su mecanismo de acción, KEYTRUDA puede causar daño fetal cuando se administra a mujeres embarazadas. En modelos animales, la vía de señalización de PD-1/PD-L1 es importante en el mantenimiento del embarazo a través de la inducción de la tolerancia inmune materna hacia el tejido fetal [ver más abajo]. Se sabe que la IgG4 humana (inmunoglobulina) cruza la placenta; por tanto, pembrolizumab tiene el potencial de transmitirse de la madre al feto en desarrollo. No hay datos humanos disponibles que proporcionen información sobre riesgo de la toxicidad embrionaria y fetal. Se debe informar a las mujeres embarazadas del riesgo potencial para el feto. Datos
Datos en animales
No se han llevado a cabo estudios de reproducción animal con KEYTRUDA para evaluar su efecto en la reproducción y desarrollo fetal, pero se realizó una evaluación de los efectos sobre la reproducción. La función principal de la vía PD-1/PD-L1 es preservar el embarazo a través del mantenimiento de la tolerancia inmune materna hacia el feto. Se ha demostrado que el bloqueo de la señalización del PD-L1 en modelos murinos gestantes interrumpe la tolerancia al feto y resulta en un incremento en la pérdida fetal; por tanto, los riesgos potenciales de administrar KEYTRUDA durante el embarazo incluye aumento de las tasas de aborto por muerte fetal. Según se reportó en la literatura, no hubo malformaciones relacionadas con el bloqueo de la señalización del PD-1 en la descendencia de estos animales; sin embargo, los desórdenes inmunomediados ocurrieron en los ratones knockout-PD-1. Con base en su mecanismo de acción, la exposición fetal a pembrolizumab puede incrementar el riesgo de desarrollar desórdenes inmunomediados o de alteración de la respuesta inmune normal.
Toxicidad embriofetal
Con base en su mecanismo de acción, KEYTRUDA puede causar daño fetal cuando se administra en mujeres embarazadas. Los modelos animales vinculan la vía de señalización PD-1/PD-L1 con el mantenimiento del embarazo a través de la inducción de la tolerancia inmune materna hacia el tejido fetal. Si este medicamento se usa durante el embarazo, o si la paciente se embaraza mientras toma este medicamento, se debe informar a la paciente del riesgo potencial para el feto. Se debe aconsejar a las mujeres con potencial reproductivo sobre el uso de anticonceptivos altamente efectivos durante el tratamiento con KEYTRUDA y durante 4 meses después de la última dosis de KEYTRUDA [ver arriba en Embarazo].
Lactancia
Resumen del riesgo
No se sabe si KEYTRUDA se excreta en leche humana. No se ha realizado ningún estudio para evaluar el impacto de KEYTRUDA sobre la producción de leche o su presencia en leche materna. Debido a que muchos medicamentos se excretan en leche humana, se debe aconsejar a las mujeres lactantes de descontinuar la lactancia durante el tratamiento con KEYTRUDA y durante 4 meses después de la dosis final.
IX. REACCIONES SECUNDARIAS Y ADVERSAS
Resumen del perfil de seguridad
Pembrolizumab se asocia más frecuentemente a reacciones adversas relacionadas con el sistema inmunitario. La mayoría de éstas, incluyendo las reacciones graves, se resolvieron después de iniciar el tratamiento médico adecuado o de suspender definitivamente el tratamiento con pembrolizumab (ver “Descripción de reacciones adversas seleccionadas” más abajo).
Se ha evaluado la seguridad de pembrolizumab en monoterapia en ensayos clínicos en 4,948 pacientes con melanoma avanzado, melanoma en estadio III resecado (tratamiento adyuvante), NSCLC, Linfoma de Hodgkin clásico, carcinoma urotelial o carcinoma de células escamosas de cabeza y cuello en cuatro dosis (2 mg/kg cada 3 semanas, 200 mg cada 3 semanas o 10 mg/kg cada 2 o 3 semanas). Las frecuencias incluidas a continuación y en la Tabla 10 se basan en todas las reacciones adversas medicamentosas notificadas, con independencia de la evaluación de causalidad por el investigador. En esta población de pacientes, la mediana del tiempo de observación fue de 7.3 meses (rango: 1 día a 31 meses) y las reacciones adversas más frecuentes con pembrolizumab fueron fatiga (34.1%), erupción (22.7%), náuseas (21.7%), diarrea (21.5%) y prurito (20.2%). La mayoría de las reacciones adversas notificadas para monoterapia fueron de intensidad de Grado 1 o 2. Las reacciones adversas más graves fueron reacciones adversas relacionadas con el sistema inmunitario y reacciones graves asociadas a la infusión (ver VII. Precauciones Generales).
Se ha evaluado la seguridad de pembrolizumab en combinación con quimioterapia en ensayos clínicos en 791 pacientes con NSCLC que recibieron 200 mg, 2 mg/kg o 10 mg/kg de pembrolizumab cada 3 semanas. Las frecuencias incluidas a continuación y en la Tabla 10 se basan en todas las reacciones adversas notificadas, con independencia de la evaluación de causalidad por el investigador. En esta población de pacientes, las reacciones adversas más frecuentes fueron náuseas (49%), anemia (48%), fatiga (38%), estreñimiento (34%), diarrea (31%), neutropenia (29%) y apetito disminuido (28%). La incidencia de reacciones adversas de Grado 3-5 fue del 67% con el tratamiento de pembrolizumab en combinación y del 66% con la quimioterapia sola.
Tabla de reacciones adversas
Las reacciones adversas observadas en ensayos clínicos de pembrolizumab en monoterapia o en combinación con quimioterapia, o notificadas con el uso de pembrolizumab tras la comercialización se incluyen en la Tabla 10. Se sabe que las reacciones adversas que se producen con pembrolizumab o quimioterapias administradas en monoterapia pueden aparecer durante el tratamiento con estos medicamentos en combinación, incluso si esas reacciones adversas no se notificaron en ensayos clínicos con el tratamiento en combinación.
Estas reacciones se presentan según el sistema de clasificación de órganos y por frecuencia. Las frecuencias se definen como: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1.000); muy raras (< 1/10.000), y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencias, se presentan las reacciones adversas en orden decreciente de gravedad.
Tabla 10: Reacciones adversas en pacientes tratados con pembrolizumab*
|
Monoterapia |
Combinación con quimioterapia |
|
|
Infecciones e infestaciones |
||
|
Frecuentes |
neumonía |
neumonía |
|
Trastornos de la sangre y del sistema linfático |
||
|
Muy frecuentes |
anemia |
neutropenia, anemia, trombocitopenia |
|
Frecuentes |
trombocitopenia, linfopenia |
neutropenia febril, leucopenia, linfopenia |
|
Poco frecuentes |
neutropenia, leucopenia, eosinofilia |
eosinofilia |
|
Raras |
púrpura trombocitopénica inmune, anemia hemolítica, aplasia eritrocitaria pura, linfohistiocitosis hemofagocítica |
|
|
Trastornos del sistema inmunológico |
||
|
Frecuentes |
reacción asociada a infusióna |
reacción asociada a infusióna |
|
Poco frecuentes |
sarcoidosis |
|
|
Frecuencia no conocida |
rechazo de trasplantes de órganos sólidos |
|
|
Trastornos endocrinos |
||
|
Muy frecuentes |
hipotiroidismob |
|
|
Frecuentes |
hipertiroidismo |
hipertiroidismo, hipotiroidismo |
|
Poco frecuentes |
hipofisitisc, tiroiditisd, insuficiencia suprarrenal |
hipofisitisc, tiroiditis, insuficiencia suprarrenal |
|
Trastornos del metabolismo y de la nutrición |
||
|
Muy frecuentes |
apetito disminuido |
apetito disminuido |
|
Frecuentes |
hiponatremia, hipocalemia, hipocalcemia |
hiponatremia, hipocalemia, hipocalcemia |
|
Poco frecuentes |
diabetes mellitus tipo 1e |
diabetes mellitus tipo 1 |
|
Trastornos psiquiátricos |
||
|
Frecuentes |
insomnio |
insomnio |
|
Trastornos del sistema nervioso |
||
|
Muy frecuentes |
cefalea |
mareo, neuropatía periférica, disgeusia, cefalea |
|
Frecuentes |
mareo, neuropatía periférica, letargia, disgeusia |
letargia |
|
Poco frecuentes |
epilepsia |
epilepsia |
|
Raras |
síndrome de Guillain-Barréf, síndrome miasténicog, meningitis (aséptica), encefalitis |
|
|
Trastornos oculares |
||
|
Frecuentes |
ojo seco |
ojo seco |
|
Poco frecuentes |
uveítish |
|
|
Raras |
síndrome de Vogt-Koyanagi- Harada |
|
|
Trastornos cardiacos |
||
|
Poco frecuentes |
derrame pericárdico, pericarditis |
derrame pericárdico, pericarditis |
|
Raras |
miocarditis |
|
|
Trastornos vasculares |
||
|
Frecuentes |
hipertensión |
hipertensión |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Muy frecuentes |
disnea, tos |
disnea, tos |
|
Frecuentes |
neumonitisi |
neumonitisi |
|
Trastornos gastrointestinales |
||
|
Muy frecuentes |
diarrea, dolor abdominalj, náuseas, vómitos, estreñimiento |
diarrea, náuseas, vómitos, estreñimiento, dolor abdominalj |
|
Frecuentes |
colitisk, boca seca |
colitisk, boca seca |
|
Poco frecuentes |
pancreatitisl |
pancreatitisl |
|
Raras |
perforación de intestino delgado |
|
|
Trastornos hepatobiliares |
||
|
Frecuentes |
hepatitism |
|
|
Poco frecuentes |
hepatitism |
|
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Muy frecuentes |
erupciónn, pruritoo |
erupciónn, alopecia, pruritoo |
|
Frecuentes |
reacciones cutáneas gravesp, eritema, vitíligoq, piel seca, alopecia, eczema, dermatitis acneiforme |
reacciones cutáneas gravesp, eritema, dermatitis acneiforme, piel seca |
|
Poco frecuentes |
queratosis liquenoider, psoriasis, dermatitis, pápula, cambios de color del pelo |
psoriasis, dermatitis, eczema, cambios de color del pelo, queratosis liquenoide, pápula, vitíligoq |
|
Raras |
necrólisis epidérmica tóxica, síndrome de Stevens-Johnson, eritema nudoso |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Muy frecuentes |
dolor musculoesqueléticos, artralgia |
dolor musculoesqueléticos, artralgia |
|
Frecuentes |
dolor en una extremidad, miositist, artritisu |
miositist, dolor en una extremidad, artritisu |
|
Poco frecuentes |
tenosinovitisv |
tenosinovitisv |
|
Trastornos renales y urinarios |
||
|
Frecuentes |
nefritisw, lesión renal aguda |
|
|
Poco frecuentes |
nefritisw |
|
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Muy frecuentes |
fatiga, astenia, edemax, pirexia |
fatiga, astenia, edemax, pirexia |
|
Frecuentes |
enfermedad de tipo gripal, escalofríos |
escalofríos, enfermedad de tipo gripal |
|
Exploraciones complementarias |
||
|
Muy frecuentes |
alanina aminotransferasa elevada, creatinina en sangre elevada |
|
|
Frecuentes |
aspartato aminotransferasa elevada, alanina aminotransferasa elevada, hipercalcemia, fosfatasa alcalina en sangre aumentada, bilirrubina elevada en sangre, creatinina en sangre elevada |
aspartato aminotransferasa elevada, hipercalcemia, fosfatasa alcalina en sangre aumentada |
|
Poco frecuentes |
amilasa elevada |
amilasa elevada, bilirrubina en sangre elevada |
* Las frecuencias de las reacciones adversas que se presentan en la Tabla 10 pueden no ser totalmente atribuibles a pembrolizumab en monoterapia, sino también incluir contribuciones de la enfermedad subyacente o de otros medicamentos usados en combinación.
Los siguientes términos representan a un grupo de acontecimientos relacionados que describen a una enfermedad más que a un acontecimiento aislado.
a. reacción asociada a infusión (hipersensibilidad a fármaco, reacción anafiláctica, reacción anafilactoide, hipersensibilidad y síndrome de liberación de citocina).
b. hipotiroidismo (mixedema).
c. hipofisitis (hipopituitarismo).
d. tiroiditis (tiroiditis autoinmune y trastorno de tiroides).
e. diabetes mellitus tipo 1 (cetoacidosis diabética).
f. síndrome de Guillain-Barré (neuropatía axonal y polineuropatía desmielinizante).
g. síndrome miasténico (miastenia gravis, incluida la exacerbación).
h. uveítis (iritis e iridociclitis).
i. neumonitis (enfermedad pulmonar intersticial).
j. dolor abdominal (molestia abdominal, dolor en la zona superior del abdomen y dolor en la zona inferior del abdomen).
k. colitis (colitis microscópica, enterocolitis y colitis autoinmune).
l. pancreatitis (pancreatitis autoinmune y pancreatitis aguda).
m. hepatitis (hepatitis autoinmunitaria y lesión hepática inducida por fármacos).
n. erupción (erupción eritematosa, erupción folicular, erupción generalizada, erupción macular, erupción maculopapular, erupción papular, erupción prurítica, erupción vesicular y erupción genital).
o. prurito (urticaria, urticaria papular, prurito generalizado y prurito genital).
p. reacciones cutáneas graves (dermatitis bullosa, dermatitis exfoliativa, eritema multiforme, erupción exfoliativa, pénfigo, necrosis de la piel, erupción cutánea tóxica y las siguientes reacciones de Grado ≥ 3: dermatosis neutropénica aguda febril, contusión, úlcera de decúbito, dermatitis psoriasiforme, erupción medicamentosa, ictericia, penfigoide, prurito, prurito generalizado, erupción, erupción eritematosa, erupción generalizada, erupción maculopapular, erupción prurítica, erupción pustular y lesión de la piel).
q. vitíligo (despigmentación de la piel, hipopigmentación de la piel e hipopigmentación del párpado).
r. queratosis liquenoide (liquen plano y liquen escleroso).
s. dolor musculoesquelético (molestia musculoesquelética, dolor de espalda, rigidez musculoesquelética, dolor torácico musculoesquelético y tortícolis).
t. miositis (mialgia, miopatía, polimialgia reumática y rabdomiólisis).
u. artritis (hinchazón articular, poliartritis y derrame articular).
v. tenosinovitis (tendinitis, sinovitis y dolor tendinoso).
w. nefritis (nefritis autoinmune, nefritis tubulointersticial y fallo renal, fallo renal agudo o lesión renal aguda con evidencia de nefritis, síndrome nefrótico).
x. edema (edema periférico, edema generalizado, sobrecarga de líquido, retención de líquidos, edema palpebral y edema de labio, edema de cara, edema localizado y edema periorbital).
Descripción de reacciones adversas seleccionadas
Los datos de las siguientes reacciones adversas relacionadas con el sistema inmunitario se basan en pacientes que recibieron pembrolizumab en cuatro dosis (2 mg/kg cada 3 semanas, 10 mg/kg cada 2 o 3 semanas o 200 mg cada 3 semanas) en ensayos clínicos (ver V. Farmacocinética y Farmacodinamia). Las directrices para el manejo de estas reacciones adversas se describen en la sección VII. Precauciones Generales.
Reacciones adversas relacionadas con el sistema inmunitario (ver VII. Precauciones Generales)
Neumonitis relacionada con el sistema inmunitario
Se produjo neumonitis en 182 (3.7%) pacientes, incluidos casos de Grado 2, 3, 4 o 5 en 78 (1.6%), 48 (1.0%), 9 (0.2%) y 7 (0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de neumonitis fue de 3.7 meses (rango, 2 días a 21.3 meses). La mediana de duración fue de 1.9 meses (rango, 1 día a 17.2+ meses). La neumonitis se produjo más frecuentemente en pacientes con antecedentes de radiación torácica previa (8.1%) que en pacientes que no recibieron radiación torácica previa (3.3%). La neumonitis condujo a la suspensión definitiva de pembrolizumab en 75 (1.5%) pacientes y se resolvió en 101 pacientes, 2 con secuelas.
En pacientes con NSCLC, se produjo neumonitis en 107 (4.9%), incluidos casos de Grado 2, 3, 4 o 5 en 39 (1.8%), 30 (1.4%), 10 (0.5%) y 9 (0.4%) pacientes, respectivamente. En pacientes con NSCLC, se produjo neumonitis en el 8.1% con antecedentes de radiación torácica previa.
Colitis relacionada con el sistema inmunitario
Se produjo colitis en 97 (2.0%) pacientes, incluidos casos de Grado 2, 3 o 4 en 28 (0.6%), 56 (1.1%) y 3 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de colitis fue de 3.8 meses (rango, 7 días a 20.2 meses). La mediana de duración fue de 1.2 meses (rango, 1 día a 8.7+ meses). La colitis condujo a la suspensión definitiva de pembrolizumab en 28 (0.6%) pacientes y se resolvió en 75 pacientes, 1 con secuelas.
Hepatitis relacionada con el sistema inmunitario
Se produjo hepatitis en 39 (0.8%) pacientes, incluidos casos de Grado 2, 3 o 4 en 7 (0.1%), 26 (0.5%) y 4 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de hepatitis fue de 2.8 meses (rango, 8 días a 21.4 meses). La mediana de duración fue de 1.1 meses (rango, 1 día a 20.9+ meses). La hepatitis condujo a la suspensión definitiva de pembrolizumab en 14 (0.3%) pacientes y se resolvió en 27 pacientes.
Nefritis relacionada con el sistema inmunitario
Se produjo nefritis en 17 (0.3%) pacientes, incluidos casos de Grado 2, 3 o 4 en 3 (0.1%), 12 (0.2%) y 1 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab en monoterapia. La mediana de tiempo hasta la aparición de nefritis fue de 5.1 meses (rango, 12 días a 12.8 meses). La mediana de duración fue de 1.8 meses (rango, 6 días a 10.5+ meses).
La nefritis condujo a la suspensión definitiva de pembrolizumab en 7 (0.1%) pacientes y se resolvió en 9 pacientes, 1 con secuelas. En pacientes con NSCLC no escamoso tratados con pembrolizumab en combinación con pemetrexed y quimioterapia basada en platino (n=488), la incidencia de nefritis fue de 1.4% (todos los Grados) con 0.8% de Grado 3 y 0.4% de Grado 4.
Endocrinopatías relacionadas con el sistema inmunitario Se produjo hipofisitis en 32 (0.6%) pacientes, incluidos casos de Grado 2, 3 o 4 en 13 (0.3%), 15 (0.3%) y 1 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de hipofisitis fue de 5.3 meses (rango, 1 día a 17.7 meses). La mediana de duración fue de 1.7 meses (rango, 3 días a 18.1+ meses). La hipofisitis condujo a la suspensión definitiva de pembrolizumab en 8 (0.2%) pacientes y se resolvió en 9 pacientes, 7 con secuelas.
Se produjo hipertiroidismo en 197 (4.0%) pacientes, incluidos casos de Grado 2 o 3 en 52 (1.1%) y 5 (0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de hipertiroidismo fue de 1.4 meses (rango, 1 día a 21.9 meses). La mediana de duración fue de 1.7 meses (rango, 4 días a 15,5+ meses). El hipertiroidismo condujo a la suspensión definitiva de pembrolizumab en 3 (0.1%) pacientes y se resolvió en 152 (77.2%) pacientes, 1 con secuelas.
Se produjo hipotiroidismo en 514 (10.4%) pacientes, incluidos casos de Grado 2 o 3 en 377 (7.6%) y 7 (0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de hipotiroidismo fue de 3.5 meses (rango, 1 día a 18.9 meses). La mediana de duración no se alcanzó (rango, 2 días a 29.9+ meses). Dos pacientes (<0.1%) discontinuaron pembrolizumab debido a hipotiroidismo. El hipotiroidismo se resolvió en 107 (20.8%) pacientes, 9 con secuelas.
Reacciones adversas cutáneas relacionadas con el sistema inmunitario
Se produjeron reacciones adversas cutáneas graves relacionadas con el sistema inmunitario en 66 (1.3%) pacientes, incluidos casos de Grado 2, 3 o 5 en 6 (0.1%), 48 (1.0%) y 1 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de reacciones cutáneas graves fue de 3.2 meses (rango, 4 días a 19.4 meses). La mediana de duración fue de 1.6 meses (rango, 1 día a 16.1+ meses). Las reacciones cutáneas graves condujeron a la suspensión definitiva de pembrolizumab en 5 (0.2%) pacientes. Las reacciones cutáneas graves se resolvieron en 46 pacientes.
Se han observado casos raros de síndrome de Stevens-Johnson y de necrólisis epidérmica tóxica, algunos de ellos con desenlace mortal (ver VII. Precauciones Generales).
X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO
No se han realizado estudios formales de interacciones medicamentosas farmacocinéticas con KEYTRUDA. Debido a que pembrolizumab es eliminado de la circulación a través de catabolismo, no se esperan interacciones medicamentosas metabólicas.
Se debería evitar el uso de corticosteroides sistémicos o inmunosupresores antes de administrar KEYTRUDA, debido a su interferencia potencial con la actividad farmacodinámica y eficacia de KEYTRUDA. Sin embargo, se pueden utilizar corticosteroides sistémicos u otros inmunosupresores después de la administración de KEYTRUDA, para tratar las reacciones adversas inmunomediadas [ver VII. Precauciones Generales].
XI. ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO
En pacientes tratados con pembrolizumab en monoterapia, el porcentaje de pacientes que experimentaron un cambio desde el estado basal hasta una anomalía de laboratorio de Grado 3 o 4 fue el siguiente: 10.8% para linfocitos disminuidos, 7.6% para sodio disminuido, 6.5% para hemoglobina disminuida, 5.2% para fosfato disminuido, 5.2% para glucosa elevada, 2.9% para fosfatasa alcalina elevada, 2.6% para AST elevada, 2.3% para ALT elevada, 2% para potasio disminuido, 1.8% para bilirrubina elevada, 1.6% para potasio elevado, 1.5% para albúmina disminuida, 1.5% para calcio elevado, 1.4% para creatinina elevada, 1.4% para plaquetas disminuidas, 1.4% para neutrófilos disminuidos, 1.2% para calcio disminuido, 0.8% para magnesio elevado, 0.6% para leucocitos disminuidos, 0.5% para glucosa disminuida, 0.2% para magnesio disminuido y 0.2% para sodio elevado.
En pacientes tratados con pembrolizumab en combinación con quimioterapia, el porcentaje de pacientes que experimentaron un cambio desde el estado basal hasta una anomalía de laboratorio de Grado 3 o 4 fue el siguiente: 23.8% para neutrófilos disminuidos, 20.2% para linfocitos disminuidos, 16.2% para hemoglobina disminuida, 14.6% para leucocitos disminuidos, 10.3% para plaquetas disminuidas, 7.9% para glucosa elevada, 7.8% para fosfato disminuido, 7.4% para sodio disminuido, 4.6% para potasio disminuido, 3.7% para ALT elevada, 3.6% para creatinina elevada, 3.5% para AST elevada, 2.9% para calcio disminuido, 2.6% para potasio elevado, 2.5% para albúmina elevada, 1.7% para calcio elevado, 1.2% para fosfatasa alcalina elevada, 0.9% para glucosa disminuida, 0.7% para bilirrubina elevada y 0.1% para sodio elevado.
XIII. DOSIS Y VÍA DE ADMINISTRACIÓN
General
Selección de Pacientes
Para el tratamiento con agente único de Cáncer de Pulmón de Células No Pequeñas
Se debe seleccionar a los pacientes con NSCLC avanzado o metastásico para el tratamiento con KEYTRUDA, con base en la presencia de expresión positiva de PD-L1 [ver IV. Indicaciones Terapéuticas, y V. Farmacocinética y Farmacodinamia - Estudios Clínicos].
Dosis Recomendada
KEYTRUDA se administra como una infusión intravenosa durante 30 minutos cada 3 semanas.
La dosis recomendada de KEYTRUDA es:
• 200 mg para NSCLC sin tratamiento previo como monoterapia o carcinoma urotelial.
• 200 mg para NSCLC en tratamiento combinado.
• 2 mg/kg para melanoma o NSCLC previamente tratado.
Al administrar KEYTRUDA como parte de un tratamiento combinado con quimioterapia, KEYTRUDA debe administrarse primero. Véase también la Información Para Prescribir de los agentes de quimioterapia administrados en tratamiento combinado.
Deberá tratarse a los pacientes con KEYTRUDA hasta la progresión de la enfermedad o toxicidad inaceptable. Se han observado respuestas atípicas (es decir, un aumento inicial transitorio en el tamaño del tumor o lesiones nuevas pequeñas dentro de los primeros meses de tratamiento, seguido por la reducción del tumor). Los pacientes estables clínicamente con evidencia inicial de progresión de la enfermedad deberían permanecer en tratamiento hasta que se confirme la progresión de la enfermedad.
Para conocer el curso máximo del tratamiento con KEYTRUDA en los estudios clínicos, por favor consulte la descripción del estudio clínico para la respectiva indicación (ver V. Farmacocinética y Farmacodinamia).
Modificaciones de la dosis
Tabla 11: Modificaciones de Dosis Recomendadas (ver VII. Precauciones Generales)
|
Reacciones adversas |
Severidad |
Modificación de la dosis |
|
Neumonitis inmunomediada |
Moderada (Grado 2) |
Suspender hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Grave o que amenaza la vida (Grado 3 o 4) o moderada recurrente (Grado 2) |
Descontinuar permanentemente |
|
|
Colitis inmunomediada |
Moderada o grave (Grado 2 o 3) |
Suspender hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Que amenaza la vida (Grado 4) o grave recurrente (Grado 3) |
Descontinuar permanentemente |
|
|
Nefritis inmunomediada |
Moderada (Grado 2) |
Suspender hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Grave o que amenaza la vida (Grado 3 o 4) |
Descontinuar permanentemente |
|
|
Endocrinopatías inmunomediadas |
Grave o que amenaza la vida (Grado 3 o 4) |
Suspender hasta que las reacciones adversas se recuperen a Grado 0-1* Para pacientes con endocrinopatía grave (Grado 3) o que amenaza la vida (Grado 4) que mejora a Grado 2 o menor y es controlada con reemplazo hormonal, puede considerarse la continuación de KEYTRUDA |
|
Hepatitis inmunomediada |
Aspartato aminotransferasa (AST) o alanina aminotransferasa (ALT) >3 a 5 veces el límite superior normal (ULN, por las siglas upper limit of normal) o bilirrubina total >1.5 a 3 veces ULN |
Suspender hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
AST o ALT >5 veces ULN o bilirrubina total >3 veces ULN |
Descontinuar permanentemente |
|
|
Para pacientes con metástasis hepáticas que inician tratamiento con elevación moderada (Grado 2) de AST o ALT, si AST o ALT incrementa ≥50% con relación a su valor basal y dura ≥1 semana |
Descontinuar permanentemente |
|
|
Reacciones cutáneas inmunomediadas o síndrome de Stevens-Johnson (SJS) o necrólisis epidérmica tóxica (TEN) |
Reacciones cutáneas graves (Grado 3) o sospecha de SJS o de TEN |
Suspender hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Reacciones cutáneas graves (Grado 4) o SJS o TEN confirmados |
Descontinuar permanentemente |
|
|
Otras reacciones adversas inmunomediadas |
Con base en la gravedad y tipo de reacción (Grado 2 o Grado 3) |
Suspender hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Miocarditis, encefalitis, o síndrome de Guillain-Barré, grave o que amenaza la vida (Grado 3 o 4) |
Descontinuar permanentemente |
|
|
Que amenaza la vida (Grado 4) o grave recurrente (Grado 3) |
Descontinuar permanentemente |
|
|
Reacciones relacionadas con la infusión |
Grave o que amenaza la vida (Grado 3 o 4) |
Descontinuar permanentemente |
Nota: los grados de toxicidad están de acuerdo con la Terminología Común para Eventos Adversos del Instituto Nacional del Cáncer, Versión 4.0 (NCI-CTCAE v.4).
* Si la dosis de los corticosteroides no puede ser reducida a ≤10 mg de prednisona o equivalente por día dentro de 12 semanas o la toxicidad relacionada con el tratamiento no se resuelve a Grado 0-1 dentro de 12 semanas después de la última dosis de KEYTRUDA, entonces debe descontinuarse permanentemente a KEYTRUDA.
Preparación y administración
• Protéjase de la luz. No se congele. No agitar.
• Esperar que el frasco ámpula de KEYTRUDA alcance la temperatura ambiente.
• Antes de la dilución, el frasco ámpula de la solución puede estar sin refrigeración (a temperatura de 25°C o por debajo de ésta) hasta por 24 horas.
• Los medicamentos parenterales se deben inspeccionar visualmente buscando partículas extrañas y alteraciones en el color antes de la administración. KEYTRUDA es una solución que va de transparente a ligeramente opalescente, de incolora a ligeramente amarilla. Deseche el frasco ámpula si se observan partículas visibles.
• Retirar el volumen requerido hasta 4 mL (100 mg) de KEYTRUDA y transferir a una bolsa de infusión intravenosa que contenga cloruro de sodio al 0.9% o glucosa al 5% (dextrosa) para preparar una solución diluida con una concentración final que varía de 1 a 10 mg/mL. Mezclar la solución diluida invirtiendo suavemente.
• No congelar la solución para infusión.
• El producto no contiene conservador. El producto diluido se debe usar inmediatamente. Si no se utiliza de inmediato, las soluciones diluidas de KEYTRUDA se pueden almacenar a temperatura ambiente durante un tiempo acumulado hasta de 6 horas. Las soluciones diluidas de KEYTRUDA también se pueden almacenar bajo refrigeración entre 2°C a 8°C; sin embargo, el tiempo total desde la dilución de KEYTRUDA hasta completar la infusión no debería exceder las 24 horas. Si se refrigera, permitir que los frascos ámpula y/o las bolsas de infusión IV alcancen la temperatura ambiente previo a su uso.
• Administrar la solución para infusión por vía intravenosa durante 30 minutos utilizando un filtro de 0.2 a 5 μm en línea o de adición, estéril, no pirogénico, de baja unión a proteínas.
• No administrar conjuntamente con otros medicamentos a través de la misma línea de infusión.
• Desechar cualquier porción no utilizada que quede en el frasco ámpula.
Pacientes Pediátricos
Aún no se establecen la seguridad y eficacia de KEYTRUDA en niños menores de 18 años.
Pacientes Geriátricos
No se reportaron diferencias generales en la seguridad o eficacia entre los pacientes de edad avanzada (65 años de edad y mayores) y pacientes más jóvenes (menores de 65 años). No es necesario ajustar la dosis en esta población.
Insuficiencia Renal
No es necesario ajustar la dosis para pacientes con insuficiencia renal leve o moderada. No se ha estudiado KEYTRUDA en pacientes con insuficiencia renal grave. Insuficiencia hepática
No es necesario ajustar la dosis para pacientes con insuficiencia hepática leve. No se ha estudiado KEYTRUDA en pacientes con insuficiencia hepática moderada o grave.
XIV. MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL
No existe información sobre la dosificación con KEYTRUDA. No se ha determinado la máxima dosis tolerada de KETYTRUDA. En estudios clínicos, los pacientes recibieron hasta 10 mg/kg con un perfil de seguridad similar al observado en pacientes que recibieron 2 mg/kg.
En caso de sobredosis, los pacientes se deberían monitorear de cerca para buscar signos y síntomas de reacciones adversas, e instaurar un tratamiento sintomático apropiado.
XII. PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD
Toxicidad Crónica
En un estudio de toxicidad de dosis repetidas de 1 mes y 6 meses se evaluó la seguridad de pembrolizumab en monos Cynomolgus a los cuales se administraron por vía IV dosis de 6, 40 o 200 mg/kg una vez a la semana en el estudio de 1 mes y una vez cada dos semanas en el estudio de 6 meses, seguido por un periodo sin tratamiento de 4 meses. No se observaron hallazgos de importancia toxicológica y el nivel sin efecto adverso observado (NOAEL, por las siglas en inglés para No Observed Adverse Effect Level) en ambos estudios fue ≥200 mg/kg, lo cual es 19 veces la exposición en humanos a la dosis clínicamente más alta analizada (10 mg/kg) y 94 veces la exposición en humanos a una dosis recomendada de 2 mg/kg.
Carcinogénesis
No se ha evaluado el potencial carcinogénico de pembrolizumab en estudios a largo plazo en animales.
Mutagénesis
No se ha evaluado el potencial genotóxico de pembrolizumab.
Reproducción
No se han realizado estudios de reproducción en animales con KEYTRUDA. La principal función de la vía PD-1/PD-L1 es preservar el embarazo al mantener la tolerancia inmune al feto. Se ha demostrado que el bloqueo de la señalización de PD-L1 en modelos murinos de gestación altera la tolerancia al feto y resulta en un incremento en las pérdidas fetales. Estos resultados indican un riesgo potencial de que la administración de KEYTRUDA durante el embarazo podría causar daño fetal, incluyendo aumento en las tasas de aborto o en el nacimiento de fetos muertos. Según se reportó en la literatura, no hubo malformaciones relacionadas con el bloqueo de la señalización del PD-1 en la descendencia de estos animales; sin embargo, los desórdenes inmunomediados ocurrieron en los ratones knockout-PD-1. Con base en su mecanismo de acción, la exposición fetal a pembrolizumab puede incrementar el riesgo de desarrollar desórdenes inmunomediados o de alteración de la respuesta inmune normal.
Desarrollo
No se han realizado estudios de toxicidad sobre el desarrollo con pembrolizumab. No se presentaron efectos notables en los órganos reproductivos masculinos y femeninos en monos, con base en los estudios de toxicidad a dosis repetidas de 1 y 6 meses.
XVIII. NOMBRE Y DOMICILIO DEL LABORATORIO
Schering-Plough, S.A. de C.V.
Av. 16 de Septiembre No. 301, Col. Xaltocan,
C.P. 16090, Xochimilco, Ciudad de México, México.
XV. PRESENTACIÓN
Caja de cartón con un frasco ámpula con 100 mg / 4 mL de Pembrolizumab e instructivos anexos (para el paciente y para el personal de salud).
XVI. RECOMENDACIONES SOBRE ALMACENAMIENTO
Consérvese en refrigeración entre 2°C y 8°C.
No se congele.
Protéjase de la luz.
Consérvese la caja bien cerrada.
No agitar.
Para condiciones de almacenamiento después de la dilución del medicamento, vea la sección XIII. Dosis y Vía de Administración.
XVII. LEYENDAS DE PROTECCIÓN
Su venta requiere receta médica.
No agitar.
El uso de este medicamento no se recomienda durante el embarazo o lactancia a menos que el beneficio supere el riesgo potencial para el feto o el lactante.
No se deje al alcance de los niños.
Medicamento de alto riesgo.
No se administre a menores de 18 años de edad.
Literatura exclusiva para médicos.
Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx;
y dpocmx@merck.com


