
KORALIX
LEFLUNOMIDA
Tableta
1 Caja, 1 Frasco(s), 30 Tabletas, 20 mg
1 Caja, 1 Envase de burbuja o celopolial, 3 Tabletas, 100 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Leflunomida 20.0 mg, 100.0 mg
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
Antirreumático modificador de enfermedad.
• Indicado para el tratamiento de la artritis reumatoide activa. Reduce los signos y síntomas, inhibe el daño estructural de las articulaciones, y mejora la función física y la calidad de vida del paciente.
• Indicado para el tratamiento de artritis psoriásica activa.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacología humana: Fármaco antirreumático modificador de la enfermedad con propiedades antiproliferativas. Reduce los signos y síntomas, y retarda la progresión de la destrucción de las articulaciones en la artritis reumatoide activa. En los estudios que se han llevado a cabo, la mayor parte de los pacientes fueron tratados concomitantemente con AINEs o corticosteroides en dosis bajas.
Farmacología animal: La leflunomida es eficaz en modelos animales con artritis, otras enfermedades autoinmunes y trasplante. Posee efectos y características inmunomoduladores/inmunosupresores, actúa como agente antiproliferativo y muestra propiedades antiinflamatorias.
En estudios “in vivo”, Leflunomida es metabolizada rápidamente y casi por completo a A771726, el cual es activo in vitro y es presumiblemente el producto medicinal activo. Cuando se administra en la fase temprana de la progresión de la enfermedad, presenta los mejores efectos protectores en modelos animales de enfermedades autoinmunes. En modelos animales de rechazo crónico injerto contra huésped, y en rechazo de injerto de órgano sólido, leflunomida ha prolongado el tiempo de rechazo o ha dado marcha atrás a las reacciones de rechazo en curso. Aunque Leflunomida sólo ha dado una débil o no-actividad analgésica o antipirética, presenta actividad antiinflamatoria. En un modelo de septicemia experimental, leflunomida no alteró la resistencia de los ratones a los patógenos bacterianos.
Mecanismo de acción: En diferentes fases del ciclo celular, el metabolito activo de la leflunomida (A771726), retarda el desarrollo del ciclo celular de las células blanco.
In vitro, después de la estimulación mitógena, el A771726 inhibe la proliferación de las células T y la síntesis de DNA. La proliferación estimulada por mitógenos de las células sanguíneas mononucleares periféricas de humanos (PBMCs) es inhibida, así como la proliferación de líneas celulares transformadas de humano y de murinos, de manera dependiente de la dosis. La actividad antiproliferativa es revertida al agregar uridina a los cultivos celulares, lo cual indica que el A771726 actúa a nivel de la vía de biosíntesis de pirimidinas. Estudios de unión con ligandos radiactivos demostraron que el metabolito activo se une e inhibe a la enzima dihidroorotato deshidrogenasa de humano (DHODH). Lo anterior sugiere que puede inhibirse la síntesis de pirimidinas en linfocitos, especialmente los que favorecen las reacciones autoinmunes, y en menor grado en otras poblaciones de células que se dividen rápidamente, in vivo y con las concentraciones de leflunomida que se alcanzaron en los pacientes tratados. Tanto in vitro como in vivo, también se ha reportado la inhibición de la actividad de la tirosina cinasa. La actividad in vitro tiene lugar sólo con concentraciones de A771726 mucho más altas que las necesarias para inhibir a la DHODH y no parece estar mediada directamente a través de la inhibición de la enzima.
Eficacia clínica:
Artritis reumatoide: En el tratamiento de la artritis reumatoide (AR), la eficacia de Leflunomida fue demostrada en tres estudios controlados mostrando una reducción en los signos y síntomas, y en la inhibición del daño estructural. La eficacia también se demostró por la mejora de la función física, en dos de los tres ensayos controlados.
En todos los estudios de monoterapia del leflunomida, una dosis de carga inicial de 100 mg por día durante tres días, sólo se utilizó 20 mg por día a partir de entonces.
Estudio US301: Estudio a dos años, controlado con placebo, aleatorizado con 482 pacientes activos con AR de al menos 6 meses de duración con leflunomida 20 mg/día (n = 182), metotrexato 7.5 mg/semana incrementándose a 15 mg/semana (n = 182) o placebo (n = 118).
Todos los pacientes recibieron ácido fólico de 1 mg dos veces al día. El análisis primario fue a las 52 semanas con tratamiento ciego a 104 semanas.
En total, 235 de los 508 pacientes tratados al azar (482 en el análisis de datos primarios y 26 pacientes adicionales), continuaron en un segundo tratamiento doble ciego por 12 meses (98 leflunomida, 101 metotrexato, 36 placebo). Leflunomida dosis continua de 20 mg/día y la dosis de metotrexato pudo ser aumentada hasta un máximo de 20 mg/semana. En total de 190 pacientes (83 leflunomida, metotrexato 80, 27 placebo) completaron 2 años de tramiento doble ciego.
Estudio MN 301/303/305: Estudio MN301, estudio controlado con placebo, aleatorizado, 358 pacientes con AR activa con leflunomida 20 mg/día (n = 133), sulfasalazina 2.0 g/día (n = 133) o placebo (n = 92).
La duración del tratamiento fue de 24 semanas. Una extensión de este estudio fue opcional a 6 meses, no placebo controlado activo, cegado continuación del MN301, resultando en 12 meses comparando con leflunomida y sulfasalazina (estudio MN303).
De los 168 pacientes que completaron 12 meses de tratamiento en MN301 y MN303, 146 pacientes (87%) entró en una extensión de 1 año sin placebo estudio activo controlado de doble tratamiento ciego activo (MN305, 60 leflunomida, con sulfasalazina 60, 26 con placebo/sulfasalazina). Los pacientes continuaron con la misma dosis diaria de leflunomida o sulfasalazina que habían estado tomando en la terminación de MN301/303. Un total de 121 pacientes (53 leflunomida, sulfasalazina 47, placebo 21/sulfasalazina) completaron dos años de tratamiento doble ciego.
Estudios MN302/304: Estudio MN302, no-controlado con placebo activo, al azar 999 pacientes con AR activa a leflunomida 20 mg/día (n=501) o metotrexato de 7.5 mg/semana aumentar a 15 mg/semana (n = 498). Suplementos de folato se utilizó en 10% de pacientes. La duración del tratamiento fue de 52 semanas.
De los 736 pacientes que completaron 52 semanas de tratamiento en el estudio MN302, 612 (83%) ingresaron en el estudio doble ciego, MN304 extensión del estudio de 1 año (292 leflunomida, 320 metotrexato). Los pacientes continuaron en la misma dosis diaria de leflunomida o metotrexato que habían estado tomando en la terminación de MN302. Hubo 533 pacientes (256 con leflunomida, metotrexato 277) que completaron 2 años de doble ciego el tratamiento.
a) Signos y síntomas de artritis reumatoide: Mediante el índice de respuesta 20 del Colegio Americano de Reumatología (ACR), compuesto de laboratorio clínico y medidas funcionales en la artritis reumatoide, se evaluaron la mejora de los signos y síntomas de la artritis reumatoide. Una respuesta de "ACR20" es la mejoría en un paciente del ≥ 20% en el dolor o molestia y en el número de articulaciones inflamadas y 3 de los siguientes 5 criterios: evaluación global del médico, evaluación global del paciente, capacidad funcional de medida [Modificado del Cuestionario de Evaluación de la Salud (MHAQ)], escala análoga visual de dolor, y la velocidad de sedimentación globular o proteína C-reactiva.
Una "respuesta final del ACR20" es un paciente que completó el estudio y respondió el ACR20 en la realización del estudio.
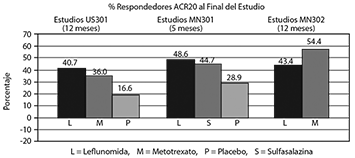
La respuesta ACR20 a las tasas de punto final con LEFLUNOMIDA fue estadísticamente significativa superior al placebo en la reducción de los signos y síntomas de la AR en el análisis primario de eficacia, la respuesta ACR20 en punto final, del estudio US301 (en el punto final primario 12 meses) y MN301 (al final del tratamiento de 6 meses) (figura 1). Las tasas de respuesta de punto final con el tratamiento LEFLUNOMIDA fueron consistentes entre los 6 y 12 meses los estudios (41-49%) en el ACR20. No se observaron diferencias consistentes entre leflunomida y metotrexato o entre leflunomida y sulfasalazina.
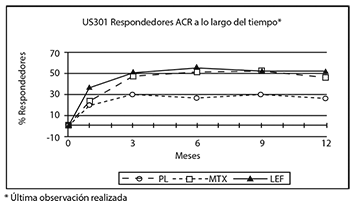
El efecto del tratamiento con LEFLUNOMIDA fue evidente por un mes, se estabilizó en 3-6 meses, y continuó a lo largo de tratamiento como se muestra en la figura 2.
Figura 1.
|
Comparaciones |
Intervalo de confianza 95% |
Valor de "p" |
|
US301 |
||
|
Leflunomida vs. placebo |
(12, 32) |
< 0.0001 |
|
Metotrexato vs. placebo |
(8, 30) |
< 0.0001 |
|
Leflunomida vs. Metotrexato |
(-4, 16) |
NS |
|
MN301 |
||
|
Leflunomida vs. placebo |
(7, 33) |
0.0026 |
|
Sulfasalazina vs. placebo |
(4, 29) |
0.0121 |
|
Leflunomida vs. sulfasalazina |
(-8, 16) |
NS |
|
MN302 |
||
|
Leflunomida vs. Metotrexato |
(-19, -7) |
< 0.0001 |
Figura 2.
• Última observación realizada.
Respuesta ACR50 y ACR70 se definen de una manera análoga a la respuesta ACR 20, usando mejoras de 50% o 70%, respectivamente (tabla 1). El cambio promedio para los componentes de los índices de respuesta ACR individuales se muestra en la tabla 2.
Tabla 1. Resumen de las tasas de respuesta del ACR*
|
Grupo de estudio y tratamiento |
ACR20 |
ACR50 |
ACR70 |
|
Estudio placebo-control |
|||
|
US301 (12 meses) |
|||
|
Leflunomida (n = 178)† |
52.2‡ |
34.3‡ |
20.2‡ |
|
Placebo (n = 118)† |
26.3 |
7.6 |
4.2 |
|
Metrotexato (n = 180)† |
45.6 |
22.8 |
9.4 |
|
MN301 (6 meses) |
|||
|
Leflunomida (n = 130)† |
54.6‡ |
33.1‡ |
10.0§ |
|
Placebo (n = 91)† |
28.6 |
14.3 |
2.2 |
|
Sulfasalazina (n = 132)† |
56.8 |
30.3 |
7.6 |
|
Estudio activo-controlado sin placebo |
|||
|
MN302 (12 meses) |
|||
|
Leflunomida (n = 495)† |
51.1 |
31.1 |
9.9 |
|
Metrotexato (n = 489)† |
65.2 |
43.8 |
16.4 |
* Intención de tratamiento (ITT) con el análisis de la última observación realizada (LOCF) técnica para pacientes con abandono temprano.
t N es el número de pacientes ITT de para quienes se disponía datos suficientes para calcular las tasas que se indican.
‡ p < 0.001 placebo vs. leflunomida.
§ p < 0.02 Estudios de leflunomida vs. placebo.
La tabla 2 muestra los resultados de los componentes de los criterios de respuesta ACR para US301, MN301 y MN302. LEFLUNOMIDA fue significativamente superior al placebo en todos los componentes de los criterios de respuesta ACR en el estudio US301 y MN301. Además LEFLUNOMIDA fue significativamente superior al placebo en la mejora de la rigidez matinal, una medida de la actividad de la enfermedad en la AR, no incluidos en los criterios de respuesta ACR. No se observaron diferencias consistentes como se demostró entre LEFLUNOMIDA y los comparadores activos.
Tabla 2. Cambio promedio en los componentes del índice de respuesta ACR*
|
Componentes |
Estudios Placebo-Control |
Estudios controlados sin placebo |
||||||
|
US301 (12 meses) |
MN301 No-US (6 meses) |
MN302 NO US (12 meses) |
||||||
|
Lefluno-mida |
Metotre-xato |
Placebo |
Lefluno-mida |
Metotre-xato |
Placebo |
Lefluno-mida |
Metotre-xato |
|
|
Número de articulaciones sensibles1 |
-7.7 |
-6.6 |
-3.0 |
-9.7 |
-8.1 |
-4.3 |
-8.3 |
-9.7 |
|
Número de articulaciones inflamadas1 |
-5.7 |
-5.4 |
-2.9 |
-7.2 |
-6.2 |
-3.4 |
-6.8 |
-9.0 |
|
Evaluación global del paciente2 |
-2.1 |
-1.5 |
0.1 |
-2.8 |
-2.6 |
-0.9 |
-2.3 |
-3.0 |
|
Evaluación global médica2 |
-2.8 |
-2.4 |
-1.0 |
-2.7 |
-2.5 |
-0.8 |
-2.3 |
-3.1 |
|
Función física/discapacidad (MHAQ/HAQ) |
-0.29 |
-0.15 |
0.07 |
-0.5 |
-0.29 |
-0.04 |
-0.37 |
-0.44 |
|
Intensidad del dolor2 |
-2.2 |
-1.7 |
-0.5 |
-2.7 |
-2 |
-0.9 |
-2.1 |
-2.9 |
|
Tasa de sedimentación eritrocitaria |
-6.26 |
-6.48 |
2.56 |
-7.48 |
-16.56 |
3.44 |
-10.12 |
-22.18 |
|
Proteína C reactiva |
-0.62 |
-0.5 |
0.47 |
-2.26 |
-1.19 |
0.16 |
-1.86 |
-2.45 |
|
No inclusión en el índice de respuesta ACR |
||||||||
|
Rigidez matutina (min) |
-101.4 |
-88.7 |
14.7 |
-93 |
-42.4 |
-6.8 |
-63.7 |
-86.6 |
* Última observación realizada; cambio negativo indica mejoría.
1 Basado en el conteo de 28 articulaciones.
2 Escala análoga visual - O = mejor; 10 = peor.
Mantenimiento del efecto: Después de completar 12 meses de tratamiento, los pacientes que continuaron el tratamiento del estudio se evaluaron por 12 meses de tratamiento en un doble ciego (periodo total de tratamiento de 2 años) en los estudios de US301, MN305 y MN304.
Las tasas de respuesta de ACR a los 12 meses se mantuvieron más de 2 años en la mayoría de los pacientes continuando un segundo año de tratamiento.
Mejora de los componentes basales individuales de los criterios de respuesta ACR, fueron mantenidos en la mayoría de los pacientes durante el segundo año de tratamiento con LEFLUNOMIDA en los tres ensayos.
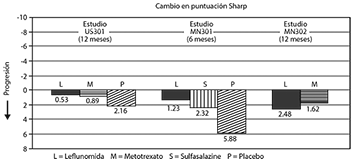
b) Inhibición del daño estructural: Por medio de la puntuación Sharp, puntuación compuesta de erosiones de rayos X y estrechamiento del espacio articular en manos, muñecas y pies, se evaluó la inhibición del daño estructural en comparación con el control.
En la figura 2 se muestra el cambio desde los puntos basales a los finales en la progresión de la enfermedad estructural, medidos por la puntuación Sharp de rayos X. LEFLUNOMIDA fue significativamente superior estadísticamente al placebo en la inhibición de la progresión de enfermedad por la escala Sharp. No se demostraron diferencias consistentes entre leflunomida y metotrexato o entre leflunomida y sulfasalizina.
Figura 3.
|
Comparación |
Intervalo de confianza 95% |
p |
|
|
US301 |
Leflunomida vs. Placebo |
(-4.0, -1.1) |
0.0007 |
|
Metotrexato vs. placebo |
(-2.6, -0.2) |
0.0196 |
|
|
Leflunomida vs. metotrexato |
(-2.3, 0.0) |
0.0499 |
|
|
MN301 |
Leflunomida vs. placebo |
(-6.2, -1.8) |
0.0004 |
|
Sulfasalazine vs. placebo |
(-6.9, 0.0) |
0.0484 |
|
|
Leflunomida vs. sulfasalazine |
(-3.3, 1.2) |
NS |
|
|
MN302 |
Leflunomida vs. metotrexato |
(-2.2, 7.4) |
NS |
c) Mejoramiento de la función física: La mejora de la función física se evaluó mediante el cuestionario de evaluación de la salud (HAQ) y el formulario de encuesta de resultados médicos de corto plazo (SF-36).
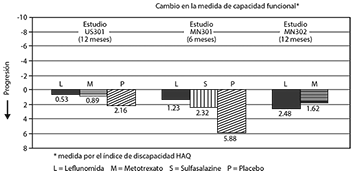
El cuestionario de evaluación de la salud (HAQ) evalúa la función física de un paciente y el grado de discapacidad. El cambio medio desde la basal en la capacidad funcional, medida por el índice de discapacidad HAQ (HAQ DI) en los 6 y 12 meses con placebo y el activo en ensayos controlados, LEFLUNOMIDA fue significativamente superior al placebo en mejorar la función física (figura 4).
Superioridad con el placebo se demostró consistentemente a través de las ocho sub-escalas HAQ DI (vestirse, levantarse, comer, caminar, higiene, alcance, agarre y actividades) en los dos estudios controlados con placebo.
Los resultados médicos del Short Form 36 (SF-36), con cualidad genérica relacionada con la salud del cuestionario de calidad de vida, se refiere además a la función física. En el US301, a los 12 meses LEFLUNOMIDA proporciona una mejoría estadísticamente significativa en comparación con el placebo en la puntuación del resumen del componente físico (PCS).
Figura 4.
|
Comparación |
Intervalo de confianza 95% |
p |
|
|
US301 |
Leflunomida vs. placebo |
(-0.58, -0.29) |
0.0001 |
|
Leflunomida vs. metotrexato |
(-0.34, -0.07) |
0.0026 |
|
|
MN301 |
Leflunomida vs. Placebo |
(-0.67, -0.36) |
<0.0001 |
|
Leflunomida vs. sulfasalazine |
(-0.33, -0.03) |
0.0163 |
|
|
MN302 |
Leflunomida vs. Metotrexato |
(0.01, 0.16) |
0.0221 |
Artritis psoriásica: Los pacientes adultos con artritis psoriásica (APs) se asignaron al azar a la leflunomida o con el placebo. La duración del tratamiento fue de 6 meses y la dosificación fue de 100 mg/día durante tres días, seguido de 20 mg/día de lefunomida para el resto del periodo. En el grupo de leflunomida, de los pacientes que fueron totalmente analizables (n = 186), 59.0% tuvo una mejoría de la artritis psoriásica de acuerdo con el criterio de respuesta del tratamiento (PsARC), de la variable principal, frente a 29.7% en el grupo placebo (p < 0.0001).
PsARC es una medida que combina una evaluación global médica, para un paciente de auto-evaluación global, en una puntuación de dolor en las articulaciones/sensibilidad y una puntuación de inflamación articular.
Mejora de PsARC se define como una disminución de ≥ 30% para las puntuaciones de las articulaciones y en ≥ 1 punto para la evaluación global. Una mejora de al menos 2 de los anteriores, uno de los cuales tuvo que ser dolor en las articulaciones/sensibilidad o puntuación de articulaciones inflamadas y sin empeoramiento en ninguna de las cuatro medidas, fue requerido para ser considerado como una respuesta (véase tabla 3).
Tabla 3. Cambio promedio en los componentes del índice de respuesta PsARC*
|
Componentes |
Placebo estudio controlado (6 meses) |
||
|
Leflunomida (n=95) |
Placebo (n=91) |
Valor p |
|
|
Dolor en las articulaciones/puntuación de sensibilidad* |
-9.1 |
-4.6 |
0.0022 |
|
Puntuación de inflamación articular |
-6.8 |
-4.2 |
0.0013 |
|
Evaluación global médica, mejora al menos en 1 categoría |
52.6% |
34.1% |
< 0.0001 |
|
Autoevaluación global del paciente, mejora al menos en 1 categoría |
31.6% |
30.8% |
0.0036 |
* Cambio negativo indica mejora.
Escala de Likert 1-5 puntos-1= muy bueno; 5= muy malo.
Los cambios en psoriasis de acuerdo al índice de intensidad y gravedad de la psoriasis (PASI) reflejan los cambios en el alcance y la gravedad de las lesiones de la psoriasis, a juzgar por eritema, descamación e infiltración. La leflunomida resultó en una mejora significativa en las puntuaciones PASI a las 24 semanas del estudio en relación al placebo, con una media de 22.4% (± 51.6) en el grupo de leflunomida en comparación con un deterioro de 2.2% (± 70,4%) en el grupo placebo (p = 0.0030). En comparación con el grupo placebo, una proporción significativamente mayor de los pacientes en el grupo de leflunomida experimentaron una reducción ≥ 50% en la puntuación PASI (PASI 50; 18.9% vs. 30.4%, p = 0.050) y ≥ 75% de reducción en la puntuación PASI (PASI 75; 7.8% vs. 17.4%, p = 0.048) respecto al valor basal.
Los eventos adversos observados en el estudio clínico en pacientes con artritis psoriásica son comparables con los eventos adversos observados en los ensayos clínicos en pacientes con artritis reumatioide.
Farmacocinética: La leflunomida es rápidamente convertida a su metabolito activo, A771726 por un metabolismo de primer paso que se lleva a cabo en la pared del intestino e hígado. En un estudio realizado en tres voluntarios sanos a los que se administró leflunomida marcada con 14C, no se detectó leflunomida intacta en plasma, orina y heces.
En otros estudios, la concentración plasmática de leflunomida intacta se detectó en raras ocasiones y fue del orden de ng/ml. El único metabolito marcado radiactivamente que se identificó en plasma fue el A771726, el cual es responsable de esencialmente toda la actividad in vivo de LEFLUNOMIDA.
Absorción: Los estudios de excreción del fármaco marcado con 14C indicaron que se absorbe, por lo menos, alrededor de 82 a 95% de la dosis. El pico de concentración plasmática del A771726 se alcanza en tiempos muy variables, por lo que puede presentarse entre 1 y 24 horas después de una sola administración de leflunomida. LEFLUNOMIDA puede administrarse con los alimentos, ya que su grado de absorción es comparable en condiciones de ayuno y después de haber tomado alimentos. Debido a que la vida media del A771726 es muy prolongada (aproximadamente 2 semanas), se requiere una dosis de carga de leflunomida de 100 mg diarios, durante 3 días, para facilitar la rápida obtención de los niveles terapéuticos (estado estable). Sin esta dosis de carga, se ha estimado que se requerirían cerca de 2 meses de tratamiento para alcanzar las concentraciones plasmáticas terapéuticas. En los estudios realizados en pacientes con artritis reumatoide a los que se administraron dosis múltiples, los parámetros farmacocinéticos del A771726 resultaron lineales en el rango de dosis de 5 a 25 mg. En estos estudios, el efecto clínico estuvo estrechamente relacionado tanto con la concentración plasmática del A771726 como con la dosis diaria de leflunomida. Con una dosis de leflunomida de 20 mg/día, la concentración plasmática promedio del A771726 en el estado estable fue de aproximadamente 35 μg/ml. En el estado estable, las concentraciones plasmáticas son de 33 a 35 veces más altas que la de una sola dosis.
Distribución: El A771726 se une ampliamente a la albúmina en el plasma humano. La fracción libre del A771726 es de aproximadamente 0.62%. La unión a proteínas del A771726 fue lineal en el rango de concentraciones terapéuticas, y apareció ligeramente reducida y muy variable en el plasma de pacientes con artritis reumatoide o insuficiencia renal crónica. La amplia unión a proteínas del A771726 puede ocasionar el desplazamiento de otros fármacos sumamente unidos. Otros estudios de interacción por unión a proteínas plasmáticas realizados in vitro con warfarina en concentraciones clínicamente relevantes, no han demostrado tal interacción. Estudios semejantes demostraron que el ibuprofeno, diclofenaco y tolbutamida no desplazan al A771726, mientras que la fracción libre de este metabolito se incrementa de 2 a 3 veces en presencia de tolbutamida. El A771726 desplaza al ibuprofeno, diclofenaco y tolbutamida, aunque la fracción libre de estos fármacos sólo se incrementa 10% a 50%. No hay evidencia de que estos efectos tengan relevancia clínica. Como resultado de la amplia unión a proteínas del A771726, el volumen aparente de distribución es bajo (aproximadamente 11 L). No hay captura preferente en eritrocitos.
Metabolismo: La leflunomida se metaboliza hasta un metabolito primario, el A771726, y varios metabolitos secundarios que incluyen a la 4-trifluorometilanilina (TFMA). La biotransformación metabólica de la leflunomida hasta el A771726, así como el subsecuente metabolismo del M1, no son controlados por una sola enzima y se presentan en las fracciones celular citosólica y microsomal. Los estudios de interacción con cimetidina (inhibidor inespecífico del citocromo P-450) y rifampicina (inductor inespecífico del citocromo P-450) han indicado que in vivo, las enzimas CYP están involucradas en menor grado en el metabolismo de la leflunomida.
Eliminación: La eliminación del metabolito activo A771726 es lenta, con una depuración aparente de aproximadamente 31 ml/h. La vida media de eliminación es de aproximadamente 2 semanas en los pacientes. Después de la administración de una dosis de leflunomida marcada radiactivamente, la radiactividad se excretó igualmente en heces, probablemente por excreción biliar y en orina. El A771726 fue detectado en orina y en heces aun después de 36 días de la administración de una sola dosis.
Los principales metabolitos que se encontraron en orina fueron glucurónidos, de leflunomida (principalmente en muestras tomadas entre 0 y 24 horas) y un ácido oxanílico derivado de A771726. El metabolito fecal primario fue el A771726.
La administración de una suspensión oral de carbón activado (polvo hecho suspensión) o colestiramina produjo un incremento rápido y significativo del grado de eliminación del A771726, así como una disminución de su concentración en plasma. Lo antes mencionado parece deberse a un mecanismo de diálisis gastrointestinal y/o a la interrupción del reciclaje enterohepático.
Farmacocinética en presencia de insuficiencia renal: La leflunomida fue administrada en dosis orales únicas de 100 mg a 3 pacientes con hemodiálisis y a 3 pacientes con diálisis peritoneal continua. En pacientes con diálisis peritoneal continua, la farmacocinética de A771726 pareció ser similar a la de voluntarios sanos. En sujetos con hemodiálisis se observó una eliminación más rápida de A771726, lo que no fue debido a la extracción del fármaco en el dializado, sino al desplazamiento de la unión a proteínas. El análisis cinético poblacional de los 6 pacientes demostró que aunque la depuración de A771726 aumenta aproximadamente dos veces, la vida media de eliminación terminal es semejante a la de sujetos sanos, ya que también se incrementa el volumen de distribución.
Farmacocinética en presencia de insuficiencia hepática: No se dispone de datos en cuanto al tratamiento de pacientes con insuficiencia hepática. El metabolito activo de la leflunomida se une ampliamente a proteínas, y se depura por metabolismo hepático y secreción biliar. Estos procesos pueden estar afectados por la insuficiencia hepática.
Efecto de la edad: La farmacocinética de leflunomida no se ha estudiado en niños ni en adolescentes. Los datos farmacocinéticos de individuos de edad avanzada (mayores de 65 años) son limitados, pero comparables a los de adultos jóvenes.
Farmacocinética en fumadores: El análisis farmacocinético poblacional de los datos obtenidos en un estudio fase III indicó que la depuración se incrementa en 38% en los fumadores, con respecto a los no fumadores; sin embargo, no se encontró ninguna diferencia en cuanto a eficacia clínica en los fumadores y no fumadores.
CONTRAINDICACIONES:
LEFLUNOMIDA no debe ser usado en:
• Pacientes con hipersensibilidad a leflunomida o a alguno de sus excipientes.
• LEFLUNOMIDA está contraindicado en mujeres embarazadas y en mujeres en edad fértil que no estén usando un método anticonceptivo confiable durante el tratamiento con leflunomida, y posteriormente, mientras las concentraciones plasmáticas del metabolito activo sean superiores a 0.02 mg/L. Debe descartarse el embarazo antes de iniciar el tratamiento con LEFLUNOMIDA.
• Inmunodeficiencia severa, afectación significativa de la médula ósea o anemia, infecciones severas no controladas, afectación severa de la función hepática, lactancia y menores de 18 años.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
LEFLUNOMIDA está contraindicado en mujeres embarazadas y en mujeres en edad fértil que no estén usando un método anticonceptivo confiable (véase Interacciones medicamentosas y de otro género) durante el tratamiento con leflunomida, y posteriormente, mientras las concentraciones plasmáticas del metabolito activo, A771726, sean superiores a 0.02 mg/L. Debe descartarse el embarazo antes de iniciar el tratamiento con LEFLUNOMIDA.
Debe indicarse a la paciente en edad fértil que informe a su médico inmediatamente si presenta algún retraso en la aparición de la menstruación o alguna otra razón que dé lugar a la sospecha de embarazo, para que se le realice una prueba de embarazo. Si la prueba de embarazo resulta positiva, el médico y la paciente deben discutir los riesgos de este estado. Es posible que disminuya el riesgo para el feto si al presentarse el primer retraso de la menstruación se instituye el procedimiento de eliminación del fármaco, con el fin de disminuir rápidamente las concentraciones sanguíneas del metabolito activo.
En las mujeres bajo tratamiento con LEFLUNOMIDA que decidan embarazarse, debe recomendarse practicar alguno de los siguientes procedimientos:
• Administrar 8 g de colestiramina 3 veces al día durante 11 días, después de suspender el tratamiento con leflunomida.
• Administrar 50 g de carbón activado 4 veces al día durante 11 días, después de suspender el tratamiento con leflunomida.
Los 11 días no necesitan ser consecutivos, a menos que se requiera descender con rapidez la concentración plasmática del A771726.
Con ambos esquemas de eliminación deberá confirmarse que la concentración plasmática del A771726 sea inferior a 0.02 mg/L, mediante dos pruebas realizadas con una diferencia de tiempo de cuando menos 14 días. Con base en los datos disponibles se ha calculado que las concentraciones plasmáticas inferiores a 0.02 mg/L (0.02 μg/ml) son de bajo riesgo.
Sin el procedimiento de eliminación del fármaco, se requieren hasta 2 años para que las concentraciones plasmáticas del A771726 sean menores a 0.02 mg/L, debido a las variaciones individuales en la eliminación del fármaco. Aun después de este tiempo, se requiere verificar que los niveles de A771726 sean menores a 0.02 mg/L en dos pruebas separadas, con un intervalo de al menos 14 días.
Si un periodo de espera de hasta aproximadamente 2 años con un método anticonceptivo eficaz es poco práctico, es aconsejable realizar profilácticamente un procedimiento de lavado.
La contracepción con anticonceptivos orales no puede ser garantizada durante el procedimiento de lavado con colestiramina o carbón activado. Se recomienda el uso alternativo de métodos anticonceptivos.
Lactancia: En estudios hechos en animales se ha visto que la leflunomida o sus metabolitos pasan a la leche materna. Sin embargo, se desconoce si en humanos LEFLUNOMIDA o sus metabolitos se excretan en la leche materna; por lo tanto, las mujeres no deben amamantar mientras estén recibiendo leflunomida , y se debe decidir si es conveniente iniciar la lactancia o el tratamiento con LEFLUNOMIDA, tomando en cuenta la importancia del fármaco para la madre.
Exposición durante el embarazo: Resultados de un estudio prospectivo en embarazo, se llevó a cabo por especialistas en información teratológica (OTIS), para estimar el riesgo de defectos congénitos y otros resultados adversos del embarazo debido a la exposición involuntaria a leflunomida durante el primer trimestre del embarazo. Embarazadas fueron reclutadas en tres grupos: mujeres con un diagnóstico de artritis reumatoide que participaron al menos con una dosis de leflunomida (n = 64), con enfermedad con ajuste con el grupo de comparación sin leflunomida durante el embarazo (n = 108), o un grupo de comparación de mujeres embarazadas sanas (n = 78).
Exposición inadvertida a la leflunomida durante el 1er trimestre de embarazo, seguida por la interrupción del procedimiento de drogas más lavado con colestiramina, dieron lugar a importantes defectos estructurales en 5.4% de los nacidos vivos en comparación con cualquiera de los grupos de comparación (4.2% en el grupo de enfermedades concordantes y 4.2% en embarazadas sanas). Los resultados de este estudio, que se interrumpió prematuramente debido a una disminución de reclutamiento, no cambia la contraindicación del uso inicial de leflunomida en el embarazo.
En particular, el estudio no se refirió a los posibles riesgos asociados con el uso de la leflunomida durante todo el periodo de desarrollo embrionario en todos los sujetos expuestos al grupo de leflunomida que abandonaron la medicación a partir del conocimiento de estar embarazada, casi todas pasaron por lo menos un curso del procedimiento de eliminación de drogas siendo la mayoría de los sujetos no expuestos a la leflunomida a más de 3 semanas después de la concepción.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las reacciones adversas relacionadas con el uso de leflunomida se han clasificado de acuerdo a su frecuencia en: comunes= 1-10% de los pacientes; no comunes = 0.1-1% de los pacientes; raras = 0.01-0.1% de los pacientes; muy raras = 0.01% o un porcentaje menor de los pacientes.
Sistema gastrointestinal y hepático:
Comunes: diarrea, náuseas, vómito, anorexia, trastornos de la mucosa oral (por ejemplo: estomatitis aftosa, úlceras orales), dolor abdominal, elevación de parámetros hepáticos (por ejemplo: transaminasas y, con menor frecuencia, gamma-glutamiltranspeptidasa, fosfatasa alcalina y bilirrubina).
Raras: hepatitis, ictericia/colestasis.
Muy raras: daño hepático severo, como insuficiencia hepática y necrosis hepática aguda, que puede poner en riesgo la vida. Pancreatitis.
Sistema cardiovascular:
Comunes: aumento de la tensión arterial.
Sistema linfático y hemático:
Comunes: leucopenia con cuenta leucocitaria > 2 x 109/L (> 2 g/L).
No comunes: anemia, trombocitopenia con cuenta plaquetaria < 100 x 109/L (< 100 g/L).
Raras: leucopenia con cuenta leucocitaria< 2 x 109/L (< 2 g/L), eosinofilia. Pancitopenia.
El uso reciente, concomitante o consecutivo, de fármacos potencialmente mielotóxicos puede ser asociado con un aumento en el riesgo de efectos hematológicos.
Muy raras : vasculitis. Debido a enfermedades subyacentes, no se ha podido establecer una relación causal.
Sistema nervioso:
Comunes: cefalea, vértigo, parestesia.
No comunes: trastornos del gusto, ansiedad.
Muy raras: neuropatía periférica.
Reacciones alérgicas, piel y apéndices:
Comunes: reacciones alérgicas moderadas (incluyendo reacciones maculopapulares y otros eritemas), prurito, eccema, resequedad de la piel, aumento de la pérdida de cabello.
No comunes: urticaria.
Muy raras : reacciones anafilácticas/anafilactoides severas. Síndrome de Stevens-Johnson (eritema multiforme mayor), necrólisis epidérmica tóxica.
En los reportes de casos recibidos hasta ahora, no se ha podido establecer una relación causal directa con el tratamiento con leflunomida, aunque no puede excluirse.
Vasculitis, incluyendo vasculitis cutánea necrotizante.
Debido a la enfermedad subyacente, una relación de causalidad no puede ser establecida.
Infecciones:
Raras: infecciones severas y sepsis, las cuales pueden poner en riesgo la vida.
La mayoría de los reportes de casos tuvieron como factor de confusión terapias inmunosupresoras concomitantes y/o enfermedades concomitantes, además de la artritis reumatoide, lo cual pudo haber predispuesto a los pacientes a la infección.
Los medicamentos con potencial inmunosupresor, como la leflunomida, pueden ocasionar que los pacientes sean más susceptibles a infecciones, incluso a infecciones oportunistas.
En los estudios clínicos, la incidencia de rinitis y bronquitis (5% vs. 2%), así como neumonía (3% vs. 0%) fue ligeramente más alta en los pacientes tratados con leflunomida, en comparación con placebo, mientras que la incidencia global de las infecciones fue comparable.
Trastornos respiratorios, torácicos y mediastinales:
Raras: enfermedad pulmonar intersticial (incluyendo neumonitis intersticial), que puede ser fatal.
Trastornos de la piel y tejido subcutáneo:
No conocidos: lupus eritematoso cutáneo, psoriasis pustulosa, o empeoramiento de la psoriasis.
Otras:
Comunes: pérdida de peso, astenia.
No comunes: hipocaliemia.
Puede presentarse hiperlipidemia leve. Las concentraciones de ácido úrico disminuyen comúnmente, debido a un efecto uricosúrico. Otros parámetros de laboratorio cuya importancia clínica no se ha establecido, pero que también pueden presentarse son: insignificante incremento de la deshidrogenasa láctica (LDH) y de la colecistocinina (CK), así como pequeña disminución en el fosfato.
Se ha reportado tendosinovitis y ruptura de tendones como eventos adversos bajo el tratamiento con leflunomida; sin embargo, no se ha establecido una relación causal.
No puede excluirse la disminución marginal (reversible) en la concentración de esperma, en la cuenta total de espermatozoides y en la motilidad progresiva rápida de los espermatozoides.
El riesgo de malignidad, particularmente trastornos linfoproliferativos, también se sabe que se incrementa con el uso de algunos fármacos inmunosupresores.
Estudios postmarketing: En un estudio multicéntrico, aleatorizado, controlado se determinó la respuesta clínica de eficacia en los pacientes que tomaron FARMEs (n = 121) con artritis reumatoide (AR) temprana tratados con leflunomida, usando los criterios ACR20 y evaluados a los 3 meses como punto final primario en dos grupos, con regímenes de tratamiento inicial (con y sin dosis de ataque). Durante el periodo de doble ciego de tres días iniciales, ambos grupos recibieron 20 mg o 100 mg de leflunomida con placebos similares . El periodo inicial fue seguido por un periodo abierto de mantenimiento de 3 meses, durante los cuales ambos grupos recibieron 20 mg de leflunomida al día. La eficacia de leflunomida fue confirmada en este estudio; sin embargo, no se observó ninguna ventaja en la población estudiada con el régimen con dosis de ataque. Al final del estudio la tasa de respuesta de ACR20 fue de 58.5% en el grupo con la dosis de ataque , contra 77.8% en el grupo sin la dosis de ataque (p = 0.025). Para los resultados de las variables secundarias (ACR50, ACR70, DAS28) no se observaron diferencias significativas en los resultados entre los dos grupos de tratamiento (p = 0.05).
Una respuesta clínica se observó durante el primer mes del tratamiento en más de la mitad de los sujetos, sin diferencia significativa a los 30 días, entre los grupos de tratamiento, para todos los criterios de eficacia. Los datos de seguridad obtenidos de ambos grupos de tratamiento fueron consistentes con el perfil de seguridad conocido de leflunomida; sin embargo, la incidencia de eventos adversos gastrointestinales y la elevación de las enzimas hepáticas tuvieron una tendencia mayor en los pacientes que recibieron la dosis de ataque con 100 mg de leflunomida.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Toxicidad aguda: Leflunomida, administrada oral e intraperitonealmente, ha sido analizada en estudios de toxicidad aguda en ratones y ratas. En los ratones, los valores de LD50 oral fluctuaron entre 200 y 500 mg/kg y en las ratas entre 100 y 250 mg/kg. Posterior a la administración intraperitoneal, los valores LD50 fueron de aproximadamente 400 mg/kg en los ratones y entre 200 y 400 mg/kg en las ratas.
Toxicidad crónica: La administración oral repetida de leflunomida a ratas y perros por hasta 6 meses de duración no reveló efectos de las dosis de 0.5 y 0.8 mg/kg/día, respectivamente. Dosis mayores causaron cambios patológicos en ratas, en especial hipoplasia de la médula ósea, trombocitopoyesis esplénica reducida, atrofia del timo, hemorragias en el tracto gastrointestinal y otros tejidos, y muerte. Con dosis de 1 mg/kg/día o mayores, se presentaron anemia y eritropoyesis extramedular esplénica.
En perros se notó reducción de los parámetros de la eritropoyesis, presencia de cuerpos de Heinz y/o cuerpos de Howell-Jolly, hemopoyesis y hemosiderosis extramedulares. Las muertes se presentaron en perros a los cuales se les administraron 8 mg/kg/día. Debido a su actividad farmacodinámica, leflunomida inhibe la proliferación y la diferenciación celular.
Consecuentemente, los efectos sobre los órganos reproductores se vieron en estudios de dosis repetida en ratones, con niveles altos de medicamento (degeneración y atrofia de testículos, próstata, vesículas seminales, con 30 mg/kg de peso corporal, y atrofia de ovarios y útero con 100 mg/kg de peso corporal). En los perros, disminución de peso de la próstata y los testículos en el grupo de altas dosis (8 mg/kg de peso corporal) en un estudio a 3 meses, fue observada.
Mutagenicidad: La leflunomida no fue mutagénica ni clastógena ante varias pruebas realizadas in vivo e in vitro. Sin embargo, la 4-trifluorometilanilina (TFMA), un metabolito secundario de la leflunomida, fue mutagénica en la prueba de Ames y en el ensayo de mutación génica de HGPRT, y fue clastógena en el ensayo in vitro para aberraciones cromosómicas en células de hámster chino. La TFMA no fue clastógena en el ensayo in vivo de micronúcleo de ratón ni en la prueba in vivo citogenética en células de médula ósea de hámster chino.
Carcinogenicidad: No se observó evidencia de carcinogenicidad en los bioensayos de dos años de duración que se realizaron en ratas a las que se administraron dosis orales de leflunomida de hasta la dosis máxima tolerada de 6 mg/kg (aproximadamente 1/40 la exposición sistémica máxima a M1 en humanos, basada en el área bajo la curva [ABC]); sin embargo, la incidencia de linfomas se incrementó en los ratones macho. Lo anterior se observó al administrar por vía oral 15 mg/kg de leflunomida (equivalente a 1.7 veces la exposición a M1 en el humano, basada en el ABC). La incidencia de adenomas broncoalveolares y carcinomas combinados se incrementó de manera dependiente de la dosis en los ratones hembra, con dosis de leflunomida de 1.5 mg/kg (aproximadamente 1/10 la exposición a M1 en el humano, basada en el ABC). Se desconoce el significado de los hechos observados en ratones con relación al uso clínico de la leflunomida.
Antigenicidad: Leflunomida no fue antigénica en la prueba de anafilaxis cutánea pasiva y sistémica activa llevada a cabo en cobayos, y estuvo exenta de propiedades sensibilizadoras.
Teratogenicidad: En ratas, la leflunomida fue teratogénica, disminuyó el peso corporal materno e incrementó la embrioletalidad, con un decremento en el peso corporal de los fetos supervivientes, cuando se administró en dosis de 15 mg/kg durante la organogénesis. La exposición sistémica de las ratas a esta dosis fue de aproximadamente 1/10 el nivel de exposición en humanos, basado en el ABC. En los conejos, la leflunomida administrada en dosis de 10 mg/kg durante la organogénesis, resultó en esternebras displásicas. El grado de exposición a esta dosis fue equivalente al nivel máximo de exposición del humano, basado en el ABC. La leflunomida en dosis de 1 mg/kg no fue teratogénica en ratas y conejos.
Cuando las ratas hembra fueron tratadas con 1.25 mg/kg de leflunomida, desde los 14 días anteriores al apareamiento hasta el término de la lactancia, se redujo de manera notable (más de 90%) la supervivencia postnatal de la progenie. El grado de exposición sistémica a 1.25 mg/kg fue aproximadamente 1/100 el nivel de exposición del humano, basado en el ABC.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Los efectos adversos pueden aumentar con el uso reciente o concomitante de sustancias hepatotóxicas (incluso alcohol), hematotóxicas o inmunosupresoras. Este hecho hay que considerarlo también cuando el tratamiento con leflunomida es seguido por otro con tales sustancias, sin un procedimiento de lavado previo de leflunomida.
En un estudio (n = 30) en el que se coadministró leflunomida (10 a 20 mg/día) con metotrexato (10 a 25 mg/semana) a pacientes con artritis reumatoide, los niveles de las enzimas hepáticas se duplicaron o triplicaron en 5 de 30 pacientes; no obstante, todos ellos se normalizaron, dos aun continuando con la administración de ambos fármacos y tres después de suspender el tratamiento con leflunomida. Se vio un incremento superior a tres veces en otros 5 pacientes; sin embargo, éstos también se normalizaron, dos aun continuando con la administración de ambos fármacos y tres después de suspender el tratamiento con leflunomida. Por lo tanto, aunque en general no es necesario un periodo de espera cuando se decide cambiar de leflunomida a metotrexato, se recomienda un monitoreo estrecho de las enzimas hepáticas en la fase inicial posterior al cambio. No se ha demostrado ninguna interacción farmacocinética entre la leflunomida (10 a 20 mg/día) y el metotrexato (10 a 25 mg/semana).
Las enzimas implicadas en el metabolismo de la leflunomida y de sus metabolitos no se conocen con exactitud. En un estudio in vivo sobre la interacción con cimetidina (inhibidor inespecífico del citocromo P-450), se demostró carencia de interacción significativa. La administración concomitante de una sola dosis de leflunomida a sujetos que recibieron dosis múltiples de rifampicina (inductor inespecífico del citocromo P-450), los niveles de A771726 produjeron un aumento de aproximadamente 40% en las concentraciones máximas, mientras que el área bajo la curva no cambió significativamente. El mecanismo de estos efectos se desconoce. Debe considerarse la posibilidad de que las concentraciones de leflunomida se incrementen con la administración de dosis múltiples, si los pacientes están recibiendo leflunomida en forma concomitante con rifampicina.
Estudios in vitro indicaron que el A771726 inhibe la actividad del citocromo P4502C9 (CYP2C9). Los fármacos metabolizados por el CYP2C9 incluyen a la fenitoína, tolbutamida, warfarina y varios AINEs. Se desconoce el significado clínico de este hecho, con respecto a la fenitoína y tolbutamida. Por otro lado, en los estudios clínicos no se observó ningún problema de seguridad cuando se coadministraron leflunomida y AINEs que son metabolizados por el CYP2C9. Ha habido reportes de casos en los que se incrementó el tiempo de protrombina cuando se coadministró leflunomida con warfarina.
La administración de colestiramina o carbón activado reduce rápidamente y en forma significativa las concentraciones plasmáticas del A771726. El mecanismo implicado parece ser la interrupción del reciclaje enterohepático y/o la diálisis gastrointestinal del A771726.
No se cuenta con datos clínicos sobre la eficacia y seguridad de la aplicación de vacunas a personas en tratamiento con leflunomida. No se recomienda vacunar con vacunas de organismos vivos. Cuando se esté considerando la administración de alguna vacuna viva después de haber suspendido el tratamiento con leflunomida, debe tomarse en cuenta la prolongada vida media de ésta.
En estudios in vivo se ha demostrado que no hay una interacción significativa entre leflunomida y anticonceptivos orales trifásicos. En un estudio en el que la leflunomida fue administrada concomitantemente con píldoras anticonceptivas orales trifásicas que contenían 30 μg de etinilestradiol a voluntarias sanas, no se redujo la actividad anticonceptiva de la píldora, y la farmacocinética del A771726 se mantuvo dentro de los rangos pronosticados.
La absorción de la leflunomida no se afecta cuando se ingieren alimentos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Elevación de parámetros hepáticos (por ejemplo: transaminasas y, con menor frecuencia, gamma glutamiltranspeptidasa , fosfatasa alcalina y bilirrubina).
Las concentraciones de ácido úrico disminuyen comúnmente debido a que LEFLUNOMIDA tiene un efecto uricosúrico. Otros parámetros de laboratorio cuya importancia clínica no se ha establecido, pero que también pueden presentarse, son: incremento insignificante de la deshidrogenasa láctica (LDH) y de la colecistocinina (CK), así como hipofosfatemia insignificante.
PRECAUCIONES GENERALES:
Generales: Debido a la prolongada vida media del metabolito activo, A771726, reacciones adversas pueden ocurrir o persistir después de la administración de leflunomida, incluso si ha sido descontinuado.
Si una reacción adversa grave a la leflunomida se produce, o si por cualquier otra razón A771726 tiene que eliminarse rápidamente del cuerpo, colestiramina o carbón activado tiene que ser iniciado como se describe en Manifestaciones y manejo de la sobredosificación o ingesta accidental y continuar/repetir cuantas veces sea necesario desde el punto de vista clínico. Para sospechas inmunológicas severas o reacciones alérgicas, puede ser necesaria la administración prolongada de colestiramina o de carbón activado de forma rápida y suficiente. Si el paciente ya está recibiendo AINEs y/o corticosteoides en dosis bajas, éstos pueden continuar después de comenzar tratamiento con leflunomida.
El uso de la leflunomida con antimaláricos utilizados en las enfermedades reumáticas (por ejemplo, cloroquina e hidroxicloroquina), D-penicilamina, azatioprina y otros fármacos inmunosupresores (por ejemplo, ciclosporina, metotrexato), no han sido suficientemente estudiados.
Hígado: Debido a que el metabolito activo de la leflunomida, A771726, se une en gran medida a proteínas y se elimina por metabolismo hepático y secreción biliar, y dado el posible riesgo de hepatotoxicidad, LEFLUNOMIDA debe administrarse con precaución en pacientes con insuficiencia hepática. Además, el uso de LEFLUNOMIDA no se recomienda en pacientes con insuficiencia hepática severa o con enfermedad hepática preexistente.
Se debe determinar la ALT (GPT) antes de iniciar el tratamiento, así como cada mes durante los primeros 6 meses de tratamiento, y posteriormente cada 6-8 semanas.
Guía para el ajuste de la dosis o suspensión según la gravedad y la persistencia de la elevación de ALT, se recomienda lo siguiente:
La confirmación de elevación de ALT entre 2 y 3 veces el límite superior de la reducción de la dosis normal de 20 a 10 mg/día puede permitir proseguir con la administración de leflunomida bajo estrecha vigilancia.
Si la ALT (GPT) se eleva entre 2 y 3 veces del límite superior del rango normal, o si se confirma elevación de ALT más de 3 veces el límite superior del rango normal leflunomida se debe interrumpir. Colestiramina o carbón activado debe ser administrado para disminuir más rápidamente los niveles de A771726.
Se han reportado casos raros de daño hepático severo, en casos aislados con consecuencias fatales, durante el tratamiento con leflunomida. La mayoría de los casos se presentaron durante los primeros 6 meses de tratamiento. A pesar de que no se ha establecido una relación causal con la leflunomida, y múltiples factores de confusión han estado presentes en la mayoría de los casos, se considera esencial seguir las recomendaciones de monitoreo mencionadas, y las descritas en Manifestaciones y manejo de la sobredosificación o ingesta accidental.
Sistema inmune y hematopoyético: En pacientes con anemia preexistente, leucopenia y/o trombo citopenia, así como en pacientes con deterioro de la función de la médula ósea o en aquellos en riesgo de supresión de la médula ósea, se incrementa el riesgo de incidencia de reacciones hematológicas.
Debe realizarse biometría hemática completa, incluyendo cuenta plaquetaria y cuenta leucocitaria diferencial, antes de iniciar el tratamiento con leflunomida, así como cada mes durante los primeros 6 meses de tratamiento, y posteriormente cada 6-8 semanas.
Debe realizarse un monitoreo hematológico frecuente (cuenta de células sanguíneas completa, incluyendo cuenta plaquetaria y cuenta leucocitaria diferencial) en:
• Pacientes con tratamiento reciente o concomitante con fármacos inmunosupresores o hematotóxicos, y cuando estos fármacos se administran como continuación de un tratamiento con leflunomida, sin un periodo de lavado.
• Pacientes con historia de anormalidades hematológicas relevantes.
• Pacientes con anormalidades hematológicas relevantes, en condiciones basales, debidas a causas diferentes a enfermedad artrítica.
Debido al riesgo teórico de inmunosupresión, aun cuando no se cuenta con experiencia clínica, la administración de leflunomida no se recomienda en pacientes con:
- Inmunodeficiencia severa (por ejemplo, SIDA).
- Deterioro significativo de la función de la médula ósea.
- Infecciones graves.
Infecciones: Los medicamentos con potencial inmunosupresor, como la leflunomida, pueden ocasionar que los pacientes sean más susceptibles a infecciones, incluso a infecciones oportunistas (véase Reacciones secundarias y adversas). Las infecciones pueden llegar a ser más severas de lo habitual y, por lo tanto, pueden ser necesarios tratamientos tempranos e intensivos.
En caso de infecciones graves puede ser necesario interrumpir el tratamiento con leflunomida e instituir el procedimiento de eliminación del fármaco, que se describe en Restricciones de uso durante el embarazo y lactancia.
Se recomienda monitorear cuidadosamente a los pacientes con reactividad a la tuberculina, debido al riesgo de reactivación de la tuberculosis.
Sistema respiratorio: Rara vez se ha reportado enfermedad pulmonar intersticial durante el tratamiento con leflunomida. Este riesgo se incrementa en pacientes con historia de enfermedad pulmonar intersticial. La enfermedad pulmonar intersticial es un trastorno potencialmente fatal, que puede ocurrir de manera aguda durante la terapia . Los síntomas pulmonares, tales como tos y disnea, pueden ser una razón para discontinuar la terapia y para investigaciones adicionales según sea apropiado.
Neuropatía periférica: Han sido reportado casos de neuropatía periférica en pacientes que están tomando LEFLUNOMIDA. La mayoría de los pacientes se recuperaron posterior a la discontinuación de LEFLUNOMIDA; sin embargo, algunos pacientes presentaron síntomas persistentes. Los pacientes mayores de 60 años de edad con terapia concomitante con medicamentos neurotóxicos y diabetes pueden incrementar el riesgo de neuropatía periférica. Si el paciente está tomando LEFLUNOMIDA y desarrolla neuropatía periférica, debe considerarse la discontinuación de LEFLUNOMIDA y realizar el procedimiento para la eliminación del fármaco como se describe en Manifestaciones y manejo de la sobredosificación o ingesta accidental.
Insuficiencia renal: No se cuenta con experiencia suficiente en pacientes con insuficiencia renal, por lo que debe tenerse precaución cuando se administra leflunomida a esta población. Debe tomarse en cuenta que el metabolito activo de la leflunomida, A771726, se une a proteínas en gran medida.
Presión sanguínea: La tensión arterial debe valorarse tanto antes de iniciar el tratamiento con leflunomida, como periódicamente durante el tratamiento.
Individuos de sexo masculino: La información disponible no sugiere que LEFLUNOMIDA pueda estar relacionado con el incremento de riesgo de toxicidad fetal mediada por individuos del sexo masculino que ingieren leflunomida. Sin embargo, no se han realizado estudios en animales para valorar este riesgo específico. Para minimizar un posible riesgo, debe discontinuarse el tramiento e instituirse el procedimiento de eliminación del fármaco que se describe en Restricciones de uso durante el embarazo y la lactancia, en los hombres que deseen procrear un hijo y que estén siendo tratados con LEFLUNOMIDA.
DOSIS Y VÍA DE ADMINISTRACIÓN:
El tratamiento con leflunomida debe ser prescrito y vigilado por un médico con experiencia en el tratamiento de enfermedades reumáticas.
El tratamiento con leflunomida para artritis reumatoide usualmente se inicia con una dosis de carga de 100 mg/día durante 3 días. La dosis de mantenimiento recomendada es 20 mg/día.
La omisión de la dosis de carga puede disminuir el riesgo de eventos adversos. Para información adicional con respecto al uso de la dosis de carga en pacientes con artritis reumatoide, véase Farmacocinética y farmacodinamia.
En caso de que la dosis de 20 mg/día no sea bien tolerada, ésta puede reducirse de acuerdo al criterio del médico.
El tratamiento con leflunomida para artritis psoriásica también se debe iniciar con una dosis de carga de 100 mg/día durante 3 días. La dosis de mantenimiento es 20 mg/día de leflunomida.
El efecto del tratamiento puede ser evidente después de 4 semanas, y puede aumentar todavía más después de hasta 4 a 6 meses de haber iniciado el tratamiento.
Por lo general, LEFLUNOMIDA se administra durante periodos prolongados.
Niños y adolescentes: Leflunomida no es recomendada en pacientes menores de 18 años de edad ya que no ha sido estudiada en este grupo.
Sujetos de edad avanzada: No se requiere ajuste de la dosis en individuos mayores de 65 años.
Alteraciones renales y hepáticas: Véase Precauciones generales.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Síntomas: Ha habido reportes de sobredosis crónica en pacientes que toman diariamente hasta 5 veces la dosis diaria recomendada de LEFLUNOMIDA, así como reportes de sobredosis aguda en niños y adultos. No hubo eventos adversos reportados en la mayoría de los reportes de casos de sobredosis. Los eventos adversos que sí se presentaron fueron consistentes con el perfil de seguridad de LEFLUNOMIDA , siendo los más frecuentes: diarrea, dolor abdominal, leucopenia, anemia y pruebas de funcionamiento hepático elevadas.
Manejo: En caso de sobredosis significativa o de toxicidad, se recomienda la administración de colestiramina o de carbón activado para acelerar la eliminación. La administración de colestiramina por vía oral a tres voluntarios sanos, en dosis de 8 g tres veces al día por 24 horas, disminuyó las concentraciones plasmáticas de A771726 en aproximadamente 40% en 24 horas y en 49-65% a las 48 horas.
La administración de una suspensión de carbón activado (polvo hecho suspensión) por vía oral o sonda nasogástrica, a razón de 50 g cada 6 horas por 24 horas, redujo la concentración plasmática del metabolito activo, A771726, en 37% en 24 horas y en 48%.
Estos procedimientos de lavado del fármaco se pueden repetir cuando clínicamente sea necesario.
Los estudios tanto de hemodiálisis como de diálisis peritoneal ambulatoria crónica indicaron que el principal metabolito de la leflunomida, A771726, no es dializable.
PRESENTACIONES:
Caja con frasco con 30 tabletas de 20 mg e instructivo anexo.
Caja con 3 tabletas de 100 mg en envase de burbuja e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 30ºC.
LEYENDAS DE PROTECCIÓN:
Léase instructivo anexo. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni en mujeres en edad fértil que no usen un método anticonceptivo confiable. No se administre durante la lactancia ni a menores de 18 años. El uso de este medicamento debe ser supervisado estrechamente por el médico tratante. Prohibida la venta fraccionada del producto. Literatura exclusiva para médicos.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Hecho en México por:
LABORATORIOS CORNE, S.A. de C.V.
Ocampo No. 167-A, Col. Las Encinas,
C.P. 66050, Escobedo, Nuevo León, México.
o
Distribuido por:
LABORATORIOS CORNE, S.A. de C.V.
Ocampo No. 167-A, Col. Las Encinas,
C.P. 66050, Escobedo, Nuevo León, México.
Distribuido por:
Laboratorios Corne, S.A. de C.V.
Oriente 171 No. 296 Bodega 3-A,
Col. Ampliación San Juan de Aragón,
C.P. 07470, Alcaldía Gustavo A. Madero,
Ciudad de México, México
Reg. Núm. 024M2017 SSA IV


