
LEFORUL
FULVESTRANT
Solución
1 Caja, 1 Jeringa(s) prellenada(s), 5 mL,
1 Caja, 2 Jeringa(s) prellenada(s), 5 mL,
FORMA FARMACÉUTICA Y FORMULACIÓN:
La jeringa prellenada contiene:
Fulvestrant 250 mg
Excipiente cbp 5 mL
INDICACIONES TERAPÉUTICAS:
Fulvestrant está indicado:
Para el tratamiento de cáncer de mama con metástasis o localmente avanzado en mujeres postmenopáusicas de cualquier edad:
- Que no han sido tratadas previamente con terapia endocrina, o
- Que hayan recibido antes tratamiento endocrino (antiestrógenos o tratamiento con inhibidores de la aromatasa), sin considerar si su estado menopáusico fue natural o se indujo en forma artificial.
Fulvestrant, en combinación con un inhibidor selectivo de las cinasas ciclinodependientes (CDK) 4/6:
En combinación con ribociclib para el tratamiento endocrino inicial o en segunda línea después de la progresión de mujeres posmenopáusicas con cáncer de mama avanzado o metastásico con receptores hormonales (RH) positivos y receptor 2 del factor de crecimiento epidérmico humano (HER2) negativo.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética:
Absorción:
Después de la administración de la inyección intramuscular de acción prolongada de fulvestrant, fulvestrant se absorbe con lentitud y las concentraciones plasmáticas máximas (Cmáx) se alcanzan después de aproximadamente 5 días. La administración del régimen de 500 mg de fulvestrant alcanza niveles de exposición en el estado estacionario, o próximos al mismo, dentro del primer mes de la administración de la dosis (media [CV]: ABC 475 [33.4%] ng/día/mL, Cmáx 25.1 [35.1%] ng/mL, Cmin 16.3 [25.9%] ng/mL, respectivamente). En estado estacionario, las concentraciones plasmáticas de fulvestrant se mantienen dentro de un intervalo relativamente estrecho con una diferencia de aproximadamente 3 veces entre las concentraciones máximas y mínimas. Después de la administración intramuscular, la exposición es aproximadamente proporcional a la dosis en el intervalo de dosis de 50 a 500 mg.
Distribución:
Fulvestrant está sujeto a una distribución extensa y rápida. El gran volumen aparente de distribución en estado estacionario (Vdss) de aproximadamente 3 a 5 l/kg sugiere que la distribución es en gran medida extravascular. Fulvestrant está altamente (99%) unido a proteínas plasmáticas. Las fracciones de lipoproteína de muy baja densidad (LMBD), lipoproteína de baja densidad (LBD) y lipoproteína de alta densidad (LAD) son los componentes de unión principales. No se realizaron estudios de interacción sobre la unión competitiva de proteínas. No se ha determinado el papel de la globulina de unión a hormonas sexuales (GBHS).
Metabolismo:
El metabolismo de fulvestrant no se ha evaluado por completo, pero implica combinaciones de varias posibles vías de biotransformación análogas a las de los esteroides endógenos. Los metabolitos identificados (incluye los metabolitos 17-cetona, sulfona, 3-sulfato, 3 y 17-glucurónido) son menos activos o muestran una actividad similar a la de fulvestrant en modelos antiestrogénicos. Los estudios que utilizan preparaciones de hígado humano y enzimas humanas recombinantes indican que CYP3A4 es la única isoenzima P450 involucrada en la oxidación de fulvestrant; sin embargo, las vías que no son P450 parecen ser más predominantes in vivo. Los datos in vitro sugieren que fulvestrant no inhibe las isoenzimas CYP450.
Eliminación: Fulvestrant se elimina principalmente en forma metabolizada. La principal vía de excreción es a través de las heces, con menos del 1% excretado en la orina. Fulvestrant tiene un alto aclaramiento, 11 ± 1.7 mL/min/kg, lo cual sugiere un alto índice de extracción hepática. La vida media terminal (t1/2) después de la administración intramuscular se rige por la tasa de absorción y se estimó en 50 días.
Poblaciones especiales:
En un análisis farmacocinético poblacional de los datos de los estudios de fase 3, no se detectaron diferencias en el perfil farmacocinético de fulvestrant con respecto a la edad (intervalo de 33 a 89 años), peso (40-127 kg) o raza. Insuficiencia renal:
La alteración leve a moderada de la función renal no influyó en la farmacocinética de fulvestrant en ningún grado clínicamente relevante.
Insuficiencia hepática:
La farmacocinética de fulvestrant se ha evaluado en un ensayo clínico de dosis única realizado en sujetos con deterioro hepático leve a moderado (clase A y B de Child-Pugh). Se utilizó una dosis alta de una formulación de inyección intramuscular de duración más corta. Hubo un aumento de aproximadamente 2.5 veces en el ABC en sujetos con deterioro hepático en comparación con sujetos sanos. En pacientes que recibieron fulvestrant, se espera que un aumento en la exposición de esta magnitud sea bien tolerado. No se evaluaron los sujetos con deterioro hepático severo (clase C de Child-Pugh).
Pacientes geriátricos: De los 484 pacientes que recibieron ribociclib en el estudio de fase III (MONALEESA 3, en el grupo de ribociclib + fulvestrant), 226 (46.7%) eran mayores o igual a 65 años y 65 (13.4%) mayores o igual a 75 años. No se han observado diferencias en la seguridad o la eficacia de Ribociclib entre tales pacientes y las pacientes más jóvenes (véase el apartado de Dosis y vía de administración).
Datos de un estudio clínico en pacientes con cáncer de mama y un análisis farmacocinético poblacional no evidenciaron efectos clínicamente relevantes sobre la exposición de ribociclib tras la coadministración de ambos fármacos.
Farmacodinamia:
Grupo farmacoterapéutico: Terapia endocrina, antagonistas hormonales y agentes relacionados, anti-estrógenos.
Código ATC: L02BA03.
Mecanismo de acción y efectos farmacodinámicos:
Fulvestrant es un antagonista competitivo del receptor de estrógeno (RE) con una afinidad comparable al estradiol. Fulvestrant bloquea las acciones tróficas de los estrógenos sin ninguna actividad agonista parcial (similar a los estrógenos). El mecanismo de acción está asociado con la regulación negativa de los niveles de proteína del receptor de estrógeno.
Los ensayos clínicos en mujeres postmenopáusicas con cáncer de mama primario han demostrado que el fulvestrant significativamente regula de manera negativa la proteína del RE en los tumores positivos al RE en comparación con el placebo. También hubo una disminución significativa en la expresión del receptor de progesterona consistente con una falta de efectos agonistas de estrógenos intrínsecos. También se ha demostrado que 500 mg de Fulvestrant regulan de manera negativa el RE y el marcador de proliferación Ki67, en mayor medida que 250 mg de Fulvestrant en tumores de mama en el contexto neoadyuvante postmenopáusico.
Se evaluó la actividad antitumoral in vivo de la combinación de ribociclib con fulvestrant en ratones inmunodeficientes que portaban xenoinjertos de tumor mamario humano ZR751 RE+ (Receptores estrogénicos positivos). La combinación de ribociclib y fulvestrant dio como resultado una inhibición completa del crecimiento tumoral.
Electrofisiología cardiaca: Se obtuvieron electrocardiogramas (ECG) en serie por triplicado después de administraciones únicas y en el estado estable para evaluar el efecto de ribociclib sobre el intervalo QTc en pacientes con cáncer avanzado. Se realizó un análisis farmacocinético-farmacodinámico en pacientes que recibieron dosis de 50 a 1200 mg de ribociclib. El análisis reveló que ribociclib prolonga, de forma dependiente de la concentración, el QTc (QTc: la medida del tiempo entre el comienzo de la onda Q y el final de la onda T). La variación media estimada del QTcF (La fórmula de corrección de Fridericia, la cual mide el acortamiento fisiológico del intervalo QT que se produce a medida que aumenta la frecuencia cardiaca) con respecto al inicio para la dosis de ribociclib 600 mg en combinación con fulvestrant fue de 23.7 ms (IC90%: 22.31, 25.08), a la Cmáx. media que se observa en el estado estable comparado con 34.7 ms (IC95%: 31.64, 37.78 en combinación con Tamoxifeno. (Véase el apartado de Precauciones generales). [IC: Intervalo de confianza].
Seguridad clínica y eficacia en el cáncer de mama avanzado: Se completó un ensayo clínico de fase 3 en 736 mujeres postmenopáusicas con cáncer de mama avanzado que tuvieron recurrencia de la enfermedad en o después de la terapia endocrina adyuvante o progresión después de la terapia endocrina para enfermedad avanzada. El estudio incluyó a 423 pacientes cuya enfermedad había recurrido o progresado durante la terapia antiestrógeno (subgrupo de AE) y 313 pacientes cuya enfermedad había recurrido o progresado durante la terapia con inhibidores de la aromatasa (subgrupo de IA). Este ensayo comparó la eficacia y seguridad de 500 mg de fulvestrant (n = 362) con 250 mg de fulvestrant (n = 374). La supervivencia libre de progresión (SLP) fue el criterio de valoración primario; los criterios de valoración de eficacia secundarios clave incluyeron la tasa de respuesta objetiva (TRO), la tasa de beneficio clínico (TBC) y la supervivencia global (SG). Los resultados de eficacia para el estudio CONFIRM se resumen en la Tabla 1.
Tabla 1. Resumen de los resultados del criterio de valoración de eficacia primario (SLP) y los criterios de valoración de eficacia secundarios clave en el estudio CONFIRM
|
Variable |
Tipo de estimación; comparación del tratamiento |
Fulvestrant 500 mg (n = 362) |
Fulvestrant 250 mg (n = 374) |
Comparación entre grupos (Fulvestrant 500 mg/fulvestrant 250 mg) |
||
|---|---|---|---|---|---|---|
|
Hazard ratio |
IC95% |
valor p |
||||
|
Pfs |
Mediana de K-M en meses; Hazard ratio |
|||||
|
Todos los pacientes |
6.5 |
5.5 |
0.80 |
0.68, 0.94 |
0.006 |
|
|
-Subgrupo AE (n = 423) |
8.6 |
5.8 |
0.76 |
0.62, 0.94 |
0.013 |
|
|
-Subgrupo IA (n = 313)un |
5.4 |
4.1 |
0.85 |
0.67, 1.08 |
0.195 |
|
|
OSb |
Mediana de K-M en meses; Hazard ratio |
|||||
|
Todos los pacientes |
26.4 |
22.3 |
0.81 |
0.69, 0.96 |
0.016c |
|
|
-Subgrupo AE (n = 423) |
30.6 |
23.9 |
0.79 |
0.63, 0.99 |
0.038c |
|
|
-Subgrupo AI (n = 313)un |
24.1 |
20.8 |
0.86 |
0.67, 1.11 |
0.241c |
|
|
Variable |
Tipo de estimación; comparación del tratamiento |
Fulvestrant 500 mg (n = 362) |
Fulvestrant 250 mg (n = 374) |
Comparación entre grupos (Fulvestrant 500 mg/fulvestrant 250 mg) |
||
|
Diferencia absoluta en % |
IC95% |
|||||
|
ORRd |
% de los pacientes con o; diferencia absoluta en% |
13.8 |
14.6 |
-0.8 |
-5.8, 6.3 |
|
|
Todos los pacientes |
||||||
|
-Subgrupo AE (n = 296) |
18.1 |
19.1 |
-1.0 |
-8.2, 9.3 |
||
|
-Subgrupo AI (n = 205)un |
7.3 |
8.3 |
-1.0 |
-5.5, 9.8 |
||
|
CBRe |
% de los pacientes con CB; diferencia absoluta en% |
|||||
|
Todos los pacientes |
45.6 |
39.6 |
6.0 |
-1.1, 13.3 |
||
|
-Subgrupo AE (n = 423) |
52.4 |
45.1 |
7.3 |
-2.2, 16.6 |
||
|
-Subgrupo IA (n = 313)un |
36.2 |
32.3 |
3.9 |
-6.1, 15.2 |
||
a. Fulvestrant está indicado en pacientes cuya enfermedad ha recurrido o progresado con una terapia antiestrógeno. Los resultados en el subgrupo de IA no son concluyentes.
b. El sistema operativo se presenta para los análisis de supervivencia finales con un 75% de madurez.
c. El valor p nominal sin ajustes por multiplicidad entre los análisis de supervivencia global inicial al 50% de madurez y los análisis de supervivencia actualizados al 75% de madurez.
d. La TRO se evaluó en pacientes que eran evaluables para la respuesta en la línea base (es decir, aquéllos con enfermedad medible en la línea base: 240 pacientes en el grupo de 500 mg de fulvestrant y 261 pacientes en el grupo de 250 mg de fulvestrant).
e. Pacientes con la mejor respuesta objetiva de respuesta completa, respuesta parcial o enfermedad estable ≥ 24 semanas.
SLP: Supervivencia libre de progresión; TRO: Tasa de respuesta objetiva; RO: Respuesta objetiva; TBC: Tasa de beneficio clínico; BC: Beneficio clínico; SG: Supervivencia global; K-M: Kaplan-Meier; IC: Intervalo de confianza; IA: inhibidor de la aromatasa; AE: Antiestrógeno.
Se realizó un estudio de fase 3, aleatorizado, doble ciego, doble simulado, multicéntrico de 500 mg de fulvestrant con respecto a 1 mg de anastrozol en mujeres postmenopáusicas con cáncer de mama con metástasis o localmente avanzado positivo al ER o positivo a PgR quienes no habían sido previamente tratadas con ninguna terapia hormonal. Un total de 462 pacientes fueron aleatorizados 1:1 de manera secuencial para recibir 500 mg de fulvestrant o 1 mg de anastrozol.
La aleatorización se estratificó por el contexto de la enfermedad (con metástasis o localmente avanzada), quimioterapia previa para enfermedad avanzada y enfermedad medible.
El criterio de valoración de eficacia primario del estudio fue que el investigador evaluó la supervivencia libre de progresión (SLP) evaluada de acuerdo con RECIST 1.1 (Criterios de evaluación de respuesta en tumores sólidos). Los criterios de valoración de eficacia secundarios clave incluyeron la supervivencia global (SG) y la tasa de respuesta objetiva (TRO).
Los pacientes inscritos en este estudio tenían una mediana de edad de 63 años (intervalo 36-90). La mayoría de los pacientes (87.0%) tenían enfermedad metastásica al inicio del estudio. El cincuenta y cinco por ciento (55.0%) de los pacientes tenía metástasis visceral en la línea base. Un total de 17.1% de los pacientes recibió un régimen de quimioterapia previa para enfermedad avanzada; el 84.2% de las pacientes tenían enfermedad medible.
En la mayoría de los subgrupos predefinidos de pacientes se observaron resultados consistentes. Para el subgrupo de pacientes con enfermedad limitada a metástasis no visceral (n=208), el HR fue 0.592 (IC95%: 0.419-0.837) para el brazo de Faslodex comparado con el brazo de anastrozol. Para el subgrupo de pacientes con metástasis visceral (n=254), el HR fue 0.993 (IC95%: 0.740-1.331) para el brazo de Faslodex comparado con el brazo de anastrozol. Los resultados de eficacia del estudio FALCON se presentan en la Tabla 2 y en la Figura 1.
Tabla 2. Resumen de los resultados del criterio de valoración de eficacia primario (SLP) y los criterios de valoración de eficacia secundarios clave (evaluación del investigador, población con intención de tratar) - Estudio FALCON
|
Fulvestrant 500 mg (n = 230) |
Anastrozol 1 mg (n = 232) |
|
|
Supervivencia libre de progresión |
||
|
Número de eventos PFS (%) |
143 (62.2%) |
166 (71.6%) |
|
PFS Hazard ratio (IC95%) y valor p |
HR 0.797 (0.637 – 0.999) p = 0.0486 |
|
|
Mediana de PFS [meses (IC95%)] |
16.6 (13.8, 21.0) |
13.8 (12.0, 16.6) |
|
Número de eventos del SO* |
67 (29.1%) |
75 (32.3%) |
|
Relación de riesgo OS (IC95%) y valor p |
HR 0.875 (0.629 – 1.217) p = 0.4277 |
|
|
ORR** |
89 (46.1%) |
88 (44.9%) |
|
La relación de probabilidades de ORR (IC95%) y el valor p |
O 1.074 (0.716 – 1.614) p = 0.7290 |
|
|
Mediana de DoR (meses) |
20.0 |
13.2 |
|
Cbr |
180 (78.3%) |
172 (74.1%) |
|
Relación de probabilidades CBR (IC95%) y valor p |
0 1.253 (0.815 – 1.932) p = 0.3045 |
|
* (31% de madurez)-no análisis final del sistema operativo.
** Para pacientes con enfermedad medible.
Figura 1. Gráfica de Kaplan-Meier de supervivencia libre de progresión (evaluación del investigador, población con intención de tratar) - Estudio FALCON
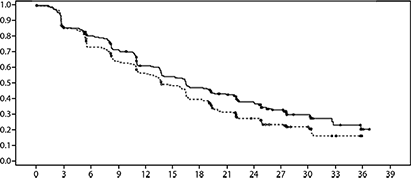
Tiempo desde la aleatorización (meses)
|
Número de pacientes en riesgo: |
Tratamiento |
LEFORUL® Fulvestrant 500 mg (N = 230) |
Anastrozol 1 mg (N = 232) |
|||||||||||
|
FUL500 |
230 |
187 |
171 |
150 |
124 |
110 |
96 |
81 |
63 |
44 |
24 |
11 |
2 |
0 |
|
ANAS1 |
232 |
194 |
162 |
139 |
120 |
102 |
84 |
60 |
45 |
31 |
22 |
10 |
0 |
0 |
Estudio CLEE011F2301 (MONALEESA-3): Ribociclib se evaluó en combinación con fulvestrant en un estudio aleatorizado, doble ciego, controlado con placebo para el tratamiento de hombres y mujeres posmenopáusicas receptores hormonales positivos (RH positivo), HER2 negativo, con cáncer de mama avanzado que no recibieron tratamiento endocrino, o que recibieron sólo una línea de tratamiento endocrino previo para enfermedad avanzada. Un total de 726 pacientes fueron asignados aleatoriamente en una proporción de 2:1 para recibir 600 mg de ribociclib + fulvestrant (n = 484) o placebo + fulvestrant (n = 242), fueron estratificados según la presencia de metástasis hepáticas y/o pulmonares [Sí (n = 351 (48.3%)) frente a No (n = 375 (51.7%))], terapia endocrina previa [A (n = 354 (48.8%)) Vs. B (n = 372 (51.2%)]. Pacientes con cáncer de mama avanzado, tratadas en primera línea (A) incluyó cáncer de mama avanzado de novo sin previo tratamiento endocrino y pacientes que recayeron después de 12 meses de finalizar el tratamiento endocrino (neo) adyuvante.
El subgrupo de pacientes tratadas en segunda línea (B) incluyó aquellos pacientes que recayeron durante el tratamiento adyuvante o en menos de 12 meses posteriores al haber finalizado el tratamiento endocrino adyuvante y aquellos pacientes que progresaron a la primera línea. Las características demográficas y patológicas iniciales fueron equilibradas y comparables entre los subgrupos estudiados. Ribociclib 600 mg o placebo se administró por vía oral durante 21 días consecutivos, seguido de 7 días sin tratamiento de la combinación con fulvestrant 500 mg administrándose intramuscularmente en el Ciclo 1: Día 1, Ciclo 1: Día 15, Ciclo 2, Día 1 y posteriormente cada 28 días.
La mediana de edad de los pacientes que participaron en el estudio era de 63 años (intervalo: 31 a 89). El 46.7% de los pacientes eran de 65 años de edad, el 13.8% de los pacientes eran de 75 años o más. Participaron pacientes: caucásicos (85.3%), asiáticos (8.7%) y de raza negra (0.7%). La mayoría de los pacientes (99.7%) presentaban un estado funcional de 0 o 1 en la escala del ECOG (Grupo Cooperativo Oncológico del Este de los EE. UU.). Los pacientes de primera y segunda línea inscritos en este estudio (de los cuales el 19.1% de los pacientes tuvieron enfermedad metastásica de novo). El 42.7% de los pacientes había recibido quimioterapia adyuvante frente al 13.1% que recibió quimioterapia neoadyuvante y el 58.5% habían recibido terapia endocrina en adyuvante frente al 1.4% que recibieron quimioterapia neoadyuvante antes del ingreso al estudio, el 21.2% de los pacientes tenían enfermedad ósea y el 60.5% padecían enfermedad visceral. Los datos demográficos y las características patológicas iniciales fueron equilibrados y comparables entre los grupos de estudio.
Análisis primario: El criterio de valoración principal del estudio se cumplió después de la observación de 361 eventos de supervivencia libre de progresión (SLP) usando (RECIST v1.1), con base a la evaluación del investigador en el conjunto de análisis completo (todos los pacientes aleatorizados) y confirmado por una evaluación central aleatoria del 40% subconjunto de imágenes por un comité de revisión independiente cegado (BIRC, por sus siglas en inglés). La mediana del tiempo de seguimiento en el momento del análisis de la SLP primaria fue de 20.4 meses.
Los análisis de SLP basados en el (BIRC, por sus siglas en inglés) respaldaron los resultados primarios de eficacia, la razón de riesgo de SLP fue de 0.492 (IC95%, 0.345 a 0.703).
Los resultados de eficacia primaria mostraron una mejoría estadísticamente significativa de la SLP en los pacientes que recibieron ribociclib + fulvestrant en comparación con los pacientes que recibieron placebo + fulvestrant en la población completa de análisis [HR] = 0.593; IC95%: 0.480-0.732, valor de p en la prueba del orden logarítmico de rangos estratificada unilateral = 4.1x10-7), con una reducción estimada del riesgo de progresión del 41% a favor del subgrupo fulvestrant + ribociclib. La mediana de la SLP fue de 20.5 meses (IC95%:18.5, 23.5) en el subgrupo de ribociclib + fulvestrant y 12.8 meses (10.9, 16.3) en el subgrupo placebo + fulvestrant. La curva de Kaplan-Meier y el diagrama de bosque de la SLP se proporcionan en la Figura 2 y 3, respectivamente.
Figura 2. Gráfico de Kaplan-Meier de la SLP basado en la evaluación de investigador del estudio MONALEESA-3 (F2301) (Conjunto de análisis c+ompleto) (límite de 30-noviembre de 2017)
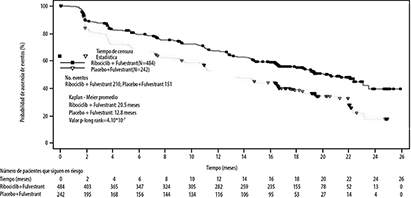
Figura 3. Diagrama de bosque de la SLP basada en la revisión del investigador- Estudio MONALEESA-3 (F2301) (Conjunto de análisis completo) [límite 03 de noviembre de 2017]


Figura 3. Diagrama de bosque de la SLP basada en la revisión del investigador- Estudio MONALEESA-3 (F2301) (Conjunto de análisis completo) [límite 03 de noviembre de 2017], continuación



La terapia endocrina previa (A vs. B) se clasifica usando datos de CRF de la siguiente manera:
A) El tratamiento sin previa medicación para enfermedad metastásica/avanzada (aBC) incluye:
i. Recaída > 12 meses después de la finalización de (neo) adyuvante ET (terapia endocrina) sin tratamiento posterior para aBC,
ii. De novo aBC (sin exposición previa a ET).
B) Recibir hasta 1 línea ET para aBC incluye:
i. Recaída dentro de los 12 meses posteriores a la finalización de la ET (neo) adyuvante, sin tratamiento posterior para una aBC, OR
ii. Recaída> 12 meses desde la finalización de la ET (neo) adyuvante y la progresión en o después de la ET posterior para una aBC, OR
iii. aBC en el momento del diagnóstico que progresó en o después de ET para un aBC sin tratamiento previo (neo) adyuvante para la enfermedad temprana.
La tasa de beneficio clínico en el brazo ribociclib+ fulvestrant y en el brazo placebo + fulvestrant se resume en la tabla 3.
Tabla 3. Resultados de eficacia MONALEESA-3 (TRG, TBC) según la evaluación del investigador (límite 03 de noviembre de 2017)
|
Análisis |
Ribociclib + Fulvestrant (%,IC95%) |
Placebo + Fulvestrant (%, IC95%) |
p-value |
|
Conjunto de análisis completo |
N = 484 |
N = 242 |
|
|
Tasa de respuesta globala |
32.4 (28.3, 36.6) |
21.5 (16.3, 26.7) |
0.000912 |
|
Tasa de beneficio clínicob |
70.2 (66.2, 74.3) |
62.8 (56.7, 68.9) |
0.020 |
|
Pacientes con enfermedad mensurable |
N = 379 |
N = 181 |
|
|
Tasa de respuesta globala |
40.9 (35.9, 45.8) |
28.7 (22.1, 35.3) |
0.003 |
|
Tasa de beneficio clínicob |
69.4(64.8, 74.0) |
59.7 (52.5, 66.8) |
0.015 |
a TRG: Tasa de respuesta global = proporción de pacientes con respuesta completa + respuesta parcial.
b TBC: tasa de beneficio clínico = proporción de pacientes con respuesta completa + respuesta parcial (+ enfermedad estable o respuesta no completa/enfermedad no progresiva ≥ 24 semanas).
El estado de salud global/(CdV) fue similar entre el subgrupo de Ribociclib + fulvestrant y el subgrupo de placebo + fulvestrant. La principal medida de calidad de vida pre especificada fue el tiempo hasta el deterioro (TTD) en el estado de salud global. El deterioro definitivo del 10% se definió como un empeoramiento del puntaje (escala de salud global EORTC QLQ-C30) en al menos 10% comparado con el valor inicial, sin mejoría posterior por encima de este umbral observado durante el periodo de tratamiento o muerte por causa alguna.
La adición de Ribociclib + fulvestrant provocó un retraso en el valor del puntaje de TTD (según escala de salud global EORTC QLQ-C30) comparado con el grupo de placebo + fulvestrant (valor de la mediana no estimable Vs. 19.4 meses, HR: 0.795 [IC95%: 0.602.1.050); p-valor 0.051.
Análisis final de la de supervivencia global (SG): Dado que la mediana de SLP para pacientes de primera línea no se había alcanzado en el momento del análisis primario, se realizó una actualización descriptiva de los resultados de eficacia primaria (SLP) en el momento del segundo análisis intermedio de la supervivencia global, los resultados actualizados de la SLP se resumen en la Tabla 4 y Figura 4 se muestra la curva de Kaplan-Meier.
Tabla 4: Resultados de eficacia primaria (F2301) (SLP) basados en la evaluación radiológica del investigador (límite 03 de junio de 2019)
|
Ribociclib + fulvestrant N = 484 |
Placebo + Fulvestrant N = 242 |
|
|
Supervivencia libre de progresión |
||
|
Mediana de SLP [meses] (IC95%) |
20.6 (18.6-24.0) |
12.8 (10.9, 16.3) |
|
Hazard ratio (IC95%) |
0.587 (0.488-0.705) |
|
Figura 4. Gráfico de Kaplan- Meier de la SLP basada en la revisión del investigador- Estudio MONALEESA-3 (F2301) (Conjunto de análisis completo) [límite 03 de junio de 2019]
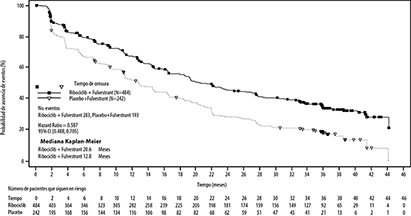
Los resultados fueron consistentes en los subgrupos de edad, quimioterapia adyuvante o neoadyuvante previa o terapia hormonal, afectación hepática y/o pulmonar y enfermedad de metástasis ósea solamente. El análisis de subgrupos basado en la terapia endocrina previa se presenta en la Tabla 5.
Tabla 5: Resultados de eficacia primaria (F2301) (SLP) para el subgrupo de terapia endocrina previa (límite 03 de junio de 2019)
|
Análisis actualizado del subgrupo (SLP) para la terapia endocrina previa (límite 03 de junio de 2019) |
||
|---|---|---|
|
Ajuste de primera línea |
Ribociclib 600 mg N = 237 |
Placebo N = 128 |
|
Número de eventos-n [%] |
112 (47.3) |
95 (74.2) |
|
Mediana de SLP [meses] (IC95%) |
33.6 (27.1, 41.3) |
19.2 (14.9, 23.6) |
|
Hazard ratio (IC95%) |
0.546 (0.415, 0.718) |
|
|
Ajuste de segunda línea o con una recaída temprana |
Ribociclib 600 mg N = 237 |
Placebo N = 109 |
|
Número de eventos-n [%] |
167 (70.5) |
95 (87.2) |
|
Mediana de SLP [meses] (IC95%) |
14.6 (12.5, 18.6) |
9.1 (5.8, 11.0) |
|
Hazard ratio (IC95%) |
0.571 (0.443, 0.737) |
|
IC = intervalo de confianza.
Ajuste de primera línea = Diagnóstico reciente de cáncer de mama avanzado de novo o recaída después de 12 meses de la finalización de la terapia endocrina (neo) adyuvante sin tratamiento para la enfermedad avanzada metastásica.
Ajuste de segunda línea o con recaída temprana = Recaída en los 12 meses posteriores a la finalización de la terapia endocrina (neo) adyuvante sin tratamiento para la enfermedad avanzada o metastásica (recaída temprana), recaída después de 12 meses desde la finalización de la terapia adyuvante (neo) con progresión posterior después de una línea de terapia endocrina para enfermedad avanzada o metastásica, ó cáncer de mama avanzado o metastásico en el momento del diagnóstico que progresó después de una línea de terapia endocrina para enfermedad avanzada sin tratamiento adyuvante previo (neo) para la enfermedad temprana.
En el segundo análisis intermedio de Supervivencia global (SG) pre especificado, el estudio cruzó el límite de detención de Lan-DeMets (O"Brien-Fleming) pre especificado, lo que demuestra una mejora estadísticamente significativa en Supervivencia Global.
Los resultados del análisis intermedio de Supervivencia global (SG) con límite 03 junio de 2019 se proporcionan en la Tabla 6 y la Figura 5.
Tabla 6. Resultados de eficacia de la Supervivencia Global (límite 03 de junio de 2019)
|
Ribociclib 600 mg |
Placebo |
|
|
Población completa de estudio |
N = 484 |
N = 242 |
|
Número de eventos-n [%] |
167 (34.5) |
108 (44.6) |
|
Mediana de SLP [meses] (IC95%) |
NE, (NE, NE) |
40 (37, NE) |
|
Hazard ratio (IC95%) |
0.724 (0568, 0924) |
|
|
p valor |
0.00455 |
|
- [1] El valor P unilateral se obtiene de la prueba de log-rank estratificada por metástasis pulmonares y/o hepáticas, terapia endocrina previa por IRT. El valor P es unilateral y se compara con un umbral de 0.01129 según lo determinado por el gasto alfa de Lan-DeMets (O’Brien-Fleming) función para un nivel de significancia general de 0.025.
- [2] La razón de riesgo se obtiene del modelo Cox PH estratificado por metástasis pulmonares y/o hepáticas, terapia endocrina previa por IRT.
NE = No estimable.
Figura 5. Gráfico de Kaplan-Meier de la Supervivencia Global-Estudio MONALEESA-3 (F2301) (Conjunto de análisis completo) [límite 03 de junio de 2019]
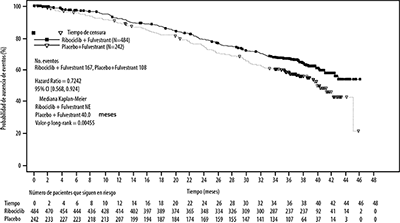
La prueba de log-rank y el modelo de Cox se estratifican por metástasis pulmonares y/o hepáticas, quimioterapia previa para enfermedad avanzada y pareja de combinación endocrina por IRT.
Los resultados de la Supervivencia Global para los subgrupos de análisis se presentan en la Figura 6, 7 y 8.
Figura 6. Gráfico de Kaplan-Meier de la Supervivencia Global de pacientes sin tratamiento previo para la enfermedad mestastásica/avanzada-Estudio MONALEESA-3 (F2301) (Conjunto de análisis completo) [límite 03 de junio de 2019]
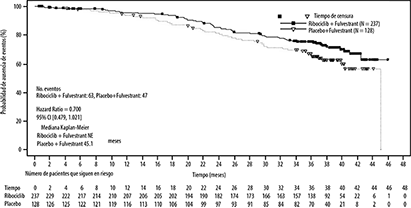
Figura 7. Gráfico de Kaplan-Meier de la Supervivencia Global de pacientes que recibieron hasta 1 línea de tratamiento para la enfermedad mestastásica/avanzada- Estudio MONALEESA-3 (F2301) (Conjunto de análisis completo) [límite 03 de junio de 2019]
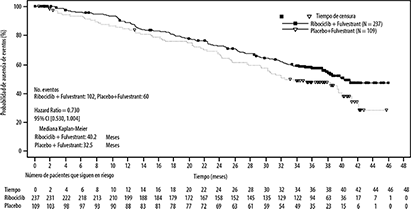
La razón del riesgo se basa en el modelo de Cox no estratificado.
Figura 8. Diagramas de bosque de la Supervivencia Global (SG) del análisis de subgrupos-Estudio MONALEESA-3 (F2301) (Conjunto de análisis completo) [límite 03 de junio de 2019]

La línea punteada no muestra ningún punto de efecto, y la línea en negrita muestra el punto de efecto del tratamiento general.
La razón de riesgo (IC95%) se basa en el modelo Cox PH estratificado por metástasis pulmonares y/o hepáticas, y la terapia endocrina previa por IRT.
Excepto: para los análisis de subgrupos relacionados con factores de estratificación (metástasis hepáticas/pulmonares y terapia endocrina previa), se utilizan modelos no estratificados.
Los subgrupos se derivan de CRF.

Además, el tiempo de progresión en la terapia de la siguiente línea o la muerte (SLP2) en pacientes en el brazo de ribociclib fue más largo comparado con los pacientes en el brazo de placebo (HR: 0.670 (IC95%: 0.542, 0.830)) en la población general del estudio. La mediana de SLP2 fue de 39.8 meses (IC95%: 32.5, NE) para el brazo de ribociclib y 29.4 meses (IC95%: 24.1, 33.1) en el brazo de placebo.
Efectos en el endometrio posmenopáusico: Los datos preclínicos no sugieren un efecto estimulante de fulvestrant en el endometrio posmenopáusico (consultar la sección Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). Un estudio de 2 semanas en voluntarias postmenopáusicas sanas tratadas con 20 microgramos por día de etinilestradiol demostró que el tratamiento previo con 250 mg de fulvestrant produjo una reducción significativa de la estimulación del endometrio postmenopáusico, en comparación con el tratamiento previo con placebo, según lo evaluado por medición por ultrasonido del espesor del endometrio.
El tratamiento neoadyuvante durante hasta 16 semanas en pacientes con cáncer de mama tratadas con 500 mg de fulvestrant o con 250 mg de fulvestrant no produjo cambios clínicamente significativos en el espesor del endometrio, lo cual indica una falta de efecto agonista. No existe evidencia de efectos endometriales adversos en las pacientes con cáncer de mama estudiadas. No existen datos disponibles sobre la morfología endometrial. En dos estudios a corto plazo (1 y 12 semanas) en pacientes premenopáusicas con enfermedad ginecológica benigna, no se observaron diferencias significativas en el espesor endometrial mediante medición por ultrasonido entre grupos de fulvestrant y placebo.
Efectos sobre el hueso: No existen datos a largo plazo sobre el efecto que ejerce fulvestrant en el hueso. El tratamiento neoadyuvante durante hasta 16 semanas en pacientes con cáncer de mama con 500 mg de fulvestrant o con 250 mg de fulvestrant no produjo cambios clínicamente significativos en los marcadores séricos de recambio óseo.
CONTRAINDICACIONES: Hipersensibilidad al principio activo o a alguno de los excipientes.
Durante el embarazo y la lactancia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Las mujeres en edad fértil: Las pacientes en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento con fulvestrant y durante 2 años después de la última dosis.
Debido a que Fulvestrant es un potente antiestrogénico, los estudios en animales han mostrado toxicidad en órganos reproductivos (véase sección Precauciones en relación a los efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Fulvestrant se ha encontrado en la leche de las ratas en niveles mucho más altos que en el plasma de las ratas. Se desconoce el riesgo potencial de Fulvestrant en los humanos, por lo que deberá evitarse su uso en mujeres embarazadas o durante la lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS: Fulvestrant 500 mg: Las categorías de la siguiente frecuencia de reacciones adversas a medicamentos (RAM´s) se calculó con base en el grupo de tratamiento con Fulvestrant 500 mg en los análisis de seguridad agrupados de CONFIRM (estudio D6997C00002), FINDER 1 (estudio D6997C00004), FINDER 2 (estudio D6997C00006) y NEWEST (estudio D6997C00003) que compararon Fulvestrant 500 mg con Fulvestant 250 mg. Las frecuencias presentadas en la siguiente tabla se basaron en todos los eventos reportados, independientemente de la evaluación de la causalidad por parte del investigador.
Tabla 7. Resumen de las reacciones adversas al medicamento observadas en los estudios clínicos de
500 mg
|
Frecuencia |
SOC |
RAM |
|
Muy común (≥ 10%) |
Trastornos generales y condiciones del lugar de administración |
Reacciones del sitio de inyección Astenia |
|
Trastornos hepatobiliares |
Elevación de las enzimas hepáticas (ALT, AST, ALP) |
|
|
Trastornos gastrointestinales |
Náuseas |
|
|
Común (≥ 1-< 10%) |
Trastornos vasculares |
Bochornos |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
|
|
Trastornos hepatobiliares |
Bilirrubina elevadaa. |
|
|
Trastornos gastrointestinales |
Vómito, diarrea |
|
|
Trastornos de metabolismo y nutrición |
Anorexia |
|
|
Trastornos de piel y tejido subcutáneo |
Salpullido |
|
|
Infecciones e infestaciones |
Infecciones del tracto urinario |
|
|
Trastornos del sistema inmune |
Reacciones de hipersensibilidad |
|
|
Poco común (≥ 0.1% y <1%) |
Trastornos hepatobiliares |
Insuficiencia hepáticab, hepatitisb, gamma-GT elevada |
|
Sangre y sistema linfático |
Disminución de la cuenta plaquetaria |
a Basado sobre cualquier cambio grado CTC desde línea base.
b El evento no se observó en estudios clínicos importantes (CONFIRM, FINDER 1, FINDER 2, NEWEST). Se ha calculado la frecuencia con un límite superior del intervalo de confianza de 95% para la estimación de punto. Esto se calcula como 3/560 (donde 560 es el número de pacientes en los principales estudios clínicos), lo cual equivale a una categoría de frecuencias de "poco común".
Sobre la base de los datos, no hay evidencia de una relación causal entre fulvestrant y eventos raros o poco frecuentes en los estudios clínicos de fulvestrant.
Fulvestrant 250 mg: Las categorías de la siguiente frecuencia de reacciones adversas a medicamentos (RAM´s) se calculó con base al grupo de tratamiento con Fulvestrant 250 mg en los análisis agrupados de seguridad de los estudios 9238IL/0020, 9238IL/0021, 9238IL/0025, D6997C00002 (CONFIRM), D6997C00004 (FINDER 1), D6997C00006 (FINDER 2) y D6997C00003 (NEWEST).
Tabla 8. Resumen de las reacciones adversas al medicamento observadas en los estudios clínicos para Fulvestrant 250 mg
|
Frecuencia |
SOC |
RAM |
|
Muy común (≥ 10%) |
Trastornos generales y condiciones del lugar de administración |
Reacciones del sitio de inyección Astenia |
|
Trastornos hepatobiliares |
Elevación de las enzimas hepáticas (ALT, AST, ALP) |
|
|
Trastornos gastrointestinales |
Náuseas |
|
|
Trastornos del sistema nervioso |
Dolor de cabeza |
|
|
Común (≥ 1-< 10%) |
Trastornos vasculares |
Bochornos |
|
Trastornos gastrointestinales |
Vómito, diarrea |
|
|
Trastornos de metabolismo y nutrición |
Anorexia |
|
|
Trastornos de piel y tejido subcutáneo |
Salpullido |
|
|
Infecciones e infestaciones |
Infecciones del tracto urinario |
|
|
Trastornos del sistema inmune |
Reacciones de hipersensibilidad |
|
|
Trastornos hepatobiliares |
Bilirrubina elevadaa |
|
|
Poco común (≥ 0.1% y <1%) |
Trastornos hepatobiliares |
Insuficiencia hepáticac, Hepatitisb, gamma-GT elevada |
|
Sangre y sistema linfático |
Disminución de la cuenta plaquetaria |
a En 4 de 7 estudios incluidos en el conjunto de análisis, pacientes en el brazo de FASLODEX® 250 mg que recibieron 2 inyecciones. En los otros 3 estudios (Estudios 9238IL/0020, 9238IL/0025 y NEWEST), los pacientes del brazo de FASLODEX® 250 mg que recibieron una inyección única y la frecuencia de reacciones en el lugar de la inyección en cada uno de estos 3 estudios fue 7.3% (16/219), 13.5 (42/310) y 5.0% (5/101), respectivamente.
b Basado sobre cualquier cambio grado CTC desde línea base.
c El evento no se observó en estudios clínicos importantes (9238IL/0020, 9238IL/0021, 9238IL/0025, CONFIRM, FINDER 1, FINDER 2, NEWEST).
Se ha calculado la frecuencia con un límite superior del intervalo de confianza de 95% para la estimación de punto. Esto se calcula como 3/1,263 (donde 1,263 es el número de pacientes en los principales estudios clínicos) lo cual equivale a una categoría de frecuencias de "poco común".
El perfil de seguridad general de ribociclib se basa en el conjunto de datos de 1065 pacientes que recibieron ribociclib en combinación con terapia endocrina (N = 483 en combinación con fulvestrant), realizados en el estudio clínicos de fase III doble ciego y controlado con placebo (MONALEESA-3) en pacientes con cáncer de mama avanzado o metastásico HER2-negativo con HER+.
En la agrupación de los estudios de fase III la mediana de duración de la exposición al tratamiento con ribociclib fue de 16.53 meses, y el 61.7% de las pacientes estuvieron expuestas al menos 12 meses.
La proporción de pacientes en las que hubo que reducir la dosis debido a eventos adversos, independientemente de la relación causal, fue del 37.3% entre las tratadas con ribociclib en los estudios clínicos de fase III, independientemente de la combinación y del 3.4% que recibieron placebo. Se notificaron retiros permanentes del tratamiento debido a eventos adversos en el 7.0% de las pacientes tratadas con ribociclib + alguna combinación y en el 2.9% de las que recibieron placebo + alguna combinación. Los eventos adversos notificados con más frecuencia como causa de retirada del tratamiento con ribociclib + alguna combinación fueron elevación de la alanina aminotransferasa (ALT) (2.0%), elevación de la aspartato aminotransferasa AST (1.4%) y vómitos (0.8%).
En el análisis agrupado de tres estudios de fase III, se informaron 21 pacientes (2.0%) de muertes por tratamiento con ribociclib + cualquier combinación frente a 16 pacientes (2.0%) de pacientes tratados con placebo + cualquier combinación de tratamiento, excluyendo las enfermedades progresivas que causan la muerte, se informaron tres causas de muerte relacionadas con el tratamiento en pacientes tratados con ribociclib + cualquier tratamiento de combinación. Causas de muerte: síndrome de dificultad respiratoria aguda 1 (0.1%), insuficiencia respiratoria aguda 1 (0.1%) y muerte súbita (en situación de hipopotasemia de Grado 3 y prolongación de QT de Grado 2, ambos reportados 10 días antes del evento) 1 (0.1%).
Las reacciones adversas (RA) más frecuentes en la agrupación de los estudios de fase III (notificadas con una frecuencia ≥ 20% y más frecuentes entre las que recibieron placebo) fueron infecciones, neutropenia, leucopenia, cefalea, tos, náuseas, fatiga, diarrea, vómitos, estreñimiento, alopecia y erupción cutánea.
Las RA de Grado 3 o 4 más frecuentes del conjunto de datos (notificadas con una frecuencia de ≥ 2% y más frecuentes entre las pacientes tratadas con ribociclib que entre las que recibieron placebo) fueron neutropenia, infecciones, leucopenia, anemia, pruebas de función hepática anormales, linfopenia, hipofosfatemia y vómitos.
Resumen tabulado de las reacciones adversas basadas en el conjunto de datos de 3 estudios clínicos de fase III: Las RA observadas en los ensayos clínicos de fase III (Tabla 8) se enumeran según la clase de órgano, aparato o sistema del MedDRA. Dentro de cada clase de órgano, aparato o sistema, las reacciones se clasifican por orden decreciente de frecuencia. En cada grupo de frecuencia, las reacciones se especifican por orden decreciente de gravedad. Además, para cada reacción adversa se indica la categoría de frecuencia correspondiente según la convención siguiente (CIOMS III): muy frecuente (≥1/10), frecuente (≥1/100 a <1/10), infrecuente (≥1/1000 a <1/100), rara (≥1/10 000 a <1/1000), muy rara (<1/10 000).
Tabla 9. Reacciones adversas observadas en el ensayo clínico de fase III
|
Reacciones adversas |
Ribociclib N = 1065 n (%) Todos los Grados |
Placebo N = 818 n (%) Todos los Grados |
Ribociclib N = 1065 n (%) Grados 3 o 4 |
Placebo N = 818 n (%) Grados 3 o 4 |
Categoría de frecuencia Todos los grados |
|---|---|---|---|---|---|
|
Infecciones e infestaciones |
|||||
|
Infecciones1 |
434 (40.8) |
245 (30.0) |
41 (3.8) |
8 (1.0) |
Muy frecuente |
|
Trastornos de la sangre y del sistema linfático |
|||||
|
Neutropenia |
785 (73.7) |
41.0 (5.0) |
624 (58.6) |
11 (1.3) |
Muy frecuente |
|
Leucopenia |
314 (29.5) |
24 (2.9) |
165 (15.5) |
4 (0.5) |
Muy frecuente |
|
Anemia |
200 (18.8) |
51 (6.2) |
30 (2.8) |
12 (1.5) |
Muy frecuente |
|
Linfopenia |
95 (8.9) |
18 (2.2) |
56 (5.3) |
5 (0.6) |
Frecuente |
|
Trombocitopenia |
95 (8.9) |
11 (1.3) |
8 (0.8) |
1 (0.1) |
Frecuente |
|
Neutropenia febril |
15 (1.4) |
2 (0.2) |
15 (1.4) |
2 (0.2) |
Frecuente |
|
Trastornos oculares |
|||||
|
Lagrimeo aumentado |
59 (5.5) |
9 (1.1) |
0 |
0 |
Frecuente |
|
Ojo seco |
54 (5.1) |
18 (2.2) |
0 |
0 |
Frecuente |
|
Trastornos del metabolismo y de la nutrición |
|||||
|
Falta de apetito |
163 (15.3) |
101 (12.3) |
6 (0.6) |
1 (0.1) |
Muy frecuente |
|
Hipocalcemia |
45 (4.2) |
14 (1.7) |
11 (1.0) |
0 |
Frecuente |
|
Hipopotasemia |
33 (3.1) |
21 (2.6) |
12 (1.1) |
5 (0.6) |
Frecuente |
|
Hipofosfatemia |
34 (3.2) |
11 (1.3) |
22 (2.1) |
7 (0.9) |
Frecuente |
|
Trastornos del sistema nervioso |
|||||
|
Cefalea |
253 (23.8) |
177 (21.6) |
5 (0.5) |
4 (0.5) |
Muy frecuente |
|
Mareos |
125 (11.7) |
83 (10.1) |
1 (1.01) |
0 |
Muy frecuente |
|
Vértigo |
46 (4.3) |
10 (1.2) |
1 (0.1) |
0 |
Frecuente |
|
Trastornos cardiacos |
|||||
|
Síncope |
19 (1.8) |
9 (1.1) |
12 (1.1) |
7 (0.9) |
Frecuente |
|
Trastornos respiratorios, torácicos y del mediastino |
|||||
|
Disnea |
132 (12.4) |
81 (9.9) |
15 (1.4) |
7 (0.9) |
Muy frecuente |
|
Tos |
218 (20.5) |
132 (16.1) |
0 |
0 |
Muy Frecuente |
|
Trastornos músculo esqueléticos y del tejido conjuntivo |
|||||
|
Dolor de espalda |
211 (19.8) |
153 (18.7) |
20 (1.9) |
7 (0.9) |
Muy frecuente |
|
Trastornos gastrointestinales |
|||||
|
Náuseas |
475 (44.6) |
219 (26.8) |
15 (1.4) |
4 (0.5) |
Muy frecuente |
|
Diarrea |
317 (29.8) |
176 (21.5) |
16 (1.5) |
5 (0.6) |
Muy frecuente |
|
Vómitos |
284 (26.7) |
128 (15.6) |
21 (2.0) |
3 (0.4) |
Muy frecuente |
|
Estreñimiento |
253 (23.8) |
129 (15.8) |
8 (0.8) |
0 |
Muy frecuente |
|
Estomatitis |
122 (11.5) |
53 (6.5) |
3 (0.3) |
1 (0.1) |
Muy frecuente |
|
Dolor abdominal2 |
182 (17.1) |
107 (13.1) |
14 (1.3) |
4 (0.5) |
Muy frecuente |
|
Disgeusia |
71 (6.7) |
36 (4.4) |
1 (0.1) |
0 |
Frecuente |
|
Dispepsia |
88 (8.3) |
35 (4.3) |
1 (0.1) |
0 |
Frecuente |
|
Trastornos hepatobiliares |
|||||
|
Hepatotoxicidad3 |
19 (1.8) |
7 (0.9) |
15 (1.4) |
4 (0.5) |
Frecuente |
|
Trastornos de la piel y del tejido subcutáneo |
|||||
|
Alopecia |
256 (24.0) |
97 (11.9) |
0 |
0 |
Muy frecuente |
|
Erupción4 |
227 (21.3) |
70 (8.6) |
10 (0.9) |
0 |
Muy frecuente |
|
Prurito |
177 (16.6) |
48 (5.9) |
3 (0.3) |
0 |
Muy frecuente |
|
Eritema |
43 (4.0) |
8 (1.0) |
2 (0.2) |
0 |
Frecuente |
|
Piel seca |
88 (8.3) |
18 (2.2) |
0 |
0 |
Frecuente |
|
Vitíligo |
16 (1.5) |
0 |
0 |
0 |
Frecuente |
|
Trastornos generales y alteraciones en el lugar de la administración |
|||||
|
Fatiga |
348 (32.7) |
249 (30.4) |
20 (1.9) |
4 (0.5) |
Muy frecuente |
|
Edema periférico |
147 (13.8) |
71 (8.7) |
1 (0.1) |
0 |
Muy frecuente |
|
Astenia |
145 (13.6) |
103 (12.6) |
7 (0.7) |
3 (0.4) |
Muy frecuente |
|
Fiebre |
139 (13.1) |
52 (6.4) |
4 (0.4) |
0 |
Muy frecuente |
|
Boca seca |
74 (6.9) |
44 (5.4) |
1 (0.1) |
0 |
Frecuente |
|
Dolor orofaríngeo |
67 (6.3) |
33 (4.0) |
0 |
0 |
Frecuente |
|
Exploraciones complementarias |
|||||
|
Pruebas de función hepática anormales5 |
184 (17.3) |
66 (8.1) |
93 (8.7) |
16 (2.0) |
Muy frecuente |
|
Aumento de la creatinina en sangre |
67 (6.3) |
15 (1.8) |
4 (0.4) |
0 |
Frecuente |
|
Prolongación del intervalo QT en el electrocardiograma |
69 (6.5) |
13 (1.6) |
13 (1.2) |
2 (0.2) |
Frecuente |
1 Infecciones: Infecciones en el tracto urinario, tracto respiratorio, gastroenteritis, sepsis (<1%).
2 Dolor abdominal: Dolor abdominal y dolor abdominal superior.
3 Hepatotoxicidad: lesión hepatocelular, lesión hepática inducida por fármacos, hepatotoxicidad, insuficiencia hepática, hepatitis autoinmunitaria (un solo caso).
4 Erupción: erupción, erupción maculopapular, prurito.
5 Pruebas de función hepática anormales: elevación de la ALT (alanina aminotransferasa), elevación de la AST (aspartato aminotransferasa), aumento de la bilirrubina sanguínea.
Tabla 10. Reacciones adversas medicamentosas derivadas de reportes de casos espontáneos y de casos reportados en la literatura
|
Trastornos de la piel y del téjido subcutáneo |
|
Necrólisis epidérmica Tóxica (NET) |
Toxicidad hepatobiliar: En los ensayos clínicos de fase III, la proporción de pacientes que presentaron reacciones adversas hepatobiliares fue mayor en los grupos de ribociclib + fulvestrant que en los del placebo + fulvestrant (23.2% y 16.5%, respectivamente), y se notificaron más reacciones adversas de grado 3 o 4 entre las pacientes que recibieron ribociclib + fulvestrant (11.4% y 5.4%, respectivamente). En el 10.4% de las pacientes tratadas con ribociclib se notificaron interrupciones transitorias de la administración, ajustes de la dosis o ambas debido a reacciones adversas hepatobiliares, que consistieron fundamentalmente en elevaciones de la alanina aminotransferasa (ALT) (6.9%) y la AST (6.1%). La proporción de pacientes en las que hubo que retirar el tratamiento con ribociclib debido a anomalías en las pruebas funcionales hepáticas (PFH) y hepatotoxicidad ocurrió en el 2.3% y el 0.4% de los pacientes, respectivamente (véase el apartado de Precauciones generales).
Sobre la base de los datos, no hay evidencia de una relación causal entre fulvestrant y eventos raros o poco frecuentes en los estudios clínicos de fulvestrant.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Toxicidad aguda: La toxicidad aguda de fulvestrant es baja. En los roedores, la dosis letal media fue mayor a 70 mg/kg después de la administración intramuscular (más de 400 veces la dosis clínica); mayor de 50 mg/kg después de la administración intravenosa y mayor de 2,000 mg/kg después de la administración oral.
Toxicidad crónica: Fulvestrant fue bien tolerado en todas las especies animales en las que se probó. En estudios de toxicidad con dosis múltiple intramuscular en ratas y perros, la actividad antiestrogénica de fulvestrant causó la mayoría de los efectos observados, sobre todo en el sistema reproductor femenino, pero también en otros órganos sensibles a las hormonas en ambos sexos. No hubo evidencia de otros efectos tóxicos sistémicos en ratas que recibieron dosis de hasta 10 mg/rata/15 días por 6 meses, ni en perros con dosis de hasta 40 mg/kg/28 días durante 12 meses.
En estudios con perros después de la administración oral e intravenosa, se observaron efectos en el sistema cardiovascular (elevaciones ligeras en el segmento ST del ECG [oral] y paro sinusal en un perro [intravenosa]), pero esto ocurrió en animales expuestos a fulvestrant a niveles mucho mayores que los registrados en pacientes (Cmáx. >15 veces) y, por lo tanto, se consideran sin importancia para la seguridad de los humanos con la dosis clínica.
Mutagenicidad: Fulvestrant no mostró potencial genotóxico.
Toxicología reproductiva: Fulvestrant mostró efectos en la reproducción y el desarrollo embrionario/fetal consistente con su actividad antiestrogénica con dosis similares a la dosis clínica. En las ratas, fulvestrant causó reducción reversible de la fertilidad femenina y la sobrevivencia embrionaria con dosis de 0.01 mg/kg/día o más, además de distocia y mayor incidencia de anormalidades fetales, incluida flexión del tarso. Las conejas que recibieron fulvestrant en dosis ≥ 1 mg/kg/día no pudieron mantener el embarazo y con dosis de hasta 0.25 mg/kg/día se observaron aumentos en el peso placentario y pérdida posterior a la implantación, pero sin efectos en el desarrollo fetal.
Carcinogenicidad: Un estudio de dos años sobre oncogenicidad en ratas (administración intramuscular) mostró aumento en la incidencia de tumores benignos de células de la granulosa ovárica en las hembras con la dosis alta, 10 mg/rata/15 días. En un estudio de dos años de oncogenicidad en ratones, la administración oral fue asociada con un aumento en la incidencia de tumores del estroma del cordón sexual (benigno y maligno) en el ovario a dosis de 150 y 500 mg/kg/día. El nivel sin efecto observado (NOEL, por sus siglas en inglés) de estos hallazgos fue de 10 mg/rata/30 días en ratas y 20 mg/kg/día en ratones, respectivamente. La inducción de estos tumores es consistente con las alteraciones en la retroalimentación endocrina, relacionadas con la farmacología en los niveles de gonadotropina, causados por el efecto antiestrogénico en los animales con ciclos hormonales. Por lo tanto, no se considera que este hallazgo tenga relevancia clínica para el uso de fulvestrant en mujeres postmenopáusicas con cáncer de mama avanzado.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Fulvestrant no inhibe en forma significativa ninguna de las isoenzimas principales de citocromo P-450 (CYP) in vitro; los resultados de una prueba farmacocinética clínica incluyeron la administración concomitante de fulvestrant con midazolam también sugiere que la dosis terapéutica de fulvestrant no tiene efectos inhibidores en CYP3A4. Adicionalmente, aunque fulvestrant puede metabolizarse por CYP3A4 in vitro, un estudio clínico con rifampicina mostró que no había cambio en la depuración de fulvestrant como resultado de la inducción de CYP3A4. Los resultados de un estudio clínico con ketoconazol, un potente inhibidor de CYP3A4, también indicaron que no hay cambios de importancia clínica en la depuración de fulvestrant. No es necesario ajustar la dosis en pacientes que al mismo tiempo reciben inductores o inhibidores de CYP3A4.
Debido a la similitud estructural de fulvestrant y estradiol, fulvestrant puede interferir con los ensayos basados en anticuerpos anti-estradiol y puede obtenerse un resultado falsamente elevado de los niveles de estradiol.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Elevación de las enzimas hepáticas (ALT, AST, ALP), bilirrubina, GGT, disminución del número de plaquetas, así como alteraciones en los niveles de estradiol.
PRECAUCIONES GENERALES: Fulvestrant debe usarse con precaución en pacientes con deterioro hepático leve a moderado.
Fulvestrant debe utilizarse con precaución en pacientes con alteración renal severa (aclaramiento de creatinina menor a 30 mL/min).
Debido a la vía de administración intramuscular, fulvestrant debe utilizarse con precaución si se trata a pacientes con diátesis hemorrágicas, trombocitopenia o pacientes que reciben tratamiento con anticoagulantes.
No existen datos a largo plazo sobre el efecto que ejerce fulvestrant en el hueso. Debido al mecanismo de acción de fulvestrant, existe un posible riesgo de osteoporosis.
Conducción y uso de máquinas: La influencia de fulvestrant sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Sin embargo, el uso de fulvestrant puede producir astenia (disminución de la fuerza muscular), por lo que, si conduce o utiliza maquinaria pesada, debe hacerlo con precaución.
Alcohol bencílico: Fulvestrant contiene 500 mg de alcohol bencílico en cada inyección, por lo que puede provocar reacciones de hipersensibilidad.
[Fulvestrant en indicación "combinación con ribociclib"]: Cuando se combina fulvestrant con ribociclib, consultar también el Resumen de las características del producto de ribociblib.
Toxicidad hepatobiliar: En los ensayos clínicos de fase III se observó elevación de transaminasas.
Se notificaron elevaciones de grado 3 o 4 de la (alanina aminotransferasa) ALT (ribociclib + fulvestrant: 9.7% vs. placebo + fulvestrant: 1.5%) y de la (aspartato aminotransferasa) AST (ribociclib + fulvestrant: 6. 7% vs. placebo + fulvestrant: 2.1%). Se reportaron aumentos de Grado 4 de la ALT (ribociclib + fulvestrant: 1.9% Vs. placebo + fulvestrant 0.1 %) y AST (ribociclib + fulvestrant: 1.1% vs. Placebo + fulvestrant: 0.1%).
En los ensayos clínicos de fase III 83.2% (89/107) de los episodios de elevación de la ALT o la AST de Grado 3 o 4 se produjeron durante los 6 primeros meses de tratamiento (véase el apartado de Reacciones secundarias y adversas). La mayoría de estas elevaciones de la ALT y la AST no se acompañaban de un aumento de la bilirrubina. Entre las pacientes con elevaciones de la ALT o la AST de Grado 3 o 4, la mediana del tiempo transcurrido hasta el comienzo de la reacción adversa fue de 85 días en el grupo tratado con ribociclib + fulvestrant. La mediana del tiempo transcurrido hasta la resolución (es decir, hasta la normalización o una neutropenia de grado ≤ 2) fue de 22 días en el grupo tratado con ribociclib + fulvestrant.
[Fulvestrant en todas las indicaciones]:
La interferencia con los ensayos de anticuerpos de estradiol:
Debido a la similitud estructural de fulvestrant y estradiol, fulvestrant puede interferir con los ensayos de estradiol basados en anticuerpos y puede resultar en niveles con incrementos falsos de estradiol.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Mujeres adultas (incluidas pacientes de edad avanzada): La dosis recomendada es de 500 mg a ser administrada por vía intramuscular con dos inyecciones de 5 mL de 250 mg cada una, aplicadas en el glúteo a intervalos de 1 mes con una dosis adicional de 500 mg administrada dos semanas después de la dosis inicial. Se recomienda administrar la inyección lentamente (1-2 minutos/inyección).
Niños: No se recomienda su empleo en niños o adolescentes, ya que no se ha establecido su seguridad y efectividad en este grupo de edad.
Pacientes con insuficiencia renal: No se recomiendan ajustes en la dosis para pacientes con depuración de creatinina mayor de 30 mL/min. No se ha hecho una evaluación adicional de la seguridad y eficacia en pacientes con depuración de creatinina menor de 30 mL/min (véase Precauciones generales).
Pacientes con insuficiencia hepática: No se recomiendan ajustes en la dosis para pacientes con categoría Child-Pugh A y B con daño hepático. El uso de Fulvestrant no ha sido evaluado en paciente Child-Pugh C y daño hepático (véase Precauciones generales y Farmacocinética y farmacodinamia).
Pacientes de edad avanzada: No se requieren ajustes en la dosis para los pacientes geriátricos.
Interacciones que requieren ajuste de dosis: No hay interacciones farmacológicas conocidas que requieran un ajuste en la dosis.
Cuando se co-administra un inhibidor selectivo de las cinasas ciclinodependientes (CDK) 4/6: La dosis recomendada es de 500 mg administrados por vía intramuscular los días 1, 15 y 29, y posteriormente una vez al mes. Consulte la información para prescripción completa del producto co-administrado.
Vía de administración: Intramuscular.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No hay experiencia de sobredosis en humanos. Los estudios en animales sugieren que no hay efectos distintos a los relacionados en forma directa o indirecta con la actividad antiestrogénica cuando se administraron dosis más altas de fulvestrant. Si ocurre una sobredosis, debe emplearse tratamiento sintomático.
PRESENTACIONES:
Caja de cartón con 1 jeringa prellenada con 5 mL y 1 aguja para inyección.
Caja de cartón con 2 jeringas prellenadas con 5 mL y 2 agujas para inyección.
Todas las presentaciones con instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 25 °C.
Consérvese la caja bien cerrada.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Si no se administra todo el producto, deséchese el sobrante. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se administre si el cierre ha sido violado. Deséchese inmediatamente después de su uso. No se use durante el embarazo ni lactancia. Su venta requiere receta médica. No se deje al alcance de los niños. Este medicamento puede afectar el estado de alerta, por lo que no deberá conducir vehículos motores ni maquinaria pesada durante su uso. Este medicamento contiene alcohol bencílico como excipiente, el cual puede causar reacciones de hipersensibilidad. Este medicamento deberá ser administrado únicamente por médicos especialistas en oncología con experiencia en quimioterapia antineoplásica.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Propiedad de:
Sandoz GmbH
Biochemiestraße 10, 6250 Kundl, Austria
Representante Legal:
SANDOZ, S.A. de C.V.
La Candelaria No. 186, Col. Atlántida,
C.P. 04370, Coyoacán, Ciudad de México, México.
Reg. Núm. 044M2021 SSA IV
22330022130274/18Ene2023/IPPA_DRA-Sandoz
®Marca Registrada


