
LITFULO
RITLECITINIB
Cápsulas
1 Caja, 1 Envase de burbuja, 30 Cápsulas, 50 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada CÁPSULA contiene:
Ritlecitinib 50 mg
Excipiente cbp 1 cápsula
INDICACIONES TERAPÉUTICAS:
Tratamiento de alopecia areata, incluida alopecia total y alopecia universal, en adultos y adolescentes de 12 años y mayores que son candidatos para el tratamiento sistémico.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacocinéticas:
Absorción:
El ritlecitinib se absorbe bien en el grado de ~89% (fa) después de la administración oral con una biodisponibilidad oral absoluta de ~64%. Las concentraciones plasmáticas máximas se alcanzan en el plazo de 1 hora.
Efecto de los alimentos:
Los alimentos no tienen un efecto clínicamente significativo en las exposiciones sistémicas de ritlecitinib y el producto se puede administrar independientemente de la ingesta de alimentos. La coadministración de una cápsula de ritlecitinib de 100 mg con una comida rica en grasas redujo la Cmáx de ritlecitinib en ~32% sin afectar el grado de absorción de ritlecitinib, ya que el ABCinf aumentó en una cantidad marginal de 11%. En los estudios clínicos, se administró ritlecitinib sin tener en cuenta las comidas (ver sección Dosis y vía de administración).
Distribución:
Después de la administración intravenosa, el volumen de distribución del ritlecitinib es alrededor de 74 L. Aproximadamente el 14% del ritlecitinib circulante está ligado a proteínas plasmáticas. La proporción de distribución sangre/plasma de ritlecitinib es de 1.62.
Biotransformación:
El metabolismo del ritlecitinib está mediado por varias isoformas de GST (GST citosólico A1/3, M1/3/5, P1, S1, T2, Z1 y MAPEG1/2/3 microsomal) y enzimas CYP (CYP3A, CYP2C8, CYP1A2 y CYP2C9), sin una vía de depuración única que contribuya a más del 25%. En un estudio radiomarcado en humanos, el ritlecitinib fue la especie circulante más prevalente (el 30.4% de la radioactividad circulante) después de la administración intravenosa, con un metabolito conjugado principal de cisteína M2 (el 16.5%), que es farmacológicamente inactivo.
Eliminación:
El ritlecitinib se elimina principalmente, mediante mecanismos de eliminación metabólica, con aproximadamente el 4% de la dosis excretada en la orina como medicamento sin cambios. Los metabolitos del ritlecitinib se excretan en la orina (el 66% de radioactividad recuperada) y en las heces (el 20%). Después de varias dosis orales, el estado de equilibrio se alcanzó aproximadamente en el Día 4 debido a la farmacocinética no estacionaria. Los parámetros farmacocinéticos en estado de equilibrio del ABCtau y la Cmáx parecieron incrementarse de manera aproximadamente proporcional a la dosis, con una vida media terminal promedio que osciló entre 1.3 y 2.3 horas.
Poblaciones especiales:
Peso corporal, sexo, genotipo, raza y edad:
El peso corporal, el sexo, el genotipo, la raza y la edad no tuvieron un efecto clínicamente significativo sobre la exposición a ritlecitinib.
Adolescentes (de 12 a < 18 años):
Con base en el análisis farmacocinético poblacional, no hubo diferencias clínicamente relevantes en las exposiciones a ritlecitinib en pacientes adolescentes en comparación con adultos.
Población pediátrica (< 12 años):
Aún no se ha establecido la farmacocinética de ritlecitinib en pacientes pediátricos menores de 12 años.
Insuficiencia renal:
El ABC24 observado en pacientes con insuficiencia renal grave (TFGe < 30 mL/min) fue un 55.2% mayor en comparación con el ABC24 en participantes emparejados con función renal normal (participantes sanos en la cohorte de función hepática normal del estudio de insuficiencia hepática) y fue de un 71% mayor en relación con el ABC24 calculada en los 1000 participantes sanos in silico con función renal normal (TFGe ≥ 90 mL/min). Esta diferencia no se considera clínicamente significativa. No se ha estudiado el ritlecitinib en pacientes con insuficiencia renal leve (TFGe 60 a < 90 mL/min) o moderada (TFGe 30 a < 60 mL/min), ya que no se espera un incremento clínicamente significativo en la exposición a ritlecitinib en estos pacientes. La TFGe y la clasificación del estado de la función renal de los participantes se realizaron mediante la fórmula de Modificación de la Dieta en Enfermedad Renal (MDER).
Con base en las consideraciones anteriores, no es necesario realizar un ajuste de la dosis en los pacientes con insuficiencia renal leve, moderada o grave. No se ha estudiado el ritlecitinib en pacientes con enfermedad renal en estado terminal (ESRD por sus siglas en inglés) o en receptores de trasplante renal (ver sección Dosis y vía de administración).
Insuficiencia hepática:
Los pacientes con insuficiencia hepática moderada (Child Pugh B) tuvieron un incremento del 18.5% en el ABC24 del ritlecitinib en comparación con los participantes con función hepática normal. No se estudió el ritlecitinib en pacientes con insuficiencia hepática leve (Child Pugh A), ya que no se espera un incremento clínicamente significativo en la exposición a ritlecitinib en estos pacientes. Con base en las consideraciones anteriores, no se necesita ningún ajuste de la dosis en pacientes con insuficiencia hepática leve o moderada. En estudios clínicos, no se ha estudiado ritlecitinib en pacientes con insuficiencia hepática grave (Child Pugh C) y no se recomienda su administración en estos pacientes (ver sección Dosis y vía de administración).
Propiedades farmacodinámicas:
Mecanismo de acción:
El ritlecitinib es un inhibidor de la Janus cinasa (JAK) 3 y también de las cinasas de la familia de la tirosina cinasa expresada en carcinoma hepatocelular (TEC). Las JAK son enzimas intracelulares que transmiten señales que surgen de las interacciones de citocinas o de receptores del factor de crecimiento en la membrana celular para influir en los procesos celulares de hematopoyesis y en el desarrollo y la función de células inmunitarias. Con su activación, las JAK fosforilan y activan los Transductores de Señal y Activadores de la Transcripción (STAT) que modulan la actividad intracelular, incluida la expresión genética. Las cinasas de la familia TEC también son enzimas intracelulares que transmiten señales que surgen de varios receptores inmunitarios que modulan la actividad intracelular, incluida la expresión genética.
El ritlecitinib inhibe de forma irreversible y selectiva la JAK3 y las cinasas de la familia TEC mediante el bloqueo del lugar de unión de trifosfato de adenosina (ATP). En entornos celulares, el ritlecitinib inhibe específicamente la fosforilación de STAT inducida por citocinas mediada por receptores dependientes de la JAK3 y evade la señalización de receptores independientemente de JAK3 (es decir, JAK1/JAK2, JAK1/TYK2, JAK2/JAK2, JAK2/TYK2). Además, el ritlecitinib inhibe la señalización de receptores inmunes dependientes de los miembros de la familia de cinasas TEC. El ritlecitinib inhibe la actividad citolítica y la producción de interferón-gamma (IFNg) en linfocitos natural killers (NK) y células T CD8+ mediante la inhibición de miembros de la familia de cinasas TEC. El tratamiento con ritlecitinib redujo los grupos peribulbares inflamatorios de células T CD3+ y células T citotóxicas CD8+, así como una reducción de linfocitos NKG2D+ NK y células T CD8+ que rodean a los folículos pilosos. Actualmente, no se conoce la relevancia de la inhibición específica de la JAK o de las enzimas de la familia TEC para la efectividad terapéutica.
Efectos farmacodinámicos:
Subconjuntos de linfocitos:
En pacientes con alopecia areata, el tratamiento con ritlecitinib se asoció con disminuciones tempranas dependientes de la dosis en los niveles absolutos de linfocitos, linfocitos T (CD3) y subconjuntos de linfocitos T (CD4 y CD8). Después de la disminución inicial, los niveles se recuperaron parcialmente y permanecieron estables hasta las 48 semanas. No se observaron cambios en los linfocitos B (CD19) en ningún grupo de tratamiento. Hubo una disminución temprana dependiente de la dosis en los linfocitos NK (CD16/56) que permaneció estable en el nivel más bajo hasta la semana 48.
Inmunoglobulinas:
En pacientes con alopecia areata, el tratamiento con ritlecitinib no se asoció con cambios clínicamente significativos en IgG, IgM o IgA hasta la semana 48, lo que indica una falta de inmunosupresión humoral sistémica.
Electrofisiología cardiaca:
A 12 veces la exposición máxima media de la dosis de 50 mg una vez al día en pacientes con alopecia areata, no hubo un efecto clínicamente significativo sobre el intervalo QTc.
Eficacia clínica y seguridad:
Se evaluó la eficacia y la seguridad del ritlecitinib en un estudio fundamental, aleatorizado, doble ciego, controlado con placebo (Estudio AA-I) en pacientes con alopecia areata de 12 años y mayores con ≥50% de pérdida de cabello en el cuero cabelludo, incluidos Alopecia Total (AT) y Alopecia Universal (AU). En este estudio también se evaluó la respuesta a la dosis de ritlecitinib. El periodo de tratamiento del estudio constaba de un periodo controlado con placebo de 24 semanas y un periodo de extensión de 24 semanas. El Estudio AA-I evaluó un total de 718 pacientes que se aleatorizaron a uno de los siguientes regímenes de tratamiento durante 48 semanas: 1) 200 mg una vez al día durante 4 semanas seguido de 50 mg una vez al día durante 44 semanas; 2) 200 mg una vez al día durante 4 semanas seguido de 30 mg una vez al día durante 44 semanas; 3) 50 mg una vez al día durante 48 semanas; 4) 30 mg una vez al día durante 48 semanas; 5) 10 mg una vez al día durante 48 semanas; 6) placebo durante 24 semanas seguido de 200 mg una vez al día durante 4 semanas y 50 mg una vez al día durante 20 semanas; o 7) placebo durante 24 semanas seguido de 50 mg durante 24 semanas. Los pacientes que completaron el estudio fundamental eran elegibles para inscribirse en un estudio abierto a largo plazo (AA-II).
La dosis recomendada de ritlecitinib es de 50 mg una vez al día y los resultados para esta dosis se analizan a continuación.
Características iniciales:
En el Estudio AA-I se evaluaron pacientes de sexo masculino o femenino de 12 años o mayores. Todos los pacientes tenían alopecia areata con ≥ 50% de pérdida de cabello en el cuero cabelludo (puntaje SALT ≥ 50) sin evidencia de recrecimiento terminal del cabello en el plazo de los 6 meses anteriores y con el episodio actual de pérdida cabello en el cuero cabelludo ≤ 10 años y sin otra causa conocida de pérdida del cabello (p. ej., alopecia androgenética). Solamente los pacientes de entre 18 y 74 años (inclusive) al momento del consentimiento informado eran elegibles para la inscripción en la UE.
Entre todos los grupos de tratamiento, el 62.1% eran de sexo femenino, el 68.0% eran caucásicos, el 25.9% eran asiáticos y el 3.8% eran de raza negra o afroamericano. La mayoría de los pacientes (el 85.4%) eran adultos (≥ 18 años) con una edad media de 33.7 años. Se inscribió a un total de 105 (el 14.6%) pacientes de 12 a < 18 años y 20 (el 2.8%) pacientes de 65 años y mayores. El puntaje medio inicial de la Herramienta de Severidad de la Alopecia (SALT, por sus siglas en inglés) osciló entre 88.3 y 93.0 entre los grupos de tratamiento; entre los pacientes sin AT/AU en el periodo inicial, el puntaje medio de SALT osciló entre 78.3 y 87.0. La mayoría de los pacientes tenía cejas (el 83.0%) y pestañas (el 74.7%) anormales en el periodo inicial entre los grupos de tratamiento. La duración mediana desde el diagnóstico de alopecia areata fue de 6.9 años y la duración mediana del episodio actual de alopecia areata fue de 2.5 años. La aleatorización se estratificó por estado de AT/AU con un 46% de pacientes clasificados como AT/AU con base en un puntaje de SALT inicial de 100.
Respuesta clínica:
La evaluación de la caída de cabello en el cuero cabelludo se basó en el puntaje de SALT. En la semana 24, una proporción significativamente mayor de pacientes tuvo una respuesta de SALT ≤ 20 (el 20% o menos de pérdida de cabello en el cuero cabelludo) con ritlecitinib 50 mg en comparación con el placebo (Tabla 1). La tasa de respuesta de SALT ≤ 20 para ritlecitinib 50 mg se incrementó aún más en la semana 48 (Tabla 1 y Figura 1). La separación estadística del placebo en la respuesta de SALT ≤ 20 ocurrió en la semana 18 para ritlecitinib 50 mg.
Una proporción significativamente mayor de pacientes tuvo una respuesta de SALT ≤ 10 (el 10% o menos de caída de cabello en el cuero cabelludo) con ritlecitinib 50 mg en comparación con el placebo en la semana 24 (Tabla 1). La tasa de respuesta de SALT ≤ 10 se incrementó aún más en la semana 48 (Tabla 1 y Figura 2).
Se observó una mejora significativa en la Impresión Global de Cambio del Paciente (PGI-C por sus siglas en inglés) para ritlecitinib 50 mg en comparación con el placebo en la semana 24, con tasas de respuesta que continuaron incrementando hasta la semana 48 (Tabla 1 y Figura 3).
Los pacientes que recibieron ritlecitinib 50 mg experimentaron una mayor mejora en los puntajes de SALT a lo largo del tiempo, según lo medido por el cambio en el puntaje de SALT a partir del valor inicial, en comparación con el placebo en la semana 24 con incrementos adicionales hasta la semana 48 (Figura 4).
Los efectos del tratamiento en los subgrupos (edad, con o sin AT/AU en el periodo inicial, sexo, raza, región, peso, duración de la enfermedad desde el diagnóstico, duración del episodio actual, tratamiento farmacológico anterior) fueron coherentes con los resultados en la población general del estudio.
Se observaron mejoras en el recrecimiento de las cejas y/o las pestañas en la semana 24 con ritlecitinib 50 mg entre los pacientes con cejas y/o pestañas anormales en el periodo inicial con incrementos adicionales observados en la semana 48 (Tabla 1).
La proporción de pacientes que informaron satisfacción con 3 aspectos de su crecimiento del cabello (cantidad, calidad y satisfacción general), según lo medido por la Satisfacción del Paciente con el Crecimiento del Cabello (P-SAT, por sus siglas en inglés), se incrementó desde la semana 4 hasta la semana 24 para ritlecitinib 50 mg con poca mejora en la satisfacción observada en el grupo de placebo.
Tabla 1. Resultados de eficacia de ritlecitinib
|
Ritlecitinib 50 mg QD |
Placebo (N = 131) |
Diferencia con respecto al Placebo |
||
|
Semana 24 |
Respuesta de |
23.4 |
1.5 |
21.9 |
|
Respuesta de |
13.7 |
1.5 |
12.2 |
|
|
Respuesta de |
49.6 |
9.2 |
40.4 |
|
|
Respuesta del |
29.0 |
4.7 |
24.3 |
|
|
Respuesta EPf |
28.9 |
5.2 |
23.7 |
|
|
% de sujetos que respondieron al tratamiento |
||||
|
Semana 48 |
Respuesta de |
43.2 |
||
|
Respuesta de |
31.2 |
|||
|
Respuesta de |
56.0 |
|||
|
Respuesta de la |
43.6 |
|||
|
Respuesta de la |
40.0 |
|||
Abreviaturas: EC = evaluación de las cejas; EP = evaluación de las pestañas; IC = intervalo de confianza; N = cantidad total de pacientes; PGI-C = impresión global de cambio del paciente; QD = una vez al día; SALT = Herramienta de severidad de la alopecia.
a Los pacientes que respondieron a la SALT ≤ 20 fueron pacientes con pérdida de cabello en el cuero cabelludo de ≤ 20%. Los puntajes de SALT oscilan entre 0 y 100 con 0 = sin pérdida de cabello en el cuero cabelludo y 100 = pérdida total del cabello en el cuero cabelludo.
b Estadísticamente significativo con ajuste por multiplicidad.
c Los pacientes que respondieron a la SALT ≤ 10 fueron pacientes con pérdida de cabello en el cuero cabelludo de ≤ 10%. Los puntajes de SALT oscilan entre 0 y 100 con 0 = sin caída de cabello en el cuero cabelludo y 100 = pérdida total del cabello en el cuero cabelludo.
d Los pacientes que respondieron a PGI-C eran pacientes con un puntaje de “mejoría moderada” o “mejoría considerable” con base en una escala de 7 puntos de “mejoría considerable” a “empeoramiento considerable”. Estadísticamente significativo con ajuste por multiplicidad.
e La respuesta de EC se define como una mejora de al menos 2 grados desde el periodo inicial o un puntaje de EC normal en pacientes con cejas anormales en el periodo inicial.
f La respuesta de la EP se define como una mejora de al menos 2 grados desde el periodo inicial o un puntaje de la EP normal en pacientes con pestañas anormales en el periodo inicial.
Figura 1. Respuesta de SALT ≤ 20 hasta la semana 48
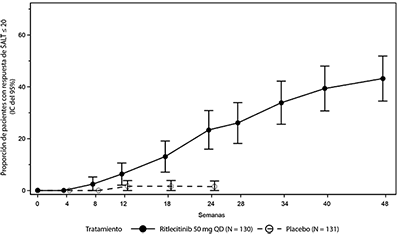
Abreviaturas: IC = intervalo de confianza; N = cantidad total de pacientes; QD = una vez al día; SALT = herramienta de severidad de la alopecia.
Figura 2. Respuesta de SALT ≤ 10 hasta la semana 48
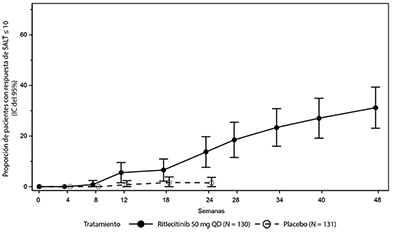
Abreviaturas: IC = intervalo de confianza; N = cantidad total de pacientes; QD = una vez al día; SALT = herramienta de severidad de la alopecia.
Figura 3. Respuesta de PGI-C hasta la semana 48
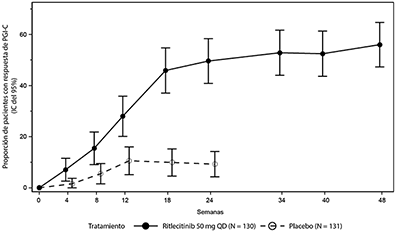
Abreviaturas: IC = intervalo de confianza; N = cantidad total de pacientes; PGI-C = impresión global de cambio del paciente; QD = una vez al día.
Figura 4. Cambio desde el periodo inicial en el puntaje SALT hasta la Semana 48
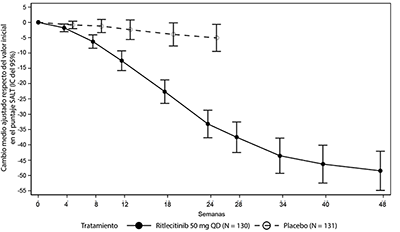
Abreviaturas: IC = intervalo de confianza; N = cantidad total de pacientes; QD = una vez al día; SALT = herramienta de severidad de la alopecia.
Población de pacientes pediátricos:
Se evaluó la eficacia y la seguridad de ritlecitinib en el Estudio AA-I que incluía a 105 pacientes que tenían entre 12 y menos de 18 años. En este estudio, los resultados en pacientes de 12 a menos de 18 años se presentan en la Tabla 2 y fueron coherentes con los resultados en la población general del estudio.
Tabla 2. Resultados de eficacia del Estudio AA-1 para pacientes de 12 a < 18 años en la semana 24
|
Ritlecitinib 50 mg QD (N = 16) |
Placebo (N = 19) |
Diferencia con respecto al Placebo (IC del 95%) |
|
|
Respuesta de SALT ≤ 20a |
25.0 |
0 |
25.0 (5.5; 49.9) |
|
Respuesta de SALT ≤ 10b |
12.5 |
0 |
12.5 (-5.9; 36.4) |
Abreviaturas: IC = intervalo de confianza; N = cantidad total de pacientes; QD = una vez al día; SALT = herramienta de severidad de la alopecia.
a Los pacientes que respondieron a la SALT ≤ 20 fueron pacientes con pérdida de cabello en el cuero cabelludo de ≤ 20%. Los puntajes de SALT oscilan entre 0 y 100 con 0 = sin pérdida de cabello en el cuero cabelludo y 100 = pérdida total de cabello en el cuero cabelludo.
b Los pacientes que respondieron a la SALT ≤ 10 fueron pacientes con pérdida de cabello en el cuero cabelludo de ≤ 10%. Los puntajes de SALT oscilan entre 0 y 100 con 0 = sin pérdida de cabello en el cuero cabelludo y 100 = pérdida total del cabello en el cuero cabelludo.
Eficacia a largo plazo después de 48 semanas:
Los pacientes elegibles en el Estudio AA-I se consideraron para la inscripción en el Estudio AA-II, lo que permitió a los pacientes extender el tratamiento con ritlecitinib pasadas las 48 semanas. Los pacientes que anteriormente recibieron ritlecitinib 50 mg una vez al día en el Estudio AA-I continuaron recibiendo ritlecitinib 50 mg una vez al día en el Estudio AA-II.
Respuesta de SALT ≤ 20:
En general, la proporción de pacientes con una respuesta de SALT ≤ 20 se incrementó durante 24 meses en pacientes tratados con ritlecitinib 50 mg en el Estudio AA-I. Además, el 28.3% de los pacientes tratados con ritlecitinib 50 mg en el Estudio AA-I que no tuvieron una respuesta de SALT ≤ 20 después de 48 semanas de tratamiento alcanzaron una respuesta de SALT ≤ 20 después de 18 meses de tratamiento.
Entre los pacientes tratados con ritlecitinib 50 mg en el Estudio AA-I que tuvieron una respuesta de SALT ≤ 20 después de 48 semanas de tratamiento e ingresaron al Estudio AA-II, el 85.4% y el 85.0% de los pacientes mantuvieron su respuesta después de 18 y 24 meses de tratamiento, respectivamente.
Respuesta de SALT ≤ 10:
En general, la proporción de pacientes con una respuesta de SALT ≤ 10 se incrementó durante 24 meses en pacientes tratados con ritlecitinib 50 mg en el Estudio AA-I. Además, el 22.0% de los pacientes tratados con ritlecitinib 50 mg en el Estudio AA-I que no tuvieron una respuesta de SALT ≤ 10 después de 48 semanas de tratamiento alcanzaron una respuesta de SALT ≤ 10 después de 18 meses de tratamiento.
Entre los pacientes tratados con ritlecitinib 50 mg en el Estudio AA-I que tuvieron una respuesta de SALT ≤ 10 después de 48 semanas de tratamiento e ingresaron al Estudio AA-II, el 91.4% y el 83.3% de los pacientes mantuvieron su respuesta después de 18 y 24 meses de tratamiento, respectivamente.
CONTRAINDICACIONES:
Pacientes con hipersensibilidad conocida a ritlecitinib o a cualquier otro ingrediente de la fórmula. Infecciones graves activas, incluida la tuberculosis, insuficiencia hepática grave, embarazo, lactancia y pacientes menores de 12 años.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Personas en edad fértil:
Se recomienda a las personas en edad fértil que empleen un método anticonceptivo efectivo durante el tratamiento y por 1 mes después de la dosis final de ritlecitinib. Considere la planificación y prevención del embarazo para las personas en edad fértil.
Embarazo:
Existen pocos datos sobre el uso del ritlecitinib en embarazos humanos. Los estudios en animales han demostrado toxicidad en el desarrollo sin efectos en exposiciones clínicamente significativas (ver sección Precauciones en relación con efecto de carcinogénesis, mutagénesis, teratogénesis y sobre fertilidad). No se recomienda consumir ritlecitinib durante el embarazo.
Lactancia:
No se dispone de datos sobre la presencia de la ritlecitinib en la leche materna humana, los efectos en el lactante ni los efectos en la producción de leche. El ritlecitinib se excretó en la leche de ratas en periodo de lactancia. No se puede excluir el riesgo a recién nacidos/lactantes y no se debe consumir ritlecitinib durante la lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad:
Las reacciones adversas al medicamento (RAM) informadas con mayor frecuencia en ≥ 2% de los pacientes tratados con ritlecitinib en los estudios controlados con placebo fueron diarrea (9.2%), acné (6.2%), urticaria (4.6%), erupción (3.8%) y mareos (2.3%).
Lista tabulada de reacciones adversas al medicamento:
Un total de 1628 pacientes recibieron tratamiento con ritlecitinib en estudios clínicos de alopecia areata, lo que representa 2085 años-paciente de exposición. Se integraron tres estudios controlados con placebo (130 participantes con 50 mg diarios y 213 participantes con placebo) para evaluar la seguridad de ritlecitinib en comparación con el placebo durante un máximo de 24 semanas después del inicio del tratamiento.
En la Tabla 3 se enlistan las reacciones adversas al medicamento observadas en los estudios clínicos de alopecia areata presentados por clasificación por órganos y sistemas. Dentro de cada clasificación por órganos y sistemas, se presentan las reacciones adversas al medicamento por orden de seriedad decreciente.
Tabla 3. Reacciones Adversas al Medicamento (RAM) por Clasificación por Órganos y Sistemas (SOC) y Categoría de Frecuencia del Consejo de Organizaciones Internacionales de Ciencias Médicas (CIOMS) Categoría de Frecuencia Enumerada en Orden Decreciente de Gravedad Médica o Importancia Clínica Dentro de Cada Categoría de Frecuencia y SOC
|
Clasificación |
Muy |
Frecuente |
Poco frecuente |
Rara |
Muy rara |
Frecuencia desconocida (no se puede calcular |
|
Infecciones e infestaciones |
Infecciones del tracto respiratorio superior, herpes zóster, foliculitis |
|||||
|
Trastornos del sistema nervioso |
Mareos |
|||||
|
Trastornos gastrointestinales |
Diarrea |
|||||
|
Trastornos de la piel y del tejido subcutáneo |
Acné, urticaria, erupción |
|||||
|
Pruebas complementarias |
Incremento de la creatinina fosfoquinasa en sangre |
Disminución del recuento de plaquetas, disminución del recuento de linfocitos |
Descripción de las reacciones adversas al medicamento seleccionadas:
Infecciones:
En los estudios controlados con placebo integrados, durante un máximo de 24 semanas, se han informado infecciones generales en el 31% de los pacientes (80.35 por 100 años-paciente) tratados con placebo y en el 33% de los pacientes (74.53 por 100 años-paciente) tratados con ritlecitinib 50 mg.
Entre todos los pacientes tratados con ritlecitinib en el análisis de seguridad integrado, incluido el estudio a largo plazo y un estudio en vitíligo, se informaron infecciones generales en el 42.4% de los pacientes (50.71 por 100 años-paciente) tratados con ritlecitinib 50 mg o más. La mayoría de las infecciones fue de severidad leve o moderada.
En los estudios controlados con placebo integrados, el porcentaje de pacientes que informaron reacciones adversas relacionadas con infecciones de herpes zóster fue del 1.5% en el grupo de 50 mg en comparación con 0 en el grupo de placebo. Todos los eventos de herpes zóster no fueron serios; 1 paciente que recibió ritlecitinib 200/50 mg (200 mg una vez al día durante 4 semanas seguido de 50 mg una vez al día) presentó un evento de infección por virus de varicela zóster que cumplió con los criterios de infección oportunista (herpes zóster multidermatómico). Entre todos los pacientes tratados con ritlecitinib en el análisis de seguridad integrado, incluido el estudio a largo plazo y un estudio en vitíligo, la tasa de herpes zóster fue de 1.17 por cada 100 años-paciente en pacientes tratados con ritlecitinib 50 mg o más.
En los estudios controlados con placebo, por hasta 24 semanas, no se informaron infecciones graves en pacientes tratados con placebo o ritlecitinib 50 mg. La tasa de infecciones graves en pacientes tratados con ritlecitinib 200/50 mg fue de 2.66 por 100 años-paciente. Entre todos los pacientes tratados con ritlecitinib en el análisis de seguridad integrado, incluido el estudio a largo plazo y un estudio en vitíligo, la tasa de infección graves en ritlecitinib 50 mg o más fue de 0.66 por 100 años-paciente.
Infecciones oportunistas:
Se informaron infecciones oportunistas de herpes zóster cutáneo multidermatómico en 1 paciente (0.50 por 100 años-paciente) tratado con ritlecitinib 200/50 mg en los estudios controlados con placebo y en 2 pacientes (0.1 por 100 años-paciente) tratados con ritlecitinib 50 mg o más en el análisis de seguridad integrado, incluido el estudio a largo plazo y un estudio en vitíligo. Los casos de herpes zóster oportunista fueron de severidad leve o moderada.
Urticaria:
En los estudios controlados con placebo, durante un máximo de 24 semanas, la tasa de urticaria fue de 8.23 por 100 años-paciente en pacientes tratados con ritlecitinib 50 mg y de 4.03 por 100 años-paciente en pacientes tratados con placebo. Entre todos los pacientes tratados con ritlecitinib 50 mg o más en el análisis de seguridad integrado, la tasa de urticaria fue de 4.10 por 100 años-paciente. El tiempo mediano hasta la aparición de un evento inicial fue de 8 semanas; la duración mediana de la urticaria fue de 7 días. La mayoría de los casos fueron de severidad leve a moderada.
Población pediátrica:
Se inscribió a un total de 181 adolescentes (12 a < 18 años) en los estudios clínicos de alopecia areata del ritlecitinib. En los estudios clínicos de alopecia areata, el perfil de seguridad observado en adolescentes fue similar al de la población adulta.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Genotoxicidad:
El ritlecitinib no fue mutagénico en el ensayo de mutagenicidad bacteriana (ensayo de Ames). El ritlecitinib no es aneugénico ni clastogénico en exposiciones iguales a 130 veces la dosis humana máxima recomendada (MRHD por sus siglas en inglés) sobre una base del ABC no unido, con base en los resultados del ensayo in vivo de micronúcleos de médula ósea de ratas.
Carcinogénesis:
No se observó evidencia de tumorigenicidad en los ratones Tg.rasH2 de 6 meses a los que se les administró ritlecitinib a exposiciones iguales a 11 veces la MRHD sobre una base de ABC no unido. En un estudio de carcinogenicidad de 2 años en ratas, se observó una mayor incidencia de timomas benignos en ratas hembra y adenomas foliculares benignos de tiroides en ratas macho después de la administración de ritlecitinib en exposiciones iguales a 29 veces la MRHD sobre una base de ABC no unido. No se observaron timomas ni adenomas foliculares tiroideos relacionados con el ritlecitinib en exposiciones iguales a 6.3 veces la MRHD sobre una base de ABC no unido.
Toxicidad reproductiva y del desarrollo:
El ritlecitinib no tuvo efectos sobre la fertilidad de las ratas hembra a exposiciones iguales a 55 veces la MRHD sobre una base de ABC no unido. Se observaron efectos sobre la fertilidad de las ratas macho (mayor pérdida previa a la implantación que dio como resultado una menor cantidad de sitios de implantación y el tamaño de la camada más bajo correspondiente en hembras no tratadas previamente apareadas con machos a los que se les administró ritlecitinib) a una exposición igual a 55 veces la MRHD sobre una base de ABC no unido. No se observaron efectos sobre la fertilidad masculina en exposiciones iguales a 14 veces la MRHD sobre una base de ABC no unido. No se observaron efectos sobre la espermatogénesis (recuentos de esperma, tasa de producción de esperma, motilidad y morfología) en ninguna dosis.
En un estudio de desarrollo embriofetal en ratas preñadas, la administración oral de ritlecitinib desde los días de gestación 6 a 17 dio como resultado malformaciones y variaciones esqueléticas fetales y pesos corporales fetales más bajos en exposiciones mayores o iguales a 49 veces el ABC no unido en la MRHD. No hubo efectos sobre el desarrollo embriofetal en exposiciones iguales a 16 veces el ABC no unido en la MRHD.
En un estudio de desarrollo embriofetal en conejas preñadas, la administración oral de ritlecitinib desde los días de gestación 7 a 19 dio como resultado pesos corporales fetales medios más bajos e incidencias más altas de malformaciones viscerales, malformaciones esqueléticas y variaciones esqueléticas en exposiciones iguales a 55 veces el ABC no unido en la MRHD. No hubo efectos sobre el desarrollo embriofetal en exposiciones iguales a 12 veces el ABC no unido en la MRHD.
En un estudio de desarrollo pre y posnatal en ratas, la administración oral de ritlecitinib desde el día 6 de gestación hasta el día 20 de lactancia dio como resultado una toxicidad del desarrollo que incluyó una supervivencia posnatal más baja, pesos corporales de las crías más bajos y retrasos secundarios del desarrollo a una exposición equivalente a 41 veces el ABC no unido en la MRHD. Las hembras criadas en la generación F1 exhibieron cantidades medias más bajas de cuerpos lúteos en exposiciones iguales a 41 veces el ABC no unido en la MRHD. No hubo efectos sobre el desarrollo pre y posnatal en exposiciones iguales a 14 veces el ABC no unido en la MRHD.
Fertilidad:
No hubo efectos sobre la fertilidad de ratas macho o hembra en exposiciones clínicamente relevantes.
Lactancia:
Después de la administración de ritlecitinib a ratas en periodo de lactancia, las concentraciones de ritlecitinib en la leche a lo largo del tiempo fueron más altas que las concentraciones en plasma, donde se determinó que la relación media entre la leche y el ABC plasmático era de 2.2.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Efectos de medicamentos coadministrados sobre la farmacocinética del ritlecitinib:
El metabolismo del ritlecitinib está mediado por varias isoformas de Glutatión S-transferasa (GST: GST citosólico A1/3, M1/3/5, P1, S1, T2, Z1 y proteínas microsómicas asociadas con la membrana involucradas en el metabolismo eicosanoide y glutatión [MAPEG]1/2/3) y enzimas CYP (CYP3A, CYP2C8, CYP1A2 y CYP2C9), sin una vía de depuración única que contribuya a más del 25%. Por lo tanto, es poco probable que los medicamentos que inhiben una vía metabólica selectiva afecten las exposiciones sistémicas de ritlecitinib. Es poco probable que los inhibidores específicos de los transportadores produzcan cambios clínicamente relevantes en la biodisponibilidad de ritlecitinib (ver sección Farmacocinética y farmacodinamia).
La coadministración de dosis múltiples de 200 mg de itraconazol, un inhibidor potente de CYP3A, incrementó el ABCinf del ritlecitinib en aproximadamente un 15%. Esto no se considera clínicamente significativo y, por lo tanto, no se requiere ajustar la dosis cuando se coadministra con inhibidores de CYP3A.
La coadministración de dosis múltiples de 600 mg de rifampicina, un inductor potente de las enzimas CYP, disminuyó el ABCinf del ritlecitinib en aproximadamente un 44%. Esto no se considera clínicamente significativo y, por lo tanto, no se requiere ajustar la dosis cuando se coadministra con inductores de las enzimas CYP.
Efectos del ritlecitinib sobre la farmacocinética de medicamentos coadministrados:
In vitro:
El ritlecitinib no es un inhibidor significativo del CYP2D6, uridina 5’ difosfo glucuronosiltransferasas (UGT) (UGT1A1, UGT1A4, UGT1A6, UGT1A9 y UGT2B7), GSTs o sulfotransferasas (SULT).
El ritlecitinib no es un inhibidor de la glucoproteína P (P-gp) ni de la bomba exportadora de sales biliares (BSEP por sus siglas en inglés) en concentraciones clínicamente significativas.
In vivo:
El ritlecitinib no produjo cambios clínicamente significativos en las exposiciones de sustratos de CYP2B6 (p. ej., efavirenz), sustratos de CYP2C (p. ej., tolbutamida) o sustratos de transportadores de aniones orgánicos (OAT) P1B1, proteína de resistencia al cáncer de mama (BCRP, por sus siglas en inglés) y OAT3 (p. ej., rosuvastatina). Según las evaluaciones de biomarcadores clínicos, el ritlecitinib no causa una inhibición clínicamente significativa de transportadores como el transportador de cationes orgánicos (OCT) 2 (creatinina sérica y urinaria, N1-metilnicotinamida [NMN]), extrusión de compuestos tóxicos y multimedicamentos (MATE) 1 (NMN), MATE2K (NMN), OAT1 (ácido piridóxico), OATP1B1 (coproporfirina 1 [CP-1]) y OATP1B3 (CP-1).
Las dosis múltiples de 50 mg de ritlecitinib una vez al día no produjeron un cambio clínicamente significativo en las exposiciones de etinilestradiol o levonorgestrel.
Las dosis múltiples de 200 mg de ritlecitinib una vez al día incrementaron el ABCinf y la Cmáx de midazolam en aproximadamente 2.7 veces y 1.8 veces, respectivamente. El ritlecitinib es un inhibidor moderado del CYP3A. Se debe tener precaución con la administración concomitante de ritlecitinib con sustratos del CYP3A, en los cuales pequeños cambios en la concentración pueden provocar reacciones adversas graves, y se deben considerar las recomendaciones de ajuste de la dosis para el sustrato de CYP3A de acuerdo con la información para prescribir aprobada del producto.
Las dosis múltiples de 200 mg de ritlecitinib una vez al día incrementaron el ABCinf y la Cmáx de la cafeína aproximadamente 2.7 veces y 1.1 veces, respectivamente. Estos incrementos en la exposición a cafeína no se consideran clínicamente relevantes. El ritlecitinib es un inhibidor moderado del CYP1A2. Se debe tener precaución con la administración concomitante de ritlecitinib con otros sustratos de CYP1A2, en los cuales pequeños cambios en la concentración pueden provocar reacciones adversas graves, y se deben considerar las recomendaciones de ajuste de dosis para el sustrato de CYP1A2 de acuerdo con la información para prescribir aprobada del producto.
La coadministración de una dosis única de 400 mg de ritlecitinib incrementó el ABCinf de sumatriptán (un sustrato de OCT1) aproximadamente 1.3 a 1.5 veces en relación con la dosis de sumatriptán administrada sola. Este incremento en la exposición de sumatriptán no se considera clínicamente relevante. Sin embargo, se debe tener precaución con la administración concomitante de ritlecitinib con sustratos de OCT1, en los cuales pequeños cambios en la concentración pueden provocar reacciones adversas graves.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Disminución del recuento de linfocitos:
Entre todos los pacientes tratados con ritlecitinib en el análisis de seguridad integrado, incluido el estudio a largo plazo y un estudio en vitíligo, ocurrió un RAL confirmado de < 500/mm3 en 1 participante (< 0.1%) tratado con ritlecitinib 50 mg. La edad pareció ser un factor de riesgo para un RAL más bajo en participantes ≥ 65 años.
Disminución del recuento de plaquetas:
En los estudios controlados con placebo, durante un máximo de 24 semanas, el tratamiento con ritlecitinib se asoció a una reducción en el recuento de plaquetas. Los efectos máximos sobre las plaquetas se observaron en el plazo de 4 semanas, después de lo cual el recuento de plaquetas permaneció estable en un nivel más bajo con el tratamiento continuado. Entre todos los pacientes tratados con ritlecitinib en el análisis de seguridad integrado, incluido el estudio a largo plazo y un estudio en vitíligo, 1 paciente (< 0.1%) tratado con ritlecitinib 50 mg o más tuvo un recuento confirmado de plaquetas < 100,000/mm3. Ningún paciente registró un recuento de plaquetas confirmado de < 75,000/mm3.
Aumentos de la creatinina fosfocinasa (CPK, por sus siglas en inglés)
En los estudios controlados con placebo, durante un máximo de 24 semanas, se informaron eventos de incremento de la CPK en sangre en 0 pacientes tratados con placebo y en 2 pacientes (el 1.5%) tratados con ritlecitinib 50 mg. No hubo informes de miopatía o rabdomiólisis.
PRECAUCIONES GENERALES:
Infecciones graves:
Se informaron infecciones graves en pacientes que recibieron el ritlecitinib. Las infecciones graves más frecuentes han sido apendicitis, infección por COVID-19 (incluida la neumonía) y sepsis. El tratamiento con ritlecitinib se debe evitar en pacientes con una infección grave activa.
Se deben considerar los riesgos y los beneficios del tratamiento en los pacientes que presentan lo siguiente:
• infecciones crónicas o recurrentes;
• que se hayan expuesto a la tuberculosis;
• antecedentes de infecciones graves u oportunistas;
• que residan en, o hayan viajado a, áreas con tuberculosis o micosis endémicas; o
• que tengan afecciones subyacentes que puedan predisponerlos a una infección.
Se debe monitorear de cerca a los pacientes a fin de determinar el desarrollo de signos y síntomas de infección durante y después del tratamiento con ritlecitinib. El tratamiento se debe interrumpir si un paciente desarrolla una infección grave u oportunista. Un paciente que presenta una nueva infección durante el tratamiento con ritlecitinib se debe someter a una evaluación diagnóstica oportuna y completa apropiada para un paciente inmunocomprometido, se debe iniciar el tratamiento antimicrobiano adecuado y se debe monitorear de cerca al paciente. El médico debe evaluar si la interrupción del tratamiento para la alopecia areata es el mejor curso de acción para el paciente en particular. Si se interrumpe el tratamiento, se puede reanudar el tratamiento con ritlecitinib una vez que se controle la infección.
Tuberculosis:
Se debe someter a los pacientes a una prueba de detección de tuberculosis (TB) antes de iniciar el tratamiento. No se debe administrar ritlecitinib en pacientes con TB activa. Se debe iniciar la terapia anti-TB antes de iniciar el tratamiento con ritlecitinib en pacientes con un nuevo diagnóstico de TB latente o TB latente no tratada anteriormente. En pacientes con una prueba de TB latente negativa, considere la terapia anti-TB antes de iniciar el tratamiento con ritlecitinib en aquellos con alto riesgo y considere la detección de pacientes con alto riesgo de TB durante el tratamiento con ritlecitinib.
Reactivación viral:
En estudios clínicos, se ha observado reactivación viral, incluidos casos de reactivación del virus del herpes (p. ej., herpes zóster) (ver sección Reacciones secundarias y adversas). Si un paciente presenta herpes zóster, se debe considerar la interrupción temporal del tratamiento hasta que el episodio se resuelva.
La detección del hepatitis viral se debe realizar según los lineamientos clínicos antes de comenzar el tratamiento con ritlecitinib. Se excluyó de los estudios clínicos a los pacientes con evidencia de infección por VIH o infección por hepatitis B o C.
Neoplasia maligna (incluido el cáncer de piel no melanoma):
Se observaron neoplasias malignas, incluido el cáncer de piel no melanoma (CPNM), en estudios clínicos con ritlecitinib. Se deben considerar los riesgos y beneficios del tratamiento con ritlecitinib antes de iniciar o continuar el tratamiento en pacientes con una neoplasia maligna conocida distinta a un CPNM o cáncer de cuello uterino tratados satisfactoriamente.
Se recomiendan exámenes periódicos de piel en los pacientes con mayor riesgo de sufrir cáncer de piel.
Eventos tromboembólicos:
Se han informado eventos de tromboembolismo venoso y arterial en pacientes que reciben ritlecitinib. Se deben considerar los riesgos y beneficios antes de iniciar el tratamiento con ritlecitinib en pacientes con alto riesgo de sufrir eventos tromboembólicos.
Hipersensibilidad:
En los estudios clínicos, se han observado reacciones tales como urticaria, erupción y reacción anafiláctica que pueden reflejar hipersensibilidad al medicamento, incluidas reacciones graves, en pacientes que reciben ritlecitinib. Si se produce una reacción de hipersensibilidad clínicamente significativa, interrumpa el tratamiento con ritlecitinib e inicie un tratamiento adecuado (ver sección Reacciones secundarias y adversas).
Anomalías hematológicas:
El tratamiento con ritlecitinib se asoció con disminuciones en linfocitos y plaquetas (ver sección Alteraciones en los resultados de pruebas de laboratorio). Antes de iniciar el tratamiento con ritlecitinib, se deben realizar los conteos de recuento absoluto de linfocitos (RAL) y plaquetas. No se debe iniciar el tratamiento con ritlecitinib en pacientes con un RAL < 500/mm3 o con un recuento de plaquetas < 100,000/mm3. Después de iniciar el tratamiento con ritlecitinib, se recomienda la interrupción o descontinuación del tratamiento en función del RAL y anormalidades en el recuento de plaquetas (ver sección Dosis y vía de Administración). Los recuentos de RAL y plaquetas se recomiendan a las 4 semanas del inicio del tratamiento con ritlecitinib y, a partir de entonces de acuerdo con el manejo terapéutico de rutina del paciente.
Vacunación:
No se dispone de datos sobre la respuesta a la vacunación en pacientes que reciben ritlecitinib. Se debe evitar la administración de vacunas de microbios vivos atenuados durante el tratamiento o inmediatamente después de iniciarlo. Antes de iniciar el tratamiento con ritlecitinib, se recomienda que todos los pacientes estén al día con todas las inmunizaciones, incluida la vacunación preventiva contra el herpes zóster, de acuerdo con los lineamientos de inmunización actuales.
Efectos sobre la capacidad para conducir y operar máquinas:
Ritlecitinib no tiene influencia conocida sobre la capacidad para conducir y operar máquinas.
Dado que existe una mayor incidencia de infecciones en personas mayores y en la población diabética en general se debe tener precaución.
Eventos neurológicos: Se ha observado distrofia axonal relacionada con ritlecitinib en estudios de el tratamiento en caso de complicaciones neurológicas inexplicables,
Lactosa: Pacientes con problemas hereditarios de intolerancia a la galactosa, deficiencia total de lactasa o las personas con problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología:
La dosis recomendada de ritlecitinib es de 50 mg una vez al día.
Se debe considerar la interrupción del tratamiento en pacientes que no han mostrado respuesta. Algunos pacientes con respuesta parcial inicial pueden mejorar posteriormente con el tratamiento continuo.
Monitoreo de laboratorio:
Tabla 4. Guía de monitoreo y mediciones de laboratorio
|
Medición de |
Guía de monitoreo |
Acción |
|
Recuento de plaquetas |
Antes de iniciar el tratamiento, 4 semanas después del inicio y posteriormente de acuerdo con el manejo terapéutico de rutina del paciente |
Se debe interrumpir el tratamiento si el recuento de plaquetas es < 50,000/mm3. |
|
Linfocitos |
El tratamiento se debe interrumpir si el RAL es < 500/mm3 y se puede reiniciar una vez que el RAL vuelva a estar por encima de este valor. |
Abreviatura: RAL = recuento absoluto de linfocitos.
Inicio del tratamiento:
No se debe iniciar el tratamiento con ritlecitinib en pacientes con un recuento absoluto de linfocitos (RAL) < 500/mm3 o un recuento de plaquetas < 100,000/mm3 (ver sección Precauciones generales).
Interrupción o suspensión del tratamiento:
Si un paciente desarrolla una infección oportunista o una infección grave, se debe interrumpir el tratamiento con ritlecitinib hasta que la infección esté controlada (ver sección Precauciones generales).
Las recomendaciones para la interrupción o suspensión del tratamiento con ritlecitinib por anormalidades hematológicas se resumen en la tabla 4.
Si se debe interrumpir el tratamiento, no se espera que una interrupción temporal del tratamiento por menos de 6 semanas dé como resultado una pérdida significativa del cabello que ha vuelto a crecer en el cuero cabelludo.
Dosis omitidas:
Si se olvida una dosis, se debe advertir a los pacientes que tomen la dosis lo antes posible, a menos que falten menos de 8 horas antes de la siguiente dosis, en cuyo caso el paciente no debe tomar la dosis olvidada. Luego, debe reanudarse la dosificación en el horario regular programado.
Poblaciones especiales:
Insuficiencia renal:
No es necesario ajustar la dosis para los pacientes con insuficiencia renal leve, moderada o grave (ver sección Farmacocinética y farmacodinamia).
No se ha estudiado la administración del ritlecitinib en pacientes con enfermedad renal en estado terminal (ESRD por sus siglas en inglés) o en pacientes con trasplantes renales.
Insuficiencia hepática:
No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve (Child Pugh A) o moderada (Child Pugh B). Ritlecitinib no se recomienda en pacientes con insuficiencia hepática grave (Child-Pugh C), (ver sección Farmacocinética y farmacodinamia).
Población de edad avanzada:
No es necesario ajustar la dosis en pacientes ≥ 65 años (ver sección Farmacocinética y farmacodinamia).
Población pediátrica:
No es necesario ajustar la dosis en pacientes de 12 a < 18 años (ver sección Farmacocinética y farmacodinamia).
No se han establecido la seguridad ni la eficacia de ritlecitinib en pacientes pediátricos menores de 12 años. No existen datos disponibles.
Método de administración:
Ritlecitinib se debe administrar por vía oral una vez al día, con o sin alimentos. Las cápsulas se deben tragar enteras y no se deben triturar, dividir ni masticar.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
El ritlecitinib se administró en estudios clínicos hasta en una dosis oral única de 800 mg. Las reacciones adversas fueron comparables con aquellas observadas en dosis más bajas y no se identificaron toxicidades específicas. Según los datos farmacocinéticos (PK) de una dosis oral única de hasta e incluyendo 800 mg en voluntarios adultos sanos, se espera que más del 90% de la dosis administrada se elimine en un plazo de 48 horas.
No existe un antídoto específico para la sobredosis con ritlecitinib. El tratamiento debe ser sintomático y de apoyo. En caso de sobredosis, se recomienda monitorear al paciente para determinar signos y síntomas de reacciones adversas (ver sección Reacciones secundarias y adversas).
PRESENTACIONES:
Caja de cartón con 30 cápsulas de 50 mg cada una en envase de burbuja e instructivo anexo.
Frasco con 28 cápsulas de 50 mg cada una y un desecante e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Almacene a no más de 25 °C.
Mantener en lugar fresco y seco.
LEYENDAS DE PROTECCIÓN:
Mantenga fuera del alcance de los niños. Bibliografía exclusiva para profesionales de la salud. No se use durante el embarazo ni la lactancia. No administrar a menores de 12 años. Este medicamento contiene lactosa, que puede producir reacciones de hipersensibilidad.
Reporte las sospechas de reacción adversa a los correos:
farmacovigilancia@cofepris.gob.mx y
MEX.AEReporting@pfizer.com y
a la línea Pfizer 800 401 2002.
PFIZER, S.A. de C.V.
Km. 63 Carretera México, Toluca,
Zona Industrial, C.P. 50140, Toluca, México, México.
Reg. Núm. 457M2023 SSA IV


