

NAXZALLA
EZETIMIBA, ROSUVASTATINA
Tabletas
1 Caja, 30 Tabletas, 10/5 mg/mg
1 Caja, 30 Tabletas, 10/10 mg/mg
1 Caja, 30 Tabletas, 10/20 mg/mg
III. FORMA FARMACÉUTICA Y FORMULACIÓN
Cada TABLETA contiene:
Ezetimiba 10 mg
Rosuvastatina cálcica equivalente a 5 mg, 10 mg, 20 mg de Rosuvastatina
Excipiente cbp 1 tableta
Ingredientes Inactivos (Lista de excipientes)
Cada tableta de NAXZALLA contiene los siguientes ingredientes inactivos: lactosa monohidratada, lauril sulfato de sodio, croscarmelosa sódica, povidona, celulosa microcristalina, crospovidona, estearato de magnesio, y manitol.
La cubierta contiene: hipromelosa, polietilenglicol 6000/macrogol 8000, talco, dióxido de titanio, óxido de hierro amarillo, y óxido de hierro rojo.
XIX. NÚMERO DE REGISTRO DEL MEDICAMENTO ANTE LA SECRETARÍA
Reg. No. 089M2020
Versión: SCCDS-MK0653H-T-122018
Tracer Number: MK0653H-MEX-2019-019806
RNC: 000002007-MEX
IV. INDICACIONES TERAPÉUTICAS
Hipercolesterolemia Primaria: NAXZALLA está indicado como tratamiento complementario a la dieta para la reducción de niveles elevados de colesterol total, colesterol de lipoproteínas de baja densidad (C-LDL), apolipoproteína B, triglicéridos, y colesterol no-HDL, y para incrementar el colesterol de lipoproteínas de alta densidad (C-HDL), en pacientes con hipercolesterolemia primaria (heterocigótica familiar y no familiar) o hiperlipidemia mixta.
Hipercolesterolemia Familiar Homocigótica: NAXZALLA está indicado para la reducción de niveles elevados de colesterol total y C-LDL en pacientes con Hipercolesterolemia Familiar Homocigótica. Los pacientes también pueden recibir tratamientos complementarios (p. ej., aféresis de LDL).
V. FARMACOCINÉTICA Y FARMACODINAMIA
CLASE TERAPÉUTICA
NAXZALLA (ezetimiba/rosuvastatina) es un producto hipolipemiante que inhibe selectivamente la absorción intestinal de colesterol y esteroles vegetales relacionados e inhibe la síntesis endógena de colesterol.
MECANISMO DE ACCIÓN
NAXZALLA: El colesterol plasmático resulta de la absorción intestinal y de la síntesis endógena. NAXZALLA contiene ezetimiba y rosuvastatina, dos compuestos hipolipemiantes con mecanismos de acción complementarios. NAXZALLA reduce los niveles elevados de colesterol total (total-C), colesterol de lipoproteínas de baja densidad (C-LDL), apolipoproteína B (Apo B), triglicéridos (TG), y colesterol no-HDL (No-C-HDL), e incrementa el colesterol de lipoproteínas de alta densidad (C-HDL) a través de la inhibición dual de la absorción y de la síntesis de colesterol.
Ezetimiba
Ezetimiba es activo y potente por vía oral, con un particular mecanismo de acción que difiere del de otras clases de compuestos reductores del colesterol (como las estatinas, los secuestradores de ácidos biliares [resinas], los fibratos y los estanoles vegetales). El objetivo molecular de la ezetimiba es el transportador de esterol, Niemann-PickC1-Like 1 (NPC1L1), el cual es responsable de la captación intestinal del colesterol y fitoesteroles.
La ezetimiba se fija al borde en cepillo de las células del intestino delgado e inhibe la absorción del colesterol, por lo que disminuye la llegada de colesterol intestinal al hígado. Como consecuencia, disminuye la reserva hepática de colesterol y aumenta la depuración de colesterol de la sangre. La ezetimiba no aumenta la excreción de ácidos biliares (como los secuestradores de ácidos biliares) ni tampoco inhibe la síntesis de colesterol en el hígado (como las estatinas).
En un estudio clínico de dos semanas en 18 pacientes hipercolesterolémicos, ezetimiba inhibió 54% la absorción intestinal de colesterol en comparación con un placebo. Al inhibir la absorción intestinal de colesterol, la ezetimiba disminuye la llegada de colesterol al hígado. Las estatinas disminuyen la síntesis hepática de colesterol, y juntos, esos dos mecanismos distintos proporcionan una disminución complementaria del colesterol. Administrado con una estatina, ezetimiba disminuye el colesterol total, el C- LDL, la Apo B, colesterol no-HDL y los triglicéridos y aumenta el C-HDL en los pacientes con hipercolesterolemia más que los mismos medicamentos solos en monoterapia. La administración de ezetimiba con fenofibrato es eficaz para mejorar el colesterol total sérico, C-LDL, Apo B, TG, C-HDL y colesterol no-HDL en pacientes con hiperlipidemia mixta.
Los estudios clínicos demuestran que las concentraciones elevadas de colesterol total, C-LDL y Apo B (la principal proteína de las LDL) favorecen la aterosclerosis humana. Además, las concentraciones disminuidas de C-HDL también se asocian con el desarrollo de aterosclerosis. Los estudios epidemiológicos han revelado que la morbilidad y la mortalidad cardiovasculares varían en proporción directa con las concentraciones de colesterol total y de C-LDL y en proporción inversa con las de C-HDL. Como las LDL, las lipoproteínas ricas en colesterol y triglicéridos, como las de muy baja densidad (VLDL), las de densidad intermedia (IDL) y sus residuos, también pueden favorecer el desarrollo de aterosclerosis.
Se hizo una serie de estudios preclínicos para determinar la selectividad de la ezetimiba para inhibir la absorción de colesterol. Ezetimiba inhibió la absorción del colesterol marcado con 14C, sin ningún efecto sobre la absorción de triglicéridos, ácidos grasos, ácidos biliares, progesterona, etinilestradiol, o las vitaminas liposolubles A y D.
Rosuvastatina
La rosuvastatina es un poderoso inhibidor selectivo y competitivo de la HMG-CoA reductasa, la enzima limitante para la conversión de la coenzima A 3-hidroxi-3-metilglutaril en mevalonato, un precursor de colesterol. Los triglicéridos (TG) y el colesterol en hígado se incorporan con apolipoproteína B (ApoB), formando una lipoproteína de muy baja densidad (VLDL) que se libera en plasma para su distribución a tejidos periféricos. Las partículas de VLDL son ricas en TG. La lipoproteína de baja densidad (LDL) rica en colesterol se forma de VLDL y es depurada principalmente a través del receptor LDL de alta afinidad en el hígado.
Rosuvastatina produce sus efectos modificadores de lípidos en dos formas: aumentando el número de receptores LDL hepáticos en la superficie celular con lo que aumenta la captación y el catabolismo de LDL e inhibiendo la síntesis hepática de VLDL, con lo cual se reduce el número total de partículas de VLDL y LDL.
La lipoproteína de alta densidad (HDL), la cual contiene ApoA-1, está involucrada, entre otras cosas, en el transporte de colesterol de los tejidos regresándolos al hígado (transporte inverso de colesterol). La participación del C-LDL en aterogénesis está bien documentada. Estudios epidemiológicos han establecido que el C-LDL y los TG altos, así como el C-HDL y ApoA-1 bajos se han asociado a un mayor riesgo de enfermedad cardiovascular. Estudios de intervención han demostrado los efectos benéficos de disminuir el índice de mortalidad y de eventos cardiovasculares (CV) al reducir el C-LDL y los TG o al aumentar el C-HDL. Datos más recientes han asociado los beneficios de los inhibidores de HMG-CoA reductasa sobre la disminución de colesterol no HDL (es decir, todo el colesterol circulante no HDL), y ApoB o la reducción en el índice ApoB/ApoA-1.
FARMACOCINÉTICA
Introducción General
NAXZALLA
NAXZALLA ha demostrado ser bioequivalente a la co-administración de las dosis correspondientes de tabletas de ezetimiba y rosuvastatina.
Absorción
Ezetimiba
Tras su administración por vía oral, la ezetimiba es absorbida rápidamente y transformada extensamente por conjugación en un glucurónido fenólico con actividad farmacológica (glucurónido de ezetimiba). El glucurónido de ezetimiba alcanza concentraciones plasmáticas medias máximas (Cmáx) en una a dos horas, y la ezetimiba en cuatro a doce horas. No se puede determinar la biodisponibilidad absoluta de la ezetimiba, por ser ésta prácticamente insoluble en medios acuosos apropiados para ser inyectados.
La administración concomitante de alimentos (altos en grasas o sin grasas) no tuvo ningún efecto sobre la biodisponibilidad de la ezetimiba administrada por vía oral en forma de comprimidos de ezetimiba con 10 mg. ezetimiba se puede administrar con o sin alimentos.
Rosuvastatina
En estudios farmacológicos en humanos, los niveles plasmáticos máximos se observaron a las 5 horas después de la dosificación por vía oral.
La exposición aumenta linealmente en proporción aproximada a la dosis. La biodisponibilidad absoluta es de 20%. Existe una mínima acumulación con la administración una vez al día.
Distribución
Ezetimiba
Se unen a las proteínas plasmáticas humanas 99.7% de la ezetimiba y 88 a 92% del glucurónido de ezetimiba.
Rosuvastatina
Rosuvastatina se une aproximadamente 90% a las proteínas plasmáticas, principalmente a la albúmina, esta unión es reversible e independiente de las concentraciones plasmáticas. El compuesto original representa más del 90% de la actividad inhibidora de HMG CoA reductasa circulante.
Metabolismo
Ezetimiba
La ezetimiba es metabolizada principalmente en el intestino delgado y en el hígado por conjugación con el ácido glucurónico (una reacción de fase II), y después es excretada con la bilis. En todas las especies estudiadas se ha observado un metabolismo oxidativo mínimo (una reacción de fase I). La ezetimiba y el glucurónido de ezetimiba son las principales formas del medicamento que se detectan en el plasma; constituyen 10 a 20% y 80 a 90%, respectivamente, del total en el plasma. Ambas formas son eliminadas lentamente del plasma, con indicios de un reciclamiento enterohepático significativo. La vida media de la ezetimiba y de su glucurónido es de 22 horas aproximadamente.
Rosuvastatina
Rosuvastatina tiene un efecto de primer paso en el hígado, el cual es el sitio primario de la síntesis de colesterol y depuración del C-LDL. Rosuvastatina sufre un metabolismo muy limitado (aproximadamente 10%), principalmente a la forma N-desmetilada.
Eliminación
Ezetimiba
Tras la administración oral de 20 mg de 14C-ezetimiba a personas, la ezetimiba total representó aproximadamente 93% de la radiactividad total en el plasma. En un periodo de diez días se recuperó aproximadamente 78% de la radiactividad en las heces y 11% en la orina. A las 48 horas, no hubo radiactividad detectable en el plasma.
Rosuvastatina
El metabolito N-desmetilado de la rosuvastatina es eliminado principalmente como medicamento inalterado en heces y el resto excretado en la orina. La vida media es de 19 horas y no aumenta con una dosis mayor.
Poblaciones Especiales
Insuficiencia Renal
Ezetimiba
En ocho pacientes con enfermedad renal grave (media de depuración de creatinina ≤ 30 mL/min/1.73 m2), después de una sola dosis de 10 mg de ezetimiba la media del ABC de ezetimiba total fue aproximadamente 1.5 veces mayor que en nueve sujetos sanos. Este resultado no se consideró clínicamente importante. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
En otro paciente de ese estudio (con trasplante renal y recibiendo varios medicamentos, incluyendo ciclosporina), la exposición a la ezetimiba total fue 12 veces mayor.
Rosuvastatina
Un estudio en sujetos con distintos grados de insuficiencia renal, enfermedad renal leve a moderada, demostró tener poca influencia en las concentraciones plasmáticas de rosuvastatina. Sin embargo, los sujetos con daño severo (CrCl <30 mL/min) tuvieron un aumento de 3 veces en la concentración plasmática, en comparación con voluntarios sanos. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Insuficiencia Hepática
Ezetimiba
Después de una sola dosis de 10 mg de ezetimiba, la media del área bajo la curva (ABC) de ezetimiba total fue aproximadamente 1.7 veces mayor en los pacientes con insuficiencia hepática leve (puntuación de Child- Pugh de 5 o 6) que en los sujetos sanos. En un estudio con dosis múltiples (10 mg diarios) durante 14 días en pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh de 7 a 9), la media del ABC de ezetimiba total fue aproximadamente cuatro veces mayor los Días 1 y 14 que en los sujetos sanos. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Rosuvastatina: En un estudio en sujetos con grados variables de insuficiencia hepática, no hubo evidencia de tener mayor exposición a la rosuvastatina a excepción de 2 sujetos con enfermedad hepática severa (Puntajes de Child-Pugh de 8 y 9). En estos sujetos, la exposición sistémica fue al menos 2 veces más alta en comparación con sujetos con menores puntajes de Child-Pugh. Ver VI. Contraindicaciones, VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Población Pediátrica
Ezetimiba
La absorción y el metabolismo de la ezetimiba son similares en los niños, los adolescentes (10 a 18 años) y los adultos. Basándose en la ezetimiba total, no hay diferencias farmacocinéticas entre los adolescentes y los adultos. No hay datos farmacocinéticos disponibles en niños menores de diez años. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Rosuvastatina
No hubo diferencias en las concentraciones plasmáticas de rosuvastatina por edad. La farmacocinética de rosuvastatina en niños y adolescentes con hipercolesterolemia familiar heterocigótica fue similar a la de voluntarios adultos. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Población Geriátrica
Ezetimiba
Las concentraciones plasmáticas de ezetimiba total son aproximadamente el doble en los pacientes de edad avanzada (de 65 años o mayores) que en pacientes más jóvenes (de 18 a 45 años). La disminución del C-LDL y el perfil de seguridad son similares entre sujetos de edad avanzada y más jóvenes tratados con ezetimiba. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Rosuvastatina
No hubo diferencias en las concentraciones plasmáticas de rosuvastatina por edad. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Raza
Ezetimiba
Basándose en un meta-análisis de estudios farmacocinéticos, no hubo diferencias farmacocinéticas entre las personas de raza negra y las de raza blanca. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Rosuvastatina
Estudios farmacocinéticos muestran una media de elevación del Área Bajo la Curva (ABC) aproximadamente del doble en sujetos asiáticos en comparación con sujetos caucásicos. Un análisis farmacocinético entre grupos caucásicos, hispanos y de raza negra o afro-caribeños, no reveló diferencias clínicamente importantes. Ver VII. Precauciones Generales, y XIII. Dosis y Vía de Administración.
Sexo
Ezetimiba
Las concentraciones plasmáticas de ezetimiba total son ligeramente mayores (menos de 20%) en las mujeres que en los hombres. La disminución del C-LDL y el perfil de seguridad son similares en los hombres y en las mujeres tratados con ezetimiba.
Rosuvastatina
No hubo diferencias en las concentraciones plasmáticas de rosuvastatina por sexo.
Polimorfismos Genéticos
Rosuvastatina
La disposición de la HMG-CoA reductasa, incluyendo rosuvastatina, involucra al Polipéptido Transportador de Aniones Orgánicos 1B1 (OATP1B1 por sus siglas en inglés) y Proteínas Resistentes al Cáncer de Mama (BCRP por sus siglas en inglés). En los pacientes con polimorfismos genéticos de Sustancias Disolutas de la Familia de Transportadores de Aniones 1B1 [SLCO1B1 por sus siglas en inglés (OATP1B1)] y/o ABCG2 (BCRP) tienen un mayor riesgo de exposición a rosuvastatina. Polimorfismos individuales de SLCO1B10 c.521CC y ABCG2 c.421AA están asociados con aproximadamente 1.6 veces más a la exposición a rosuvastatina (ABC) ó 2.4 veces la exposición más alta, respectivamente, en comparación con los genotipos SLCO1B1 c.521TT o ABCG2 c.421CC.
ESTUDIOS CLÍNICOS
En estudios clínicos controlados, NAXZALLA (como la co-administración de ezetimiba y rosuvastatina) redujo significativamente los niveles de colesterol total (total-C), colesterol de lipoproteínas de baja densidad (C- LDL), apolipoproteína B (Apo B), triglicéridos (TG), y colesterol no-HDL, e incrementó el colesterol de lipoproteínas de alta densidad (C-HDL) en pacientes con hipercolesterolemia.
Hipercolesterolemia Primaria
NAXZALLA
Adición de Ezetimiba a Tratamiento en Curso con Rosuvastatina (Estudios de Titulación).
En un estudio multicéntrico, aleatorizado, doble ciego, de 6 semanas, con comparador activo (ACTE), 440 sujetos con riesgo moderadamente alto/alto de cardiopatía coronaria con niveles de colesterol LDL que no lograban alcanzar su meta de C-LDL (100 mg/dL [<2.6 mmol/L] o 70 mg/dL [<1.8 mmol/L] dependiendo de las características basales) fueron estratificados a tratamiento con 5 mg o 10 mg de rosuvastatina durante 4-5 semanas. Los pacientes fueron entonces aleatorizados para recibir ya sea el doble de su dosis rosuvastatina (a 10 mg o 20 mg) o la adición de 10 mg de ezetimiba su tratamiento con rosuvastatina (5 o 10 mg), equivalente a NAXZALLA 10/5 o 10/10.
Los pacientes que tomaron dosis de ezetimiba y rosuvastatina equivalentes a NAXZALLA 10/5 o 10/10 alcanzaron reducciones significativamente mayores de C-LDL en comparación a los pacientes en quienes se duplicó su dosis inicial de rosuvastatina (a 10 mg o 20 mg) (p <0.001). La diferencia media en LS (mínimos cuadrados, por sus siglas en inglés, least square) en C-LDL desde el nivel basal hasta el final del estudio fue de -20.96% cuando se añadieron 10 mg de ezetimiba a rosuvastatina y -5.71% cuando la dosis original de rosuvastatina se duplicó (información agrupada a través de los estratos de 5 mg y 10 mg de rosuvastatina). La diferencia debida al tratamiento en la media de LS fue de -15.25% con un intervalo de confianza (CI, por sus siglas en inglés, confidence interval) de 95% (-19.89, -10.60) (Tabla 1).
Tabla 1: Análisis de Cambio Porcentual desde Nivel Basal en Colesterol LDL (mg/dL) en el Punto Final del Estudio Después de 6 Semanas de Tratamiento
|
Tratamiento |
N |
Medición Basal (mg/dL) Media ± SD |
Punto final (mg/dL) Media ± SD |
Media de LS (95% CI) |
Diferencia en Media de LS (95% CI) |
Valor de P |
|
General |
||||||
|
NAXZALLA (10/5 o 10/10 mg) |
219 |
103.9 ± 25.4 |
80.7 ± 32.3 |
-21.0 (-24.3, -17.6) |
-15.3 (-19.9, -10.6) |
<0.001 |
|
Rosuvastatina (10 o 20 mg) |
217 |
100.2 ± 24.4 |
92.9 ± 26.5 |
-5.71 (-9.05, -2.38) |
||
|
Estrato I (Pacientes no en meta de C-LDL con 5 mg de rosuvastatina) |
||||||
|
NAXZALLA 10/5 mg |
98 |
106.7 ± 23.5 |
85.9 ± 31.4 |
-18.0 (-22.69, -13.15) |
-12.3 (-18.9, -5.67) |
<0.001 |
|
Rosuvastatina 10 mg |
96 |
102.4 ± 23.4 |
95.4 ± 23.3 |
-5.61 (-10.4, -0.79) |
||
|
Estrato II (Pacientes no en meta de C-LDL con 10 mg de rosuvastatina) |
||||||
|
NAXZALLA 10/10 mg |
121 |
101.6 ± 26.7 |
76.8 ± 32.6 |
-23.7 (-28.3, -19.1) |
-17.5 (-23.9, -11.0) |
<0.001 |
|
Rosuvastatina 20 mg |
121 |
98.4 ± 25.2 |
90.9 ± 28.8 |
-6.28 (-10.9, -1.69) |
||
Los pacientes que tomaron las dosis de ezetimiba y rosuvastatina equivalentes a NAXZALLA 10/5 o 10/10 redujeron significativamente su colesterol total, non-C-HDL y Apo B, en comparación con la duplicación de su dosis basal de rosuvastatina (p <0.001) y resultó en una proporción significativamente mayor de pacientes que alcanzaron su meta de C-LDL en comparación con duplicar la dosis basal de rosuvastatina (10 mg o 20 mg) (59.4% vs. 30.9%; p <0.001), en donde la meta de C-LDL fue <100mg/dL (<2.6 mmol/L) para pacientes con riesgo moderadamente alto/alto de cardiopatía coronaria (CHD, por sus siglas en inglés, coronary heart disease) sin enfermedad vascular aterosclerótica (AVD, por sus siglas en inglés, atherosclerotic vascular disease) y <70 mg/dL (<1.8 mmol/L) para pacientes con muy alto riesgo de CHD con AVD. Además, hubo una proporción de pacientes significativamente mayor que alcanzó la meta de C-LDL de <70 mg/dL (<1.8 mmol/L), independientemente del estatus de riesgo, en pacientes que recibieron NAXZALLA, en comparación con los que recibieron una duplicación de la dosis basal de rosuvastatina (43.8% vs. 17.5%; p <0.001) (Ver Figura 1).
Figura 1a. Logro de Metas Pre-especificadas de C- LDL Después de 6 Semanas de Tratamiento
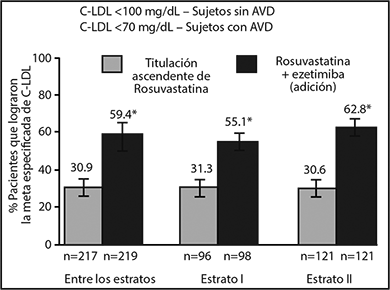
Figura 1b. Logro de C-LDL <70 mg/dL Después de 6 Semanas de Tratamiento
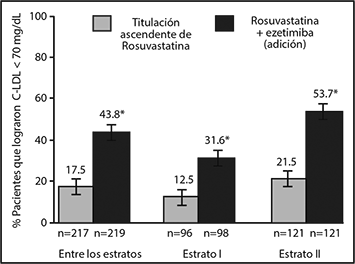
Entre los estratos = todos los pacientes en adición de 10 mg de ezetimiba a rosuvastatina (5 mg, 10 mg) versus todos los pacientes en titulación ascendente de rosuvastatina (10 mg, 20 mg)
Estrato I = adición de 10 mg de ezetimiba a 5 mg de rosuvastatina versus 10 mg de rosuvastatina
Estrato II = adición de 10 mg de ezetimiba a 10 mg de rosuvastatina versus 20 mg de rosuvastatina.
* p<0.001.
Estudio de Eficacia y Seguridad de la Combinación de Dosis Fija Ezetimiba/Rosuvastatina
En un estudio multi-céntrico, de 8 semanas, doble ciego, Fase 3, 412 sujetos coreanos hipercolesterolémicos fueron aleatorizados con una combinación de dosis fija de 10 mg de ezetimiba más rosuvastatina a dosis de 5 mg, 10 mg o 20 mg o monoterapia con rosuvastatina a dosis de 5 mg, 10 mg o 20 mg.
Los pacientes que tomaron la combinación de dosis fija de ezetimiba más rosuvastatina alcanzaron reducciones significativamente mayores de C-LDL en comparación con el conjunto de pacientes tratados con rosuvastatina en monoterapia, pool entre las distintas dosis (p<0.0001), y en comparación con cada dosis (p≤0.01) (Tabla 2).
Tabla 2: Cambio porcentual desde Medición Basal en Colesterol LDL (mg/dL) a la Semana 8 Resultado por ANCOVA del Set de Análisis Completo
|
C-LDL |
Rosuvastatina |
Ezetimiba/Rosuvastatina |
|
Efecto Global |
||
|
Media de LS (Error Standard, ES) |
-49.38 (1.86) |
-59.10 (1.83) |
|
Diferencia de la media de LS (ES) |
-9.73 (1.57) |
|
|
CI 95% para Diferencia de la media de LS |
(-12.82, -6.63) |
|
|
Valor de p |
<0.0001 |
|
|
Rosuvastatina 5 mg |
||
|
Media de LS (Error Standard, ES) |
-42.45 (3.29) |
-55.96 (3.26) |
|
Diferencia de la media de LS (ES) |
-13.52 (2.77) |
|
|
CI 95% para Diferencia de la media de LS |
(-19.00, -8.04) |
|
|
Valor de p |
<0.0001 |
|
|
Rosuvastatina 10 mg |
||
|
Media de LS (Error Standard, ES) |
-51.89 (3.13) |
-58.57 (3.03) |
|
Diferencia de la media de LS (ES) |
-6.67 (2.56) |
|
|
CI 95% para Diferencia de la media de LS |
(-11.75, -1.60) |
|
|
Valor de p |
0.0103 |
|
|
Rosuvastatina 20 mg |
||
|
Media de LS (Error Standard, ES) |
-53.81 (2.95) |
-62.82 (2.91) |
|
Diferencia de la media de LS (ES) |
-9.01 (2.53) |
|
|
CI 95% para Diferencia de la media de LS |
(-14.01, -4.02) |
|
|
Valor de p |
0.0005 |
|
En el pool de todas las dosis, la combinación de dosis fija de ezetimiba más rosuvastatina redujo los niveles de colesterol total, colesterol no-HDL, apolipoproteína B, y triglicéridos más que rosuvastatina en monoterapia.
Además, en el pool de todas las dosis, el tratamiento con dosis fija de ezetimiba/rosuvastatina resultó en una mayor proporción de sujetos que alcanzaron la meta de C-LDL en comparación con rosuvastatina en monoterapia (Tabla 3). Los resultados para comparaciones por cada dosis se muestran en la Tabla 4.
Tabla 3: Proporción de Sujetos que Lograron metas de C-LDL (Set de Análisis Completo) Población General
|
Ezetimiba/Rosuvastatina (n=203) |
Rosuvastatina (n=204) |
|
|
Total de pacientes que lograron meta de C-LDL, n (%) |
191 (94.1)† |
176 (86.3) |
† valor de p <0.05 por la prueba de Cochran–Mantel–Haenszel, con los factores de riesgo de CHD definidos de acuerdo con el National Cholesterol Education Program Adult Treatment Panel III.
Tabla 4: Proportion of Subjects Achieving C-LDL goals (Full Analysis Set) By Treatment Group
|
Ezetimiba/ Rosuvastatina 10/5 mg |
Rosuvastatina 5 mg |
Ezetimiba/ Rosuvastatina 10/10 mg |
Rosuvastatina 10 mg |
Ezetimiba/ Rosuvastatina 10/20 mg |
Rosuvastatina 20 mg |
|
|
Total de pacientes que lograron meta de C-LDL, n (%) |
65 (97.0)† |
50 (73.5) |
62 (91.2) |
63 (94.0) |
64 (94.1) |
63 (91.3) |
† valor de p <0.05 por la prueba de Cochran–Mantel–Haenszel, son los factores de riesgo de CHD definidos de acuerdo con el National Cholesterol Education Program Adult Treatment Panel III.
Ezetimiba
Monoterapia:
En dos estudios multicéntricos, doble ciego, controlados con placebo, de 12 semanas en 1,719 pacientes con hipercolesterolemia primaria, 10 mg de ezetimiba redujeron significativamente total-C (-13%), C-LDL (-19%), Apo B (-14%), TG (-8%), y no-C-HDL (-17%) e incrementaron C-HDL (+3%) en comparación con placebo. La reducción en C-LDL fue consistente entre edad, sexo, raza, y C-LDL basal.
Rosuvastatina
Estudio Controlado con Activo:
Rosuvastatina se comparó con los inhibidores de HMG-CoA reductasa atorvastatina, simvastatina, y pravastatina en un estudio multicéntrico, abierto, de búsqueda de dosis de 2,240 pacientes con hiperlipidemia o dislipidemia mixta. Después de la aleatorización, los pacientes fueron tratados durante 6 semanas con una dosis única diaria de rosuvastatina, atorvastatina, simvastatina, o pravastatina (Figura 2).
Figura 2. Cambio Porcentual en C-LDL por dosis de Rosuvastatina, Atorvastatina, Simvastatina, y Pravastatina a la Semana 6 en Pacientes con Hiperlipidemia o Dislipidemia Mixta
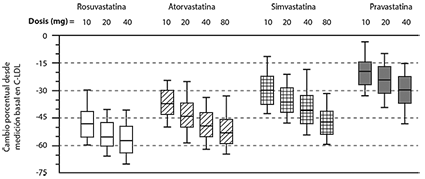
Los diagramas de caja son una representación de los valores de los percentiles 25, 50, y 75, representando los bigotes los valores de los percentiles 10 y 90. C-LDL promedio basal: 189 mg/dL
Hipercolesterolemia Familiar Homocigótica (HoFH)
Ezetimiba
Se realizó un estudio para evaluar la eficacia de ezetimiba en el tratamiento de HoFH (por las siglas en inglés para homozygous familial hypercholesterolemia). Este estudio doble ciego, aleatorizado, de 12 semanas enroló a 50 pacientes con un diagnóstico clínico y/o genotípico de HoFH, con o sin aféresis de LDL concomitante, que ya estaban recibiendo atorvastatina o simvastatina (40 mg). Los pacientes fueron aleatorizados a uno de tres grupos de tratamiento, atorvastatina o simvastatina (80 mg), 10 mg de ezetimiba administrados con atorvastatina o simvastatina (40 mg), o 10 mg de ezetimiba administrados con atorvastatina o simvastatina (80 mg). Ezetimiba, administrada con atorvastatina (40 u 80 mg) o simvastatina (40 u 80 mg), produjo una reducción de C-LDL de 21% desde nivel basal en comparación con incrementar la dosis de la monoterapia con simvastatina o atorvastatina de 40 a 80 mg, lo cual resultó en una reducción de C-LDL de 7% desde nivel basal. En aquellos tratados con ezetimiba más 80mg de atorvastatino o con ezetimiba más 80mg de simvastatina, el C-LDL se redujo en 27%.
Rosuvastatina
En un estudio abierto, de titulación forzada, se evaluó la respuesta de pacientes con HoFH (n=40, 8-63 años) a 20 a 40 mg de rosuvastatina titulada en un intervalo de 6 semanas. En la población general, la reducción promedio desde el nivel basal en C-LDL promedio fue de 22%. Alrededor de un tercio de los pacientes se beneficiaron de incrementar su dosis de 20 mg a 40 mg, con disminución adicional de LDL de más de 6%. En los 27 pacientes con al menos una reducción de 15% en C-LDL, la reducción media en C-LDL fue de 30% (mediana 28% de reducción). Entre 13 pacientes con una reducción de C-LDL <15%, 3 no tuvieron cambio o un incremento en C-LDL. Se observaron reducciones en C-LDL de 15% o mayores en 3 de 5 pacientes con estatus conocido de receptor negativo.
VI. CONTRAINDICACIONES
NAXZALLA está contraindicado en:
• pacientes con hipersensibilidad a ezetimiba, rosuvastatina, o cualquier de los ingredientes inactivos,
• pacientes con enfermedad hepática activa,
• en el embarazo, en periodo de lactancia y en mujeres con potencial de concebir que no utilicen medidas anticonceptivas adecuadas.
VII. PRECAUCIONES GENERALES
Efectos en el Músculo-Esquelético:
Ezetimiba: En estudios clínicos, no hubo exceso de miopatía o rabdomiólisis asociados con ezetimiba comparado con el grupo de control relacionado (placebo o estatina sola). Sin embargo, la miopatía y la rabdomiólisis son reacciones adversas conocidas de las estatinas y otros fármacos hipolipemiantes. En estudios clínicos, la incidencia de aumento de la creatina-fosfocinasa en más de 10 veces el límite superior normal fue de 0.2% con ezetimiba contra 0.1% con placebo, y 0.1% con ezetimiba administrado junto con una estatina contra 0.4% con la estatina sola.
En la experiencia después de la comercialización, se han reportado casos de miopatía y rabdomiólisis independientemente de la causalidad. La mayoría de los pacientes que desarrollaron rabdomiólisis estaban tomando una estatina antes de iniciar con ezetimiba. Sin embargo, la rabdomiólisis ha sido reportada muy raramente con la monoterapia con ezetimiba y muy raramente al agregar ezetimiba a medicamentos con asociación conocida con un mayor riesgo de rabdomiólisis. A todos los pacientes que inicien tratamiento con ezetimiba se les debe notificar del riesgo de miopatía y decirles que reporten lo más pronto posible cualquier dolor muscular, hipersensibilidad o debilidad muscular inexplicables. Ezetimiba, así como cualquier estatina que el paciente esté tomando al mismo tiempo, debe ser inmediatamente descontinuado si se sospecha o diagnostica miopatía. La presencia de estos síntomas y un nivel de creatina-fosfocinasa mayor a 10 veces el límite superior normal indican miopatía.
En un estudio clínico en el que más de 9,000 pacientes con enfermedad renal crónica fueron distribuidos al azar para recibir ezetimiba 10 mg combinado con simvastatina 20 mg al día (n = 4,650) o combinado con placebo (n = 4,620) (mediana de seguimiento 4.9 años), la incidencia de miopatía / rabdomiólisis fue de 0.2% para ezetimiba combinado con simvastatina y 0.1% para placebo. (Ver IX. Reacciones Secundarias y Adversas).
Rosuvastatina
Como con otros inhibidores de la HMG-CoA reductasa, se han reportado efectos en el músculo esquelético, por ejemplo, mialgia, miopatía y raramente rabdomiólisis en pacientes tratados con rosuvastatina. Al igual que con otros inhibidores de la HMG-CoA reductasa, la frecuencia de reportes post-comercialización para rabdomiólisis es mayor con la dosis más alta. En aquellos pacientes que desarrollen cualquier signo o síntoma indicativo de miopatía, se deberán determinar los niveles de creatina-fosfocinasa. La administración de rosuvastatina debe descontinuarse si los niveles de creatina-fosfocinasa son considerablemente elevados (mayor a 10 veces el límite superior normal) o si se diagnostica o sospecha miopatía.
Se han notificado casos muy raros de miopatía necrotizante mediada por el sistema inmune, clínicamente caracterizada por debilidad muscular proximal persistente y elevación en la creatinina cinasa sérica durante el tratamiento o después de la interrupción del tratamiento con estatinas, incluyendo rosuvastatina. Pruebas neuromusculares y serológicas adicionales pueden ser necesarias. Puede requerirse tratamiento con agentes inmunosupresores.
En estudios con rosuvastatina no hubo evidencia de aumento de efectos musculo-esqueléticos cuando se administró rosuvastatina con otro tratamiento concomitante. Sin embargo, se ha observado mayor incidencia de miositis y miopatía en pacientes que reciben otros inhibidores de la HMG-CoA reductasa junto con ciclosporina, derivados de ácido fíbrico, incluyendo gemfibrozilo, ácido nicotínico, antimicóticos azoles y antibióticos macrólidos.
El riesgo de miopatía durante el tratamiento con rosuvastatina puede incrementarse con la administración concurrente de productos que contengan elbasvir/grazoprevir (ver X. Interacciones Medicamentosas y de Otro Género).
Se han registrado reportes de miopatía y/o rabdomiólisis con el uso de inhibidores de la HMG-CoA reductasa co-administrados con daptomicina. Se debe tener precaución cuando se prescribe un inhibidor de la HMG- CoA reductasa junto con daptomicina; ésto debido a que cualquiera de estos dos fármacos puede causar miopatía y/o rabdomiólisis cuando se administran solos. Se debe considerar la suspensión temporal de NAXZALLA en pacientes en tratamiento con daptomicina (ver X. Interacciones Medicamentosas y de Otro Género).
NAXZALLA
NAXZALLA debe prescribirse con precaución en pacientes con factores predisponentes a miopatía, por ejemplo, disfunción renal, edad avanzada e hipotiroidismo, o en situaciones donde se pueda tener aumento en los niveles plasmáticos (Ver X. Interacciones Medicamentosas y de Otro Género). NAXZALLA debe suspenderse temporalmente en cualquier paciente con una condición seria aguda sugerente de miopatía o con predisposición a desarrollar desde insuficiencia renal secundaria a rabdomiólisis (por ejemplo, sepsis, hipotensión, cirugía mayor, traumatismos, trastornos metabólicos graves, alteraciones endocrinas y/o desórdenes electrolíticos o en status epilepticus).
Anormalidades en Enzimas Hepáticas
Ezetimiba
En estudios controlados en pacientes a los que se les co-administraron ezetimiba y una estatina se han observado aumentos sucesivos de las transaminasas (al triple o más del límite superior de sus valores normales). (Ver IX. Reacciones Secundarias y Adversas).
Se deben hacer pruebas de funcionamiento hepático al iniciar el tratamiento. (Ver IX. Reacciones Secundarias y Adversas).
En un estudio clínico controlado en el que más de 9,000 pacientes con enfermedad renal crónica fueron distribuidos al azar para recibir ezetimiba 10 mg combinado con simvastatina 20 mg al día (n = 4,650) o combinado con placebo (n = 4,620) (mediana de seguimiento 4.9 años), la incidencia de incrementos consecutivos de transaminasas (≥3 veces el límite superior normal) fue de 0.7% para ezetimiba combinado con simvastatina y 0.6% para placebo. (Ver IX. Reacciones Secundarias y Adversas).
NAXZALLA
Como con otros inhibidores de la HMG-CoA reductasa, NAXZALLA debe utilizarse con precaución en pacientes que consuman cantidades excesivas de alcohol y/o que tengan antecedentes de enfermedad hepática.
Uso con otros medicamentos
Fenofibrato
Si se sospecha colelitiasis en un paciente que recibe NAXZALLA y fenofibrato, están indicados estudios de la vesícula biliar y deberá considerarse un tratamiento hipolipemiante alternativo [ver la Información Para Prescribir de fenofibrato y ácido fenofíbrico].
Otros fibratos
No se ha estudiado la co-administración de ezetimiba con fibratos diferentes a fenofibrato. Por lo tanto, no se recomienda la co-administración de NAXZALLA y fibratos (diferentes a fenofibrato). (Ver X. Interacciones Medicamentosas y de Otro Género).
Anticoagulantes
Si NAXZALLA se agrega al tratamiento con warfarina, a otro anticoagulante cumarínico, o a fluindiona, el INR (por sus siglas en inglés para International Normalized Ratio) debe ser monitoreado apropiadamente. (Ver X. Interacciones Medicamentosas y de Otro Género).
Ciclosporina
Se debe tener cuidado cuando se administre ezetimiba en pacientes tratados con ciclosporina. Las concentraciones de ciclosporina deben ser monitoreadas en pacientes que tomen ezetimiba y ciclosporina (Ver X. Interacciones Medicamentosas y de Otro Género).
Diabetes Mellitus
Rosuvastatina
Como con otros inhibidores de la HMG-CoA reductasa, se ha observado incremento en los niveles de HbA1c y glucosa sérica en pacientes tratados con rosuvastatina y en algunos casos este aumento puede exceder el umbral para el diagnóstico de diabetes mellitus, principalmente en pacientes que ya están en alto riesgo de desarrollar diabetes. (Ver. IX. Reacciones Secundarias y Adversas, XI. Resultados Anormales de Pruebas de Laboratorio).
Uso en poblaciones especiales
Raza
Rosuvastatina
Estudios farmacocinéticos muestran un incremento en la concentración plasmática de rosuvastatina en sujetos asiáticos en comparación con sujetos caucásicos. (Ver V. Farmacocinética y Farmacodinamia).
NAXZALLA
La dosis de NAXZALLA debe ajustarse en pacientes asiáticos. (Ver XIII. Dosis y Vía de Administración).
Uso Pediátrico
Ezetimiba
La seguridad y la eficacia de ezetimiba co-administrado con simvastatina en pacientes de 10 a 17 años de edad con hipercolesterolemia familiar heterocigótica, han sido evaluadas en estudios clínicos controlados en niños y niñas que tenían al menos un año de estar menstruando. Los pacientes adolescentes tratados con ezetimiba y hasta 40 mg/día de simvastatina tuvieron un perfil de experiencias adversas similar al de los adultos tratados con ezetimiba y simvastatina. En un estudio controlado, no hubo efecto detectable en el crecimiento o en la maduración sexual de las y los adolescentes, o algún efecto en la duración del ciclo menstrual de las adolescentes (Ver V. Farmacocinética y Farmacodinamia). Ezetimiba no ha sido estudiado en pacientes menores de 10 años de edad o en niñas pre-menárquicas.
Rosuvastatina
La evaluación del crecimiento lineal (altura), peso, IMC (índice de masa corporal), y características secundarias de maduración sexual mediante clasificación Tanner, en pacientes pediátricos que tomaron rosuvastatina se limitó a un periodo de un año. (Ver V. Farmacocinética y Farmacodinamia).
NAXZALLA
El tratamiento con NAXZALLA no se recomienda en población pediátrica. Ver XIII. Dosis y Vía de Administración.
Uso Geriátrico
NAXZALLA
Los pacientes de edad avanzada pueden tener un mayor riesgo de miopatía y se debe tener precaución cuando se prescribe NAXZALLA en pacientes de edad avanzada (ver más arriba en Efectos en el Músculo-Esquelético).
Insuficiencia Renal
NAXZALLA
Se requiere ajuste de dosis de NAXZALLA en pacientes con insuficiencia renal grave (CLcr<30 mL/min/1.73m2). (Ver V. Farmacocinética y Farmacodinamia, y XIII. Dosis y Vía de Administración).
Insuficiencia Hepática
NAXZALLA
NAXZALLA está contraindicado en pacientes con enfermedad hepática activa. Debido a que se desconocen los efectos de la mayor exposición a la ezetimiba en los pacientes con insuficiencia hepática moderada o grave, no se recomienda el tratamiento con NAXZALLA en pacientes con insuficiencia hepática moderada (puntaje Child-Pugh de 7 a 9) o grave (puntaje Child-Pugh >9). (Ver V. Farmacocinética y Farmacodinamia, VI. Contraindicaciones, y XIII. Dosis y Vía de Administración
Efectos en la habilidad para conducir u operar maquinaria
Rosuvastatina
Pruebas farmacológicas no revelaron evidencia de efecto sedante con rosuvastatina. Por su perfil de seguridad, no se espera que rosuvastatina afecte la capacidad de manejar u operar maquinaria.
VIII. RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA
Embarazo
La aterosclerosis es un proceso crónico y la descontinuación de los fármacos hipolipemiantes durante el embarazo debería tener poco impacto sobre el desenlace del tratamiento a largo plazo de la hipercolesterolemia primaria.
Ezetimiba
No hay datos clínicos sobre la administración de ezetimiba a mujeres embarazadas. Los estudios en animales sobre la administración de ezetimiba sola no indican efectos perjudiciales directos o indirectos sobre el embarazo, desarrollo embrionario y fetal, parto o desarrollo post-natal (ver XII. Precauciones En Relación Con Efectos De Carcinogénesis, Mutagénesis, Teratogénesis Y Sobre La Fertilidad). Sin embargo, se debe tener precaución cuando se prescriba ezetimiba a una mujer embarazada.
Cuando se administró ezetimiba con lovastatina, simvastatina, pravastatina o atorvastatina a ratas gestantes en estudios de desarrollo embrio-fetal, no se observaron efectos teratogénicos. En conejas gestantes se observó una incidencia baja de malformaciones esqueléticas (ver XII. Precauciones En Relación Con Efectos De Carcinogénesis, Mutagénesis, Teratogénesis Y Sobre La Fertilidad).
Rosuvastatina
No se ha establecido la seguridad de rosuvastatina durante el embarazo. Las mujeres con potencial de concebir deben utilizar medidas anticonceptivas adecuadas (ver VI. Contraindicaciones.
NAXZALLA
NAXZALLA está contraindicado durante el embarazo.
Lactancia
Ezetimiba
Los estudios en ratas han mostrado que la ezetimiba es excretada en la leche. No se sabe si también es excretada en la leche humana, por lo que no se debe administrar ezetimiba a mujeres lactantes, a menos que el beneficio potencial justifique el posible riesgo para el lactante.
Rosuvastatina
No se ha establecido la seguridad de rosuvastatina durante la lactancia. (ver VI. Contraindicaciones).
NAXZALLA
NAXZALLA está contraindicado en madres en periodo de lactancia. Debido al potencial de reacciones adversas graves en el lactante amamantado, las mujeres en periodo de lactancia no deben de tomar NAXZALLA.
IX. REACCIONES SECUNDARIAS Y ADVERSAS
Experiencia de Estudios Clínicos:
NAXZALLA
Se ha evaluado la seguridad de NAXZALLA (como la co-administración de ezetimiba y rosuvastatina equivalente a NAXZALLA) en un estudio clínico con más de 220 pacientes. NAXZALLA fue generalmente bien tolerado.
Se reportaron las siguientes experiencias adversas poco comunes (≥1/1,000, <1/100) relacionadas con el fármaco, en pacientes que tomaron NAXZALLA:
Trastornos gastrointestinales:
Poco comunes: distensión abdominal; dolor abdominal; estreñimiento; boca seca; náusea
Trastornos de piel y tejido subcutáneo:
Poco comunes: dermatitis alérgica; eczema
Trastornos musculoesqueléticos y del tejido conectivo:
Poco comunes: artralgia; mialgia
Combinación de Dosis Fija de Ezetimiba/Rosuvastatina:
Se reportaron las siguientes experiencias adversas comunes (≥1/100, <1/10) o poco comunes (≥1/1,000, <1/100) relacionadas con el fármaco en pacientes que tomaban una combinación de dosis fija de ezetimiba/rosuvastatina en un estudio clínico con más de 200 pacientes coreanos.
Trastornos gastrointestinales:
Poco común: náusea
Trastornos de piel y tejido subcutáneo:
Común: prurito
Poco común: erupción
Exploraciones Complementarias:
Poco común: incremento en ALT
Otras Experiencias de Estudios Clínicos y Experiencia Post-Comercialización:
Las siguientes reacciones adversas adicionales han sido reportadas en estudios clínicos o en el uso post- comercialización para ezetimiba (con o sin alguna estatina) o rosuvastatina:
Trastornos de sangre y sistema linfático: trombocitopenia
Trastornos del sistema inmune: reacciones de hipersensibilidad, incluyendo anafilaxia, angioedema, erupción cutánea, y urticaria
Trastornos endocrinos: diabetes mellitus
Trastornos del metabolismo y nutrición: disminución del apetito
Trastornos psiquiátricos: depresión, trastornos del sueño (incluyendo insomnio y pesadillas)
Trastornos del sistema nervioso: mareo, cefalea, parestesia, pérdida de memoria
Trastornos vasculares: bochornos, hipertensión
Trastornos respiratorios, torácicos, y mediastinales: tos
Trastornos gastrointestinales: diarrea, dispepsia, flatulencia, gastritis, enfermedad por reflujo gastroesofágico, pancreatitis
Trastornos hepatobiliares: ictericia, hepatitis, colelitiasis, colecistitis, incremento en transaminasas hepáticas
Trastornos de piel y tejido subcutáneo: eritema multiforme, urticaria
Trastornos musculoesqueléticos y del tejido conectivo: dolor de espalda, espasmos musculare, debilidad muscular, miopatía (incluyendo miositis) y rabdomiólisis que ocasionalmente se asoció a deterioro de la función renal (ver VII. Precauciones Generales), dolor de cuello, dolor en extremidades, miopatía necrotizante inmunomediada
Trastornos del Sistema reproductivo y mamas: ginecomastia
Trastornos generales y condiciones en el sitio de administración: astenia, dolor de pecho, fatiga, dolor, edema periférico
Exploraciones Complementarias: incremento de AST, incremento de la creatinina-fosfocinasa sanguínea, incremento de gamma-glutamiltransferasa, pruebas de función hepática anormales
Al igual que con otros inhibidores de la HMG-CoA reductasa, la incidencia de reacciones adversas relacionadas al fármaco tiende a aumentar con dosis altas.
Otros efectos:
En un estudio clínico controlado a largo plazo se demostró que rosuvastatina no tuvo efectos dañinos sobre lentes intraoculares.
En pacientes tratados con rosuvastatina no hubo deterioro en la función adrenocortical.
X. INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO
NAXZALLA
No se observó una interacción farmacocinética clínicamente significativa cuando se co-administró ezetimiba con rosuvastatina.
Múltiples mecanismos pueden contribuir a las interacciones potenciales con inhibidores de la HMG-CoA reductasa. Los fármacos o productos herbolarios que inhiben ciertas rutas enzimáticas y/o del transportador (p.ej., OATP1B) pueden incrementar las concentraciones plasmáticas de rosuvastatina y puede conducir a un incremento en el riesgo de miopatía/rabdomiólisis.
Consulte la información para prescribir de todos los medicamentos utilizados concomitantemente para obtener información adicional acerca de las interacciones potenciales con rosuvastatina y/o el potencial de alteraciones enzimáticas o del transportador y posibles ajustes a la dosis y esquemas de tratamiento.
Rosuvastatina
Efecto de la coadministración de medicamentos sobre rosuvastatina:
In vitro e in vivo, los datos indican que la rosuvastatina no tiene interacciones clínicamente significativas con el citocromo P-450 (como un sustrato, inhibidor o inductor). La rosuvastatina es un sustrato para ciertas proteínas incluyendo el transportador de la captación hepática del Polipéptido Transportador de Aniones Orgánicos 1B1 (OATP1B1) y el transportador de flujo de salida de Proteínas Resistentes al Cáncer de Mama (BCRP). La administración concomitante de rosuvastatina con medicamentos que son inhibidores de estas proteínas transportadoras puede resultar en aumento de las concentraciones plasmáticas de rosuvastatina y un mayor riesgo de miopatía (ver tabla 5, VII. Precauciones Generales, y XIII. Dosis y Vía De Administración).
Inhibidores de la Proteína de Resistencia al Cáncer de Mama (BCRP): La administración concomitante de productos que son inhibidores de la BCRP (por sus siglas en inglés para breast cancer protein resistant) (p.ej., elbasvir y grazoprevir) puede conducir a un incremento en las concentraciones plasmáticas de rosuvastatina; por lo tanto, la dosis de NAXZALLA no deberá exceder de 10/10 mg una vez al día en pacientes que reciban medicación concomitante con productos que contienen elbasvir o grazoprevir (ver VII. Precauciones Generales, y XIII. Dosis y Vía De Administración).
Daptomicina: El riesgo de miopatía y/o rabdomiólisis puede incrementarse por la administración concomitante de inhibidores de la HMG-CoA reductasa y daptomicina (ver VII. Precauciones Generales).
Tabla 5. Efecto de la coadministración de los medicamentos sobre la exposición de rosuvastatina (ABC, en orden decreciente de magnitud) de los estudios clínicos publicados
|
Régimen de dosis de medicamentos interactuantes |
Régimen de dosis de rosuvastatina |
Cambio de ABC en rosuvastatina |
|
Ciclosporina 75 mg dos veces al día a 200 mg dos veces al día por 6 meses |
10 mg una vez al día por 10 días |
7.1 veces ↑ |
|
Atazanavir 300 mg/ritonavir 100 mg una vez al día por 8 días |
10 mg dosis única |
3.1 veces ↑ |
|
Grazoprevir 200 mg y elbasvir 50 mg una vez al día por 11 días |
10 mg, dosis única |
2.3 veces ↑ |
|
Lopinavir 400 mg/ritonavir 100 mg dos veces al día por 17 días |
20 mg una vez al día por 7 días |
2.1 veces ↑ |
|
Gemfibrozilo 600 mg dos veces al día por 7 días |
80 mg dosis única |
1.9 veces ↑ |
|
Eltrombopag 75 mg una vez al día por 10 días |
10 mg dosis única |
1.6 veces ↑ |
|
Darunavir 600 mg/ritonavir 100 mg dos veces al día por 7 días |
10 mg una vez al día por 7 días |
1.5 veces ↑ |
|
Tipranavir 500 mg/ritonavir 200 mg dos veces al día por 11 días |
10 mg dosis única |
1.4 veces ↑ |
|
Dronedarona 400 mg dos veces al día |
No disponible |
1.4 veces ↑ |
|
Itraconazol 200 mg una vez al día por 5 días |
10 u 80 mg, dosis única |
1.4 veces ↑ |
|
Ezetimiba 10 mg una vez al día por 14 días |
10 mg una vez al día por 14 días |
1.2 veces ↑ |
|
Fosamprenavir 700 mg/ritonavir 100 mg dos veces al día por 8 días |
10 mg dosis única |
↔ |
|
Aleglitazar 0.3 mg por 7 días |
40 mg por 7 días |
↔ |
|
Silimarin 140 mg tres veces al día por 5 días |
10 mg dosis única |
↔ |
|
Fenofibrato 67 mg tres veces al día por 7 días |
10 mg por 7 días |
↔ |
|
Rifampicina 450 mg una vez al día por 7 días |
20 mg dosis única |
↔ |
|
Ketoconazol 200 mg dos veces al día por 7 días |
80 mg dosis única |
↔ |
|
Fluconazol 200 mg una vez al día por 11 días |
80 mg dosis única |
↔ |
|
Eritromicina 500 mg cuatro veces al día por 7 días |
80 mg dosis única |
28% ↓ |
|
Baicalin 50 mg tres veces al día por 14 días |
20 mg dosis única |
47% ↓ |
Interacciones que requieren ajustes de dosis de rosuvastatina (ver tabla 5):
Cuando sea necesario la coadministración de rosuvastatina con otros medicamentos que aumentan la exposición a rosuvastatina, las dosis de rosuvastatina deben ajustarse. Iniciar con una dosis de 5 mg de rosuvastatina una vez al día, si el aumento esperado en la exposición Área Bajo la Curva (ABC) es de aproximadamente 2 veces o más. La dosis máxima diaria de rosuvastatina debe ajustarse de manera que la exposición esperada de rosuvastatina no supere una dosis diaria de 40 mg administrada sin que haya interacción con medicamentos, por ejemplo, una dosis de 5 mg de rosuvastatina con ciclosporina (7.1 veces mayor en la exposición), una dosis de 10 mg de rosuvastatina en combinación con ritonavir/atazanavir (3.1 veces mayor) y una dosis de 20 mg de rosuvastatina con gemfibrozilo (1.9 veces mayor).
Otros medicamentos que interactúan:
Antiácidos:
La administración simultánea de rosuvastatina con una suspensión antiácida que contenga hidróxido de aluminio y magnesio da como resultado una disminución en la concentración plasmática de rosuvastatina aproximadamente 50%. Este efecto se mitigó cuando el antiácido se administró 2 horas después en rosuvastatina. La importancia clínica de esta interacción no se ha estudiado.
Efecto de rosuvastatina cuando se administra concomitantemente con otros medicamentos
Warfarina
La farmacocinética de warfarina no se afecta considerablemente después de la coadministración con rosuvastatina. Sin embargo, al igual que otros inhibidores de HMG-CoA reductasa, la coadministración de rosuvastatina y warfarina puede dar como resultado un aumento en INR o tiempos de coagulación, en comparación con warfarina sola. En pacientes que reciben antagonistas de la vitamina K, se recomienda el monitoreo de INR, tanto al inicio como al término de la terapia con rosuvastatina o después de un ajuste de la dosis.
Fenofibratos o derivados del ácido fíbrico:
Aunque no se ha observado ninguna interacción farmacocinética entre rosuvastatina y fenofibrato, se puede presentar una interacción farmacodinámica. Gemfibrozil, fenofibrato y otros ácidos fíbricos, incluyendo el ácido nicotínico, pueden aumentar el riesgo de miopatía cuando se administran de manera concomitante con inhibidores de la HMG-CoA reductasa (ver VII. Precauciones Generales) .
Ciclosporina:
La coadministración de rosuvastatina con ciclosporina dio como resultado cambios no significativos en la concentración plasmática de ciclosporina.
Otros medicamentos:
No hubo interacciones clínicamente significativas con anticonceptivos orales, digoxina, ezetimibe o fenofibrato.
En estudios clínicos rosuvastatina se coadministró con agentes antihipertensivos, antidiabéticos y terapia de reemplazo hormonal. Estos estudios no produjeron ninguna evidencia de interacciones adversas clínicamente significativas.
Ezetimiba
Los estudios preclínicos han mostrado que la ezetimiba no induce las enzimas metabolizadoras de medicamentos del citocromo P450. No se ha observado ninguna interacción de importancia clínica entre la ezetimiba y medicamentos que se sabe que son metabolizados por los citocromos P450 1A2, 2D6, 2C8, 2C9 y 3A4 o por la N-acetiltransferasa.
La co-administración de ezetimiba no tuvo ningún efecto sobre la farmacocinética de la dapsona, dextrometorfano, digoxina, anticonceptivos orales (etinilestradiol y levonorgestrel), glipicida, tolbutamida o midazolam. La co-administración de cimetidina no tuvo ningún efecto sobre la biodisponibilidad de la ezetimiba.
Antiácidos: La administración concomitante de antiácidos disminuyó la tasa de absorción de la ezetimiba, pero no modificó su biodisponibilidad. Esta disminución de la tasa de absorción de la ezetimiba no se considera clínicamente importante.
Colestiramina: La administración concomitante de colestiramina disminuyó 55% aproximadamente la media del área bajo la curva de concentración de la ezetimiba total (ezetimiba + glucurónido de ezetimiba). Esta interacción puede hacer que sea menor la disminución adicional de C-LDL debida a la co-administración de ezetimiba y colestiramina.
Ciclosporina: En un estudio realizado con ocho pacientes después de trasplante renal y depuración de creatinina >50 mL/min que recibían dosis estables de ciclosporina, la administración de una dosis única de 10 mg de ezetimiba incrementó 3.4 veces (rango de 2.3 a 7.9 veces) la media del área bajo la curva de ezetimiba, en comparación con los datos reportados en otro estudio realizado con sujetos sanos control (n=17). En otro estudio, un paciente con trasplante renal con insuficiencia renal grave (depuración de creatinina de 13.2 mL/min/1.73 m2) que recibió múltiples medicamentos, incluyendo ciclosporina, tuvo una concentración 12 veces mayor de ezetimiba en comparación con los pacientes del grupo control.
En un estudio cruzado de dos periodos en doce sujetos sanos, la administración diaria de 20 mg de ezetimiba durante 8 días con una dosis de 100 mg de ciclosporina en el Día 7, produjo un incremento promedio de 15% en el ABC de la ciclosporina (rango 10% de disminución a 51% de aumento) comparado con una dosis única de 100 mg de ciclosporina sola (ver VII. Precauciones Generales).
Fibratos: La seguridad y eficacia de ezetimiba co-administrada con fenofibrato han sido evaluadas en un estudio clínico (ver V. Farmacocinética y Farmacodinamia, y IX. Reacciones Secundarias Y Adversas); la co- administración de ezetimiba con otros fibratos no ha sido estudiada. Los fibratos pueden aumentar la excreción de colesterol en la bilis y producir así colelitiasis. En un estudio preclínico en perros, la ezetimiba aumentó el contenido de colesterol de la bilis (ver XII. Precauciones En Relación Con Efectos De Carcinogénesis, Mutagénesis, Teratogénesis Y Sobre La Fertilidad). Aunque se desconoce la importancia de este resultado preclínico para los seres humanos, no se recomienda la co-administración de ezetimiba y fibratos (que no sean fenofibrato) hasta que se estudie su uso en pacientes.
Fenofibrato: En un estudio de farmacocinética, la administración concomitante de fenofibrato incrementó la concentración total de ezetimiba aproximadamente 1.5 veces. Este incremento no se considera clínicamente significativo.
Gemfibrozilo: En un estudio de farmacocinética, la administración concomitante de gemfibrozilo incrementó la concentración total de ezetimiba aproximadamente 1.7 veces. Este incremento no se considera clínicamente significativo. No están disponibles datos clínicos.
Estatinas: No se observó ninguna interacción farmacocinética de importancia clínica cuando se co-administró ezetimiba con atorvastatina, simvastatina, pravastatina, lovastatina, fluvastatina o rosuvastatina.
Anticoagulantes: En un estudio clínico de 12 hombres adultos sanos, la administración concomitante de ezetimiba (10 mg una vez al día) no tuvo un efecto significativo en la biodisponibilidad de los tiempos de warfarina o protrombina. Ha habido reportes después de la comercialización de incrementos en el INR en pacientes que han agregado ezetimiba a warfarina o fluindiona. La mayoría de estos pacientes también tomaban otros medicamentos (ver VII. Precauciones Generales).
XI. ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO
Ezetimiba
En estudios clínicos controlados en monoterapia, la incidencia de aumentos clínicamente importantes de las transaminasas séricas (ALT y/o AST al triple o más del límite superior de sus valores normales, consecutivos) fue similar con ezetimiba (0.5%) y con placebo (0.3%). En los estudios en los que se co-administraron ezetimiba y una estatina, la incidencia fue de 1.3% con la co-administración y de 0.4% con la estatina sola. Generalmente esos aumentos de las transaminasas fueron asintomáticos, no se asociaron con colestasis, y volvieron a los valores normales al suspender o al continuar el tratamiento. (Ver VII. Precauciones Generales).
Los aumentos clínicamente importantes de la creatinina-fosfocinasa (≥10 veces el límite superior normal) observados en pacientes tratados con ezetimiba administrado en monoterapia o en combinación con una estatina fueron similares a los aumentos observados con placebo o con la estatina sola, respectivamente.
Rosuvastatina: Como con otros inhibidores de HMG-CoA reductasa, se ha observado un aumento relacionado con la dosis en las transaminasas hepáticas y creatinina-cinasa en un pequeño número de pacientes que recibieron rosuvastatina. También se ha observado un aumento de la HbA1c en pacientes tratados con rosuvastatina (ver VII. Precauciones Generales). Se han observado resultados anormales en pruebas de análisis de orina (prueba con tira reactiva positiva para proteinuria) en un pequeño número de pacientes que han recibido rosuvastatina y otros inhibidores de HMG-CoA reductasa. La proteína detectada fue principalmente de origen tubular. En la mayoría de los casos, la proteinuria disminuye o desaparece espontáneamente al continuar la terapia y no es predictiva de enfermedad renal aguda o progresiva.
XIII. DOSIS Y VÍA DE ADMINISTRACIÓN
Vía de Administración: Oral
General
El paciente debe estar bajo una dieta hipolipemiante apropiada y deberá continuar con esta dieta durante el tratamiento con NAXZALLA. La dosis deberá individualizarse de acuerdo con el nivel basal de LDL-C, el objetivo de tratamiento recomendado, y la respuesta del paciente. NAXZALLA se puede administrar como una dosis única en cualquier momento del día, con o sin alimentos.
Adultos
Hipercolesterolemia Primaria
La dosis inicial recomendada de NAXZALLA es de 10 mg/10 mg. Para pacientes de poblaciones especiales está disponible la dosis inicial de NAXZALLA de 10 mg/5 mg en caso de que se requiera. El rango de dosis de NAXZALLA es de 10 mg/5 mg a 10 mg/20 mg una vez al día. Al iniciar el tratamiento o al cambiar de otro tratamiento con un inhibidor de HMG-CoA reductasa, deberá utilizarse primero la dosis inicial apropiada de NAXZALLA, y solo entonces titularla de acuerdo con la respuesta del paciente y las metas de tratamiento individualizado. Si se requiere, puede hacerse ajuste de la dosis en intervalos de 2 a 4 semanas
Para pacientes con hipercolesterolemia severa (incluyendo hipercolesterolemia familiar heterocigótica) o aquéllos con metas agresivas de lípidos, puede considerarse una dosis inicial de NAXZALLA de 10 mg/20 mg.
Dosis en Pacientes con Hipercolesterolemia Familiar Homocigótica
La dosis inicial usual de NAXZALLA en pacientes con hipercolesterolemia familiar homocigótica es de 10 mg/20 mg una vez al día.
Poblaciones Especiales
Pacientes Pediátricos
No se recomienda el tratamiento con NAXZALLA. (Ver V. Farmacocinética y Farmacodinamia, y VII. Precauciones Generales)
Pacientes Geriátricos
No se requiere ajustar la dosis en pacientes de edad avanzada. (Ver V. Farmacocinética y Farmacodinamia, y VII. Precauciones Generales)
Insuficiencia Renal
No se requiere ajustar la dosis para pacientes con insuficiencia renal de leve a moderada. Para pacientes con insuficiencia renal grave (depuración de creatinina <30 mL/min/1.73 m2), la dosis de NAXZALLA no debe exceder de 10 mg/10 mg una vez al día. (Ver V. Farmacocinética y Farmacodinamia, y VII. Precauciones Generales).
Insuficiencia Hepática
No se requiere ajustar la dosis en pacientes con insuficiencia hepática leve (puntaje Child-Pugh de 5 a 6). No se recomienda el tratamiento con NAXZALLA en pacientes con disfunción hepática moderada (puntaje Child- Pugh de 7 a 9) o grave (puntaje Child-Pugh >9). (Ver V. Farmacocinética y Farmacodinamia, VI. Contraindicaciones y VII. Precauciones Generales).
Dosificación en Pacientes Asiáticos
Se debe considerar una dosis inicial de NAXZALLA de 10 mg/5 mg en pacientes asiáticos. Un incremento en las concentraciones plasmáticas de rosuvastatina se ha observado en sujetos asiáticos. Se debe considerar este aumento en la exposición sistémica cuando se trata a sujetos asiáticos que no tengan su hipercolesterolemia apropiadamente controlada con dosis de NAXZALLA de hasta 10 mg/20 mg/día. (Ver V. Farmacocinética y Farmacodinamia, y VII. Precauciones Generales).
Tratamientos concomitantes
Co-administración con Secuestradores de Ácidos Biliares: La dosificación de NAXZALLA debería ocurrir ya sea ≥2 horas antes o ≥4 horas después de la administración de un secuestrador de ácidos biliares.
La rosuvastatina, un componente de NAXZALLA, es un sustrato de diversas proteínas transportadoras (por ejemplo, OATP1B1 y BCRP). El riesgo de miopatía (incluida la rabdomiólisis) se incrementa cuando se administra NAXZALLA de manera concomitante con ciertos medicamentos que pueden aumentar la concentración plasmática de rosuvastatina debido a interacciones con estas proteínas transportadoras (por ejemplo, ciclosporina y ciertos inhibidores de la proteasa, que incluyen combinaciones de ritonavir con atazanavir, lopinavir, y / o tipranavir (ver VII. Precauciones Generales, y X. Interacciones Medicamentosas Y De Otro Género). Siempre que sea posible, se deben considerar medicamentos alternativos y si es necesario, considerar la suspensión temporal del tratamiento con NAXZALLA. En situaciones en donde es inevitable la administración conjunta de estos medicamentos con NAXZALLA, el beneficio y el riesgo del tratamiento concomitante y los ajustes de dosis de NAXZALLA se deben considerar cuidadosamente (Ver X. Interacciones Medicamentosas Y De Otro Género).
Co-administración de elbasvir o grazoprevir
En pacientes que toman elbasvir o grazoprevir, la dosis de NAXZALLA no debe exceder de 10 mg/10 mg una vez al día (ver VII. Precauciones Generales, y X. Interacciones Medicamentosas Y De Otro Género)
Co-administración de Antiácidos
Al tomar NAXZALLA con antiácidos que contengan la combinación de hidróxido de aluminio y magnesio, el antiácido deberá tomarse al menos 2 horas después de la administración de NAXZALLA.
Polimorfismos genéticos
Se ha demostrado que los genotipos de las sustancias disolutas de la familia de transportadores aniones 1B1 (SLCO1B1) (del polipéptido transportador de aniones orgánicos 1B1 [OATP1B1]) c.521CC y ABCG2 (de las proteínas resistentes al cáncer de mama [BCRP]) c.421AA están asociadas a un incremento en la exposición a rosuvastatina (ABC) en comparación con SLCO1B1 c.521TT y ABCG2 c.421CC. Para los pacientes que se sabe que tienen el genotipo c.521CC o c.421AA, se recomienda una dosis diaria máxima de NAXZALLA de 10 mg/20 mg. (ver VII. Precauciones Generales, y X. Interacciones Medicamentosas Y De Otro Género).
XIV. MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL
NAXZALLA: No puede recomendarse un tratamiento específico para la sobredosis con NAXZALLA. En el caso de una sobredosis, deberán emplearse medidas sintomáticas y de soporte.
Ezetimiba: En estudios clínicos, la administración de 50 mg diarios de ezetimiba a 15 personas sanas hasta por 14 días, de 40 mg diarios a 18 pacientes con hipercolesterolemia primaria hasta por 56 días, y de 40 mg diarios a 27 pacientes con sitosterolemia homocigótica por 26 semanas fue generalmente bien tolerada.
Se han reportado pocos casos de sobredosificación con ezetimiba, la mayoría no se ha asociado con reacciones adversas. Las reacciones adversas reportadas no han sido graves.
Rosuvastatina
La hemodiálisis no mejora significativamente la depuración de rosuvastatina.
XII. PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD
Ezetimiba
Carcinogénesis
En los estudios de dos años en ratones y ratas la ezetimiba no fue cancerígena.
Mutagénesis
Ezetimiba no fue genotóxica en una serie de pruebas in vivo e in vitro.
La ezetimiba y sus combinaciones con atorvastatina, simvastatina, pravastatina o lovastatina no fueron genotóxicas en una serie de pruebas in vivo e in vitro.
Reproducción
La ezetimiba no afectó la fertilidad de las ratas macho o hembra.
Desarrollo Fetal
La ezetimiba no fue teratogénica en las ratas ni en los conejos, y no tuvo ningún efecto sobre el desarrollo fetal o post-natal.
La administración concomitante de ezetimiba y estatinas no fue teratogénica en las ratas. En conejas gestantes se observó una incidencia baja de malformaciones esqueléticas (fusión de esternebras, fusión y disminución del número de vértebras caudales) cuando se les administró ezetimiba (1,000 mg/kg, 146 o más veces mayor que la exposición humana a la dosificación de 10 mg diarios, basándose en el ABC0-24h de ezetimiba total) con lovastatina (2.5 y 25 mg/kg), simvastatina (5 y 10 mg/kg), pravastatina (25 y 50 mg/kg) o atorvastatina (5, 25 y 50 mg/kg). La exposición a la forma farmacológicamente activa de la estatina varió entre 1.4 (atorvastatina) y 547 (lovastatina) veces mayor que la exposición humana con 10 mg diarios (simvastatina o atorvastatina) o con 20 mg diarios (lovastatina y pravastatina), basándose en el ABC0-24h.
Rosuvastatina
Los datos preclínicos no revelan riesgos especiales para humanos con base en estudios convencionales de farmacología de seguridad, toxicidad con dosis repetidas, toxicidad genética, potencial carcinogénico y toxicidad reproductiva.
XVIII. NOMBRE Y DOMICILIO DEL LABORATORIO
SCHERING-PLOUGH, S.A. de C.V.
Av. 16 de Septiembre No. 301 Col. Xaltocan
C.P. 16090, Xochimilco,
Ciudad de México, México
XV. PRESENTACIÓN
Caja con 30 tabletas de 10 mg/5 mg, 10 mg/10 mg y 10 mg/20 mg e instructivo anexo.
XVI. RECOMENDACIONES SOBRE ALMACENAMIENTO
Consérvese a no más de 30°C.
Consérvese la caja bien cerrada.
Protéjase de la luz.
XVII. LEYENDAS DE PROTECCIÓN
Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni lactancia. Este producto contiene lactosa. Literatura exclusiva para profesionales de la salud.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
dpocmx@merck.com


