
NEULASTIM - Solución
Sustancia(s):
- Pegfilgrastim
Presentaciones:
- 1 Caja, 1 Jeringa(s) prellenada(s), 6/0.6 mg/ml
- 1 Caja, 1 Jeringa(s) prellenada(s), 0.64 ml, 6/0.6 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
La jeringa prellenada contiene:
Pegfilgrastim 6 mg
Vehículo cbp 0.6 mL
Filgrastim de origen ADN recombinante expresado en Escherichia coli, conjugado con polietilenglicol (PEG).
INDICACIONES TERAPÉUTICAS: Reducción de la duración de la neutropenia, reducción en la incidencia de neutropenia febril y reducción en la incidencia de infección manifestada por neutropenia febril en pacientes con tumores malignos tratados con quimioterapia citotóxica (con excepción de leucemia mieloide crónica y síndromes mielodisplásicos).
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Inmunoestimulantes, factor estimulante de colonias de granulocitos.
Código ATC: L03AA13.
El factor estimulante de colonias de granulocitos (FEC-G) humano es una glucoproteína que regula la producción y liberación de neutrófilos desde la médula ósea. Pegfilgrastim es un conjugado covalente del FEC-G recombinante humano (r-metHuFEC-G) con una molécula de polietilenglicol (PEG) de 20 kd. Pegfilgrastim es una forma de duración sostenida de filgrastim como consecuencia de una menor depuración renal. Pegfilgrastim y filgrastim presentan el mismo mecanismo de acción, causando un aumento marcado de los neutrófilos en la sangre periférica dentro de las 24 horas de su administración, con elevaciones mínimas en los monocitos y/o linfocitos. Al igual que filgrastim, los neutrófilos producidos en respuesta a pegfilgrastim presentan una funcionalidad normal o mejorada como demuestran las pruebas de quimiotaxis y de función fagocítica. Al igual que otros factores de crecimiento hematopoyéticos, el FEC-G ha demostrado in vitro propiedades estimuladoras sobre las células endoteliales humanas. El FEC-G puede promover el crecimiento in vitro de las células mieoloides, incluyendo las células tumorales, y pueden observarse efectos similares en algunas células no mieloides in vitro.
En los dos ensayos clínicos principales, aleatorizados, doble ciego, en pacientes con cáncer de mama en etapas de alto riesgo II-IV tratadas con quimioterapia mielosupresora consistente en doxorrubicina y docetaxel, el uso de pegfilgrastim, como dosis única una vez por ciclo, redujo la duración de la neutropenia y la incidencia de neutropenia febril de forma similar a la observada con la administración diaria de filgrastim (con una mediana de 11 días de administración). De acuerdo con la literatura, en ausencia de soporte con factor de crecimiento, este régimen ocasiona una duración media de la neutropenia grado 4, de 5 a 7 días, y un 30% a 40% de incidencia de neutropenia febril. En un ensayo (n = 157) en el que se usó una dosis fija de 6 mg de pegfilgrastim, la duración media de la neutropenia grado 4 para el grupo tratado con pegfilgrastim fue de 1.8 días comparado con 1.6 días del grupo tratado con filgrastim (0.23 días de diferencia, IC 95% -0.15, 0.63). Durante el ensayo completo, el porcentaje de neutropenia febril fue del 13% en los pacientes tratados con pegfilgrastim comparado con 20% de los pacientes tratados con filgrastim (diferencia del -7%, IC 95%, -19%, 5%). En un segundo ensayo (n = 310) en el que se usó una dosis ajustada según el peso (100 mcg/kg), la duración media de la neutropenia grado 4 en el grupo tratado con pegfilgrastim fue de 1.7 días, comparado con 1.8 días en el grupo tratado con filgrastim (diferencia de 0.03 días, IC 95%, -0.36, 0.30). El porcentaje total de neutropenia febril fue de 9% en los pacientes tratados con pegfilgrastim y de 18% en los pacientes tratados con filgrastim (diferencia del -9%, IC 95%, -16.8%, -1.1%).
Un estudio controlado con placebo, doble ciego en pacientes con cáncer de mama, evaluó el efecto de pegfilgrastim sobre la incidencia de neutropenia febril posterior a la administración de un régimen de quimioterapia y estuvo asociado con una tasa de neutropenia febril de 10% a 20% (docetaxel 100 mg/m2 cada 3 semanas durante 4 ciclos). Novecientos veintiocho (928) pacientes fueron aleatorizados para recibir, ya sea una dosis única de pegfilgrastim o de placebo, aproximadamente 24 horas (día 2) después de la quimioterapia en cada ciclo. La incidencia de neutropenia febril fue significativamente más baja para los pacientes tratados con pegfilgrastim que los que recibieron placebo (1% versus 17%, p ≤ 0.001, respectivamente). La incidencia de hospitalización y uso de agentes antimicrobianos, intravenosos en asociación con un diagnóstico clínico de neutropenia febril fue significativamente más baja en el grupo tratado con pegfilgrastim que en el grupo que recibió placebo (1% versus 14%, p < 0.001; y 2% contra 10%, p < 0.001, respectivamente).
Un estudio pequeño (n=83), aleatorizado, doble ciego de fase II, en pacientes tratados con quimioterapia contra la leucemia mieloide aguda de nueva aparición, comparó la administración de pegfilgrastim (dosis única de 6 mg) contra filgrastim, administrados durante la quimioterapia de inducción. La mediana del tiempo hasta la recuperación de la neutropenia grave fue de 22 días en ambos grupos de tratamiento. El resultado a largo plazo no fue estudiado (ver Precauciones Generales).
En un estudio fase II (n = 37), multicéntrico, aleatorizado y abierto en pacientes pediátricos con sarcoma, que recibieron I00 μg/kg de pegfílgrastim tras un ciclo de quimioterapia con vincristina, doxorrubicina y ciclofosfamida (VAdriaC/IE), se observó una mayor duración de la neutropenia grave (neutrófilos < 0.5 x 109/L) en niños más jóvenes entre 0-5 años (8.9 días), comparado con niños de mayor edad, entre 6-11 años y entre 12-21 años (6 días y 3.7 días, respectivamente) y adultos. Adicionalmente, se observó mayor incidencia de neutropenia febril en niños más jóvenes entre 0-5 años (75%) comparado con niños de mayor edad entre 6-11 años y entre 12-21 años (70% y 33%, respectivamente) y adultos (ver Reacciones secundarias y adversas y Propiedades farmacocinéticas).
Propiedades farmacocinéticas: Tras una administración única subcutánea de pegfilgrastim, la concentración sérica máxima ocurre de 16 a 120 horas después de la administración y las concentraciones séricas de pegfilgrastim se mantienen durante el periodo de neutropenia posterior a la quimioterapia mielosupresora. La distribución de pegfilgrastim se limitó al compartimiento plasmático. La eliminación de pegfilgrastim no es lineal con respecto a la dosis, la depuración sérica de pegfilgrastim disminuye con el aumento de la dosis. Pegfilgrastim parece eliminarse principalmente por depuración mediada por los neutrófilos (> 99%), que se saturan a altas dosis. Consistente con un mecanismo de depuración autorregulado, la concentración sérica de pegfilgrastim disminuye rápidamente al comenzar la recuperación de los neutrófilos (ver Figura 1).
Figura 1. Perfil de la mediana de la concentración sérica de pegfilgrastim y la Cuenta Absoluta de Neutrófilos (CAN) en pacientes tratados con quimioterapia después de la administración de una única inyección de 6 mg
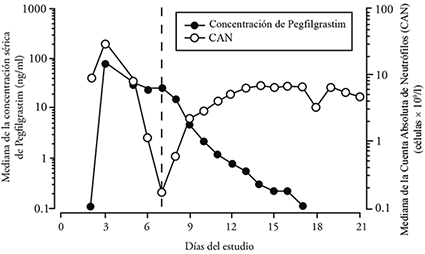
Debido al mecanismo de depuración regulado por los neutrófilos, no se espera que la farmacocinética de pegfilgrastim se vea afectada en casos de insuficiencia renal o hepática En un estudio ahierto de dosis única (n = 31). los diferentes estadios de la insuficiencia renal, incluyendo insuficiencia renal crónica en etapa terminal, no tuvieron impacto sobre la farmacocinética del pegfilgrastim.
En voluntarios sanos, la farmacocinética de pegfilgrastim fue comparable cuando se administraba por vía subcutánea a través de una jeringa prellenada manual, en comparación con la administración a través del on-body injector (OBI).
Pacientes de la tercera edad: Los escasos datos disponibles indican que la farmacocinética de pegfilgrastim en personas de la tercera edad (> 65 años) es similar a la de los adultos.
Población pediátrica: La seguridad y farmacocinética de pegfilgrastim fueron estudiadas en 37 pacientes pediátricos con sarcoma que recibieron 100 mcg/kg de pegfilgrastim tras completar la quimioterapia con VAdriaC/IE. El grupo de menor edad (0 a 5 años, presentó una media de exposición más alta a pegfilgrastim (ABC) (± Desviación Estándar) (47.9 ± 22.5 mcg• hr/mL) que los niños de mayor edad entre 6 a 11 años y entre 12 a 21 años (22.0 ± 13.1 mcg• hr/mL y 29.3 ± 23.2 mcg• hr/mL, respectivamente) (ver Propiedades farmacodinárnicas). Con la excepción del grupo de edad más joven (0-5 años), la media en la ABC en pacientes pediátricos fue similar a la de pacientes adultos con cáncer de mama de alto riesgo en etapas II-IV que recibieron 100 mcg/kg de pegfilgrastim tras completar el tratamiento con doxorrubicina/docetaxel (ver Reacciones secundarias y adversas y Propiedades farmacodinámicas).
CONTRAINDICACIONES: Hipersensibilidad al principio activo o alguno de los excipientes: acetato sódico, sorbitol (E420), polisorbato 20, agua para preparaciones inyectables.
NEULASTIM está contraindicado en el embarazo, lactancia o en niños menores de 18 años.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: Se carece de datos o existe información limitada sobre el uso de pegfilgrastim en mujeres embarazadas. Estudios en animales han mostrado toxicidad sobre la reproducción (ver Precauciones en relación con efectos en carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). El uso de pegfilgrastim no se recomienda durante el embarazo y en mujeres en edad fértil que no utilicen anticonceptivos. Lactancia: No hay información suficiente sobre la excreción de pegfilgrastim/metabolitos en la leche humana, no se puede excluir un riesgo para los recién nacidos/lactantes. Debe tomarse una decisión sobre suspender la lactancia o suspender/abstenerse de la terapia con pegfilgrastim considerando el beneficio de la lactancia para el niño y el beneficio de la terapia para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad: Las reacciones adversas que se reportaron con mayor frecuencia fueron dolor óseo (muy frecuente [≥ 1/10]) y dolor musculoesquelético (frecuente [≥ 1/100 a < 1/10]). Por lo general, el dolor óseo es de intensidad leve a moderada, transitorio y puede controlarse en la mayoría de los pacientes con analgésicos estándar.
Las reacciones de hipersensibilidad, que incluyen erupción cutánea, urticaria, angioedema, disnea, eritema, enrojecimiento e hipotensión, ocurrieron durante el tratamiento inicial o subsecuente con pegfilgrastim (poco frecuente [≥ 1/1,000 a < 1/100]). Las reacciones alérgicas graves, incluyendo anafilaxia, pueden ocurrir en pacientes que reciben pegfilgrastim (poco frecuente) (ver Precauciones generales).
El Síndrome de Extravasación Capilar, que puede poner en riesgo la vida si el tratamiento se retrasa, ha sido reportado con baja frecuencia (≥ 1/1,000 a < 1/100) en pacientes con cáncer que se sometieron a quimioterapia y posterior a la administración de factores estimulantes de colonias de granulocitos (ver Precauciones generales y Descripción de las reacciones adversas seleccionadas).
La esplenomegalia, generalmente asintomática, es poco frecuente.
La esplenomegalia, generalmente asintomática, es poco frecuente.
La ruptura esplénica, incluyendo algunos casos con desenlace fatal, ha sido reportada con baja frecuencia, posterior a la administración de pegfilgrastim (ver Precauciones generales).
Se han reportado reacciones adversas pulmonares poco frecuentes, incluyendo neumonía intersticial, edema pulmonar, infiltrados pulmonares y fibrosis pulmonar. Con poca frecuencia, estos casos resultaron en insuficiencia respiratoria o Síndrome de Dificultad Respiratoria ARDS, por sus siglas en inglés, que puede asociarse con mortalidad (ver Precauciones generales).
Se han informado casos aislados de crisis drepanocíticas en pacientes con rasgos de células falciformes o drepanocitosis (poco frecuente en pacientes con anemia falciforme) (ver Precauciones generales).
Tabla de resumen de reacciones adversas: Los datos en la siguiente tabla describen las reacciones adversas informadas en ensayos clínicos y de informes espontáneos. Dentro de cada grupo de frecuencia, se presentan los efectos no deseables en orden decreciente de gravedad.
|
MedDRA por órgano o sistema |
Reacciones adversas |
|||
|---|---|---|---|---|
|
Muy frecuente (≥ 1/10) |
Frecuente (≥ 1/100 a < 1/10) |
Poco frecuente (≥ 1/1,000 a < 1/100) |
Raro (≥ 1/10,000 a < 1/1,000) |
|
|
Neoplasias benignas, malignas y sin especificar (incluidos quistes y pólipos) |
Síndrome mielodisplásico1 Leucemia mieloide agudal |
|||
|
Trastornos de la sangre y del sistema linfático |
Trombocitopenia1 Leucocitosis1 |
Anemia de células falciformes con crisis drepanocítica2 Esplenomegalia2; Ruptura esplénica2 |
||
|
Trastornos del sistema inmunitario |
Reacciones de hipersensibilidad; Anafilaxia |
|||
|
Trastornos del metabolismo y de la nutrición |
Elevaciones en el ácido úrico |
|||
|
Trastornos del sistema nervioso |
Cefalea1 |
|||
|
Trastornos vasculares |
Síndrome de extravasación capilar1 |
Aortitis |
||
|
Trastornos mediastinales, torácicos y respiratorios |
Síndrome de Dificultad Respiratoria Aguda2; Reacciones adversas pulmonares (neumonía intersticial, edema pulmonar, infiltrados pulmonares y fibrosis pulmonar) Hemoptisis |
Hemoragia pulmonar |
||
|
Trastornos gastrointestinales |
Náusea1 |
|||
|
Trastornos de la piel y del tejido subcutáneo |
Dermatitis de contacto1 |
Síndrome de Sweet (dermatosis febril aguda)1,2 Vasculitis cutánea1,2 |
Síndrome de Stevens-Johnson |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Dolor óseo |
Dolor musculoesquelético (mialgia, artralgia, dolor en extremidades, dolor de espalda, dolor musculoesquelético, dolor de cuello) |
||
|
Trastornos renales y urinarios |
Glomerulonefritis2 |
|||
|
Trastornos generales y en el sitio de administración |
Dolor en el sitio de la inyección1 Reacción en el sitio de aplicación1 Dolor torácico no cardiaco |
Reacciones en el sitio de la inyección2 |
||
|
Investigación |
Elevaciones en lactato deshidrogenasa y fosfatasa alcalina1; Elevaciones transitorias en pruebas de función hepática para ALT o AST1 |
|||
1 Ver la sección, Descripción de las reacciones adversas seleccionada".
2 Esta reacción adversa fue identificada mediante la vigilancia posterior a la comercialización, pero no observada en estudios clínicos controlados aleatorizados en adultos. La categoría de frecuencia se estimó a partir de un cálculo estadístico con base en 1,576 pacientes que recibieron NEULASTIM en nueve estudios clínicos aleatorizados.
Descripción de las reacciones adversas seleccionadas:
Se han reportado casos poco frecuentes de síndrome de Sweet, aunque en algunos casos las neoplasias malignas hematológicas subyacentes pueden tener un papel.
Se han informado eventos poco frecuentes de vasculitis cutánea en pacientes tratados con pegfilgrastim. El mecanismo de la vasculitis en los pacientes que recibieron pegfilgrastim es desconocido.
Se produjeron reacciones en el sitio de inyección, incluyendo eritema (poco frecuente) así como dolor en el sitio de la inyección (frecuente) en el tratamiento inicial o posterior con pegfilgrastim.
Con el uso del on-body injector (OBI) se han reportado reacciones en la zona de aplicación (que incluyen eventos como hemorragia y dolor en la zona de aplicación, malestar en la zona de aplicación, equimosis y eritema en la zona de aplicación).
Con el uso del on-body injector (OBI) se ha reportado dermatitis de contacto y reacciones locales en la piel, como erupciones, prurito y urticaria, indicando una posible reacción de hipersensibilidad al adhesivo.
Se han reportado casos frecuentes de leucocitosis (cuenta de Glóbulos Blancos > 100 x 109/L) (ver Precauciones Generales).
Elevaciones leves a moderadas, reversibles, en ácido úrico, fosfatasa alcalina y lactato deshidrogenasa, sin efectos clínicos asociados fueron poco frecuentes; en pacientes que recibieron NEULASTIM después de la quimioterapia citotóxica.
Se observaron náuseas y cefalea frecuentemente en pacientes que recibieron quimioterapia.
Se observaron elevaciones poco frecuentes en pruebas de función hepática para alanina aminotransferasa (ALT) o aspartato aminotransferasa (AST) en pacientes posterior a recibir pegfilgrastim después de la quimioterapia citotóxica. Estas elevaciones son transitorias y retornan a valores basales.
En un estudio epidemiológico realizado en pacientes con cáncer de mama y de pulmón, se ha observado un aumento del riesgo de SMD/LMA luego del tratamiento con NEULASTIM junto con quimioterapia o radioterapia (ver Precauciones generales).
Se informaron casos comunes de trombocitopenia.
Los casos de síndrome de extravasación capilar se han reportado en el entorno post-comercialización con el uso de factor estimulante de colonias de granulocitos. Por lo general, esto se presentó en pacientes con enfermedades malignas avanzadas, sepsis, recibiendo diferentes fármacos de quimioterapia o sometidos a aféresis (ver Precauciones generales).
Población pediátrica:
La experiencia en niños es limitada. Una mayor frecuencia de reacciones adversas graves en niños pequeños de 0 a 5 años (92%) se ha observado en comparación con niños mayores de 6 a 11 y 12 a 21 años, respectivamente (80% y 67%) y adultos. La reacción adversa más común informada fue dolor óseo (ver Propiedades farmacodinámicas y Propiedades farmacocinéticas).
Reporte de sospecha de reacciones adversas:
Es importante informar sobre las sospechas de reacciones adversas posterior de un producto farmacéutico. Esto permite el monitoreo continuo del balance riesgo-beneficio del producto farmacéutico. Se solicita a los profesionales de la salud que cualquier sospecha de reacción adversa se reporte directamente a farmacovigilancia@cofepris.gob.mx y a farmacovigilanciamx@amgen.com
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogenicidad: Según se ha demostrado, ciertas células malignas expresan receptores para el factor estimulante de colonias de granulocitos (FEC-G). La posibilidad de que el pegfilgrastim pueda actuar como un factor de crecimiento para cualquier tipo de tumor no puede ser excluida. El potencial carcinogénico del pegfilgrastim no ha sido evaluado en estudios a largo plazo con animales. Un estudio de toxicidad de 6 meses de duración, en el cual se inyectaron dosis subcutáneas de hasta 1,000 mcg/kg de pegfilgrastim (aproximadamente hasta 23 veces la dosis recomendada para humanos) en ratas una sola vez por semana, no se identificó la presencia de lesiones precancerosas o cancerosas.
Mutagenicidad: No se han llevado a cabo estudios de mutagenicidad.
Teratogenicidad y fertilidad: No se observaron efectos adversos en las crías de ratas preñadas tratadas con pegfilgrastim por vía subcutánea, pero en los conejos, pegfilgrastim ha demostrado causar toxicidad embrio-fetal (pérdida del embrión) a dosis acumulativas de aproximadamente 4 veces la dosis recomendada en humanos, las cuales no se han visto en conejas preñadas que fueron expuestas a la dosis recomendada para humanos. En estudios en ratas, se demostró que pegfilgrastim puede atravesar la placenta. Los estudios en ratas indicaron que el desarrollo reproductivo, la fertilidad, ciclo estrogénico, días entre el apareamiento y el coito no fueron afectados por pegfilgrastim subcutáneo. Se desconoce la relevancia de estos hallazgos en humanos.
Datos preclínicos de seguridad:
Los datos preclínicos procedentes de estudios convencionales de toxicidad a dosis repetidas mostraron los efectos farmacológicos esperados, incluyendo incremento en el recuento de leucocitos, hiperplasia mieloide en la médula ósea, hematopoyesis extramedular y aumento en el tamaño del bazo.
Fertilidad:
Pegfilgrastim no afectó el desempeño reproductivo o la fertilidad en ratas macho o hembra con dosis semanales acumuladas aproximadamente 6 a 9 veces superiores a la dosis humana recomendada (con base en el área de superficie corporal).
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Debido a la potencial sensibilidad a la quimioterapia citotóxica de las células mieloides en rápida división, pegfilgrastim debe administrarse por lo menos 24 horas después de la administración de la quimioterapia citotóxica. En los estudios clínicos, NEULASTIM® ha sido administrado de forma segura 14 días antes de la quimioterapia. La administración simultánea de NEULASTIM con fármacos quimioterapéuticos no ha sido evaluada en pacientes. En modelos animales, la administración simultánea de NEULASTIM y 5-fluouracilo (5-FU) u otros antimetabolitos han demostrado potenciar la mielosupresión.
En los ensayos clínicos no se ha investigado específicamente las posibles interacciones con otros factores de crecimiento hematopoyéticos y con citocinas.
No se ha investigado específicamente la posibilidad de interacción con el litio, que también estimula la liberación de los neutrófilos. No hay evidencia de que dicha interacción sea nociva.
La seguridad y eficacia de NEULASTIM no han sido evaluadas en pacientes tratados con fármacos quimioterapéuticos con acción mielosupresora retardada p. ej. nitrosoureas.
No se han realizado estudios específicos de interacciones o metabolismo, sin embargo, los ensayos clínicos no han indicado ninguna interacción entre NEULASTIM y cualquier otro medicamento.
Incompatibilidades: Este producto no debe ser mezclado con otros fármacos, particularmente con soluciones de cloruro de sodio.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Se recomienda el seguimiento con biometrías hemáticas completas durante la terapia con pegfilgrastim.
PRECAUCIONES GENERALES:
Trazabilidad: Con el fin de mejorar la trazabilidad de los factores estimuladores de colonias de granulocitos (FEC-Gs), el nombre comercial del producto administrado debe estar claramente registrado en el expediente del paciente.
Datos clínicos limitados sugieren un efecto comparable entre pegfilgrastim y filgrastim sobre el tiempo hasta la recuperación en casos de neutropenia grave en pacientes con leucemia mieloide aguda (LMA) de nueva aparición (ver Farmacocinética y farmacodinamia: Propiedades farmacoinámicas). Sin embargo, los efectos a largo plazo de pegfilgrastim no han sido establecidos en LMA; por lo tanto, NEULASTIM deberá ser utilizado con precaución en esta población de pacientes.
El FEC-G (factor estimulante de colonias de granulocitos) puede promover la proliferación de células mieloides, in vitro y es posible observar efectos similares sobre ciertas células no mieloides in vitro.
La seguridad y eficacia de pegfilgrastim no han sido investigadas en pacientes con síndrome mielodisplásico, con leucemia mieloide crónica ni en pacientes con LMA secundaria; por lo tanto, dicho fármaco no debe ser usado en estos pacientes. Se debe tener particular cuidado al distinguir el diagnóstico de la transformación blástica de la leucemia mieloide crónica respecto de la LMA.
La seguridad y eficacia de la administración de pegfilgrastim en pacientes adultos < 55 años con LMA de nueva aparición con citogenética t(15;17) no se han determinado.
No se ha investigado la seguridad ni la eficacia de pegfilgrastim en pacientes tratados con quimioterapia a altas dosis. Este producto no debe ser usado para incrementar la dosis de la quimioterapia citotóxica, más allá de los regímenes de dosificación establecidos.
Eventos adversos pulmonares: Se han reportado reacciones adversas pulmonares, en particular neumonía intersticial posterior a la administración de FEC-G. Pacientes con antecedentes recientes de infiltrados pulmonares o neumonía pueden estar en mayor riesgo (ver Reacciones secundarias y adversas).
La aparición de signos pulmonares tales como tos, fiebre y disnea, en asociación con signos radiológicos de infiltración pulmonar y deterioro de la función pulmonar, junto con un aumento en el recuento de neutrófilos pueden ser los signos preliminares del Síndrome de Dificultad Respiratoria progresiva del adulto (ARDS, por sus siglas en inglés). En estos casos, se deberá suspender la administración de pegfilgrastim, a discreción del médico, y administrar el tratamiento apropiado (ver Reacciones secundarias y adversas).
Glomerulonefritis: Se ha reportado glomerulonefritis en pacientes que reciben filgrastim y pegfilgrastim. En general, los eventos de glomerulonefritis se resuelven después de la reducción o suspensión de la dosis de filgrastim y pegfilgrastim. Se recomienda el monitoreo del análisis de orina.
Síndrome de extravasación capilar: Se ha reportado síndrome de extravasación capilar después de la administración del factor estimulante de colonias de granulocitos, y está caracterizado por hipotensión, hipoalbuminemia, edema y hemoconcentración. Pacientes quienes desarrollan síntomas de síndrome de extravasación capilar deben monitorearse de manera cercana y recibir un tratamiento sintomático estándar, que pudiera incluir la necesidad de cuidados intensivos (ver Reacciones secundarias y adversas).
Esplenomegalia y ruptura esplénica: Se han reportado casos generalmente asintomáticos de esplenomegalia y casos de ruptura esplénica incluyendo algunos con desenlace fatal, posterior a la administración de pegfilgrastim (ver Reacciones secundarias y adversas). Por lo tanto, el tamaño del bazo deberá ser monitoreado cuidadosamente (ej. examen clínico, ultrasonido). Se debe considerar el diagnóstico de ruptura esplénica en pacientes que reportan dolor abdominal en cuadrante superior izquierdo o dolor de hombro.
Trombocitopenia y anemia: El tratamiento únicamente con pegfilgrastim no evita la trombocitopenia ni la anemia debidas a la administración de dosis completas de quimioterapia mielosupresora en el esquema prescrito. Se recomienda controlar regularmente la cuenta de plaquetas y el hematocrito. Se debe tener cuidado especial cuando se administran agentes solos o en combinación, los cuales se sabe que causan trombocitopenia severa.
Síndrome mielodisplásico y leucemia mieloide aguda en pacientes con cáncer de mama y de pulmón:
En el contexto del estudio observacional post-comercialización, pegfilgrastim, en conjunto con quimioterapia y/o radioterapia, se ha asociado al desarrollo del síndrome mielodisplásico (SMD) y con leucemia mieloide aguda (LMA) en pacientes con cáncer de mama y de pulmón (ver Reacciones secundarias y adversas). Se debe monitorizar a los pacientes con cáncer de mama y de pulmón en busca de signos y síntomas de SMD/LMA.
Posibles fallas del dispositivo: Se han informado pérdidas de dosis o dosis incompletas en pacientes que reciben NEULASTIM a través del on-body injector (OBl) debido a que el dispositivo no haya funcionado según lo previsto. En caso de una pérdida de una dosis o dosis incompleta, los pacientes pueden presentar un mayor riesgo de eventos, tales como neutropenia, neutropenia febril y/o infección, contrario a si la dosis se hubiera administrado correctamente. Indique a los pacientes que utilizan el OBI que deben notificar a su profesional de la salud de inmediato, con el fin de determinar la necesidad de una dosis de sustitución de NEULASTIM, si sospechan que el dispositivo pudo no haber funcionado según lo previsto.
Anemia de células falciformes: Crisis de células falciformes se han asociado con el uso de pegfilgrastim en pacientes con anemia drepanocítica o enfermedad de células falciformes (ver Reacciones secundarias y adversas). Por lo tanto, los médicos deberán tener precaución cuando prescriben pegfilgrastim en pacientes con anemia drepanocítica o enfermedad de células falciformes, se deben monitorear los parámetros clínicos y de laboratorio apropiados y estar atentos a una posible asociación de este fármaco con el incremento en el tamaño del bazo y presencia de crisis de vaso-oclusiva.
Leucocitosis: Se han observado recuentos de glóbulos blancos (WBC, por sus siglas en inglés) de 100 x 109/L o mayores en menos del 1% de los pacientes que reciben pegfilgrastim. No se ha reportado ningún efecto adverso directamente atribuible a este grado de leucocitosis. Tal elevación de glóbulos blancos es transitoria, por lo general, observada 24 a 48 horas después de la administración y coincide con los efectos farmacodinámicos de este medicamento. En concordancia con los efectos clínicos y el potencial de leucocitosis, debe realizarse un recuento de leucocitos a intervalos regulares durante la terapia. Si la cuenta de leucocitos excede 50 x 109/L después del nadir esperado, el fármaco debe suspenderse inmediatamente.
Hipersensibilidad: Se ha reportado hipersensibilidad, incluyendo reacciones anafilácticas, que ocurren en el tratamiento inicial o subsecuente en pacientes tratados con pegfilgrastim. Suspender permanentemente pegfilgrastim en pacientes con hipersensibilidad clínicamente significativa. No administrar pegfilgrastim a pacientes con antecedentes de hipersensibilidad a pegfilgrastim o filgrastim. Si se produce una reacción alérgica grave, deberá administrarse una terapia apropiada, con seguimiento cercano del paciente durante varios días.
Síndrome de Stevens-Johnson: Rara vez se ha notificado síndrome de Stevens-Johnson (SSJ), que puede poner en peligro la vida o llegar a ser mortal, en asociación con el tratamiento con pegfilgrastim. Si el paciente ha desarrollado SSJ con el uso de pegfilgrastim, no deberá reiniciarse el tratamiento con pegfilgrastim en este paciente en ningún momento.
Inmunogenicidad: Como con todas las proteínas terapéuticas, existe un potencial de inmunogenicidad. La tasa de generación de anticuerpos contra pegfilgrastim es generalmente baja. Los anticuerpos de unión ocurren de acuerdo con lo esperado con todos los biológicos; sin embargo, no han estado asociados con actividades neutralizantes hasta el momento.
Aortitis: Se ha reportado aortitis después de la administración de FEC-G en sujetos sanos y en pacientes con cáncer. Los síntomas presentados incluyeron fiebre, dolor abdominal, malestar, dolor de espalda y aumento de marcadores inflamatorios (ej. proteína c-reactiva y conteo de leucocitos). En la mayoría de los casos la aortitis fue diagnosticada por TC y resuelta generalmente después de retirar el FEC-G (ver Reacciones secundarias y adversas).
Alergias a acrílicos: El on-body injector (OBI) utiliza un adhesivo acrílico. En pacientes que presentan reacciones a los adhesivos acrílicos, el uso de este producto puede provocar una reacción significativa.
Otras precauciones: La seguridad y eficacia de NEULASTIM para la movilización de células progenitoras sanguíneas en pacientes o donadores sanos no ha sido evaluada de manera adecuada.
La cubierta del aguja de la jeringa prellenada podría contener goma natural seca (un derivado del látex), lo que podría provocar reacciones alérgicas.
El incremento en la actividad hematopoyética en la médula ósea como respuesta a la terapia con factor de crecimiento se ha asociado con hallazgos transitorios de positividad ósea observables por imagen. Esto debe considerarse cuando se interpretan los resultados de imagen.
Sorbitol: Se debe considerar el efecto aditivo de los productos que contienen sorbitol (o fructosa) administrados de manera concomitante y la ingesta dietética de sorbitol ( o fructosa).
Sodio: Este medicamento contiene menos de 1 mmol (23 mg) de sodio por cada dosis de 6 mg, es decir, en esencia está “exento de sodio”.
Poblaciones especiales:
Población pediátrica: No se ha establecido aún la seguridad y eficacia de NEULASTIM en niños. Los datos actualmente disponibles están descritos en las secciones Reacciones secundarias y adversas, Farmacocinética y farmacodinamia: Propiedades y efectos y, Farmacocinética; sin embargo, no se puede hacer una recomendación posológica.
Pacientes con insuficiencia renal: No se recomienda modificar la dosis en pacientes con insuficiencia renal, incluidos aquellos con enfermedad renal terminal.
Habilidad para manejar y utilizar máquinas: Pegfilgrastim tiene un efecto nulo o insignificante en la capacidad para manejar y utilizar máquinas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Subcutánea
Dosis:
Adultos (≥ 18 años): La dosis recomendada de NEULASTIM es de 6 mg (una jeringa prellenada) por cada ciclo de quimioterapia, administrada por vía subcutánea, aproximadamente 24 horas después de la quimioterapia citotóxica.
Forma de administración:
NEULASTIM se administra por vía subcutánea a través de una jeringa prellenada de dosis única para administración manual o para administración con el on-body injector (OBI), que se empaca en conjunto con una jeringa prellenada de dosis única.
Instrucciones especiales para el profesional de la salud con respecto a la jeringa prellenada:
Las inyecciones administradas de manera manual se deben administrar en el muslo, abdomen, la parte superior del brazo o los glúteos.
Instrucciones especiales para el profesional de la salud con respecto al on-body Injector:
Un profesional de la salud debe llenar el on-body injector (OBI) con NEULASTIM con la ayuda de la jeringa prellenada, y, luego, colocar el OBI sobre la piel del paciente (abdomen o parte posterior del brazo). La parte posterior del brazo sólo puede ser utilizada si hay un cuidador disponible para supervisar el estado del OBI. Aproximadamente, 27 horas después que se colocó el OBI en la piel del paciente, NEULASTIM se administrará durante aproximadamente 45 minutos. Un profesional de la salud debe colocar el OBI el mismo día que la administración de la quimioterapia citotóxica, siempre que el OBI administre el NEULASTIM no menos de 24 horas después de la administración de la quimioterapia citotóxica.
La jeringa prellenada empacada en conjunto sólo debe ser utilizada con el OBI. La jeringa prellenada contiene solución adicional para compensar la pérdida de líquido durante la administración a través del OBI. Si la jeringa prellenada empacada en conjunto se utiliza para la inyección subcutánea manual, el paciente recibirá una sobredosis. Si la jeringa prellenada de dosis única para administración manual se utiliza con el OBI, el paciente puede recibir menos dosis de la recomendada.
No utilice el OBI para administrar ningún otro medicamento, excepto la jeringa prellenada con NEULASTIM empacada en conjunto con el OBI.
El OBI se debe aplicar en piel intacta y no irritada, en el brazo o en el abdomen.
Se puede producir una pérdida de una dosis debido a una falla en el OBI o a una fuga. Si el paciente no se administra una dosis, se debe administrar una nueva dosis mediante la jeringa prellenada de dosis única para administración manual, lo antes posible después de la detección.
Consulte el Instructivo destinado para el profesional de la salud en relación con el OBI para ver información completa sobre la administración.
El tratamiento con NEULASTIM debe ser iniciado y supervisado por un médico con experiencia en oncología y/o hematología.
Consejos para los pacientes en relación con la administarción a través del on-body injector:
Recomiende a los pacientes que eviten actividades como viajar, conducir u operar maquinaria pesada durante las 26-29 horas posteriores a la aplicación del on-body injector (OBI), esto incluye el periodo de administración de 45 minutos más una hora después de la administración. Los pacientes deben tener un cuidador cerca para la primera administración.
Refiera al paciente a la información sobre la administración de la dosis escrita en el Instructivo destinado para el Usuario. Proporcione capacitación a los pacientes para asegurarse de que comprendan cuándo comenzará la administración de la dosis de NEULASTIM y cómo supervisar el OBI para verificar que se administre la dosis completa. Asegúrese de que los pacientes comprenden cómo identificar signos de mal funcionamiento del OBI (ver Posibles fallas del dispositivo). Indique a los pacientes que utilizan el OBI que deben notificar a su profesional de la salud de inmediato, con el fin de determinar la necesidad de una dosis de reemplazo de NEULASTIM, si sospechan que el dispositivo puede no haber funcionado según lo previsto (ver Posibies fallas del dispositivo).
Instrucciones de uso y manipulación:
NEULASTIM jeringa prellenada es de un solo uso. Si no se administra todo el producto deséchese el sobrante. NEULASTIM es una solución estéril, sin conservadores.
Antes de usar NEULASTIM comprobar que la solución esté visualmente libre de partículas. Solamente deben inyectarse las soluciones que sean transparentes e incoloras.
La agitación excesiva puede producir el agregamiento de NEULASTIM haciéndolo biológicamente inactivo.
Deje que la jeringa prellenada para administración manual y la jeringa prellenada envasada junto con el on-body injector para administración automática alcancen la temperatura ambiente durante 30 minutos antes de utilizarlas.
La eliminación del medicamento no utilizado y de todos los materiales de desecho se realizará de acuerdo con la normativa local.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Se han administrado dosis únicas de 300 mcg/kg por vía subcutánea a un número limitado de voluntarios sanos y a pacientes con cáncer de pulmón de células no pequeñas sin que se hayan presentado efectos adversos graves.
Los eventos adversos fueron similares a los exhibidos por los sujetos tratados con dosis más bajas de pegfilgrastim.
PRESENTACIONES:
Caja con una jeringa prellenada con 6 mg/0.6 mL con o sin protección de aguja, con o sin envase de burbuja e instructivo anexo.
Caja con una jeringa prellenada con 0.64 mL (6 mg/0.6 mL) con un inyector corporal (on-body injector, OBI) con envase de burbuja e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese en refrigeración de (2 °C a 8 °C).
NEULASTIM puede dejarse a temperatura ambiente (que no supere los 30 °C) durante un único periodo máximo de hasta 72 horas. Todo el NEULASTIM que haya permanecido a temperatura ambiente durante más de 72 horas debe desecharse.
La jeringa prellenada para uso con el on-body injector puede ser expuesta a temperatura ambiente por no más de 36 horas previas al llenado del on-body injector.
No se congele. No se agite fuertemente.
Mantener en la caja para proteger de la luz.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Deseche cualquier porción no utilizada. No administre si el empaque ha sido violado. No se use en embarazo y lactancia, ni en menores de 18 años. No se deje al alcance de los niños. Deséchese inmediatamente después de su uso. Protéjase de la luz. Si no se administra todo el producto deséchese el sobrante. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se agite fuertemente. Su venta requiere receta médica.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx y farmacovigilanciamx@amgen.com
Titular del Registro Sanitario:
Amgen Manufacturing Limited
Carr. 31, Km 24.6 Juncos, PR, EUA.
Representante Legal:
AMGEN MÉXICO, S.A. de C.V.
Av. Vasco de Quiroga, No. 3000,
Piso 4, Col. Santa Fe, C.P. 01210,
Álvaro Obregón, Ciudad de México, México.
Reg. Núm. 061M2006 SSA IV
®Marca Registrada