
ONCASPAR - Solución
Sustancia(s):
- Pegaspargasa
Presentaciones:
- 1 Vial(es), 3750 U.I.,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada vial contiene:
|
Pegaspargasa |
3750 UI |
|
Excipientes c.s. 5 ml. |
|
INDICACIONES TERAPÉUTICAS: Leucemia linfoblástica aguda (ALL, por sus siglas en inglés) de primera línea.
ONCASPAR está indicado como componente de un régimen de múltiples agentes de quimioterapia para el tratamiento de primera línea de pacientes con ALL.
Leucemia linfoblástica aguda e hipersensibilidad a la asparaginasa.
ONCASPAR está indicado como un componente de un régimen de múltiples agentes de quimioterapia para el tratamiento de pacientes con leucemia linfoblástica aguda (ALL) e hipersensibilidad a las formas nativas de L-asparaginasa.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética: Las evaluaciones de farmacocinética se basaron en un ensayo enzimático que midió la actividad de asparaginasa. Las evaluaciones de farmacocinética en suero se evaluaron en 34 pacientes pediátricos recientemente diagnosticados con riesgo estándar de leucemia linfoblástica aguda (ALL) en el estudio 1 después de la administración intramuscular de 2,500 UI/m2. La vida media de eliminación de ONCASPAR fue de aproximadamente 5.8 días durante la fase de inducción. Se observaron similares vidas medias de eliminación durante la intensificación tardía 1 y la intensificación tardía 2. Se observaron concentraciones > 0.1 UI/ml en más del 90% de las muestras de pacientes tratados con ONCASPAR durante la inducción, la intensificación tardía 1 y la intensificación tardía 2 durante aproximadamente 20 días.
En 3 estudios de farmacocinética, 37 pacientes con recidiva de leucemia linfoblástica aguda (ALL) recibieron ONCASPAR a 2,500 UI/m2 por vía intramuscular cada 2 semanas. La vida media de ONCASPAR en plasma fue de 3.2 ± 1.8 días en 9 pacientes que eran previamente hipersensibles a la L-asparaginasa de E. coli nativa y 5.7 ± 3.2 días en 28 pacientes no hipersensibles. El área bajo la curva de concentración plasmática-tiempo (AUC, por sus siglas en inglés) fue de 9.5 ± 4.0 UI/ml al día en los pacientes previamente hipersensibles y de 9.8 ± 6.0 UI/ml al día en los pacientes no hipersensibles.
Farmacodinamia: En el estudio 1 se evaluó la farmacodinamia en 57 pacientes pediátricos recientemente diagnosticados con riesgo estándar de leucemia linfoblástica aguda (ALL) que recibieron 3 dosis intramusculares de ONCASPAR (2,500 UI/m2), cada una durante la inducción y 2 fases de tratamiento de intensificación tardía.
La actividad farmacodinámica se evaluó a través de una serie de mediciones de asparagina en suero (n = 57) y de líquido cefalorraquídeo (CSF, por sus siglas en inglés) (n = 50). Los datos de depleción de asparagina se presentan en la sección de Estudios Clínicos (véase Estudios clínicos [5.4]).
Mecanismo de acción: Se cree que el mecanismo de acción de ONCASPAR se basa en la eliminación selectiva de las células leucémicas debido a la depleción de asparagina en plasma. Algunas células leucémicas no tienen la capacidad de sintetizar asparagina debido a la falta de asparagina sintetasa y son dependiente de la fuente exógena de asparagina para la supervivencia. La depleción de asparagina, que resulta del tratamiento con la enzima L-asparaginasa, mata las células leucémicas. No obstante, las células normales son menos afectadas por la depleción debido a su capacidad de sintetizar la asparagina.
Leucemia linfoblástica aguda (ALL) de primera línea: La seguridad y eficacia de ONCASPAR se evaluó en un estudio abierto, multicéntrico, aleatorizado, controlado con activo (estudio 1). En este estudio, 118 pacientes pediátricos de 1 a 9 años de edad con leucemia linfoblástica aguda (ALL) de riesgo estándar sin tratamiento previo fueron aleatorizados 1:1 a ONCASPAR o a L-asparaginasa de E. coli como parte de la terapia de combinación. ONCASPAR se administró por vía intramuscular a una dosis de 2,500 UI/m2 en el día 3 de la fase de inducción de 4 semanas y en el día 3 en cada una de las 2 fases de intensificación tardía de 8 semanas. Se administró L-asparaginasa de E. coli nativa por vía intramuscular a una dosis de 6,000 UI/m2 3 veces a la semana por 9 dosis durante la inducción y por 6 dosis durante cada fase de intensificación tardía.
La determinación primaria de eficacia se basó en la demostración de depleción similar de asparagina (magnitud y duración) en los brazos de ONCASPAR y de L-asparaginasa de E. coli. El objetivo especificado fue el logro de la depleción de asparagina a una concentración en suero ≤ 1 µM. La proporción de pacientes con este nivel de depleción fue similar entre los 2 brazos del estudio durante todas las 3 fases de tratamiento en los puntos temporales especificados en el protocolo.
En todas las fases de tratamiento, las concentraciones de asparagina en suero en un plazo de 4 días de la primera dosis de asparaginasa en la fase de tratamiento y siguieron bajas durante aproximadamente 3 semanas para los brazos tanto de ONCASPAR como de L-asparaginasa de E. coli. Las concentraciones de asparagina en suero durante la fase de inducción se muestran en la figura 1. Los patrones de depleción de asparagina en suero en las 2 fases de intensificación tardía son similares para el patrón de depleción de asparagina en suero en la fase de inducción.
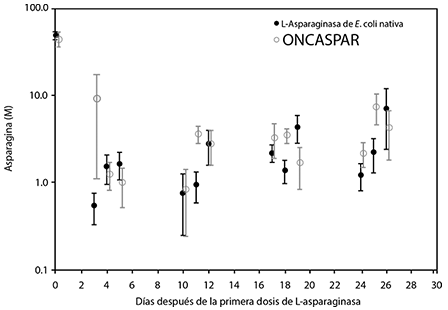
Figura 1. media (± error estándar)
de las concentraciones de asparagina en suero
durante la fase de inducción del estudio 1.
Nota: ONCASPAR (2,500 UI/m2 por vía intramuscular) se administró en el día 3 de la fase de inducción de 4 semanas. Se administró en el día 3 de la fase de inducción de 4 semanas. L-asparaginasa de E. coli nativa (6,000 UI/m2 por vía intramuscular) se administró 3 veces a la semana por 9 dosis durante la inducción. Las concentraciones de asparagina CSF se determinaron en 50 pacientes durante la fase de inducción. Asparagina CSF disminuyó de una concentración promedio previa al tratamiento de 3.1 µM a 1.7 µM en el día 4 ± 1 y de 1.5 µM a 25 ± 1 días después de la administración de ONCASPAR. Estos hallazgos fueron similares a aquellos observados en el brazo de tratamiento de L-asparaginasa de E. coli nativa.
Aunque la sobrevida sin eventos (EFS, por sus siglas en inglés) de 3 años para ONCASPAR y los brazos del estudio de L-asparaginasa de E. coli nativa fueron similares en el rango del 80%, el estudio 1 no estaba diseñado para evaluar las diferencias en las tasas de EFS.
Pacientes con leucemia linfoblástica aguda (ALL) hipersensibles a asparaginasa: Se evaluó la seguridad y eficacia de ONCASPAR en 4 estudios abiertos que reclutaron a un total de 42 pacientes con leucemia aguda con múltiples recidivas (39 [93%] con ALL) con antecedentes de reacción alérgica clínica previa a asparaginasa. La hipersensibilidad a asparaginasa se definió mediante una historia de exantema sistémico, urticaria, broncoespasmo, edema laríngeo, hipotensión, o eritema local, urticaria, o edema > 2 cm durante al menos 10 minutos después de la administración de L-asparaginasa de E. coli. Todos los pacientes recibieron ONCASPAR a una dosis de 2,000 o 2,500 UI/m2 administrada por vía intramuscular o intravascular cada 14 días. Los pacientes recibieron ONCASPAR como agente único o en combinación con quimioterapia de múltiples agentes. La tasa de respuesta de reinducción fue del 50% (intervalo de confianza del 95%: 35% al 65%), con base en el 36% de remisiones completas y el 14% remisiones parciales. Los resultados fueron similares a las tasas de respuesta global reportadas para los pacientes con leucemia linfoblástica aguda (ALL) que recibieron quimioterapia de reinducción que contenía L-asparaginasa de E. coli nativa de segunda línea. La actividad antitumoral también se observó con el agente único ONCASPAR. Se observaron tres respuestas (una remisión completa y dos remisiones parciales) en nueve pacientes adultos y pediátricos con recidiva de ALL e hipersensibilidad a L-asparaginasa de E. coli nativa.
CONTRAINDICACIONES:
• Antecedentes de reacciones alérgicas graves a ONCASPAR.
• Antecedentes de trombosis grave con la terapia previa de L-asparaginasa.
• Antecedentes de pancreatitis con la terapia previa de L-asparaginasa.
• Antecedentes de eventos hemorrágicos graves con la terapia previa de L-asparaginasa.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Embarazo categoría C. No se han conducido estudios de reproducción animal con ONCASPAR. Tampoco se sabe si ONCASPAR puede provocar daño fetal cuando se administra a mujeres embarazadas o puede afectar la capacidad de reproducción. ONCASPAR se deberá administrar a mujeres embarazadas solo si es claramente necesario.
Madres lactantes: Se desconoce si ONCASPAR se excreta en la leche humana. Debido a que muchos medicamentos se excretan en la leche humana y debido al potencial de reacciones adversas graves de ONCASPAR en bebés lactantes, se deberá tomar una decisión para suspender la lactancia o el medicamento, tomando en cuenta la importancia del medicamento para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las siguientes reacciones adversas graves se describen con mayor detalle en otras secciones:
• Anafilaxia y reacciones alérgicas graves (véase Precauciones generales).
• Trombosis grave (véase Precauciones generales).
• Pancreatitis (véase Precauciones generales)
• Intolerancia a la glucosa (véase Precauciones generales).
• Coagulopatía (véase Precauciones generales).
• Hepatotoxicidad y función hepática anormal (véase Precauciones generales).
Las reacciones adversas más comunes con ONCASPAR son reacciones alérgicas (incluida anafilaxia), hiperglucemia, pancreatitis, trombosis del sistema nervioso central (CNS, por sus siglas en inglés), coagulopatía, hiperbilirrubinemia, y transaminasas elevadas.
Se ha reportado hiperlipidemia (hipercolesterolemia e hipertrigliceridemia) en pacientes expuestos a ONCASPAR.
Experiencia en ensayos clínicos: Debido a que los ensayos clínicos se conducen bajo condiciones ampliamente variables, las tasas de reacciones adversas observadas no se pueden comparar de forma directa en otros ensayos clínicos y pueden no reflejar las tasas observadas en la práctica clínica.
Leucemia linfoblástica aguda (ALL) de primera línea: Los datos presentados a continuación se derivan de 2 estudios en pacientes con leucemia linfoblástica aguda (ALL) de riesgo estándar que recibieron ONCASPAR como un componente de la quimioterapia de múltiples agentes de primera Línea. El estudio 1 fue un estudio aleatorizado (1:1), controlado con activo que reclutó a 118 pacientes, con una mediana de edad de 4.7 años (1.1 a 9.9 años) de quienes el 54% eran varones y el 65% blancos, 14% hispanos, 8% negros, 8% asiáticos y el 6% otros. De los 59 pacientes en el estudio 1 que fueron aleatorizados a ONCASPAR, 48 pacientes (81%) recibieron todos tres dosis planeadas de ONCASPAR, seis (10%) recibieron dos dosis, 4 (7%) recibieron una dosis, y un paciente (2%) no recibió el tratamiento asignado. El estudio 2 se encuentra en curso, es de diseño multifactorial en el que todos los pacientes recibieron ONCASPAR como componente de varios agentes de quimioterapia de múltiples agentes; los datos interinos de seguridad se encuentran disponibles para 2,770 pacientes. Los participantes del estudio tenían una mediana de edad de 4 años (1 a 10 años), y el 55% eran varones, 68% blancos, 18% hispanos, 4% negros, 3% asiáticos, y 7% otros. Según el protocolo, el calendario de ONCASPAR varía por brazo de tratamiento, con dosis intermitentes de ONCASPAR hasta de 10 meses.
En el estudio 1, se recopiló información detallada de seguridad para reacciones adversas previamente especificadas, identificadas como reacciones adversas inducidas por asparaginasa y para las reacciones adversas no hemáticas de grado 3 y 4 en conformidad con los criterios de toxicidad y complicación del Grupo Oncológico Infantil (Children"s Cancer Group-CCG). La incidencia por paciente, por brazo de tratamiento, para aquellas reacciones adversas seleccionadas que se presentaron con una intensidad de grado 3 o 4, se presentan en la tabla 1 siguiente:
Tabla 1
Estudio 1: Incidencia por paciente de reacciones adversas seleccionadas grado 3 y 4
|
ONCASPAR (n = 58) |
L-asparaginasa de E. coli nativa (n = 59) |
|
|
Pruebas hepáticas anormales |
3 (5%) |
5 (8%) |
|
Transaminasas elevadas1 |
2 (3%) |
4 (7%) |
|
Hiperbilirrubinemia |
1 (2%) |
1 (2%) |
|
Hiperglucemia |
3 (5%) |
2 (3%) |
|
Trombosis del sistema nervioso central |
2 (3%) |
2 (3%) |
|
Coagulopatía2 |
1 (2%) |
3 (5%) |
|
Pancreatitis |
1 (2%) |
1 (2%) |
|
Reacciones alérgicas clínicas a la asparaginasa |
1 (2%) |
0 |
1 Aspartato aminotransferasa, alanina aminotransferasa.
2 Tiempo de protrombina o tiempo parcial de tromboplastina prolongado; o hipofibrinogenemia.
Se recopilaron datos de seguridad en el estudio 2 solo de las toxicidades no hemáticas de grado 3 y 4, criterios comunes de toxicidad del Instituto Nacional del Cáncer (National Cancer Institute-NCI CTC), versión 2.0. En este estudio, la incidencia por paciente para las siguientes reacciones adversas se presentó durante los cursos de tratamiento en los que los pacientes recibieron ONCASPAR fue: transaminasas elevadas, 11%; coagulopatía, 7%; hiperglucemia, 5%; trombosis o hemorragia del sistema nervioso central (CNS), 2%; pancreatitis, 2%; reacción alérgica clínica, 1%; e hiperbilirrubinemia, 1%. Hubo 3 defunciones debidas a la pancreatitis.
Leucemia linfoblástica aguda (ALL) previamente tratada: Se obtuvo información de reacciones adversas de cinco ensayos clínicos que reclutaron a un total de 174 pacientes con recidiva de leucemia linfoblástica aguda (ALL) que recibieron ONCASPAR como agente único o en combinación con la quimioterapia de múltiples agentes. El perfil de toxicidad de ONCASPAR en pacientes con recidiva de ALL previamente tratada es similar a la reportada arriba, a excepción de las reacciones alérgicas clínicas (véase Tabla 2). Las reacciones adversas más comunes de ONCASPAR fueron reacciones alérgicas clínicas, transaminasas elevadas, hiperbilirrubinemia, y coagulopatías. Los eventos adversos serios más comunes debidos al tratamiento de ONCASPAR fueron trombosis (4%), hiperglucemia que requiere de terapia de insulina (3%), y pancreatitis (1%).
Reacciones alérgicas: Las reacciones alérgicas incluyen las siguientes: broncoespasmo, hipotensión, edema laríngeo, eritema o inflamación local, exantema sistémico, y urticaria.
Leucemia linfoblástica aguda (ALL) de primera línea: Entre los 58 pacientes tratados con ONCASPAR, reclutados en el estudio 1, se reportaron reacciones alérgicas clínicas en dos pacientes (3%). Un paciente presentó una reacción alérgica grado 1 y el otro, ronchas grado 3; ambas se presentaron durante la primera fase de intensificación tardía del estudio (véase Tabla 2).
Leucemia linfoblástica aguda (ALL) previamente tratada: Entre 62 pacientes con recidiva de leucemia linfoblástica aguda (ALL) y reacciones de hipersensibilidad previas a asparaginasa, 35 pacientes (56%) tenían antecedentes de reacciones alérgicas clínicas a la L-asparaginasa de Escherichia coli nativa, y 27 pacientes (44%) tenían antecedentes de reacciones alérgicas clínicas a la L-asparaginasa tanto de E. coli nativa como de Erwinia nativa. Veinte (32%) de estos 62 pacientes presentaron reacciones alérgicas clínicas a ONCASPAR (véase Tabla 2).
Entre 112 pacientes con leucemia linfoblástica aguda (ALL) sin reacciones de hipersensibilidad previas a asparaginasa, 11 pacientes (10%) presentaron reacciones alérgicas clínicas a ONCASPAR (véase Tabla 2).
Tabla 2
Incidencia de reacciones alérgicas clínicas, en general y por grado de intensidad
|
Estatus del paciente |
Grado de toxicidad, n (%) |
||||
|
1 |
2 |
3 |
4 |
Total |
|
|
Pacientes previamente hipersensibles (n = 62) |
7 (11) |
8 (13) |
4 (6) |
1 (2) |
20 (32) |
|
Pacientes sin hipersensibilidad (n = 112) |
5 (4) |
4 (4) |
1 (1) |
1 (1) |
11 (10) |
|
Primera línea (n = 58) |
1 (2) |
0 |
1 (2) |
0 |
2 (3) |
Inmunogenia: Como con todas las proteínas terapéuticas, existe potencial de inmunogenia, definida como el desarrollo de anticuerpos de unión o neutralizantes al producto.
En el estudio 1, los pacientes tratados con ONCASPAR fueron evaluados para evidencia de anticuerpos de unión usando un método de ensayo de inmunoabsorción ligado a enzimas (ELISA, por sus siglas en inglés). La incidencia de la formación de anticuerpos "de título elevado" especificados en el protocolo fue del 2% en la inducción (n = 48), del 10% en la intensificación tardía 1 (n = 50), y del 11% en la intensificación tardía 2 (n = 44). Existe información insuficiente para determinar si el desarrollo de anticuerpos está asociado con un aumento en el riesgo de reacciones alérgicas clínicas, farmacocinética alterada, o pérdida de la eficacia antileucémica.
La detección de la formación de anticuerpos es altamente dependiente de la sensibilidad y especificidad del ensayo, y la incidencia observada de positividad de anticuerpos en un ensayo puede ser influida por varios factores, incluidos el manejo de la muestra, medicamentos concomitantes, y enfermedad subyacente. Por lo tanto, la comparación de la incidencia de anticuerpos a ONCASPAR con la incidencia de anticuerpos a otros productos puede ser confusa.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
• No se han conducido estudios de carcinogenia a largo plazo en animales con ONCASPAR.
• No se han conducido estudios relevantes que aborden el potencial mutagénico. ONCASPAR no mostró ningún efecto mutagénico cuando se analizó contra cepas de Salmonella typhimurium en el ensayo de Ames.
• No se han realizado estudios sobre el deterioro de la fertilidad.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se han conducido estudios de interacción medicamentosa entre ONCASPAR y otros medicamentos.
Uso pediátrico: (Véase la sección de Leucemia linfoblástica aguda [ALL] de primera línea).
Uso geriátrico: Los estudios clínicos de ONCASPAR no incluyeron cantidades suficientes de pacientes de 65 años de edad ni mayores para determinar si ellos responden de forma diferente a los pacientes más jóvenes.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No hay estudios de interacción.
PRECAUCIONES GENERALES:
Anafilaxia y reacciones alérgicas graves: Se puede presentar anafilaxia y reacciones alérgicas graves en pacientes que reciben ONCASPAR. El riesgo de reacciones alérgicas graves es más alto en pacientes con hipersensibilidad conocida a otras formas de L-asparaginasa. Observe a los pacientes durante una hora después de la administración de ONCASPAR en un ámbito con equipo de resucitación y otros agentes necesarios para tratar la anafilaxia (por ejemplo, epinefrina, oxígeno, esteroides intravenosos, y antihistamínicos). Suspenda ONCASPAR en pacientes con reacciones alérgicas graves.
Trombosis: Se pueden presentar eventos trombóticos graves, incluida la trombosis del seno sagital en pacientes que reciban ONCASPAR. Suspenda ONCASPAR en pacientes con eventos trombóticos graves.
Pancreatitis: Se puede presentar pancreatitis en pacientes que reciban ONCASPAR. Evalúe a los pacientes con dolor abdominal para evidencia de pancreatitis. Suspenda ONCASPAR en pacientes con pancreatitis.
Intolerancia a la glucosa: Se puede presentar intolerancia a glucosa en pacientes que reciban ONCASPAR. En algunos casos, la intolerancia a la glucosa es irreversible. Monitorice la glucosa en suero.
Coagulopatía: Se puede presentar aumento en el tiempo de protrombina, aumento en el tiempo parcial de tromboplastina, e hipofibrinogenemia en pacientes que reciben ONCASPAR. Monitorice los parámetros de coagulación al inicio y de forma periódica durante y después del tratamiento. Inicie el tratamiento con plasma fresco congelado para reemplazar los factores de coagulación en pacientes con coagulopatía grave o sintomática.
Hepatotoxicidad y función hepática anormal: Puede ocurrir hepatotoxicidad y función hepática anormal, incluidas las elevaciones de AST (SGOT), ALT (SGPT), fosfatasa alcalina, bilirrubina (directa e indirecta), y depresión de la albúmina sérica, y del fibrinógeno en plasma. Realice la monitorización correspondiente.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis recomendada: La dosis recomendada de ONCASPAR es de 2,500 UI/m2 por vía intramuscular o intravenosa. ONCASPAR se deberá administrar con una frecuencia no mayor a cada 14 días.
Instrucciones para la administración: Cuando ONCASPAR se administra por vía intramuscular, el volumen en un solo sitio de inyección se deberá limitar a 2 ml. Si el volumen que se administrará es > 2 ml, se deberán emplear múltiples sitios de inyección. ONCASPAR no contiene conservadores. Utilice una dosis por vial; descarte el producto no utilizado.
Cuando se administre por vía intravenosa, ONCASPAR se deberá administrar durante un periodo de 1 a 2 horas en una inyección de 100 ml de cloruro de sodio al 0.9%, a través de una infusión que se encuentre en marcha. Después de que se diluya la solución para uso intravenoso, la solución se deberá utilizar de forma inmediata. Si el uso inmediato no es posible, la solución diluida se deberá almacenar refrigerada entre 2°C y 8°C (36°F y 46°F). El almacenamiento después de la dilución no deberá exceder 48 horas desde el momento de la preparación hasta la conclusión de la administración. Proteja las bolsas de infusión de la luz solar directa.
Preparación y precauciones para el manejo:
No administre ONCASPAR si el medicamento se ha:
• Congelado.
• Almacenado a temperatura ambiental de 15°C a 25°C (59°F a 77°F) durante más de 48 horas.
• Agitado o se ha agitado vigorosamente (véase Recomendaciones de almacenamiento).
Los productos medicinales parenterales se deberán inspeccionar de forma visual para materia particulada, turbidez, o decoloración antes de la administración, siempre que la solución y el envase lo permitan. Si alguno de éstos está presente, descarte el vial.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Tres pacientes recibieron 10,000 UI/m2 de ONCASPAR como infusión intravenosa. Un paciente presentó una ligera elevación en las enzimas hepáticas. Un segundo paciente desarrolló exantema 10 minutos después del inicio de la infusión, que se controló con la administración de un antihistamínico y reduciendo la velocidad de infusión. Un tercer paciente no presentó ninguna reacción adversa.
PRESENTACIONES:
ONCASPAR es suministrado como solución estéril en viales de uso único de tipo I que contienen 3,750 UI de L-asparaginasa por 5 ml de solución.
ONCASPAR es suministrado como solución estéril isotónica clara, incolora, sin conservadores, en solución salina amortiguada con fosfato, pH 7.3. Cada mililitro contiene 750 ± 150 UI de pegaspargasa, fosfato de sodio dibásico, USP (5.58 mg), fosfato de sodio monobásico, USP (1.20) y cloruro de sodio, USP (8.50 mg) en agua para inyección, USP.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Almacene ONCASPAR en refrigeración entre 2°C y 8ºC (36°F a 46°F). No agite ni congele el producto. Proteja de la luz. No utilice ONCASPAR después de la fecha de caducidad en el vial.
LEYENDAS DE PROTECCIÓN:
Medicamento de alto riesgo. Su venta requiere receta. No lo deje al alcance de los niños. Literatura exclusiva para médicos. No se administre si la solución no es transparente, si contiene partículas suspendidas o sedimento. No se administre si el cierre fue violado. No agite. Todo producto medicinal no utilizado o material de desecho se deberá descartar en conformidad con los requisitos locales. ONCASPAR deberá ser prescrito y administrado por médicos y profesionales de la salud con experiencia en el uso de productos antineoplásicos. Solo se deberá administrar en un ámbito hospitalario en donde haya disponible el equipo de resucitación adecuado.
Reporte las reacciones adversas al correo electrónico:
farmacovigilancia@cofepris.gob.mx.
Hecho en E.U.A. por:
Sigma-Tau PharmaSource, Inc.
6925 Guion Road, Indianapolis, IN 46268
Titular del Reconocimiento como Producto Huérfano e importado por:
Baxalta México, S. de R.L. de C.V.
Av. Paseo de la Reforma No. 483, Piso 28,
Col. Cuauhtémoc, C.P. 06500,
Deleg. Cuauhtémoc, Ciudad de México, México.
Importado y distribuido por:
BAXALTA MÉXICO, S. de R.L. de C.V.
Kilómetro 12.5 de Vía Gustavo Baz Prada,
Edificio 1, Bodega 7, Almacén 8,
Col. San Pedro Barrientos, C.P. 54010,
Tlalnepantla de Baz, México, México.
Reg. Núm. 163300EL870072, SSA

