
OZEMPIC - Solución inyectable
Sustancia(s):
- Semaglutida
Presentaciones:
- 1 Caja, 1 Pluma precargada, 1.5 ml, 1.34 mg/ml
- 1 Caja, 1 Pluma precargada, 3 ml, 1.34 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
Solución para inyección en pluma precargada
Cada ml contiene:
Semaglutida 1.34 mg
Vehículo cbp 1 ml
Análogo humano del péptido similar al glucagón tipo 1 (GLP-1) de origen ADN recombinante expresado en Saccharomyces cerevisiae.
Ozempic® 0.25 mg, 0.5 mg/dosis:
Un ml de solución contiene 1.34 mg de semaglutida. Una pluma precargada contiene 2 mg de semaglutida en 1.5 ml de solución.
Ozempic® solución inyectable de 0.25 mg ó 0.5 mg/dosis en pluma precargada.
Ozempic® 1 mg/dosis:
Un ml de solución contiene 1.34 mg de semaglutida. Una pluma precargada contiene 4 mg de semaglutida en 3 ml de solución.
Ozempic®, solución inyectable de 1 mg/dosis en pluma precargada.
*Denominación Común Internacional (DCI).
INDICACIONES TERAPÉUTICAS:
Ozempic® está indicado como un complemento de la dieta y el ejercicio en adultos con diabetes mellitus tipo 2 como:
• Monoterapia.
• Terapia de combinación con otros medicamentos para el tratamiento de la diabetes.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades Farmacocinéticas:
Semaglutida tiene propiedades farmacocinéticas compatibles con una administración semanal con una vida media de eliminación de aproximadamente 1 semana.
Absorción:
La concentración máxima se alcanzó entre 1 y 3 días después de la dosis.
El estado estacionario se alcanzó después de 4-5 semanas de la administración una vez a la semana. En pacientes con diabetes tipo 2, las concentraciones medias en estado estacionario tras la administración subcutánea de 0.5 mg y 1 mg de semaglutida fueron de aproximadamente 16 nmol/l y 30 nmol/l, respectivamente.
Para las dosis de 0.5 mg y 1 mg la exposición a semaglutida aumentó de forma proporcional a la dosis.
Asímismo, se logró una exposición similar con la administración de semaglutida subcutánea en el abdomen, el muslo o la parte superior del brazo.
La biodisponibilidad absoluta de semaglutida subcutánea fue del 89%.
Distribución:
El volumen medio de distribución de semaglutida tras la administración subcutánea en pacientes con diabetes tipo 2 fue de aproximadamente 12.5 l. Semaglutida se encontraba ampliamente unido a albúmina en plasma (>99%).
Metabolismo/biotransformación:
Semaglutida se metaboliza a través de proteólisis de su estructura peptídica y a través de beta-oxidación secuencial de la cadena lateral del ácido graso.
Eliminación:
Las principales vías de excreción de los productos relacionados con semaglutida fueron a través de la orina y las heces. Aproximadamente 3% de la dosis se excretó en forma de semaglutida intacta en orina.
En pacientes con diabetes tipo 2, el aclaramiento de semaglutida fue de 0.05 l/hora aproximadamente. Con una vida media de eliminación aproximada de una semana, semaglutida permanecerá en la circulación durante un tiempo de 5 semanas después de la última dosis.
Poblaciones especiales:
No es necesario ningún ajuste de dosis de semaglutida basado en la edad, género, raza, origen étnico, peso corporal, deterioro renal o hepático. Los efectos de factores intrínsecos en la farmacocinética de semaglutida se muestran en la figura 1.
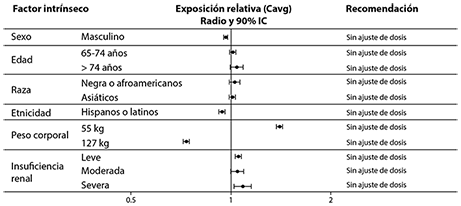
La exposición relativa de semaglutida (Cavg) en relación con el perfil del sujeto de referencia: mujer no-hispana/no latina, blanca, menor a 65 años, peso corporal de 85 kg, con función renal normal. El modelo de población farmacocinética (PK) también incluyó el sitio de inyección y dosis de mantenimiento como covariables. Las categorías de estudio de peso corporal (127 y 55 kg) representan los percentiles 5% y 95% en el conjunto de datos. Las dosis de semaglutida de 0.5 mg y 1 mg proporcionan adecuada exposición sistémica sobre el rango de peso corporal de 40-198 kg evaluado en los estudios clínicos.
Abreviaturas: Cavg: promedio de concentración de semaglutida. IC: Intervalo de confianza.
Figura 1. Impacto de los factores intrínsecos en la exposición de semaglutida
Edad:
La edad no tuvo efecto sobre la farmacocinética de semaglutida, según los resultados de los estudios de fase 3a realizados, que incluyeron a pacientes de 20-86 años de edad.
Género:
El género no tuvo efecto sobre la farmacocinética de semaglutida.
Raza:
Raza (blanca, negra o afroamericana, asiática) no tuvo efecto sobre la farmacocinética de semaglutida.
Grupo étnico:
La etnia (hispana o latina) no tuvieron ningún efecto sobre la farmacocinética de semaglutida.
Peso corporal:
El peso corporal tiene efecto en la exposición de semaglutida. Cuanto mayor es el peso corporal menor es la exposición. Las dosis de 0.5 mg y 1 mg de semaglutida proporcionan una exposición sistémica adecuada en el rango de peso corporal de 40-198 kg evaluado en los estudios clínicos.
Insuficiencia renal:
La insuficiencia renal no tuvo ningún efecto clínicamente significativo sobre la farmacocinética de semaglutida. Esto fue demostrado comparando los efectos de una dosis única de semaglutida de 0.5 mg en pacientes con diversos grados de insuficiencia renal (leve, moderada, grave o en pacientes en diálisis) vs. sujetos con función renal normal. Los datos de los estudios de fase 3a realizados confirmaron esto mismo en sujetos con diabetes tipo 2 e insuficiencia renal (Figura 1), aunque la experiencia en pacientes con enfermedad renal en etapa terminal fue limitada.
Insuficiencia hepática:
La insuficiencia hepática no tuvo ningún efecto en la exposición de semaglutida. La farmacocinética de semaglutida se evaluó en pacientes con diversos grados de insuficiencia hepática (leve, moderada, grave) comparados con sujetos con función hepática normal en un estudio en el que se utilizó dosis única de 0.5 mg de semaglutida.
Pediatría:
Semaglutida no se ha estudiado en pacientes pediátricos.
Farmacodinamia:
Propiedades farmacodinámicas:
Clase farmacológica agonista del receptor del péptido similar al glucagón tipo 1 (GLP-1 por sus siglas en inglés). Código ATC: A10BJ06.
Mecanismo de acción:
Semaglutida es un análogo de GLP-1 con un 94% de homología de secuencia con el GLP-1 humano. Semaglutida actúa como un agonista del receptor de GLP-1 que se une de forma selectiva al receptor de GLP-1 (el objetivo del GLP-1 humano o nativo) y lo activa.
El GLP-1 es una hormona fisiológica que desempeña diversas funciones en la regulación de la glucosa y el apetito, así como en el sistema cardiovascular. Los efectos sobre la glucosa y el apetito son específicamente mediados a través de los receptores de GLP-1 en el páncreas y el cerebro. Semaglutida trabaja a niveles farmacológicos disminuyendo la glucosa en sangre y reduciendo el peso mediante una combinación de efectos que se describen a continuación.
Semaglutida reduce la glucosa en sangre a través de un mecanismo en el que estimula la secreción de insulina y disminuye la secreción de glucagón, ambos de manera dependiente de la glucosa. Por lo tanto, cuando la glucosa en sangre es alta, se estimula la secreción de insulina y se inhibe la secreción de glucagón. El mecanismo de reducción de la glucosa en sangre también implica un retraso leve en el vaciamiento gástrico en la fase postprandial temprana. Durante la hipoglucemia, semaglutida disminuye la secreción de insulina y no altera la secreción de glucagón.
Semaglutida reduce el peso corporal y masa grasa corporal mediante la disminución del aporte calórico. El mecanismo implica reducción general del apetito, que incluye un aumento de la saciedad y reducción del apetito así como un mejor control de la ingesta y la disminución de la ansiedad por comer. La resistencia a la insulina también se reduce, probablemente a través de la reducción del peso corporal. Además, semaglutida reduce la preferencia por alimentos altos en grasas. En estudios con animales, semaglutida es captada en regiones cerebrales específicas e incrementa las señales clave en saciedad y disminuye las señales clave de apetito. Utilizando secciones aisladas en el tejido cerebral, semaglutida activa las neuronas relacionadas con saciedad e inhibe las neuronas relacionadas con el apetito.
Los receptores de GLP-1 se expresan también en el corazón, sistema vascular, sistema inmune y los riñones donde pueden mediar efectos cardiovasculares y microvasculares; se ha observado disminución en la presión arterial sistólica, y reducción en marcadores inflamatorios seleccionados observados en estudios clínicos.
En comparación con el GLP-1 humano o nativo, semaglutida tiene una vida media prolongada de alrededor de 1 semana, por lo que es adecuada para su administración subcutánea una vez por semana. El mecanismo principal de prolongación de acción, es la unión a la albúmina, que da como resultado una disminución del aclaramiento renal y la protección contra la degradación metabólica. Además, semaglutida se estabiliza contra la degradación por la enzima DPP-4.
Semaglutida tuvo un efecto benéfico sobre lípidos plasmáticos, la presión arterial sistólica disminuida y la reducción de la inflamación en estudios clínicos.
En estudios en animales, semaglutida atenúa el desarrollo de la aterosclerosis al prevenir la progresión de la placa aórtica y reducir la inflamación en la placa.
Datos farmacodinámicos:
Todas las evaluaciones farmacodinámicas se realizaron transcurridas 12 semanas de tratamiento (incluido el escalamiento de dosis) en estado estacionario con 1 mg de semaglutida una vez por semana.
Glucosa en ayuno y postprandial:
Semaglutida reduce las concentraciones de glucosa en ayuno y postprandial. En pacientes con diabetes tipo 2, el tratamiento con 1 mg de semaglutida, en comparación con placebo, logró reducciones en la glucosa en términos de cambio absoluto respecto al valor de referencia (mg/dl) y reducción relativa (%) en los valores de glucosa en ayuno (29 mg/dl; 22%), glucosa postprandial a las 2 horas (74 mg/dl; 37%), concentración media de glucosa en 24 horas (30 mg/dl; 22% reducción) y fluctuación de la glucosa postprandial durante 3 comidas (11-20 mg/dl) en comparación con placebo (ver figura 2).
Semaglutida redujo la glucosa en ayuno después de la administración de la primera dosis.
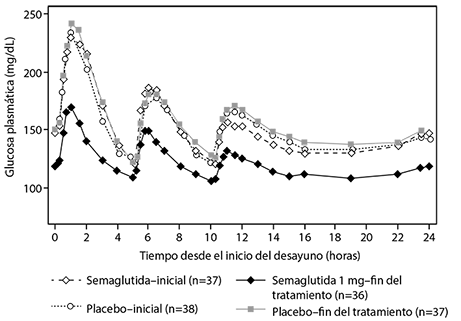
Figura 2. Media de los perfiles de glucosa plasmática en 24 horas (comidas estandarizadas) en pacientes con diabetes tipo 2 antes (línea basal) y después de 12 semanas de tratamiento con semaglutida o placebo.
Función de las células beta y secreción de la insulina:
Semaglutida mejora la función de las células beta. Semaglutida, en comparación con el placebo, mejoró la respuesta a la insulina durante la primera y segunda fase con un aumento que triplicó y duplicó ésta, respectivamente, cuando se administró un bolo intravenoso de glucosa, e incrementó la capacidad máxima secretora de las células beta después de una prueba de estimulación de la arginina en pacientes con diabetes tipo 2. Además, el tratamiento con semaglutida incrementó las concentraciones de insulina en ayuno comparadas con placebo.
Secreción de glucagón:
Semaglutida disminuye las concentraciones de glucagón en ayuno y postprandial. En pacientes con diabetes tipo 2, en comparación con placebo, semaglutida logró las siguientes reducciones relativas de glucagón: glucagón en ayuno (8-21%), respuesta de glucagón postprandial (14- 15%) y concentración media de glucagón a las 24 horas (12%).
Secreción de glucagón e insulina dependiente de la glucosa:
Semaglutida disminuyó las concentraciones elevadas de glucosa en sangre mediante la estimulación de la secreción de insulina y la disminución de la secreción de glucagón de un modo dependiente de la glucosa. Con semaglutida, la tasa de secreción de insulina en pacientes con diabetes tipo 2 fue comparable a la de los sujetos sanos (ver figura 3).
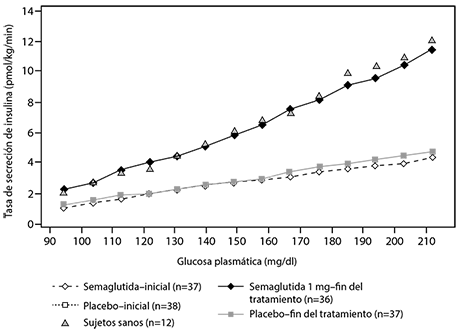
Figura 3 Media de la tasa de secreción de insulina frente a la concentración de glucosa en pacientes con diabetes tipo 2 durante la infusión de glucosa gradual antes (inicial) y después de 12 semanas de tratamiento con semaglutida o con placebo y en sujetos sanos no tratados.
Durante la hipoglucemia inducida, en comparación con placebo, semaglutida no alteró las respuestas contrarreguladoras del aumento de glucagón y tampoco afectó a la disminución del péptido C en pacientes con diabetes tipo 2.
Vaciamiento gástrico:
Semaglutida causó un ligero retraso del vaciamiento gástrico en la fase postprandial temprana, reduciendo así la velocidad a la que la glucosa aparece en la circulación postprandialmente.
Peso corporal y composición corporal:
Se observó una mayor reducción en el peso corporal con semaglutida en relación con otros tratamientos comparativos (placebo, sitagliptina, exenatida ER e insulina glargina) (sección 5. propiedades farmacodinámicas. Peso corporal). La pérdida de peso con semaglutida fue predominantemente para tejido graso con pérdida de masa grasa que era 3 veces mayor que la pérdida de masa magra.
Apetito, ingesta de calórica y elecciones alimentarias:
Semaglutida comparada con placebo disminuyó la ingesta calórica de 3 comidas consecutivas a voluntad en un 18-35%. Este hecho se vio favorecido por otros efectos de semaglutida como la supresión del apetito en ayuno y postprandial, un mejor control de la ingesta, una disminución en la ansiedad por comer y una preferencia relativamente menor por alimentos ricos en grasas.
Lípidos en ayuno y postprandial:
Semaglutida comparada con placebo, redujo las concentraciones de triglicéridos y colesterol de lipoproteínas de muy baja densidad (VLDL) en ayuno en 12% y 21% respectivamente. Los triglicéridos y colesterol VLDL postprandiales en respuesta a una comida alta en grasas se redujeron en >40%.
Electrofisiología cardiaca (QTc)
El efecto de semaglutida sobre la repolarización cardiaca se evaluó en un estudio exhaustivo de QTc. Semaglutida no prolongó los intervalos QTc en dosis supraterapéuticas (hasta 1.5 mg en estado estacionario).
Estudios clínicos:
Información sobre la eficacia y seguridad clínica:
Tanto la mejora del control glucémico como la reducción de morbilidad y mortalidad cardiovascular son una parte integral del tratamiento de la diabetes tipo 2.
La eficacia y seguridad de Ozempic® 0.5 mg y 1 mg administrado una vez por semana, se evaluaron en seis estudios clínicos controlados de fase 3a. De estos, cinco estudios (SUSTAIN 1-5) el objetivo principal fue la evaluación de la eficacia glucémica, mientras que un estudio (SUSTAIN 6) tuvo como objetivo primario evaluar el resultado en desenlaces cardiovasculares. Adicionalmente, dos estudios de la fase 3 se realizaron con Ozempic® en pacientes japoneses. Los estudios incluyeron en total 8,124 pacientes aleatorizados con diabetes tipo 2 (4,792 tratados con Ozempic®).
Un estudio adicional que incluyó 1,201 pacientes, se realizó para comparar la eficacia y seguridad de Ozempic® 0.5 mg y 1 mg una vez por semana versus dulaglutida 0.75 y 1.5 mg una vez por semana, respectivamente.
El tratamiento con Ozempic® demostró reducciones sostenidas, estadísticamente superiores y clínicamente significativas en la reducción de hemoglobina glucosilada (HbA1C) (ver figura 4) y peso corporal en un periodo de hasta 2 años en comparación con el placebo y el tratamiento control activo (sitagliptina, insulina glargina, exenatida ER y dulaglutida).

Figura 4. (%) de hemoglobina glucosilada -Cambio estimado desde el inicio al final del tratamiento en SUSTAIN 1-7, monoterapia japonesa y combinación de HGO* Japón estudios (semaglutida gris oscuro de 0.5 mg, semaglutida 1 mg negro, tratamiento competidores blanco y placebo gris claro. *Hipoglucemiantes Orales.
La eficacia de Ozempic® no se vio afectada por la edad, género, raza, grupo étnico, índice de masa corporal al inicio (IMC), peso corporal (kg) al inicio, duración de la diabetes y nivel de deterioro de la función renal.
Información sobre la eficacia y seguridad clínica:
SUSTAIN 1 - Monoterapia:
En un estudio doble ciego, controlado con placebo, a 30 semanas. Se aleatorizaron 388 pacientes inadecuadamente controlados con dieta y ejercicio para recibir Ozempic® 0.5 mg u Ozempic® 1 mg una vez por semana o placebo.
Los pacientes tenían edad promedio de 54 años y duración media de la diabetes tipo 2 de 4.2 años. Hubo 64% de pacientes blancos, 8% eran de raza negra o afroamericanos y 21% eran asiáticos. Por grupo étnico, el 30% de los pacientes (n = 115) eran hispanos o latinos. La media de IMC fue de 33 kg/m2.
La monoterapia con Ozempic® 0.5 mg y 1 mg una vez por semana durante 30 semanas resultó en reducciones estadísticamente superiores en la prueba de hemoglobina glucosilada (HbA1C) y el peso corporal en comparación con el placebo (véase tabla 1).
Tabla 1. Resultados de los estudios de una a 30 semanas de monoterapia (SUSTAIN 1)
|
Ozempic® 0.5 mg |
Ozempic® 1 mg |
Placebo |
|
|
Población (n) con intención de tratar (ITT por sus siglas en inglés) |
128 |
130 |
129 |
|
Hemoglobina glucosilada (HbA1c) (%) |
|||
|
Basal (media) |
8.1 |
8.1 |
8.0 |
|
Cambio del inicio a la semana 30 |
-1.5 |
-1.6 |
0 |
|
Diferencia del placebo [IC del 95%] |
-1.4 [-1.7; -1.1]ª |
-1.5 [-1.8; -1.2]ª |
- |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) <7% |
74b |
72b |
25 |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) ≤ 6.5% |
59b |
60b |
13 |
|
GPA (mg/dl) |
|||
|
Basal (media) |
174 |
179 |
174 |
|
Cambio del inicio a la semana 30 |
-45 |
-42 |
-10 |
|
Diferencia del placebo [IC del 95%] |
-35 [-45; -26]b |
-32 [-42; -23]b |
- |
|
Peso corporal (kg) |
|||
|
Basal (media) |
89.8 |
96.9 |
89.1 |
|
Cambio del inicio a la semana 30 |
-3.7 |
-4.5 |
-1.0 |
|
Diferencia del placebo [IC del 95%] |
-2.7 [-3.9; -1.6]a |
-3.6 [-4.7; -2.4]a |
- |
|
(%) de pacientes que lograron una pérdida de peso ≥ 5% |
37b |
45b |
7 |
|
(%) de pacientes que lograron una pérdida de peso ≥ 10% |
8c |
13c |
2 |
ª p <0.0001 (por ambos lados) por superioridad, ajuste por multiplicidad basado en las pruebas jerárquicas de la hemoglobina glucosilada (HbA1c) y el peso corporal.
b p <0.0001 de diferencia del tratamiento, no ajustado por multiplicidad.
c p <0.05 de diferencia del tratamiento, no ajustado por multiplicidad.
SUSTAIN 2 - Ozempic® vs sitagliptina ambos en combinación con 1-2 medicamentos antidiabéticos (metformina y/o tiazolidinedionas):
En un estudio doble ciego de 56 semanas, 1,231 pacientes fueron aleatorizados para recibir Ozempic® 0.5 mg o Ozempic® 1 mg una vez por semana o sitagliptina 100 mg una vez al día, todos en combinación con metformina (94%) y/o tiazolidinedionas (6%). Los pacientes tenían una edad promedio de 55 años y una duración media de la diabetes tipo 2 de 6.6 años. Hubo 68% de pacientes blancos, 5% de raza negra o afroamericanos y 25% asiáticos. Por grupo étnico, el 17% de los pacientes (n = 209) eran hispanos o latinos. La media del IMC fue de 32 kg/m2.
El tratamiento con Ozempic® 0.5 mg y 1 mg, una vez por semana durante 56 semanas, resultó en reducciones estadísticamente superiores en la hemoglobina glucosilada (HbA1C) y el peso corporal en comparación con sitagliptina (véase tabla 2). De una media basal de 132.6 mmHg, el tratamiento con Ozempic® 0.5 mg y 1 mg redujo significativamente la presión arterial sistólica en comparación con sitagliptina (-5.1 mmHg; -5.6 mmHg vs -2.3 mmHg, p<0.05). No hubo cambios en la presión arterial diastólica.
Tabla 2. Resultados del estudio de 56 semanas de Ozempic® en comparación con sitagliptina (SUSTAIN 2)
|
Ozempic® 0.5 mg |
Ozempic® 1 mg |
Sitagliptina 100 mg |
|
|
Población (n) con intención de tratar (ITT por sus siglas en inglés) |
409 |
409 |
407 |
|
(%) de hemoglobina glucosilada (HbA1c) |
|||
|
Basal (media) |
8.0 |
8.0 |
8.2 |
|
Cambio del inicio a la semana 56 |
-1.3 |
-1.6 |
-0.5 |
|
Diferencia de sitagliptina [IC del 95%] |
-0.8 [-0.9; -0.6]a |
-1.1 [-1.2; 0.9]a |
- |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) <7% |
69b |
78b |
36 |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) ≤6.5% |
53b |
66b |
20 |
|
GPA (mg/dl) |
|||
|
Basal (media) |
168 |
167 |
173 |
|
Cambio del inicio a la semana 56 |
-37 |
-47 |
-20 |
|
Diferencia del sitagliptina [IC del 95%] |
-18 [-23; --12]b |
-27 [-32; -22]b |
|
|
Peso corporal (kg) |
|||
|
Basal (media) |
89.9 |
89.2 |
89.3 |
|
Cambio del inicio a la semana 56 |
-4.3 |
-6.1 |
-1.9 |
|
Diferencia de sitagliptina [IC del 95%] |
-2.3 [-3.1; -1.6]a |
-4.2 [-4.9; 3.5]a |
- |
|
(%) de pacientes que lograron una pérdida de peso ≥ 5% |
46b |
62b |
18 |
|
(%) de pacientes que lograron una pérdida de peso ≥ 10% |
13b |
24b |
3 |
ª p <0.0001 (por ambos lados) por superioridad, ajuste por multiplicidad basado en las pruebas jerárquicas de la hemoglobina glucosilada (HbA1c) y el peso corporal.
b p <0.0001 para diferencia del tratamiento, no ajustado por multiplicidad.

Figura 5. Cambio promedio en el (%) de hemoglobina glucosilada (HbA1c) y en el peso corporal (kg) desde el inicio del estudio hasta la semana 56 (SUSTAIN 2).
SUSTAIN 7 - Ozempic® vs dulaglutida ambos en combinación con metformina:
En un estudio de 40 semanas abierto, 1,201 pacientes con metformina fueron aleatorizados a cualquiera de los cuatro, Ozempic® 0.5 mg, Ozempic® 1 mg, dulaglutida 0.75 mg o dulaglutida 1.5 mg; todos una vez por semana. El estudio comparó las dosis 0.5 mg de semaglutida con 0.75 mg de dulaglutida y las dosis de 1 mg de semaglutida con 1.5 mg de dulaglutida. Los pacientes tenían una edad promedio de 56 años y una duración media de la diabetes tipo 2 de 7.4 años. Hubo 77% de pacientes blancos, 6% de negros o afroamericanos y 16% eran asiáticos. Por grupo étnico, el 11% de los pacientes (n= 138) eran hispanos o latinos. La media del IMC fue de 33.5 kg/m2.
Tabla 3 Resultados del estudio a la semana 40 de Ozempic® versus dulaglutida (SUSTAIN 7)
|
Ozempic® 0.5 mg |
Ozempic® 1 mg |
Dulaglutida 0.75 mg |
Dulaglutida 1.5 mg |
|
|
Población (n) con intención de tratar (ITT por sus siglas en inglés) |
301 |
300 |
299 |
299 |
|
(%) de hemoglobina glucosilada (HbA1c) |
||||
|
Basal (media) |
8.3 |
8.2 |
8.2 |
8.2 |
|
Cambio desde el inicio a la semana 40 |
-1.5 |
-1.8 |
-1.1 |
-1.4 |
|
Diferencia de dulaglutida [IC del 95%] |
-0.4 [-0.6; -0.2]a,c |
-0.4 [-0.6; -0.2]a,d |
- |
- |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) <7% |
68b |
79b |
52 |
67 |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) ≤ 6.5% |
49b |
67b |
34 |
47 |
|
GPA (mg/dl) |
||||
|
Basal (media) |
176 |
177 |
174 |
172 |
|
Cambio del inicio a la semana 40 |
-39 |
-51 |
-34 |
-40 |
|
Diferencia del dulaglutida [IC del 95%] |
-65 [-11; 0]b,c |
-110 [-16; ]b,d |
||
|
Peso corporal (kg) |
||||
|
Basal (media) |
96.4 |
95.5 |
95.6 |
93.4 |
|
Cambio desde el inicio a la semana 40 |
-4.6 |
-6.5 |
-2.3 |
-3.0 |
|
Diferencia de dulaglutida [IC del 95%] |
-2.3 [-3.0; 1.5]a,c |
-3.5 [-4.3; -2.8]a,d |
- |
|
|
(%) de pacientes que lograron una pérdida de peso ≥ 5% |
44b |
63b |
23 |
30 |
|
(%) de pacientes que lograron una pérdida de peso ≥ 10% |
14b |
27b |
3 |
8 |
ª p <0.0001 (por ambos lados) por superioridad, ajuste por multiplicidad basado en las pruebas jerárquicas de la hemoglobina glucosilada (HbA1c) y el peso corporal.
b p <0.001 para diferencia del tratamiento, no ajustado por multiplicidad.
c Ozempic® 0.5 mg vs. dulaglutida 0.75 mg.
d Ozempic® 1 mg vs. dulaglutida 1.5 mg.

Figura 6 Cambio promedio en el (%) de hemoglobina glucosilada (HbA1c) y en el peso corporal (kg) desde el inicio a la semana 40.
SUSTAIN 3 - Ozempic® vs exenatida ER ambos en combinación con metformina o metformina con sulfonilurea:
En un estudio abierto de 56 semanas, se aleatorizaron 813 pacientes con sólo metformina (49%), metformina con sulfonilurea (45%) u otros (6%) para recibir Ozempic® 1 mg o exenatida ER 2.0 mg una vez por semana. Los pacientes tenían una edad promedio de 57 años y una duración media de la diabetes tipo 2 de 9 años. Hubo 84% de pacientes blancos, 7% de negros o afroamericanos y 2% eran asiáticos. Por grupo étnico, el 24% de los pacientes (n=197) eran hispanos o latinos. La media del IMC fue de 34 kg/m2.
El tratamiento con Ozempic® 1 mg una vez por semana durante 56 semanas, resultó en reducciones estadísticamente superiores en la hemoglobina glucosilada (HbA1C) y el peso corporal en comparación con exenatida ER de 2.0 mg.
Tabla 4 Resultados del estudio de 56 semanas de Ozempic® versus exenatida ER (SUSTAIN 3)
|
Ozempic® 1 mg |
Exenatida ER 2.0 mg |
|
|
Población (n) con intención de tratar (ITT por sus siglas en inglés) |
404 |
405 |
|
(%) de hemoglobina glucosilada (HbA1c) |
||
|
Basal (media) |
8.4 |
8.3 |
|
Cambio desde inicio a la semana 56 |
-1.5 |
-0.9 |
|
Diferencia de exenatida ER [IC del 95%] |
-0.6 [-0.8; -0.4]a |
- |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) <7% |
67b |
40 |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) ≤ 6.5% |
47b |
22 |
|
GPA (mg/dl) |
||
|
Basal (media) |
191 |
188 |
|
Cambio del inicio a la semana 56 |
-51 |
-36 |
|
Diferencia de exenatida ER [IC del 95%] |
-15 [-22; -8]b |
- |
|
Peso corporal (kg) |
||
|
Basal (media) |
96.2 |
95.4 |
|
Cambio desde inicio a la semana 56 |
-5.6 |
-1.9 |
|
Diferencia de exenatida ER [IC del 95%] |
-3.8 [-4.6; -3.0]a |
- |
|
(%) de pacientes que lograron una pérdida de peso ≥ 5% |
52b |
17 |
|
(%) de pacientes que lograron una pérdida de peso ≥ 10% |
21b |
4 |
ª p <0.0001 (por ambos lados) por superioridad, ajuste por multiplicidad basado en las pruebas jerárquicas de la hemoglobina glucosilada (HbA1c) y el peso corporal.
b p <0.0001 para diferencia del tratamiento, no ajustado por multiplicidad.
SUSTAIN 4 - Ozempic® vs insulina glargina ambos en combinación con 1-2 medicamentos antidiabéticos orales (monoterapia con metformina o metformina y sulfonilurea):
En un estudio clínico abierto de 30 semanas, se aleatorizaron 1,089 pacientes para recibir Ozempic® 0.5 mg una vez por semana, Ozempic® 1 mg una vez por semana o insulina glargina una vez al día, además del tratamiento existente con metformina (48%) o metformina y sulfonilurea (51%).
Pacientes en insulina glargina iniciaron con 10 U inyectado una vez al día. La dosis diaria promedio de insulina al final del estudio fue 29 U por día.
Los pacientes tenían una edad promedio de 57 años y una duración media de la diabetes tipo 2 de 8.6 años, 77% eran blancos, 9% eran negros o afroamericanos y 11% eran asiáticos. Por grupo étnico, el 20% de los pacientes (n=213) eran hispanos o latinos. La media del IMC fue de 33 kg/m2.
El tratamiento con Ozempic® 0.5 mg y 1 mg una vez por semana durante 30 semanas resultó estadísticamente superior en reducción de la hemoglobina glucosilada (HbA1C) y el peso corporal en comparación con insulina glargina (ver tabla 4). La proporción de pacientes que reportaron episodios hipoglucémicos graves o con glucosa en sangre confirmada (<56 mg/dl) fue menor con Ozempic® 0.5 mg (4.4%), y Ozempic® 1 mg (5.6%) en comparación con insulina glargina (10.6%).
Más pacientes en Ozempic® 0.5 mg (47%) y Ozempic® 1 mg (64%) alcanzaron una hemoglobina glucosilada (HbA1C) <7% sin presentar hipoglucemia sintomática grave o con glucosa sanguínea confirmada y sin aumento de peso en comparación con insulina glargina (16%).
Tabla 5. Resultados del estudio de 30 semanas de Ozempic® comparado con insulina glargina (SUSTAIN 4)
|
Ozempic® 0.5 mg |
Ozempic® 1 mg |
Insulina glargina |
|
|
Población (n) con intención de tratar (ITT por sus siglas en inglés) |
362 |
360 |
360 |
|
(%) de hemoglobina glucosilada (HbA1c) |
|||
|
Basal (media) |
8.1 |
8.2 |
8.1 |
|
Cambio desde el inicio a la semana 30 |
-1.2 |
-1.6 |
-0.8 |
|
Diferencia de insulina glargina [IC del 95%] |
-0.4 [-0.5; -0.2]a |
-0.8 [-1.0; -0.7]a |
- |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) <7% |
57b |
73b |
38 |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) ≤ 6.5% |
37b |
54b |
18 |
|
GPA (mg/dl) |
|||
|
Basal (media) |
172 |
179 |
174 |
|
Cambio desde el inicio a la semana 30 |
-37 |
-49 |
-38 |
|
Diferencia de insulina glargina [IC del 95%] |
1 [-4; 7] |
-11 [-17; --5]b |
- |
|
Peso corporal (kg) |
|||
|
Basal (media) |
93.7 |
94.0 |
92.6 |
|
Cambio desde inicio a la semana 30 |
-3.5 |
-5.2 |
+1.2 |
|
Diferencia de insulina glargina [IC del 95%] |
-4.6 [-5.3; -4.0]a |
-6.3 [-7.0; -5.7]a |
- |
|
(%) de pacientes que lograron una pérdida de peso ≥ 5% |
37b |
51b |
4 |
|
(%) de pacientes que lograron una pérdida de peso ≥ 10% |
7b |
16b |
1 |
ª p <0.0001 (por ambos lados) por superioridad, ajuste por multiplicidad basado en las pruebas jerárquicas de la hemoglobina glucosilada (HbA1c) y el peso corporal.
b p <0.001 para diferencia del tratamiento, no ajustado por multiplicidad.
SUSTAIN 5 - Ozempic® vs placebo en combinación con insulina basal:
En un estudio clínico doble ciego de 30 semanas, se aleatorizaron 397 pacientes inadecuadamente controlados con insulina basal con o sin metformina para recibir Ozempic® 0.5 mg o 1 mg de Ozempic® una vez por semana o placebo. Pacientes con hemoglobina glucosilada (HbA1C) ≤8.0% a la selección, redujeron la dosis de insulina en un 20% al comienzo del estudio para reducir el riesgo de hipoglucemia.
Los pacientes tenían una edad promedio de 59 años y una duración media de la diabetes tipo 2 de 13 años, 78% eran blancos, 5% eran negros o afroamericanos y 17% eran asiáticos. Por grupo étnico, el 12% de los pacientes (n=46) eran hispanos o latinos. La media del IMC fue de 32 kg/m2.
El tratamiento con Ozempic® 0.5 mg y 1 mg resultó en una reducción estadísticamente superior de hemoglobina glucosilada (HbA1C) después de 30 semanas de tratamiento comparado a placebo. Episodios sintomáticos de hipoglucemia graves o con glucosa sanguínea confirmada (GS) no fueron significativamente diferentes entre Ozempic® y el placebo. La proporción de pacientes reportando episodios de hipoglucemia sintomática ( <56 mg/dl) fue mayor con Ozempic® comparado con placebo al inicio con hemoglobina glucosilada (HbA1C) <8%.
Tabla 6. Resultados del estudio de 30 semanas de Ozempic® en combinación con insulina basal con o sin metformina (SUSTAIN 5)
|
Ozempic® 0.5 mg |
Ozempic® 1 mg |
Placebo |
|
|
Población (n) con intención de tratar (ITT por sus siglas en inglés) |
132 |
131 |
133 |
|
(%) de hemoglobina glucosilada (HbA1c) |
|||
|
Basal (media) |
8.4 |
8.3 |
8.4 |
|
Cambio desde el inicio a la semana 30 |
-1.4 |
-1.8 |
-0.1 |
|
Diferencia del placebo [IC del 95%] |
-1.4 [-1.6; -1.1]a |
-1.8 [-2.0; -1.5]a |
- |
|
(%) de pacientes que alcanzaron la hemoglobina glucosilada (HbA1c) <7% |
61b |
79b |
11 |
|
(%) de pacientes que obtuvieron una hemoglobina glucosilada (HbA1c) ≤ 6.5% |
41b |
61b |
5 |
|
GPA (mg/dl) |
|||
|
Basal (media) |
161 |
153 |
154 |
|
Cambio desde el inicio a la semana 30 |
-29 |
-42 |
-9 |
|
Diferencia del placebo [IC del 95%] |
-21 [-31; --10]c |
-34 [-45; --23]b |
- |
|
Peso corporal (kg) |
|||
|
Basal (media) |
92.7 |
92.5 |
89.9 |
|
Cambio desde el inicio a la semana 30 |
-3.7 |
-6.4 |
-1.4 |
|
Diferencia del placebo [IC del 95%] |
-2.3 [-3.3; -1.3]a |
-5.1 [-6.1; -4.0]a |
- |
|
(%) de pacientes que lograron una pérdida de peso ≥ 5% |
42b |
66b |
11 |
|
(%) de pacientes que lograron una pérdida de peso ≥ 10% |
9c |
26b |
3 |
ª p <0.0001 (por ambos lados) por superioridad, ajuste por multiplicidad basado en las pruebas jerárquicas de la hemoglobina glucosilada (HbA1c) y el peso corporal.
b p <0.001 para diferencia del tratamiento, no ajustado por multiplicidad.
c p <0.05 para diferencia del tratamiento, no ajustado por multiplicidad.
Combinación con la monoterapia de sulfonilurea:
En SUSTAIN 6 (ver subsección reducción de enfermedad cardiovascular), un subgrupo de monoterapia con sulfonilurea fue evaluado en la semana 30. Había 123 pacientes en monoterapia con sulfonilurea al inicio del estudio. La hemoglobina glucosilada (HbA1C) al inicio del estudio fue de 8.2%, 8.4% y 8.4% para Ozempic® 0.5 mg, Ozempic® 1 mg y placebo, respectivamente. En la semana 30, el cambio en la hemoglobina glucosilada (HbA1C) fue de -1.6%, -1.5% y 0.1 % para Ozempic® 0.5 mg, Ozempic® 1 mg y placebo respectivamente.
Combinación con premezcla de insulina ± 1-2 ADOs (Antidiabéticos Orales):
En SUSTAIN 6 (ver subsección reducción de enfermedades cardiovasculares), un subgrupo de insulina pre-mezclada (con o sin 2 ADOs) fue evaluado en la semana 30. Había 867 pacientes con una pre-mezcla de insulina al inicio del estudio. La hemoglobina glucosilada (HbA1C) al inicio del estudio fue de 8.8%, 8.9% y 8.9% para Ozempic® 0.5 mg, Ozempic® 1 mg y placebo, respectivamente. En la semana 30, el cambio en la hemoglobina glucosilada (HbA1C) fue de - 1.3%, -1.8% y -0.4% para Ozempic® 0.5 mg, Ozempic® 1 mg y placebo respectivamente.
Proporción de pacientes que alcanzaron los objetivos de hemoglobina glucosilada (HbA1C):
Hasta 79% de los pacientes lograron los objetivos del tratamiento de una hemoglobina glucosilada (HbA1C) <7% y la proporción de pacientes fue significativamente mayor con Ozempic® en comparación con pacientes que recibieron sitagliptina, exenatida ER, insulina glargina, dulaglutida y placebo (véase tabla 1 a 6).
La proporción de pacientes que lograron una hemoglobina glucosilada (HbA1C) inferior al 7% sin hipoglucemia sintomática grave o con glucosa sanguínea confirmada y sin ganancia de peso fue significativamente mayor con Ozempic® 0.5 mg y 1 mg (hasta un 66% y 74%, respectivamente) en comparación con pacientes que recibieron sitagliptina (27%), exenatida ER (29%), insulina glargina (16%) y dulaglutida 0.75 mg (44%) y 1.5 mg (58%).
Peso corporal:
Ozempic® 1 mg, utilizado como monoterapia o en combinación con 1-2 medicamentos resultaron en reducciones estadísticamente superiores en el peso corporal de más de 6.5 kg, comparado con pacientes que recibieron placebo, sitagliptina, exenatida ER, insulina glargina o dulaglutida (tabla 1 a 5). La reducción en el peso corporal se mantuvo hasta 2 años (Figura 7).
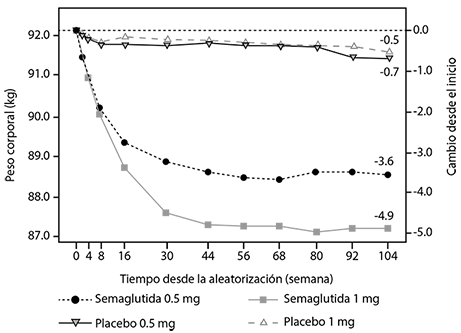
Figura 7. Cambio medio en el peso corporal (kg) en el tiempo en SUSTAIN 6.
Después de un año de tratamiento, se logró una pérdida de peso ≥5% y ≥10% en más sujetos con Ozempic® 0.5 mg (46% y 13%) y 1 mg (hasta un 62% y 24%) en comparación el comparador activo sitagliptina y exenatida ER (hasta 18% y hasta al 4%) (Tablas 1 a 5).
En el estudio de 40 semanas versus dulaglutida una pérdida de peso ≥5% y ≥10% fue alcanzada por más sujetos con Ozempic® 0.5 mg (44% y 14%) en comparación con dulaglutida 0.75 mg (23% y 3%) y Ozempic® 1 mg (hasta un 63% y 27%) en comparación con dulaglutida 1.5 mg (30% y 8%).
En el estudio cardiovascular (SUSTAIN 6), se logró una pérdida de peso ≥5% y ≥10% para más sujetos con Ozempic® 0.5 mg (36% y 13%) y 1 mg (47% y el 20%) en comparación con el placebo 0.5 mg (18% y 6%) y placebo 1 mg (19% y 7%).
Glucosa plasmática en ayuno e incrementos postprandiales:
Ozempic® 0.5 mg y 1 mg mostraron reducciones significativas en los niveles de glucosa plasmática en ayuno de hasta 50 mg/dl y reducciones en los incrementos postprandiales en las tres comidas diarias (diferencia entre valores antes de la comida y después de la comida de tres alimentos) de hasta 22 mg/dl (adicionalmente vea sección 5 Datos Farmacodinámicos).
Resistencia a la insulina y función de células beta:
La función de células beta medida por la evaluación del modelo de homeostasis para la función de células beta (HOMA-B) y la resistencia a la insulina medida por la evaluación del modelo de homeostasis para la resistencia a la insulina (HOMA-IR) mejoró en general con Ozempic® 0.5 mg y 1 mg (además ver sección 5 Datos Farmacodinámicos).
Lípidos:
En general, se observó mejoría en el perfil de lípidos plasmáticos en ayuno con Ozempic® a través de los estudios, sobre todo para el grupo de 1 mg (adicionalmente ver sección 5 Datos Farmacodinámicos).
Reducción de enfermedad cardiovascular:
En un estudio doble ciego a 104 semanas (SUSTAIN 6), se aleatorizaron 3,297 pacientes con diabetes tipo 2 con alto riesgo cardiovascular para recibir Ozempic® 0.5 mg o 1 mg una vez por semana adicional al tratamiento estándar (cardiovascular y diabetes) o placebo 0.5 mg o placebo 1 mg adicional al tratamiento estándar (cardiovascular y diabetes) con seguimiento de 2 años. En total 98.0% de los pacientes completó el estudio y, al final de éste se conocía el estado vital del 99.6% de los pacientes.
La población de estudio fue distribuida por edad: 1,598 pacientes (48.5%) ≥65 años, 321 (9.7%) ≥75 años y 20 (0.6%) ≥85 años. Se incluyeron 2,358 pacientes con función renal normal o insuficiencia renal leve, 832 con insuficiencia renal moderada y 107 con insuficiencia renal grave o insuficiencia renal en etapa terminal. Fueron 61% varones, la edad promedio fue de 65 años y la media del IMC de 33 kg/m2. La media de duración de la diabetes fue de 13.9 años.
El criterio de valoración principal fue el tiempo transcurrido desde la aleatorización hasta la primera aparición de un evento adverso cardiovascular mayor (MACE por sus siglas en inglés): muerte cardiovascular, infarto del miocardio no fatal o accidente cerebrovascular no fatal. El criterio de valoración secundario fue el tiempo transcurrido desde la aleatorización hasta la ocurrencia del primer desenlace cardiovascular compuesto extendido, definido como MACE, revascularización (coronaria y/o periférica), angina inestable que requiriera hospitalización u hospitalización por insuficiencia cardiaca. El número total de episodios de los componentes del criterio de valoración principal MACE fue de 254, de los que 108 (6.6%) ocurrieron en el grupo de Ozempic® y 146 (8.9%) en el grupo de placebo.
El tratamiento con Ozempic® logró una reducción del riesgo en un 26% en el criterio de valoración principal compuesto de muerte por causas cardiovasculares, infarto del miocardio no fatal o accidente cerebrovascular no fatal. Esta reducción del riesgo en el criterio de valoración principal compuesto se debió a una disminución significativa (39%) en la tasa de accidente cerebrovascular no fatal y una disminución no significativa (26%) en el infarto del miocardio no fatal sin diferencia significativa en la muerte cardiovascular (ver Figura 8).
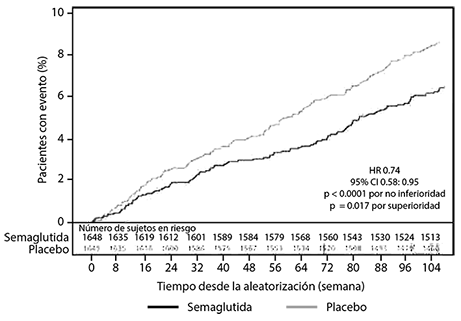
Figura 8. Estimado de Kaplan- Meier del tiempo desde la primera aparición de un resultado compuesto: muerte cardiovascular, infarto del miocardio no fatal o accidente cerebrovascular no fatal (SUSTAIN 6).
El riesgo de una revascularización coronaria o periférica compuesta se redujo significativamente, mientras que el riesgo de angina inestable que requiriera hospitalización y el riesgo de hospitalización por insuficiencia cardiaca no se redujeron de manera significativa. Vea la figura 9 para ver los resultados en desenlace primario y secundario cardiovascular
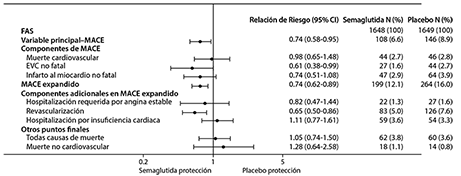
Figura 9. Diagrama de efectos (Forest Plot): análisis de cada tipo de evento cardiovascular (SUSTAIN 6).
Los resultados microvasculares se componen por 158 nuevos eventos o empeoramiento de los eventos de nefropatía. El cociente de riesgo para el tiempo de nefropatía (nueva aparición de macroalbuminuria persistente, duplicación persistente de la creatinina sérica, necesidad de terapia de reemplazo renal continua y la muerte debida a enfermedad renal) fue de 0.64 [0.46; 0.88] guiado por macroalbuminuria persistente de nueva aparición.
Se observó una reducción significativa y sostenida de hemoglobina glucosilada (HbA1C) desde el inicio a la semana 104 con Ozempic® 0.5 mg y 1 mg versus placebo 0.5 mg y 1 mg, adicional al estándar de tratamiento (-1.1 y -1.4 vs -0.4 y -0.4, respectivamente).
Presión arterial:
Se observaron reducciones significativas en la presión arterial sistólica media cuando Ozempic® 0.5 mg (3.5 - 5.1 mmHg) y Ozempic® 1 mg (5.4 - 7.3 mmHg) se usaron en combinación con medicamentos antidiabéticos orales o insulina basal. Para la presión arterial diastólica, no hubo diferencias significativas entre Ozempic® y comparadores.
Inmunogenicidad:
De acuerdo con las propiedades potencialmente inmunogénicas de los medicamentos proteicos y peptídicos, los pacientes pueden desarrollar anticuerpos después del tratamiento con semaglutida. La proporción de sujetos que resultaron positivos para anticuerpos anti-semaglutida en cualquier momento post-inicio fue baja (1-2%) y ningún sujeto tenía anticuerpos neutralizantes anti-semaglutida o anticuerpos anti-semaglutida con efecto neutralizante de GLP-1 endógeno al final del estudio.
CONTRAINDICACIONES:
Hipersensibilidad al principio activo o alguno de los excipientes (Fosfato disódico dihidratado, Propilenglicol, Fenol, Ácido clorhídrico, Hidróxido de sodio, Agua para fabricación de inyectables).
Embarazo y Lactancia:
En niños y adolescentes menores de 18 años, Síndrome de Neoplasia endócrina, multiple Tipo 2, Pacientes con antecedentes personales o familiares de carcinoma medular de tiroides.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Los estudios en animales han mostrado toxicidad reproductiva (ver sección 5). Existen datos limitados del uso de semaglutida en mujeres embarazadas. Por lo tanto, semaglutida no debe usarse durante el embarazo. Se recomienda el uso de anticonceptivos a las mujeres en edad fértil cuando se tratan con semaglutida. Si una paciente desea quedar embarazada o si se produce un embarazo, debe suspenderse la administración de semaglutida. Semaglutida debe interrumpirse al menos 2 meses antes de un embarazo planificado debido a la larga vida media (ver sección 5 Datos Farmacodinámicos). Madres Lactando/Lactancia:
En ratas lactantes, semaglutida fue excretada en leche. El riesgo de amamantar a un niño no puede ser excluido. No se puede excluir un riesgo para un niño amamantado. Semaglutida no debe usarse durante la lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad:
En 8 estudios fase 3a, 4,792 pacientes fueron expuestos a Ozempic® solo o en combinación con otros medicamentos reductores de glucosa. La duración del tratamiento varió de 30 semanas a 2 años.
Las reacciones adversas reportadas con más frecuencia en los estudios clínicos fueron trastornos gastrointestinales, que incluyen náuseas, diarrea y vómito. En general, estas reacciones fueron leves o moderadas en gravedad y de corta duración.
Lista tabulada de reacciones adversas:
La Tabla 6 enumera las reacciones adversas identificadas en los estudios de fase 3a en pacientes con diabetes tipo 2 (que se describen con más detalle en la sección 5. Farmacodinamia). Estudios clínicos. Información sobre la seguridad y eficacia. Las frecuencias de las reacciones adversas se basan en un grupo de estudios de fase 3a que excluyeron el estudio de resultados cardiovasculares (ver el texto a continuación de la tabla para obtener información adicional).
Las reacciones adversas se enlistan a continuación por clase de órgano del sistema y frecuencia absoluta. Las frecuencias se definen como: muy frecuentes: (≥1 / 10); común: (≥1 / 100 a <1/10); poco común (≥1 / 1,000 a <1/100); raras: (≥1 / 10,000 a <1 / 1,000); y muy raras: (<1 / 10,000). Dentro de cada agrupación de frecuencia, las reacciones adversas se presentan en orden de gravedad decreciente.
Tabla 6. Reacciones adversas para estudios controlados fase 3a a
|
MedDRA Clasificación por órganos y sistemas Clase |
Muy común |
Común |
Poco común |
Raro |
|
Trastornos del sistema inmune |
Reacción anafiláctica |
|||
|
Trastornos del metabolismo y nutrición |
Hipoglucemiaª cuando se usa con insulina o sulfonilurea |
Hipoglucemiaª cuando se usa con otros ADOs Disminución del apetito |
||
|
Trastornos del sistema nervioso |
Mareos |
Disgeusia |
||
|
Trastornos oculares |
Complicaciones de retinopatía diabéticab |
|||
|
Trastornos cardiacos |
Aumento de frecuencia cardiaca |
|||
|
Trastornos gastrointestinales |
Náusea Diarrea |
Vómito Dolor abdominal Distensión abdominal Estreñimiento Dispepsia Gastritis Enfermedad por reflujo gastroesofágico Eructos Flatulencia |
||
|
Trastornos hepatoblliares |
Colelitiasis |
|||
|
Trastornos generales y afecciones en el sitio de administración |
Fatiga |
Reacciones en el sitio de la inyección |
||
|
Investigación |
Aumento de la lipasa Aumento de la amilasa Pérdida de peso |
ª Hipoglucemia definida como grave (que requiere la asistencia de otra persona) o sintomática en combinación con glucosa sanguinea <56 mg/dl.
b Las complicaciones de la retinopatía diabética son un compuesto de: necesidad de fotocoagulación retiniana, necesidad de tratamiento con agentes intravítreos, hemorragia vítrea, aparición de ceguera relacionada con la diabetes. Frecuencia basada en el estudio de los resultados cardiovasculares.
2 años de los resultados cardiovasculares y estudios de seguridad:
En una población de alto riesgo cardiovascular el perfil de reacciones adversas fue similar a la observada en los otros estudios fase 3a descritos en la sección 5 Propiedades Farmacodinámicas. Información sobre eficacia y seguridad clínica).
Descripción de las reacciones adversas seleccionadas:
Hipoglucemia:
No se observó ningún episodio de hipoglucemia grave cuando Ozempic® fue utilizado como monoterapia. La hipoglucemia grave fue observada principalmente cuando Ozempic® fue utilizado con una sulfonilurea (1.2% de los sujetos, 0.03 eventos/paciente al año) o insulina (1.5% de los sujetos, 0.02 eventos/paciente al año). Se observaron muy pocos episodios graves (0.1% de los sujetos, 0.001 eventos/pacientes al año) con Ozempic® en combinación con otros antidiabéticos orales a excepción de las sulfonilureas.
Eventos adversos gastrointestinales:
La náusea ocurrió en 17.0% y 19.9% de pacientes cuando son tratados con Ozempic® 0.5 mg y 1 mg respectivamente, diarrea en 12.2% y 13.3% y vómitos en 6.4% y 8.4%. La mayoría de los eventos fueron de leves a moderados en cuanto a gravedad y de corta duración. Los eventos condujeron a la interrupción del tratamiento en 3.9% y 5.9% de los sujetos. Los eventos se reportaron con mayor frecuencia durante los primeros meses en tratamiento.
Complicaciones de retinopatía diabética:
En un estudio clínico de 2 años que incluyó 3,297 pacientes con diabetes tipo 2 de larga duración, alto riesgo cardiovascular y nivel de glucosa en sangre no controlado adecuadamente.En este estudio, los episodios adjudicados de complicaciones de la retinopatía diabética ocurrieron en más pacientes tratados con Ozempic® (3.0%) que en los que recibieron placebo (1.8%). El aumento de riesgo absoluto de complicaciones de la retinopatía diabética fue mayor entre los pacientes con antecedentes de retinopatía diabética al inicio del estudio. En los pacientes que no tenían un historial documentado de retinopatía diabética, el número de eventos fue similar para Ozempic® y placebo.
En otros estudios clínicos de hasta 1 año de duración en los que participaron 4,807 pacientes con diabetes tipo 2, se informaron eventos adversos relacionados con la retinopatía diabética en proporciones similares de sujetos tratados con Ozempic® (1.7%) y tratamientos comparadores (2.0%).
Discontinuación debido a un evento adverso:
La incidencia de discontinuación del tratamiento debido a eventos adversos fue de 8.7% para los pacientes tratados con 1 mg de Ozempic®. Los eventos adversos más frecuentes, llevando a la descontinuación fueron gastrointestinales.
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas del medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Los datos preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas o genotoxicidad.
Los tumores de células C tiroideas no letales observados en roedores son un efecto de clase para los agonistas del receptor GLP-1. En estudios de carcinogenicidad de 2 años en ratas y ratones, semaglutida causó tumores de células C tiroideas a exposiciones clínicamente relevantes. No se observaron otros tumores relacionados con el tratamiento. Los tumores de células C de roedores son causados por un mecanismo no genotóxico específico del receptor GLP-1 al que los roedores son particularmente sensibles. La relevancia para los humanos se considera baja, pero no se puede excluir por completo.
En estudios de fertilidad en ratas, semaglutida no afectó el rendimiento de apareamiento ni la fertilidad masculina. En ratas hembras, se observó un aumento en la duración del ciclo estral y una pequeña reducción en los cuerpos lúteos (ovulaciones) a dosis asociadas con la pérdida de peso corporal materna.
En estudios de desarrollo embriofetal en ratas, semaglutida causó embriotoxicidad por debajo de las exposiciones clínicamente relevantes. Semaglutida causó reducciones marcadas en el peso corporal materno y reducciones en la supervivencia y el crecimiento embrionarios. En los fetos, se observaron malformaciones esqueléticas y viscerales importantes, incluidos los efectos en los huesos largos, las costillas, las vértebras, la cola, los vasos sanguíneos y los ventrículos cerebrales. Las evaluaciones mecanísticas indicaron que la embriotoxicidad implicaba un deterioro mediado por el receptor GLP-1 del suministro de nutrientes al embrión a través del saco vitelino de rata. Debido a las diferencias de especies en la anatomía y función del saco vitelino y debido a la falta de expresión del receptor GLP-1 en el saco vitelino de primates no humanos, se considera que este mecanismo no es relevante para los humanos.
En estudios de toxicidad del desarrollo en conejos y monos cynomolgus, se observó una mayor pérdida de embarazo y una incidencia ligeramente mayor de anormalidades fetales en exposiciones clínicamente relevantes. Los hallazgos coincidieron con una marcada pérdida de peso corporal materna de hasta 16%. Se desconoce si estos efectos están relacionados con la disminución del consumo de alimentos maternos como un efecto directo de GLP-1.
Crecimiento y desarrollo postnatal fueron evaluados en monos cynomolgus. Los bebés fueron levemente más pequeños al momento del parto, pero se recuperaron durante el periodo de lactancia.
En ratas jóvenes, semaglutida provocó una maduración sexual retardada tanto en hombres como en mujeres. Estas demoras no tuvieron ningún impacto sobre la fertilidad y la capacidad reproductiva de ningún sexo, ni sobre la capacidad de las mujeres para mantener el embarazo.
Fertilidad:
Se desconoce el efecto de semaglutida sobre la fertilidad en humanos. En el caso de las ratas semaglutida no afectó a la fertilidad de los machos. En el caso de ratas hembras, se observó un aumento de la duración del ciclo estral y una ligera disminución del número de ovulaciones asociadas con pérdida de peso corporal materno.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Estudios in vitro han demostrado muy bajo potencial de semaglutida para inhibir o inducir las enzimas CYP y para inhibir transportadores del principio activo.
El retraso del vaciamento gástrico con semaglutida puede influir en la absorción de medicamentos orales administrados concomitantemente. Se estudió el efecto potencial de semaglutida en la absorción de medicamentos administrados conjuntamente por vía oral en estudios en exposición de estado estacionario de 1 mg de semaglutida.
No se observó ninguna interacción clínicamente relevante fármaco-fármaco con semaglutida (Figura 10) basado en los medicamentos evaluados. Por lo tanto, ningún ajuste de dosis es necesario cuando es administrado conjuntamente con semaglutida.

La exposición relativa en términos de ABC y Cmáx para cada medicamento cuando se administra con semaglutida con respecto a sin semaglutida. Se evaluaron medicamentos anticonceptivos orales (etinilestradiol/levonorgestrel) en estado estacionario. La warfarina (S-warfarina / R-warfarina), la digoxina y la atorvastatina se evaluaron después de una sola dosis.
Abreviaturas: ABC : área bajo la curva. Cmáx: concentración máxima. IC: intervalo de confianza.
Figura 10. Impacto de semaglutida en la exposición de los medicamentos administrados conjuntamente por vía oral.
Uso de anticonceptivos orales:
No se prevé que semaglutida disminuya la eficacia de los anticonceptivos orales, ya que semaglutida no modificó de una forma clínicamente significativa la exposición general de etinilestradiol ni de levonorgestrel tras la administración conjunta de un medicamento anticonceptivo oral combinado (0.03 mg de etinilestradiol/0.15 mg de levonorgestrel) y semaglutida. La exposición de etinilestradiol no se vio afectada; se observó un aumento del 20% en la exposición al levonorgestrel en estado estacionario. La Cmáx no se vio afectada por ninguno de los dos compuestos.
Atorvastatina:
Semaglutida no modificó la exposición general de la atorvastatina después de la administración de una dosis única de atorvastatina (40 mg). La Cmáx de atorvastatina disminuyó en un 38%. Esto fue determinado como no clínicamente significativo.
Digoxina:
Semaglutida no modificó la exposición general ni la Cmáx de digoxina después de la administración de una dosis única de digoxina (0.5 mg).
Metformina:
Semaglutida no modificó la exposición general ni la Cmáx de metformina después de la administración de 500 mg dos veces al día durante 3.5 días.
Warfarina:
Semaglutida no modificó la exposición general o la Cmáx de R- y S- de warfarina después de una dosis única de warfarina (25 mg) y los efectos farmacodinámicos de la warfarina, determinados por la International Normalized Ratio (INR por sus siglas en inglés) no se vieron afectados de una forma clínicamente significativa.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
No se encontraron diferencias clínicas relevantes entre semaglutida y competidores para los parámetros de laboratorio de hematología o bioquímica (incluyendo calcitonina).
Los niveles séricos de lipasa y amilasa incrementaron con semaglutida en un modo dosis dependiente al igual que fue descrito con otras terapias basadas en incretinas. Tras el incremento inicial de lipasa y amilasa, los niveles de actividad no mostraron cambios hasta por 2 años, como se mostró en SUSTAIN 6 (CVOT). En la ausencia de otros hallazgos clínicos o síntomas de pancreatitis, la elevación de niveles de lipasa y amilasa observados con semaglutida no predice un desarrollo tardío de pancreatitis. Por lo tanto, en la ausencia de otros signos o síntomas de pancreatitis, la observación de elevación de lipasa y amilasa no deben ser considerados una preocupación de seguridad.
De acuerdo a las propiedades potencialmente inmunogénicas de los medicamentos que contienen proteínas o péptidos, los pacientes pueden desarrollar anticuerpos tras el tratamiento con semaglutida. La proporción de pacientes con un resultado positivo en el análisis de anticuerpos antisemaglutida en cualquier punto temporal posterior al inicio del ensayo fue baja (1-2%) y, al final del ensayo, ningún paciente presentó anticuerpos neutralizantes antisemaglutida ni anticuerpos antisemaglutida con efecto neutralizante del GLP-1 endógeno, no hubo efecto en los niveles de exposición a semaglutida, HbA1C o perfil de seguridad de semaglutida.
La función renal por eGFR (tasa de filtración glomerular estimada) disminuyó con semaglutida de una manera dosis independiente: alrededor del 50% de los pacientes con semaglutida (0.5 mg y 1.0 mg) y 48% del grupo comparador, el eGFR diminuyó de la basal en un 10 a 25%. El eGFR también disminuyó con los comparadores activos como sitagliptina, insulina glargina y ADO‘s en la fase individual de los estudios de fase 3ª (misma magnitud que semaglutida), esto sugiere que la disminución neta de eGFR no estuvo relacionada con las propiedades de medicamentos basados en incretinas.
PRECAUCIONES GENERALES:
Ozempic® no debe utilizarse en pacientes con diabetes mellitus tipo 1 o para el tratamiento de la cetoacidosis diabética.
Ozempic® no es un sustituto de la insulina.
Efectos gastrointestinales:
El uso de agonistas del receptor de GLP-1 puede asociarse con reacciones adversas gastrointestinales. Esto se debe considerar cuando se trata a un paciente con función renal deteriorada por los efectos como náusea, vómito y diarrea los cuales pueden causar deshidratación, lo cual podría incrementar el deterioro de la función renal.
Pancreatitis aguda:
Se ha observado pancreatitis aguda con el uso de agonistas del receptor de GLP-1. Los pacientes deben ser informados sobre los síntomas característicos de la pancreatitis aguda. Si se sospecha pancreatitis aguda, debe suspenderse el tratamiento con Ozempic®; si se confirma pancreatitis, Ozempic® no debe reiniciarse. Se debe tener precaución en pacientes con un historial de pancreatitis.
En la ausencia de otros signos y síntomas de pancreatitis aguda, la elevación de enzimas pancreáticas por sí solas no son predictivas de pancreatitis aguda.
Hipoglucemia:
Los pacientes tratados con Ozempic® en combinación con una sulfonilurea o con insulina pueden tener un mayor riesgo de hipoglucemia. El riesgo de hipoglucemia puede disminuirse reduciendo la dosis de sulfonilurea o insulina, cuando se inicia el tratamiento con Ozempic®.
Retinopatía diabética:
La rápida mejora en el control de la glucosa se ha asociado con un empeoramiento temporal de la retinopatía diabética. El control glucémico a largo plazo disminuye el riesgo de la retinopatía diabética. Los pacientes con historia de retinopatía diabética deben ser monitoreados por empeoramiento y tratados según las guías clínicas.
Insuficiencia cardiaca:
No existe experiencia terapéutica en pacientes con insuficiencia cardiaca congestiva clase IV de acuerdo a la asociación del corazón de Nueva York (NYHA por sus siglas en inglés).
Efectos sobre la capacidad para manejar y uso de maquinaria:
Ozempic® no tiene influencia alguna sobre la capacidad para conducir y utilizar maquinaria. Cuando se utiliza en combinación con una sulfonilurea o insulina, se debe advertir a los pacientes tomar precauciones para evitar la hipoglucemia al conducir y utilizar máquinas.
Incompatibilidades:
Las sustancias añadidas a Ozempic® pueden causar la degradación de semaglutida. Ozempic® no debe mezclarse con otros medicamentos, p.e. fluidos de infusión.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Subcutánea.
Dosis:
La dosis inicial de Ozempic® es de 0.25 mg, una vez por semana. Después de 4 semanas, la dosis se debe incrementar a 0.5 mg una vez por semana. Transcurridas al menos 4 semanas con una dosis de 0.5 mg una vez por semana, la dosis puede incrementar a 1 mg una vez por semana, para lograr así una nueva mejora del control glucémico.
La dosis de Ozempic® 0.25 mg no es una dosis de mantenimiento.
Ozempic® puede ser usado como monoterapia o como terapia de combinación con uno o más medicamentos antidiabéticos (ver sección 5 Propiedades Farmacodinámicas. Información sobre la eficacia y seguridad clínica).
Cuando Ozempic® se agrega a la terapia existente de metformina y/o tiazolidinediona, la dosis actual de metformina y/o tiazolidinediona puede continuar sin cambios.
Cuando Ozempic® se agrega a la terapia existente de sulfonilurea o insulina, se debe considerar una disminución de la dosis de sulfonilurea o de la insulina para reducir el riesgo de hipoglucemia (ver sección 7 Precauciones generales).
El uso de Ozempic® no requiere llevar a cabo un autocontrol glucémico de la glucosa en sangre. El autocontrol glucémico puede realizarse cuando Ozempic® se utiliza junto con sulfonilureas o insulina para permitir el ajuste de la dosis de estos medicamentos.
Poblaciones especiales:
Personas de edad avanzada (≥ 65 años de edad):
No se requiere ajuste de dosis basado en la edad.
Género:
No se requiere ajuste de dosis basado en la edad.
Raza y grupo étnico:
No se requiere ajuste de dosis basado en la raza y grupo étnico.
Pacientes con insuficiencia hepática:
No se recomienda ajustar la dosis para pacientes con insuficiencia hepática (ver sección 5 Propiedades Farmacocinéticas). La experiencia con el uso de semaglutida en pacientes con insuficiencia hepática grave es limitada. Se debe tener precaución al tratar a estos pacientes con semaglutida.
Pacientes con insuficiencia renal:
No se recomienda ajustar la dosis para pacientes con insuficiencia renal. La experiencia con el uso de semaglutida en pacientes con insuficiencia renal en etapa terminal es limitada. Se debe tener precaución al tratar a estos pacientes con semaglutida (ver sección 5 Propiedades Farmacocinéticas).
Niños y adolescentes (Contraindicado):
Ozempic está contraindicado en niños y adolescentes menores de 18 años. La seguridad y eficacia de Ozempic® en niños y adolescentes menores de 18 años no han sido estudiadas.
Método de administración:
Ozempic® debe ser administrado una vez por semana a cualquier hora del día, con o sin alimentos.
Ozempic® debe ser inyectado por vía subcutánea en el abdomen, el muslo o en la parte superior del brazo. El sitio y el momento de la inyección pueden cambiarse sin ajustar la dosis. Ozempic® no debe ser administrado por vía intravenosa o intramuscular. Para mayores instrucciones sobre la administración, consulte la sección 16 Recomendaciones de almacenamiento.
El día de la administración semanal se puede cambiar si es necesario siempre que el tiempo entre dos dosis sea por lo menos de 2 días (>48 horas).
Dosis omitida:
Si se omite una dosis, debe ser administrada tan pronto como sea posible y dentro de los 5 días después de la dosis que se omitió. Si han pasado más de 5 días, debe saltarse la dosis omitida y la siguiente dosis debe ser administrada en el día como fue regularmente programada. En cada caso, los pacientes pueden entonces volver a su horario regular de dosificación de una vez por semana.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Se han informado sobredosis de hasta 4 mg en una sola dosis, y hasta 4 mg en una semana en estudios clínicos. El evento adverso más comúnmente informado fue náuseas. Todos los pacientes se recuperaron sin complicaciones.
No existe un antídoto específico para la sobredosis con Ozempic®. En caso de sobredosis, se debe iniciar un tratamiento de soporte apropiado de acuerdo con los signos y síntomas clínicos del paciente. Puede ser necesario un periodo prolongado de observación y tratamiento para estos síntomas, teniendo en cuenta la larga vida media de Ozempic® de aproximadamente 1 semana (ver sección 5. Propiedades Farmacodinámicas. Datos Farmacodinámicos) .
PRESENTACIONES:
Caja de cartón con una pluma precargada con 1.5 mL (1.34 mg/mL) de solución, incluye 6 agujas desechables NovoFine® Plus (Reg. 625C94 SSA).
Caja de cartón con una pluma precargada con 3 mL (1.34 mg/mL) de solución, incluye 4 agujas desechables NovoFine® Plus (Reg. 625C94 SSA).
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese la caja bien cerrada.
Antes de su uso: Consérvese en refrigeración (2ºC a 8ºC). No congelar.
Después del primer uso el medicamento, se conserva hasta 6 semanas a no más de 30ºC o en refrigeración (2°C a 8°C). Una vez transcurrido este periodo, el producto deberá desecharse. No congelar y no usar si el producto ha sido congelado. Proteger de la luz excesiva y el calor. Remueva siempre la aguja inmediatamente después de cada inyección y guarde su pluma sin la aguja puesta, para prevenir bloqueo de agujas, contaminación, infecciones, fuga de la solución y dosificación incorrecta.
LEYENDAS DE PROTECCIÓN:
Precauciones para la eliminación:
Se debe advertir al paciente que deseche la aguja de inyección después de cada inyección de acuerdo con los requisitos locales.
Otra información de manejo:
La pluma de Ozempic® debe ser utilizada por una sola persona.
Ozempic® no debe usarse si no aparece transparente e incoloro, o casi incoloro.
Ozempic® no debe usarse si se ha congelado.
La pluma de Ozempic® está diseñada para utilizarse con las agujas desechables NovoFine® Plus (Reg. Núm. 625C94 SSA).
Literatura exclusiva para médicos. No se deje al alcance y a la vista de los niños. No se administre si el cierre ha sido violado.
No se administre en niños y adolescentes menores de 18 años ni en embarazo y lactancia. Su venta requiere receta médica.
Reporte Ias reacciones adversas aI correo: farmacovigilancia@cofepris.gob.mx
Titular del Registro:
Novo Nordisk A/S
Novo Allé
Bagsvaerd 2880, Dinamarca
Representante Legal:
NOVO NORDISK MÉXICO, S.A. de C.V.
Homero 1500 piso 3,
Col. Polanco , C.P. 11560,
Miguel Hidalgo,
Ciudad de México, México
Reg. Núm. 096M2019 SSA IV
®Marca Registrada

