
PERGOVERIS 150 IU/75IU
FOLITROPINA ALFA, LUTROPINA ALFA
Solución inyectable
1 Caja,1 Frasco ámpula con liofilizado y diluyente,
1 Caja,3 Frasco ámpula con liofilizado y diluyente,
1 Caja,10 Frasco ámpula con liofilizado y diluyente,
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula con liofilizado contiene:
|
Folitropina alfa (r-hFSH) (equivalente a 11 microgramos) |
150 UI. |
|
Lutropina alfa (r-hLH) (equivalente a 3.0 microgramos) |
75 UI. |
El frasco ámpula con diluyente contiene:
|
Agua estéril para uso inyectable |
1 mL |
La solución reconstituida contiene 150 UI r-hFSH y 75 UI r-hLH por mililitro. Folitropina alfa y lutropina alfa son producidas por ingeniería genética en células de ovario de hámster chino (CHO).
INDICACIONES TERAPÉUTICAS: PERGOVERIS® está indicado para la estimulación del desarrollo folicular en mujeres con deficiencia severa de LH y FSH.
PERGOVERIS® también está indicado para estimulación ovárica controlada en pacientes con insuficiencia ovárica (reserva ovárica disminuida) que se traduce en una respuesta subóptima. En los estudios clínicos las respondedoras subóptimas han sido identificadas como sigue:
1) < 7 folículos preovulatorios u ovocitos; y/ó
2) Dosis altas de FSH (≥ 3000 UI por ciclo); y/ó.
3) Edad materna avanzada (≥ 35 años).
PERGOVERIS es usado en pacientes que se someten a técnicas de reproducción asistida (TRA) tales como la fertilización in vitro (FIV), inyección intracitoplasmática de espermatozoides (ICSI), transferencia intratubárica de gametos (GIFT) y transferencia intratubárica de cigotos (ZIFT).
PERGOVERIS® no debe ser utilizado en pacientes con falla ovárica.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas: PERGOVERIS® es una preparación de hormona folículo estimulante humana recombinante (folitropina alfa, r-hFSH) y hormona luteinizante humana recombinante (lutropina alfa, r-hLH). La preparación se produce por la técnica de ADN recombinante en células de Ovario de Hámster Chino CHO transgénicas.
La FSH está involucrada principalmente con el inicio de la foliculogénesis ejerciendo su efecto sobre las células de la granulosa del folículo ovárico en desarrollo, mientras que la LH tiene un papel importante en mejorar la producción tardía del estradiol folicular, y la inducción de la maduración folicular, la ovulación durante su pico de actividad, y el mantenimiento del cuerpo lúteo, por lo tanto, ayudar a la implantación temprana del embarazo.
La FSH y el estradiol inducen receptores de la LH en la superficie de las células de la granulosa durante el desarrollo folicular. En las células de la teca, la acción de la LH proporciona el sustrato andrógeno para las células de la granulosa. Los andrógenos se transforman en estrógenos por medio del sistema enzimático de la aromatasa en las células de la granulosa. Por tanto, en ausencia de la LH, la FSH puede inducir el crecimiento folicular pero poca producción de estradiol. Sin la producción adecuada de estradiol, el ambiente clínico necesario para promover la fertilidad, la secreción de moco cervical, el crecimiento endometrial y la formación de un cuerpo lúteo funcional en respuesta a la hCG, se ve afectada.
Se ha demostrado en estudios clínicos la eficacia de la combinación de la folitropina alfa y la lutropina alfa en mujeres con hipogonadismo hipogonadotrópico.
En la estimulación del desarrollo folicular en mujeres anovulatorias deficientes de LH y de FSH, el efecto primario resultante de la administración de lutropina alfa es un incremento en la secreción de estradiol por parte de los folículos, el crecimiento de los cuales es estimulado por la FSH.
En ensayos clínicos, las pacientes con severa deficiencia de FSH y LH fueron definidas por un nivel sérico endógeno < 1.2 UI/l medidas en un laboratorio central. Sin embargo, debe ser tomado en cuenta que hay variaciones entre las medidas de LH realizadas en diferentes laboratorios.
Se investigó, en un estudio clínico de mujeres con hipogonadismo hipogonadotrópico y concentraciones endógenas séricas de LH menores a 1.2 UI/L, la dosis apropiada de r-hLH (lutropina alfa). Una dosis de 75 UI diarias de r-hLH (en combinación con 150 UI de folitropina alfa [r-hFSH]) resultó en un desarrollo folicular y una producción de estrógeno adecuadas. Una dosis de 25 UI diarias de r-hLH (en combinación con 150 UI de folitropina alfa) resultó en un desarrollo folicular insuficiente. Por tanto, la administración de menos de un frasco ámpula de PERGOVERIS® diario puede proporcionar muy poca actividad de LH para asegurar un desarrollo folicular adecuado.
Aunque la mayoría de las pacientes sometidas a TRA se benefician de la monoterapia con r-hFSH, estudios publicados han demostrado el efecto benéfico del co-tratamiento con r-hLH en subpoblaciones de pacientes con respuesta inadecuada a tratamiento con r-hFSH sola (respondedores subóptimos a r-hFSH). El agregar r-hLH tiene como objetivo el incremento de la sensibilidad ovárica a la r-hFSH, promover la secreción de E2 por parte del folículo preovulatorio, resultando en crecimiento endometrial, y promover la luteinización posterior de los folículos, resultando en niveles normales de progesterona en la fase lútea.
Propiedades farmacocinéticas: La folitropina alfa y la lutropina alfa han mostrado el mismo perfil farmacocinético que la folitropina alfa y la lutropina alfa de manera separada. No hay interacción farmacocinética de lutropina alfa con folitropina alfa cuando son administradas de manera simultánea.
Folitropina alfa: Después de la administración subcutánea, la biodisponibilidad absoluta es de alrededor del 70%. Después de la administración repetida, la folitropina alfa se acumula 3 veces alcanzando un estado estacionario en 3-4 días. En mujeres cuya secreción endógena de gonadotropina está suprimida, la foIitropina alfa ha mostrado estimular efectivamente el desarrollo folicular y la esteroidogénesis, a pesar de niveles no medibles de LH.
Después de la administración intravenosa, la folitropina alfa es distribuida al espacio de líquido extracelular con una vida media inicial de alrededor de 2 horas y se elimina del cuerpo con una vida media terminal de alrededor de un día. El volumen de distribución de estado estacionario y de depuración total son de 10.1 y 0.6 L/h, respectivamente.
Un octavo de la dosis de folitropina alfa se excreta por la orina.
Lutropina alfa: Después de la administración subcutánea de PERGOVERIS, la máxima concentración sérica es alcanzada después de aproximadamente 4 a 16 horas. La biodisponibilidad absoluta promedio de PERGOVERIS siguiendo una sola inyección subcutánea (a una dosis mucho más alta para permitir la cuantificación adecuada, esto es, 10,000 UI) a voluntarias femeninas sanas es de 56 ± 23%, apoyada por el método de inmunoensayo.
Después de una administración intravenosa de 300 UI de PERGOVERIS, una fase de distribución rápida de aproximadamente 1 hora y una vida media terminal (t½) de aproximadamente 11 horas fue observada para la r-hLH. El volumen de distribución en el estado estacionario (Vss) fue de aproximadamente 10 l. El tiempo medio de residencia (MRT, por sus siglas en inglés) fue de aproximadamente 6 horas.
La lutropina alfa muestra una farmacocinética lineal, evaluada por el AUC, la que es directamente proporcional a la dosis administrada. La farmacocinética de la lutropina alfa después de una administración única y repetida de PERGOVERIS son comparables y la proporción de acumulación de lutropina alfa es mínima. No hay interacción farmacocinética con folitropina alfa cuando se administra de manera simultánea.
Después de una administración subcutánea de PERGOVERIS, la r-hLH es eliminada del cuerpo con una vida media terminal de alrededor de 18 horas. La depuración total del cuerpo es de aproximadamente 2 a 3 l/h siendo menos del 5 por ciento de la dosis excretada por vía renal sin cambio.
CONTRAINDICACIONES:
PERGOVERIS está contraindicado en pacientes con:
• Hipersensibilidad a las sustancias activas folitropina alfa y lutropina alfa o a cualquiera de los excipientes.
• En caso de tumores del hipotálamo y de la glándula pituitaria.
• Crecimiento ovárico o quiste debido a síndrome de ovario poIiquístico o de etiología desconocida.
• Hemorragias ginecológicas de etiología desconocida.
• Carcinoma ovárico, uterino o mamario.
PERGOVERIS no debe usarse cuando no se puede obtener una respuesta efectiva, como:
• Falla ovárica (primaria y secundaria).
• Malformaciones de los órganos sexuales incompatibles con el embarazo.
• Tumores fibroides del útero incompatibles con el embarazo.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No hay indicación para el uso de PERGOVERIS durante el embarazo.
Información en un número limitado de embarazos expuestos indican que no hay reacciones adversas de las gonadotropinas sobre el embarazo, desarrollo fetal o embrionario, parto o desarrollo postnatal después de una estimulación ovárica controlada. No se ha observado efecto teratogénico de PERGOVERIS en estudios con animales.
En caso de exposición durante el embarazo, los datos clínicos no son suficientes para excluir el efecto teratogénico de PERGOVERIS.
Lactancia: PERGOVERIS no está indicado durante la lactancia; la información clínica no es suficiente para excluir un efecto teratogénico de PERGOVERIS.
REACCIONES SECUNDARIAS Y ADVERSAS: Durante el tratamiento con este medicamento, diferentes reacciones adversas pueden presentarse. Conforme a su frecuencia, las reacciones adversas pueden ser divididas en: muy raras (< 1/10,000 casos), raras (> 1/10,000 a < 1/1,000), poco comunes (≥ 1/1,000 a < 1/100), comunes (≥ 1/100 a < 1/10) y muy comunes (≥ 1/10).
|
Trastornos generales y condiciones en el sitio de administración |
Muy común |
Reacciones en el sitio de inyección leves a severas (dolor, enrojecimiento, tumefacción, hematomas y/o irritación en el sitio de la inyección). |
|
Trastornos del Sistema Nervioso |
Muy común |
Cefalea. |
|
Trastornos del Aparato Reproductivo y mamas |
Muy común |
Quistes ováricos. |
|
Común |
Dolor en las mamas, dolor pélvico, síndrome de hiperestimulación ovárica (SHO) leve a moderado (Incluyendo la sintomatología asociada). |
|
|
Poco comunes |
SHO grave (incluyendo la sintomatología asociada). |
|
|
Raras |
Torsión ovárica, como complicación de SHO grave. |
|
|
Alteraciones del Aparato Gastrointestinal |
Común |
Dolor abdominal y síntomas gastrointestinales tales como náusea, vómito, diarrea, distensión abdominal, incomodidad abdominal. |
|
Alteraciones vasculares |
Muy raras |
Tromboembolismo, asociado conmúnmente con SHO grave. |
|
Trastornos respiratorios torácicos y mediastinales |
Muy raras |
En pacientes con asma-exacerbación o empeoramiento del asma. |
|
Trastornos del Sistema Inmune |
Muy raras |
Reacciones de hipersensibilidad leves a severas, incluyendo reacciones anafilácticas y shock. |
Cualquier efecto no deseable durante el tratamiento con PERGOVERIS debe ser reportado oportunamente al médico tratante.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Pérdida del embarazo: La incidencia de fracaso del embarazo por aborto involuntario o de un aborto es mayor en pacientes sometidas a estimulación del desarrollo folicular para inducir la ovulación que en la población normal.
Embarazo ectópico: Las mujeres con antecedentes de enfermedad tubárica presentan riesgo de embarazo ectópico, si el embarazo es obtenido por concepción espontánea o con tratamientos de fertilidad. La prevalencia de embarazo ectópico después de FIV es del 2 al 5%, en comparación con el 1 al 1.5% en la población general.
Neoplasias del sistema reproductor: Han habido reportes de neoplasias ováricas y del aparato reproductor, tanto benignas como malignas, en mujeres que han pasado por múltiples regímenes de medicamentos para tratamiento de la infertilidad. Todavía no se ha establecido si el tratamiento con gonadotropina incrementa o no el riesgo de estos tumores en mujeres infértiles.
Malformaciones congénitas: La prevalencia de malformaciones congénitas después de TRA puede ser ligeramente mayor que después de embarazos espontáneos. Esto se cree que es debido a diferencias en las características de los padres (como edad de la madre, características del esperma) y embarazos múltiples.
Genotoxicidad: Las series completas de análisis de toxicología genética después de la exposición in vitro (células bacterianas y mamíferos) y la administración in vivo (análisis de micronúcleos) no revelan ningún potencial para la r-hFSH y r-hLH de causar daño genético.
Carcinogenicidad: Con respecto a la carcinogenicidad, no se han realizado estudios in vivo ni con PERGOVERIS o cada una de las hormonas recombinantes individuales ya que para productos de origen biotecnológico estos estudios son generalmente considerados como inapropiados. Además, debido a la similitud esencial de r-hFSH y r-hLH a las gonadotropinas humanas nativas, no se anticipa algún riesgo carcinogénico del uso terapéutico de PERGOVERIS.
Toxicidad de la Reproducción: Los resultados de un conjunto completo de experimentos de toxicidad reproductivo realizados en ratas y conejos con las sustancias individuales (r-hFSH y h-LH) fueron consistentes con las consecuencias predecibles del desbalance hormonal inducido por la administración extemporánea de altas dosis de potentes gonadotropinas que afectan la fertilidad, implantación, el parto y la viabilidad fetal. Sin embargo, no se ha observado teratogenicidad.
Ningún efecto adverso aparente fue observado en todos los estudios con ratas a dosis de 5 UI/kg/día de cada gonadotropina individual.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: PERGOVERIS no debe administrarse como mezcla con otros medicamentos, en la misma inyección, excepto con la folitropina alfa.
Ninguna otra interacción medicamentosa clínicamente significativa ha sido reportada durante la terapia con PERGOVERIS.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No conocidas.
PRECAUCIONES GENERALES: PERGOVERIS contiene sustancias gonadotrópicas potentes capaces de causar reacciones adversas de leves a severas, y debe ser indicada sólo por médicos que están realmente familiarizados con problemas de infertilidad y su manejo.
PERGOVERIS no está recomendado en técnicas de baja complejidad tales como inseminación intra-uterina.
No se use en senescencia ovárica.
Antes de comenzar el tratamiento, la infertilidad de la pareja debe ser evaluada en forma apropiada y las contraindicaciones supuestas para el embarazo deben ser evaluadas. En particular, las pacientes deben ser evaluadas para lo siguiente: hipotiroidismo, deficiencia adenocortical, hiperprolactinemia. Un tratamiento específico adecuado debe ser proporcionado.
La terapia con gonadotropina requiere un compromiso de tiempo determinado por parte de los médicos y profesionales de la salud, así como de la disponibilidad de instalaciones de monitoreo apropiadas. En mujeres, el uso seguro y efectivo de PERGOVERIS requiere el monitoreo de la respuesta ovárica con ultrasonido, solo o preferentemente en combinación con la medición de niveles de estradiol sérico de manera regular. Puede existir un cierto grado de variabilidad interpaciente en la respuesta a la administración de r-hFSH/r-hLH, con una pobre respuesta a r-hFSH/r-hLH en algunas pacientes.
Debe usarse la dosis efectiva más baja en relación al objetivo del tratamiento.
Las pacientes con porfiria o antecedentes familiares de porfiria deben ser monitoreadas de cerca durante el tratamiento con PERGOVERIS. El deterioro o una primera aparición de esta condición puede requerir el cese del tratamiento.
PERGOVERIS contiene menos de 1 mmol de sodio (23 mg) por dosis, esto lo hace esencialmente "libre de sodio".
Las pacientes que están bajo estimulación de crecimiento folicular están bajo un mayor riesgo de desarrollar hiperestimulación ovárica en vistas de una posible respuesta estrogénica excesiva y un desarrollo folicular múltiple.
En estudios clínicos, la lutropina alfa en combinación con la folitropina alfa, ha demostrado incrementar la sensibilidad ovárica a las gonadotropinas. Si un incremento en la dosis de FSH es considerado, el ajuste de las dosis debe ser preferentemente a intervalos de 7-14 días y preferentemente con incrementos de 37.5-75 UI utilizando una preparación de folitropina alfa con registro.
Síndrome de Hiperestimulación Ovárica (SHO): Un cierto grado de crecimiento ovárico es un efecto esperado de la estimulación ovárica controlada. Es visto más frecuentemente en mujeres con síndrome ovárico poliquístico y generalmente remite sin tratamiento.
A diferencia de un crecimiento ovárico no complicado, el SHO es una condición que puede manifestarse por sí misma con grados crecientes de severidad. Comprende crecimiento ovárico marcado, altos niveles séricos de esteroides sexuales y un incremento en la permeabilidad vascular lo cual puede resultar en una acumulación de líquido en las cavidades peritoneal, pleural y raramente en la cavidad pericárdica.
Manifestaciones leves de la SHO incluyen dolor abdominal, malestar y distensión abdominal y crecimiento de los ovarios. Un SHO moderado puede adicionalmente presentarse con náusea, vómito, evidencia de ascitis por ultrasonido y marcado crecimiento ovárico.
Un SHO severo adicionalmente incluye síntomas tales como crecimiento ovárico severo, ganancia de peso, disnea u oliguria. La evaluación clínica puede revelar signos tales como hipovolemia, hemoconcentración, desequilibrio electrolítico, ascitis, derrames pleurales ó insuficiencia respiratoria aguda. Muy raramente, un SHO severo puede estar complicado por torsión ovárica ó eventos tromboembólicos, tales como embolismo pulmonar o choque isquémico o infarto al miocardio.
Los factores de riesgo independientes para el desarrollo de un SHO incluyen una edad joven y disminución en la masa corporal, síndrome de ovario poliquístico, altas dosis de gonadotropinas exógenas, absolutamente altos o rápidamente incrementados niveles de estradiol sérico y episodios previos de SHO, gran número de folículos ováricos en desarrollo y gran número de ovocitos recuperados en los ciclos de TRA.
La adherencia a la dosis recomendada de PERGOVERIS®, a la dosis de FSH y al régimen de administración pueden minimizar el riesgo de hiperestimulación ovárica. El monitoreo de ciclos de estimulación por ultrasonido así como las mediciones de estradiol son recomendadas para identificar factores de riesgo tempranamente.
Existe evidencia para sugerir que la hCG juega un papel clave en el disparo del SHO y que el síndrome puede ser más severo y más prolongado si el embarazo ocurre. Además, si ocurren signos de hiperestimulación ovárica, se recomienda que la hCG se retire y que la paciente sea advertida de abstenerse del coito o usar métodos anticonceptivos por al menos 4 días. Cómo el SHO puede progresar rápidamente (dentro de las 24 horas) o en varios días convertirse en un evento médico serio, las pacientes deben ser seguidas durante al menos 2 semanas después de la administración de hCG .
El SHO leve o moderado por lo general se resuelve espontáneamente. Si se presenta un SHO severo, se recomienda que el tratamiento con gonadotropina sea interrumpido si es que aún continúa y que la paciente sea hospitalizada para iniciar una terapia apropiada.
Embarazos múltiples: En las pacientes sometidas a la inducción de la ovulación, la incidencia de embarazos y partos múltiples son más elevadas que el embarazo natural. La mayoría de los embarazos múltiples son gemelares. Los embarazos múltiples, especialmente de alto orden fetal, llevan a un mayor riesgo de resultados adversos maternos y perinatales.
Para minimizar el riesgo de embarazo múltiple de alto orden fetal, se recomienda un control cuidadoso de la respuesta ovárica.
En las pacientes sometidas a procedimientos de TRA el riesgo de embarazo múltiple está relacionado principalmente con el número de embriones transferidos, su calidad y la edad de la paciente.
Eventos Tromboembólicos: En mujeres con enfermedad tromboembólica en curso o reciente, o en mujeres con factores de riesgo generalmente reconocidos para eventos tromboembólicos, tales como historia familiar o personal, el tratamiento con gonadotropinas puede favorecer el incremento en el riesgo de la gravedad u ocurrencia de tales eventos. En estas mujeres, los beneficios de la administración de gonadotropina necesita ser evaluada contra el riesgo. Debe ser notado, sin embargo, que por sí mismo el embarazo puede también llevar a un incremento en el riesgo de eventos tromboembólicos.
Las pacientes deben ser advertidas de los riesgos potenciales antes de iniciar el tratamiento. Cuando el riesgo de SHO o embarazos múltiples ha sido asumido, se debe considerar la interrupción del tratamiento.
Los médicos deben ser informados en relación a las reacciones alérgicas y otras medicinas tomadas por la paciente antes de iniciar el tratamiento con PERGOVERIS.
Efectos en la capacidad para manejar o usar maquinaria: No se ha realizado ningún estudio sobre los efectos en la capacidad para manejar y usar maquinaria.
Edad Avanzada: No hay indicación relevante para el uso de PERGOVERIS en la población de edad avanzada. La seguridad y eficacia de PERGOVERIS en pacientes de edad avanzada no han sido establecidas.
Pacientes con daño hepático o renal: La seguridad, eficacia y farmacocinética de PERGOVERIS en pacientes con deterioro renal o daño hepático no han sido establecidas.
DOSIS Y VÍA DE ADMINISTRACIÓN: PERGOVERIS está planeado para administración subcutánea. El polvo debe reconstituirse con el solvente proporcionado inmediatamente antes de su uso.
Para uso único: Cualquier medicamento sin usar o material desechable debe ser dispuesto conforme a los requerimientos locales.
El tratamiento con PERGOVERIS debe ser iniciado bajo supervisión de un médico experimentado en el tratamiento de desórdenes de fertilidad.
Deficiencia de LH y FSH: Un régimen recomendado comienza con un frasco ámpula de PERGOVERIS (150 UI de r-hFSH + 75 UI de r-hLH) diario. Dado que estas pacientes son amenorreicas y tienen una secreción de estrógenos endógenos baja, el tratamiento puede iniciar en cualquier momento.
El tratamiento debe adecuarse a la respuesta individual de la paciente evaluada por medio de la medición del tamaño de los folículos por ultrasonido y de la respuesta estrógeno.
Si se considera apropiado un aumento de dosis de r-hFSH, el ajuste de dosis debe preferiblemente ser en intervalos de 7 a 14 días y preferiblemente en incrementos de 37.5-75 Ul usando una preparación de folitropina alfa registrada.
Puede ser aceptable extender la duración de la estimulación en cualquier ciclo hasta por 5 semanas. PERGOVERIS® puede mezclarse con folitropina alfa y co-administrarse como una sola inyección.
Cuando se obtiene una respuesta óptima, una sola inyección de 5,000 UI a 10,000 UI ó 250 mcg de hCG debe administrarse de 24 a 48 horas después de la última inyección de PERGOVERIS. Se recomienda a la paciente tener coito el día de, y al día siguiente de la administración de la hCG. Se debe considerar el soporte de la fase lútea dado que la falta de sustancias con actividad luteotrópica (LH/hCG) después de la ovulación puede llevar a una falla prematura del cuerpo lúteo.
Si se obtiene una respuesta excesiva, se debe detener el tratamiento y suspender la aplicación de hCG. El tratamiento se debe recomenzar en el próximo ciclo a una dosis de r-hFSH menor que la del ciclo previo.
Respuesta subóptima en TRA: El tratamiento recomendado comienza con 300 UI de r-hFSH diarios durante los primeros 5 a 7 días del tratamiento seguido de dos frascos ámpula de PERGOVERIS (300 UI de r-hFSH y 150 UI de r-hLH) comenzando los días 6 a 8 de la estimulación ovárica controlada (EOC). Alternativamente, el tratamiento puede iniciar con la administración diaria de dos frascos ámpula de PERGOVERIS (300 UI de r-hFSH y 150 UI de r-hLH), iniciado en el primer día de EOC seguido de supresión de la función pituitaria.
El tratamiento debe continuarse hasta lograr un desarrollo folicular adecuado (evaluado mediante el monitoreo de las concentraciones séricas de estrógeno y revisión con ultrasonido), con ajustes de dosis de r-hFSH conforme a la respuesta de la paciente. Si se considera apropiado un incremento de dosis de r-hFSH, la dosis total diaria de r-hFSH usualmente no debe exceder de 450 UI.
Una vez que se ha logrado un desarrollo folicular adecuado, la hCG debe administrarse para inducir la maduración folicular final en preparación para la recuperación de los ovocitos. La administración de hCG debe suspenderse en casos en los que los ovarios están anormalmente crecidos el último día de la terapia. Esto debe reducir la posibilidad de desarrollar SHO. Si se obtiene una respuesta excesiva, se debe detener el tratamiento y suspender la hCG. El tratamiento debe recomenzar en el próximo ciclo a una dosis menor que la del ciclo previo.
Recomendaciones para pacientes en caso de autoadministración de la medicina: La autoadministración de PERGOVERIS sólo debe realizarse por parte de pacientes bien motivadas, adecuadamente entrenadas y con acceso a supervisión por parte de algún experto.
La primera inyección de PERGOVERIS debe realizarse bajo supervisión médica directa.
Por favor lea cuidadosamente las siguientes instrucciones:
1) Lave sus manos. Es importante que sus manos y los artículos que use estén tan limpios como sea posible.
2) Ensamble y disponga en una superficie limpia todo lo que necesita:
• Un frasco ámpula con polvo de PERGOVERIS.
• Un frasco ámpula de solvente.
• Dos torundas con alcohol.
• Una jeringa.
• Una aguja para reconstitución y una aguja fina para inyección subcutánea.
• Contenedor de objetos punzocortantes.
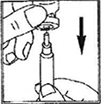 3) Retire la tapa protectora del frasco ámpula de solvente. Ajuste la aguja de reconstitución a la jeringa e introduzca algo de aire a la aguja jalando el émbolo hasta aproximadamente la marca de 1 mL. Posteriormente, inserte la aguja en el frasco ámpula, presione el émbolo para sacar el aire, gire el frasco ámpula hacia abajo y extraiga cuidadosamente todo el solvente. Coloque cuidadosamente la jeringa en la superficie de trabajo cuidando no tocar la aguja.
3) Retire la tapa protectora del frasco ámpula de solvente. Ajuste la aguja de reconstitución a la jeringa e introduzca algo de aire a la aguja jalando el émbolo hasta aproximadamente la marca de 1 mL. Posteriormente, inserte la aguja en el frasco ámpula, presione el émbolo para sacar el aire, gire el frasco ámpula hacia abajo y extraiga cuidadosamente todo el solvente. Coloque cuidadosamente la jeringa en la superficie de trabajo cuidando no tocar la aguja.
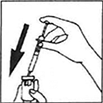 4) Prepare la solución de inyección: Retire la tapa protectora del frasco ámpula con polvo de PERGOVERIS®, tome la jeringa e inyecte lentamente el solvente en el frasco ámpula de polvo. Gire cuidadosamente sin retirar la jeringa. No agite. Después de que se disuelva el polvo (que usualmente ocurre inmediatamente), revise que la solución resultante es clara y no contiene ninguna partícula. Gire el frasco ámpula hacia abajo, extraiga cuidadosamente la solución de nuevo a la jeringa.
4) Prepare la solución de inyección: Retire la tapa protectora del frasco ámpula con polvo de PERGOVERIS®, tome la jeringa e inyecte lentamente el solvente en el frasco ámpula de polvo. Gire cuidadosamente sin retirar la jeringa. No agite. Después de que se disuelva el polvo (que usualmente ocurre inmediatamente), revise que la solución resultante es clara y no contiene ninguna partícula. Gire el frasco ámpula hacia abajo, extraiga cuidadosamente la solución de nuevo a la jeringa.
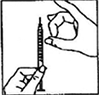 5) Cambie la aguja por la aguja fina y retire cualquier burbuja de aire. Si ve burbujas de aire en la jeringa, sostenga la jeringa con la aguja apuntando hacia arriba y golpee cuidadosamente la jeringa hasta que todo el aire se acumule en la parte superior. Presione el émbolo hasta que las burbujas de aire salgan.
5) Cambie la aguja por la aguja fina y retire cualquier burbuja de aire. Si ve burbujas de aire en la jeringa, sostenga la jeringa con la aguja apuntando hacia arriba y golpee cuidadosamente la jeringa hasta que todo el aire se acumule en la parte superior. Presione el émbolo hasta que las burbujas de aire salgan.
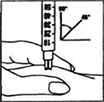 6) Inyecte inmediatamente la solución: Su médico o enfermera ya debe haberle aconsejado dónde inyectar (e.g., estómago, frente de la cadera). Limpie el área escogida con una torunda con alcohol. Pellizque firmemente la piel e inserte la aguja en un ángulo de 45° a 90° usando un movimiento como de dardo. Inyecte bajo la piel, como se le enseñó. No inyecte directamente en una vena. Inyecte la solución presionando cuidadosamente el émbolo. Tómese el tiempo que necesite para inyectar toda la solución. Retire inmediatamente la aguja y limpie la piel con una torunda con alcohol usando un movimiento circular.
6) Inyecte inmediatamente la solución: Su médico o enfermera ya debe haberle aconsejado dónde inyectar (e.g., estómago, frente de la cadera). Limpie el área escogida con una torunda con alcohol. Pellizque firmemente la piel e inserte la aguja en un ángulo de 45° a 90° usando un movimiento como de dardo. Inyecte bajo la piel, como se le enseñó. No inyecte directamente en una vena. Inyecte la solución presionando cuidadosamente el émbolo. Tómese el tiempo que necesite para inyectar toda la solución. Retire inmediatamente la aguja y limpie la piel con una torunda con alcohol usando un movimiento circular.
7) Deseche todos los artículos usados: Una vez que acabe su inyección, deseche inmediatamente todas las agujas y frascos de vidrio vacíos en el contenedor de objetos punzocortantes proporcionado. Cualquier solución sin usar debe desecharse.
8) Si usa más PERGOVERIS del que debería: Los efectos de una sobredosis de PERGOVERIS son desconocidos; sin embargo, uno podría esperar que se dé un síndrome de hiperestimulación ovárica, que se describe en las secciones Reacciones secundarias y adversas y Precauciones generales. Sin embargo, esto sólo ocurrirá si se administra hCG.
9) Si olvida la aplicación de PERGOVERIS, no administre una doble dosis para reponer una dosis olvidada, póngase en contacto con su médico.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Los efectos de una sobredosis de PERGOVERIS son desconocidos. Sin embargo, uno puede esperar que ocurra el síndrome de hiperestimulación ovárica, que se describe a detalle en las secciones Reacciones secundarias y adversas y Precauciones generales.
El tratamiento está dirigido a los síntomas.
PRESENTACIONES: Cajas con 1, 3 o 10 frascos ámpula con liofilizado y con 1, 3 o 10 frascos ámpula con diluyente.
Es probable que no todos los tamaños de los empaques sean comercializados.
RECOMENDACIONES SOBRE ALMACENAMIENTO: No almacenar a más de 30°C.
Almacenar en el empaque original para protegerlo de la luz.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. Manténgase alejado del alcance de los niños.
Hecho en Suiza por:
Merck Serono, S.A.
Zone lndustrielle de l"Ouriettaz, Aubone, Suiza
Acondicionado por
ATUSA
Ruta 8 Km 17.5, Montevideo, Uruguay
Distribuido por:
MERCK, S.A. de C.V.
Calle 5 No. 7
Fracc. Industrial Alce Blanco, C.P. 53370
Naucalpan de Juárez, México
Reg. Núm. 137M2012, SSA IV


