
PRADAXAR
DABIGATRÁN
Cápsulas
1 Caja, 10 Cápsulas, 75 mg
1 Caja, 10 Cápsulas, 110 mg
1 Caja, 30 Cápsulas, 75 mg
1 Caja, 30 Cápsulas, 110 mg
1 Caja, 30 Cápsulas, 150 mg
1 Caja, 60 Cápsulas, 75 mg
1 Caja, 60 Cápsulas, 110 mg
1 Caja, 60 Cápsulas, 150 mg
1 Caja, 10 Cápsulas, 150 mg
1 Caja, 20 Cápsulas, 75 mg
1 Caja, 20 Cápsulas, 110 mg
1 Caja, 20 Cápsulas, 150 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada CÁPSULA contiene:
Dabigatrán etexilato mesilato equivalente a 75 mg, 110 mg o 150 mg de dabigatrán etexilato
Excipiente cbp 1 cápsula
INDICACIONES TERAPÉUTICAS:
PRADAXAR® está indicado para:
• Prevención de eventos tromboembólicos venosos (ETV) en pacientes que han sido sometidos a cirugía ortopédica mayor.
• Prevención de evento vascular cerebral (EVC), embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular.
• Tratamiento de trombosis venosa profunda (TVP) aguda y/o embolia pulmonar (EP) y prevención de muerte relacionada.
• Prevención de trombosis venosa profunda (TVP) recurrente y/o embolia pulmonar (EP) y prevención de muerte relacionada.
FARMACOCINÉTICA Y FARMACODINAMIA:
Grupo farmacéutico: Inhibidor oral directo de la trombina.
Código ATC: B01 AE07-etexilato de dabigatrán.
El etexilato de dabigatrán es un profármaco de molécula pequeña que no muestra ninguna actividad farmacológica. Después de la administración oral, el etexilato de dabigatrán se absorbe rápidamente y se transforma en dabigatrán mediante hidrólisis catalizada por esterasas en plasma y en el hígado. El dabigatrán es un potente inhibidor directo de la trombina, competitivo y reversible y es el principal principio activo en plasma.
Dado que la trombina (serina proteasa) permite la conversión de fibrinógeno a fibrina durante la cascada de coagulación, su inhibición impide el desarrollo de trombos. El dabigatrán también inhibe la trombina libre, la trombina unida a fibrina y la agregación plaquetaria inducida por trombina.
Los estudios in vivo y ex vivo realizados con animales han demostrado la eficacia antitrombótica y la actividad anticoagulante del dabigatrán después de la administración intravenosa, así como también posterior a la administración oral en diversos modelos animales de trombosis.
Existe una estrecha correlación entre las concentraciones de dabigatrán en plasma y el grado del efecto anticoagulante.
Dabigatrán prolonga el tiempo de tromboplastina parcial activado (TTPa), el tiempo de coagulación de ecarina (ECT), y el tiempo de trombina (TT).
Ensayos clínicos en prevención primaria de tromboembolismo venoso (TEV) después de una cirugía mayor de reemplazo de articulación: En 2 ensayos clínicos grandes, aleatorios, de grupos paralelos, doble ciego y de confirmación de dosis, los pacientes sometidos a cirugía ortopédica mayor electiva (uno para cirugía de reemplazo de rodilla y uno para cirugía de reemplazo de cadera) recibieron etexilato de dabigatrán 75 mg ó 110 mg en las 1-4 horas posteriores a la cirugía, seguidos después por 150 o 220 mg una vez al día, habiéndose asegurado la hemostasia o 40 mg de enoxaparina el día anterior a la cirugía y después una vez al día diariamente.
En el ensayo RE-MODEL (reemplazo de rodilla), el tratamiento se administró durante 6-10 días y en el ensayo RE-NOVATE (reemplazo de cadera), durante 28-35 días. Se trataron un total de 2076 pacientes (rodilla) y 3494 (cadera), respectivamente.
Los resultados del estudio de rodillas (RE-MODEL) con respecto al criterio de valoración primaria, total, incluyendo TEV asintomático, más la mortalidad por cualquier causa mostraron que el efecto antitrombótico de ambas dosis de etexilato de dabigatrán fueron estadísticamente no inferiores al de enoxaparina. De manera similar, el TEV total, incluyendo el asintomático y la mortalidad por cualquier causa, constituyeron el criterio de valoración principal para el estudio de caderas (RE-NOVATE). Nuevamente el etexilato de dabigatrán en ambas dosis de una vez al día no fue estadísticamente inferior respecto a enoxaparina 40 mg al día.
Adicionalmente, en un tercer ensayo aleatorio, de grupos paralelos, doble ciego (RE-MOBILIZE), pacientes sometidos a cirugía electiva total de rodilla recibieron etexilato de dabigatrán 75 mg o 110 mg en un lapso de 6-12 horas después de la cirugía seguidos posteriormente por 150 mg y 220 mg una vez al día. La duración del tratamiento fue de 12-15 días. En total 2615 pacientes fueron escogidos aleatoriamente y 2596 fueron tratados. La dosificación de comparación de enoxaparina fue 30 mg dos veces al día de acuerdo con la etiqueta para los EEUU. En el ensayo RE-MOBILIZE no se estableció la no inferioridad. No hubo diferencias estadísticas en sangrado entre los comparadores.
Adicionalmente, se evaluó un estudio de fase II, aleatorio, de grupos paralelos, doble ciego, controlado con placebo en pacientes japoneses en el cual se administró etexilato de dabigatrán en dosis de 110 mg, 150 mg y 220 mg el día siguiente después de la cirugía electiva de reemplazo total de rodilla. El estudio japonés mostró una relación evidente de respuesta a la dosis en cuanto a la eficacia de etexilato de dabigatrán y un perfil de sangrado tipo placebo.
En el estudio RE-MODEL y el RE-NOVATE la aleatorización al medicamento de estudio respectivo se hizo antes de la cirugía y en RE-MOBILIZE y el ensayo japonés controlado con placebo, la aleatorización al medicamento de estudio respectivo se realizó después de la cirugía. Esto vale la pena observarse especialmente en la evaluación de seguridad de estos ensayos. Por esta razón, los ensayos se agrupan en ensayos aleatorios antes y después de la cirugía en la Tabla 1.
Los datos para el criterio de valoración TEV mayor y mortalidad relacionada con TEV y criterios de valoración principales de sangrado que fueron adjudicados se muestran en la Tabla 1 a continuación. TEV fue definido como la incidencia compuesta de trombosis venosa profunda y el embolismo pulmonar.
Tabla 1. Análisis de TEV mayor y mortalidad relacionada con TEV durante el periodo de tratamiento en los estudios de cirugía ortopédica RE-MODEL y RE-NOVATE
|
Ensayo |
Etexilato de dabigatrán 220 mg |
Etexilato de dabigatrán 150 mg |
Enoxaparina 40 mg |
|
RE-NOVATE (cadera)1 |
|||
|
N |
909 |
888 |
917 |
|
Incidencias (%) |
28 (3.1) |
38 (4.3) |
36 (3.9) |
|
Diferencias en riesgo vs. enoxaparina (%) |
-0.8 |
0.4 |
|
|
IC 95% |
-2.5, 0.8 |
-1.5, 2.2 |
|
|
Razón de riesgo respecto a enoxaparina |
0.78 |
1.09 |
|
|
IC 95% |
0.48, 1.27 |
0.70, 1.70 |
|
|
RE-MODEL (rodilla)1 |
|||
|
N |
506 |
527 |
511 |
|
Incidencias (%) |
13 (2.6) |
20 (3.8) |
18 (3.5) |
|
Diferencias en riesgo vs. enoxaparina (%) |
-1.0 |
0.3 |
|
|
IC 95% |
-3.1, 1.2 |
-2.0, 2.6 |
|
|
Razón de riesgo respecto a enoxaparina |
0.73 |
1.08 |
|
|
IC 95% |
0.36, 1.47 |
0.58, 2.01 |
|
|
RE-MOBILIZE (rodilla)2 |
Enoxaparina 60 mg |
||
|
N |
618 |
656 |
668 |
|
Incidencias (%) |
21 (3.4) |
20 (3.0) |
15 (2.2) |
|
Diferencias en riesgo vs. enoxaparina (%) |
1.2 |
0.8 |
|
|
IC 95% |
(-0.7, 3.0) |
(-0.9, 2.5) |
|
|
Razón de riesgo respecto a enoxaparina |
1.51 |
1.36 |
|
|
IC 95% |
(0.79, 2.91) |
(0.70, 2.63) |
|
|
Estudio japonés de rodilla2 |
|||
|
Placebo |
|||
|
N |
102 |
113 |
104 |
|
Incidencias (%) |
0 |
2 (1.8) |
6 (5.8) |
|
Diferencias en riesgo vs. Placebo (%) |
-5.8 |
-4.0 |
|
|
IC 95% |
(-10.3, - 1.3) |
(-9.1, 1.1) |
|
1 Estudios de selección aleatoria pre-operatoria.
2 Estudios de selección aleatoria post-operatoria.
Ensayos clínicos en prevención de eventos vasculares cerebrales (EVC) y embolismo sistémico en pacientes con fibrilación auricular: La evidencia clínica para la eficacia del etexilato de dabigatrán se deriva del estudio RE-LY (Randomized Evaluation of Long-term anticoagulant therapy) [Evaluación aleatoria de terapia anticoagulante a largo plazo] un estudio multicéntrico, multinacional, aleatorio, de grupos paralelos que comparaba dos dosis cegadas de etexilato de dabigatrán (110 mg dos veces al día y 150 mg dos veces al día) con warfarina en fase abierta en pacientes con fibrilación auricular en riesgo moderado a elevado de evento vascular cerebral o embolismo sistémico. El principal objetivo en este estudio era determinar si el dabigatrán era no-inferior a la warfarina para reducir la incidencia del criterio de valoración compuesto, EVC y eventos embólicos sistémicos (EES).
En el estudio RE-LY, un total de 18113 pacientes fueron aleatorizados, con una media de edad de 71.5 años y un puntaje medio de CHADS2 de 2.1. La población tenía proporciones aproximadamente iguales de pacientes con puntaje de CHADS2 de 1, 2 y ≥ 3. La población de pacientes era 64% de varones, 70% caucásicos y 16% asiáticos. RE-LY tuvo una mediana de tratamiento de 20 meses con etexilato de dabigatrán administrado como dosis fija sin monitoreo de la coagulación. Además de la fibrilación auricular no valvular documentada (FA) por ejemplo FA persistente o paroxística, los pacientes tenían uno de los siguientes factores adicionales de riesgo para EVC:
• EVC previo, ataque isquémico transitorio, o embolismo sistémico.
• Fracción de eyección del ventrículo izquierdo < 40%.
• Insuficiencia cardiaca sintomática, ≥ NYHA Clase 2.
• Edad ≥ 75 años.
• Edad ≥ 65 años asociada con uno de los siguientes; diabetes mellitus, enfermedad arterial coronaria (EAC) o hipertensión.
Las enfermedades concomitantes de los pacientes en este ensayo incluyeron hipertensión 79%, diabetes 23% y EAC 28%. 50% de la población de pacientes era virgen a antagonistas de la vitamina K (AVK), que se define como una exposición total durante la vida menor a 2 meses. 32% de la población nunca había sido expuesta a AVK. Para aquellos pacientes aleatorizados a warfarina, el tiempo en el intervalo terapéutico (INR 2.0 a 3.0) para el ensayo fue una mediana del 67%. Los medicamentos concomitantes incluyeron el ácido acetilsalicílico (AAS) (25% de los sujetos lo usaron al menos 50% del tiempo en estudio), clopidogrel (3.6%), AAS + clopidogrel (2%), AINEs (6.3%) beta-bloqueadores (63.4%), diuréticos (53.9%), estatinas (46.4%), inhibidores de la ECA (44.6%), bloqueadores del receptor de la angiotensina (26.1%), hipoglucemiantes orales (17.5%), insulina (5.2%), digoxina (29.4%), amiodarona (11.3%)diltiazem (8.9%), verapamilo (5.4%), e inhibidores de la bomba de protones (17.8%).
Para el criterio de valoración principal, el EVC y el embolismo sistémico, no se identificaron subgrupos (es decir, edad, peso, género, función renal, origen étnico, etc.) con una razón de riesgo diferente en comparación con la warfarina.
Este estudio demostró que el etexilato de dabigatrán, en dosis de 110 mg dos veces al día, no es inferior a la warfarina en la prevención de EVC y el embolismo sistémico en sujetos con fibrilación auricular, con una reducción en el riesgo de hemorragia intracraneal y de sangrado total. La dosis más elevada de 150 mg dos veces al día, reduce significativamente el riesgo de eventos vasculares cerebrales isquémicos y hemorrágicos, muerte vascular, hemorragia intracraneal y sangrado total en comparación con la warfarina. La dosis menor de dabigatrán tuvo un riesgo significativamente menor de sangrado mayor comparado con la warfarina.
La Figura 1 y las tablas 2-6 presentan los detalles de los resultados clave.
Tabla 2. Análisis del primer episodio de EVC o embolismo sistémico (criterio de valoración principal) durante el periodo del estudio en el RE-LY (grupo aleatorizado)
|
Etexilato de dabigatrán 150 mg dos veces al día |
Etexilato de dabigatrán 110 mg dos veces al día |
Warfarina |
|
|
Sujetos aleatorizados |
6,076 |
6,015 |
6,022 |
|
EVC y/o EES |
|||
|
Incidencias (%) |
135 (1.12) |
183 (1.54) |
203 (1.72) |
|
Razón de riesgo instantáneo respecto a warfarina (IC 95%) |
0.65 (0.52, 0.81) |
0.89 (0.73, 1.09) |
|
|
Superioridad del valor p |
p = 0.0001 |
p = 0.2721 |
|
% se refiere a la tasa de eventos anuales.
Figura 1. Curva Kaplan-Meier para el estimado de tiempo para el primer EVC o embolismo sistémico
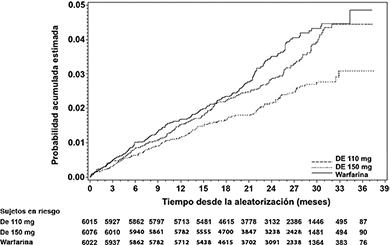
Tabla 3. Análisis del primer episodio de EVC isquémico o hemorrágico durante el periodo de estudio en RE-LY (grupo aleatorizado)
|
Etexilato de dabigatrán 150 mg dos veces al día |
Etexilato de dabigatrán 110 mg dos veces al día |
Warfarina |
|
|
Sujetos aleatorizados |
6,076 |
6,015 |
6,022 |
|
EVC |
|||
|
Incidencias (%) |
123 (1.02) |
171 (1.44) |
187 (1.59) |
|
Razón de riesgo instantáneo vs. warfarina (IC 95%) |
0.64 (0.51, 0.81) |
0.91 (0.74, 1.12) |
|
|
Valor-p |
0.0001 |
0.3553 |
|
|
EES |
|||
|
Incidencias (%) |
13 (0.11) |
15 (0.13) |
21 (0.18) |
|
Razón de riesgo instantáneo vs. warfarina (IC 95%) |
0.61 (0.30, 1.21) |
0.71 (0.37, 1.38) |
|
|
Valor-p |
0.1582 |
0.3099 |
|
|
EVC isquémico |
|||
|
Incidencias (%) |
104 (0.86) |
152 (1.28) |
134 (1.14) |
|
Razón de riesgo instantáneo vs. warfarina (IC 95%) |
0.76 (0.59, 0.98) |
1.13 (0.89, 1.42) |
|
|
Valor-p |
0.0351 |
0.3138 |
|
|
EVC hemorrágico |
|||
|
Incidencias (%) |
12 (0.10) |
14 (0.12) |
45 (0.38) |
|
Razón de riesgo instantáneo vs. warfarina (IC 95%) |
0.26 (0.14, 0.49) |
0.31 (0.17, 0.56) |
|
|
Valor-p |
< 0.0001 |
< 0.0001 |
|
% se refiere a la tasa de eventos anuales.
Tabla 4. Análisis de supervivencia por todas las causas y cardiovasculares durante el periodo de estudio en el estudio RE-LY (grupo aleatorizado)
|
Etexilato de dabigatrán 150 mg dos veces al día |
Etexilato de dabigatrán 110 mg dos veces al día |
Warfarina |
|
|
Sujetos aleatorizados |
6076 |
6015 |
6022 |
|
Mortalidad por todas las causas |
|||
|
Incidencias (%) |
438 (3.64) |
446 (3.75) |
487 (4.13) |
|
Cociente de riesgo frente a la warfarina (IC 95%) |
0.88 (0.77, 1.00) |
0.91 (0.80, 1.03) |
|
|
Valor-p |
0.0517 |
0.1308 |
|
|
Mortalidad vascular |
|||
|
Incidencias (%) |
274 (2.28) |
289 (2.43) |
317 (2.69) |
|
Cociente de riesgo frente a la warfarina (IC 95%) |
0.85 (0.72, 0.99) |
0.90 (0.77, 1.06) |
|
|
Valor-p |
0.0430 |
0.2081 |
|
% se refiera a la tasa de eventos anuales.
El beneficio clínico neto medido por el criterio de valoración clínico compuesto de EVC, embolismo sistémico, embolismo pulmonar (EP), infarto agudo al miocardio (IM), muertes vasculares y sangrados mayores fue valorado y se presenta como parte de la Tabla 5. Las tasas de eventos anuales para los grupos de etexilato de dabigatrán fueron menores comparadas con el grupo de warfarina. La reducción del riesgo para este criterio de valoración combinado fue del 8% y 10% para los grupos de tratamiento con etexilato de dabigatrán 110 mg dos veces al día y 150 mg dos veces al día. Otros componentes evaluados incluyeron todas las hospitalizaciones, las cuales tuvieron menos hospitalizaciones estadísticamente significativas con etexilato de dabigatrán 110 mg dos veces al día comparadas con warfarina (reducción del riesgo de 7%, IC 95% 0.87, 0.99, p = 0.021).
Tabla 5. Otras mediciones evaluadas
|
Etexilato de dabigatrán 150 mg dos veces al día |
Etexilato de dabigatrán 110 mg dos veces al día |
Warfarina |
|
|
Sujetos aleatorizados |
6076 |
6015 |
6022 |
|
EVCI/EES/muerte |
|||
|
Incidencias (%) |
520 (4.32) |
577 (4.85) |
613 (5.20) |
|
Razón de riesgo instantáneo vs. Warfarina (IC 95%) |
0.83 (0.74. 0.93) |
0.93 (0.83, 1.04) |
|
|
Valor-p |
0.0015 |
0.2206 |
|
|
EVC/EES/EP/IM/muerte/sangrado mayor (beneficio clínico neto) (BCN) |
|||
|
Incidencias (%) |
850 (7.06) |
863 (7.25) |
925 (7.84) |
|
Razón de riesgo instantáneo vs. Warfarina (IC 95%) |
0.90 (0.82, 0.99) |
0.92 (0.84, 1.01) |
|
|
Valor-p |
0.0287 |
0.0849 |
|
|
Embolismo pulmonar (EP) |
|||
|
Incidencias (%) |
18 (0.15) |
14 (0.12) |
12 (0.10) |
|
Razón de riesgo instantáneo vs. Warfarina (IC 95%) |
1.41 (0.71, 3.06) |
1.16 (0.54, 2.51) |
|
|
Valor-p |
0.2980 |
0.7076 |
|
|
Infarto al miocardio (IM) (incluyendo infarto silencioso) |
|||
|
Incidencias (%) |
97 (0.81) |
98 (0.82) |
75 (0.64) |
|
Razón de riesgo instantáneo vs. Warfarina (IC 95%) |
1.27 (0.94, 1.71) |
1.29 (0.96, 1.75) |
|
|
Valor-p |
0.1240 |
0.0929 |
|
Tabla 6. Pruebas de función hepática.
En el estudio RE-LY, las anomalías potenciales de las pruebas de función hepática (PFH) se presentaron con una incidencia comparable o menor en los pacientes tratados con etexilato de dabigatrán vs. Warfarina
|
Etexilato de dabigatrán 150 mg dos veces al día N (%) |
Etexilato de dabigatrán 110 mg dos veces al día N (%) |
Warfarina N (%) |
|
|
Total tratados |
6059 (100.0) |
5983 (100.0) |
5998 (100.0) |
|
ALT o AST |
106 (1.7) |
118 (2.0) |
125 (2.1) |
|
ALT o AST |
45 (0.7) |
36 (0.6) |
50 (0.8) |
|
ALT o AST > 3xULN + Bilirrubina |
14 (0.2) |
11 (0.2) |
21 (0.4) |
El estudio de extensión RE-LY (RELY-ABLE) brindó información adicional de seguridad para un amplio grupo de pacientes que continuaban con la misma dosis de dabigatrán etexilato según fue asignado en el estudio RE-LY. Los pacientes eran elegibles para el estudio RELY-ABLE si no habían discontinuado permanentemente la medicación de estudio al momento de realizar la visita final del estudio RE-LY. Los pacientes enrolados continuaron recibiendo la misma dosis doble-ciego de dabigatrán etexilato asignado aleatoriamente en RE-LY, por hasta 43 meses de seguimiento luego de RE-LY (seguimiento promedio total RE-LY + RELY-ABLE, 4.5 años). Hubo 5897 pacientes enrolados representando 49% de los pacientes asignados aleatoriamente para recibir dabigatrán etexilato en RE-LY y 86% del total de pacientes elegibles para el estudio RELY-ABLE.
Durante los 2.5 años adicionales de tratamiento en RELY-ABLE, con una exposición máxima de hasta 6 años (total de exposición en RELY + RELY-ABLE), se confirmó el perfil de seguridad a largo plazo de dabigatrán etexilato en ambas dosis. No se observaron nuevos hallazgos de seguridad.
Las tasas de resultados de eventos, incluyendo sangrado mayor y otros eventos de sangrado fueron consistentes con los reportados en el estudio RELY-ABLE.
Además del estudio RE-LY, un estudio no intervencionista internacional (GLORIA-AF), recopiló prospectivamente (en su segunda fase) datos de seguridad y eficacia en pacientes con diagnóstico reciente de fibrilación auricular no valvular tratados con dabigatrán etexilato en un entorno real. El estudio incluyó 4,859 pacientes tratados con dabigatrán etexilato (dosificación según la práctica clínica local y el rótulo local; 55% tratados con 150 mg 2 veces al día, 43% tratados con 110 mg dos veces al día, 2% tratados con 75 mg dos veces al día). A los pacientes se les hizo un seguimiento durante 2 años. Los valores medios de los puntajes CHADS2 y HAS-BLED fueron 1.9 y 1.2 respectivamente, en comparación con un valor medio de los puntajes CHADS2 y HAS-BLED de 2.1 y 1.3 en el estudio RE-LY, respectivamente. El tiempo promedio de seguimiento durante el tratamiento fue de 18.3 meses. Se produjo sangrado importante en 0.97 por 100 pacientes-año. Se informó sangrado que pone en riesgo la vida en 0.46 por 100 pacientes-año, hemorragia intracraneana en 0.17 por 100 pacientes-año y sangrado gastrointestinal en 0.60 por 100 pacientes-año. Se presentó accidente cerebrovascular en 0.65 por 100 pacientes-año.
Asimismo, en un estudio no intervencionista [Graham DJ et al., Circulation. 2015] realizado en más de 134 000 pacientes de edad avanzada con fibrilación auricular no valvular en los Estados Unidos (que aportó más de 37 500 pacientes-año de tiempo de seguimiento durante el tratamiento) se asoció al dabigatrán etexilato (84% de pacientes tratados con 150 mg dos veces al día, 16% de pacientes tratados con 75 mg dos veces al día) con una reducción estadísticamente significativa del riesgo de accidente cerebrovascular isquémico (razón de riesgos instantáneos 0.80; intervalo de confianza del 95% [lC] 0.67-0.96), de hemorragia intracraneana (razón de riesgos instantáneos 0.34; lC 0.26-0.46) y de mortalidad (razón de riesgos instantáneos 0.86; IC 0.77-0.96), y con un aumento del riesgo de sangrado gastrointestinal (razón de riesgos instantáneos 1.28; IC 1.14-1.44) en comparación con la warfarina. No se observó una diferencia significativa en el caso de sangrado importante (razón de riesgos instantáneos 0.97; IC 0.88-1.07).
Estas observaciones en entornos reales resultan compatibles con el perfil de seguridad y eficacia establecido para el dabigatrán etexilato en esta indicación.
Manejo de síntomas gastrointestinales: En un estudio exploratorio se evaluó la eficacia de dos estrategias de manejo de síntomas gastrointestinales (SGl): la administración de PRADAXAR® dentro de los 30 minutos posteriores a una de las comidas principales o administrar pantoprazol en un régimen de 40 mg diarios.
En total, n = 1067 pacientes tratados con PRADAXAR® ingresaron en el estudio; 117 pacientes desarrollaron SGI y fueron aleatorizados a uno de los dos tratamientos.
Ambas estrategias de manejo iniciales (la administración de PRADAXAR® luego de una comida o administrar pantoprazol 40 mg diarios) brindaron un alivio completo de los SGI primarios en más del 55% de los pacientes que informaron SGI (administración de PRADAXAR® después de una comida: 55.9%; pantoprazol: 67.2%).
Como estrategia individual de manejo de los SGI, la administración de pantoprazol en un régimen de 40 mg diarios permitió una completa resolución de los síntomas en el 67.2% de los pacientes luego de 4 semanas de tratamiento, en tanto que la administración de PRADAXAR® tras una de las comidas principales se tradujo en la resolución completa de los síntomas en el 55.9 % de los pacientes. Luego de 1 semana de tratamiento, se logró una completa resolución de los síntomas en el 51.7% de los pacientes que recibieron pantoprazol frente a un 39.0% en el caso de los pacientes que tomaron PRADAXAR® luego de una comida.
En los pacientes que al finalizar las 4 semanas no tenían una respuesta completa a la estrategia inicial debía implementarse además una estrategia alternativa (= estrategias combinadas) durante 4 semanas más.
Se informó una efectividad completa o parcial luego de 4 semanas de las estrategias de manejo combinadas (8 semanas, tratamiento total) en 12 de 14 (85.7%) pacientes que tomaron PRADAXAR® después de una comida en la primera parte del estudio y en 12 de 15 (80.0%) pacientes que tomaron pantoprazol en la primera parte del estudio.
Por último, 92 (78.6%) pacientes (79 con eficacia completa y 13 con eficacia parcial), experimentaron resultados positivos utilizando las dos estrategias de gestión de los SGI, 45 en el grupo que al que se le administró PRADAXAR® después de la comida (39 con eficacia completa y 6 con eficacia parcial) y 47 en el grupo de Pantoprazol (40 con eficacia completa y 7 con eficacia parcial.
Pacientes sometidos a ablación con catéter de la fibrilación auricular: Se llevó a cabo un estudio exploratorio, aleatorizado, prospectivo, abierto, multicéntrico con adjudicación central y ciega de los criterios de valoración (RE-CIRCUIT) en 704 pacientes que estaban bajo tratamiento anticoagulante estable. En el estudio se compararon 150 mg de dabigatrán etexilato ininterrumpido administrado dos veces al día con warfarina ajustada según INR en la ablación con catéter de la fibrilación auricular persistente o paroxística. De los 704 pacientes enrolados, 317 fueron sometidos a ablación de la fibrilación auricular sin interrumpir el tratamiento con dabigatrán y 318 fueron sometidos a ablación de la fibrilación auricular sin interrumpir el tratamiento con warfarina. A todos los pacientes se les realizó una ecocardiografía transesofágica (ETE) antes de la ablación con catéter. El criterio de valoración primario (sangrado importante adjudicado de acuerdo con el criterio de la ISTH) se presentó en 5 (1.6 %) pacientes del grupo de dabigatrán etexilato y en 22 (6.9 %) pacientes del grupo de warfarina (diferencia de riesgo -5.3 %; IC 95 % -8.4, -2.2; P = 0,0009). No hubo ningún evento de accidente cerebrovascular/embolia sistémica/ataque isquémico transitorio (AIT) (criterio de valoración compuesto) en el grupo de dabigatrán etexilato, y hubo un evento (AIT) en el grupo de warfarina desde el momento de la ablación y hasta 8 semanas postablación. La tasa de incidencia compuesta de ESG y de eventos de tromboembolia (accidente cerebrovascular/embolia sistémica/AIT) fue menor en el grupo de dabigatrán etexilato (5 [1.6 %] versus 23 [7.2 %] pacientes). En este estudio exploratorio quedó demostrado que el dabigatrán etexilato estuvo asociado con una reducción significativa desde el punto de vista estadístico y clínicamente relevante de la tasa de ESG en comparación con la warfarina ajustada según INR, y no hubo diferencias en la incidencia de accidente cerebrovascular o de embolia sistémica en el contexto de la ablación.
Pacientes que fueron sometidos a una intervención coronaria percutánea con colocación de stent: Se llevó a cabo un estudio aleatorizado, prospectivo, abierto con adjudicación ciega de los criterios de valoración (PROBE) (Fase IIIb) para evaluar el tratamiento doble con dabigatrán etexilato (110 mg o 150 mg dos veces al día) más clopidogrel o ticagrelor (antagonista del receptor P2Y12) versus el tratamiento triple con warfarina (ajustada a un INR de 2.0-3.0) más clopidogrel o ticagrelor y aspirina en 2725 pacientes con fibrilación auricular no valvular sometidos a una ICP (PCI) con colocación de stent (RE-DUAL PCI). Los pacientes fueron aleatorizados al tratamiento doble con dabigatrán etexilato 110 mg 2 veces al día, al tratamiento doble con dabigatrán etexilato 150 mg 2 veces al día o al tratamiento triple con warfarina. Los pacientes de edad avanzada que viven fuera de los Estados Unidos (≥ 80 años de edad en todos los países, ≥ 70 años de edad en Japón) fueron aleatorizados al grupo de tratamiento doble con dabigatrán etexilato 110 mg o al grupo de tratamiento triple con warfarina. El criterio de valoración primario consistió en una combinación de hemorragia mayor según la definición de la ISTH o en un evento de hemorragia menor clínicamente relevante.
La incidencia del criterio de valoración primario fue de 15.4% (151 pacientes) en el grupo de tratamiento doble con dabigatrán etexilato 110 mg en comparación con 26.9% (264 pacientes) en el grupo de tratamiento triple con warfarina (HR 0.52; IC 95% 0.42; 0.63; P < 0.0001 para la no inferioridad y P < 0.0001 para la superioridad) y de 20.2% (154 pacientes) en el grupo de tratamiento doble con dabigatrán etexilato 150 mg en comparación con 25.7% (196 pacientes) en el correspondiente grupo de tratamiento triple con warfarina (HR 0.72; IC 95% 0.58; 0.88; P < 0.0001 para la no inferioridad y P = 0.002 para la superioridad). Como parte del análisis descriptivo, los eventos de sangrado importante de la trombólisis en el infarto de miocardio (Thrombolysis In Myocardial Infarction, TIMI) fueron menores en ambos grupos de tratamiento doble con dagibatrán etexilato que en el grupo de tratamiento triple con warfarina: 14 eventos (1.4%) en el grupo de tratamiento doble con dabigatrán etexilato 110 mg en comparación con 37 eventos (3.8%) en el grupo de tratamiento triple con warfarina (HR 0.37; IC 95% 0.20; 0.68; P = 0.002) y 16 eventos (2.1%) en el grupo de tratamiento doble con dabigatrán etexilato 150 mg en comparación con 30 eventos (3.9%) en el correspondiente grupo de tratamiento triple con warfarina (HR 0.51; IC 95% 0.28; 0.93; P = 0.03). Ambos grupos de tratamiento doble con dabigatrán etexilato presentaron menores tasas de hemorragias intracraneales en comparación con las del correspondiente grupo de tratamiento triple con warfarina: 3 eventos (0.3%) en el grupo de tratamiento doble con dabigatrán etexilato 110 mg en comparación con 10 eventos (1.0%) en el grupo de tratamiento triple con warfarina (HR 0.30; IC 95% 0.08; 1.07; P = 0.06) y 1 evento (0.1%) en el correspondiente grupo de tratamiento triple con warfarina (HR 0.12; IC 95% 0.02; 0.98; P = 0.047). La incidencia del criterio de valoración compuesto para eficacia de muerte, eventos tromboembólicos (infarto de miocardio, accidente cerebrovascular o embolia sistémica) o revascularización no planificada en los dos grupos de tratamiento doble con dabigatrán etexilato combinados fue no inferior a la del grupo de tratamiento triple con warfarina (13.7% versus 13.4%, respectivamente; HR 1.04; IC 95%: 0.84; 1.29; P = 0.0047 para la no inferioridad). No hubo diferencias estadísticas en los componentes individuales de los criterios de valoración de la eficacia entre cualquiera de los grupos de tratamiento doble con dabigatrán etexilato y el tratamiento triple con warfarina. En este estudio quedó demostrado que el tratamiento doble, con dabigatrán etexilato y un antagonista del receptor P2Y12, redujo significativamente el riesgo de sangrado versus el tratamiento triple con warfarina, con un criterio de valoración compuesto de eventos tromboembólicos no inferior en pacientes con fibrilación auricular sometidos a una ICP con colocación de stent.
Ensayos clínicos en el tratamiento de trombosis (TVP) venosa aguda y/o embolismo pulmonar (EP) y prevención de muerte relacionada: La evidencia clínica obtenida a partir de dos estudios multicéntricos, aleatorizados, doble ciego, de grupos paralelos, replicados, RE-COVER y RE-COVER II ha demostrado que dabigatrán etexilato es un tratamiento seguro y efectivo para la TVP y/o la EP. En estos estudios se comparó dabigatrán etexilato (150 mg dos veces al día) con warfarina (valor objetivo de RIN 2.0-3.0) en pacientes con TVP aguda y/o EP. El objetivo primario de estos estudios fue determinar si dabigatrán era no inferior a la warfarina en la reducción de la ocurrencia del criterio de valoración primario, que fue el criterio de valoración compuesto de TVP y/o EP sintomática recurrente y muertes relacionadas dentro del periodo de tratamiento agudo de 6 meses.
En los estudios RE-COVER y RE-COVER II combinados, un total de 5153 pacientes fueron aleatorizados y 5107 de ellos recibieron tratamiento. Los eventos índice en el nivel basal fueron: TVP -68.5%, EP -22.2%, EP y TVP -9.1%. Los factores de riesgo más frecuentes fueron antecedentes de TVP y/o EP -21.5%, cirugía/traumatismo -18.1%, insuficiencia venosa -17.6% e inmovilización prolongada -14.6%. Las características basales de los pacientes fueron: media de edad 54.8 años, sexo masculino 59.5%, raza caucásica 86.1%, raza asiática 11.8%, raza negra 2.1%. Las comorbilidades incluyeron: hipertensión 35.5%, diabetes mellitus 9.0%, arteriopatía coronaria 6.8% y úlcera gástrica o duodenal 4.1%.
La duración del tratamiento con la dosis fija de dabigatrán fue 174.0 días sin monitoreo de la coagulación. En el caso de los pacientes aleatorizados a warfarina, la mediana del tiempo dentro del rango terapéutico (RIN 2.0 a 3.0) fue 60.6%. Los medicamentos concomitantes incluyeron vasodilatadores 28.5%, reguladores del sistema renina-angiotensina 24.7%, hipolipemiantes 19.1%, betabloqueantes 14.8%, bloqueadores del canal de calcio 9.7%, AINES 21.7%, aspirina 9.2%, antiplaquetarios 0.7%, inhibidores de la P-gp 2.0% (verapamilo -1.2% y amiodarona -0.4%).
Dos estudios en pacientes con TVP aguda y/o EP tratados inicialmente durante un mínimo de 5 días con tratamiento parenteral, el estudio RE-COVER y el estudio RE-COVER II, demostraron que el tratamiento con dabigatrán etexilato administrado en dosis de 150 mg dos veces al día era no inferior al tratamiento con warfarina (valores p para la no inferioridad: RE-COVER p < 0.0001, RE-COVER II p = 0.0002). Los eventos de sangrado (ESG, ESG/ESCR y sangrado de cualquier tipo) fueron significativamente más bajos en los pacientes que recibieron dabigatrán etexilato 150 mg dos veces al día en comparación con aquellos que recibieron warfarina.
Figura 2. Tiempo para el primer ETV adjudicado y muerte relacionada con ETV adjudicada hasta el final del periodo después del tratamiento para los estudios agrupados RE-COVER y RE-COVER II
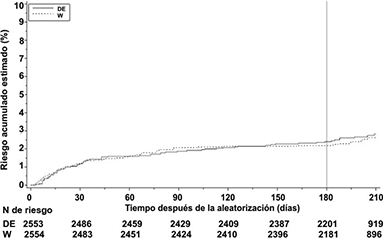
Tabla 7. Análisis de los criterios de valoración de eficacia primario y secundario (ETV es un compuesto de TVP y/o EP) hasta el final del periodo posterior al tratamiento para los estudios agrupados RE-COVER y RE-COVER II
|
Etexilato de dabigatrán 150 mg |
Warfarina |
|
|
RE-COVER/RE-COVER II agrupado |
||
|
Pacientes, n (%) |
2,553 (100.0) |
2,554 (100.0) |
|
ETV sintomático recurrente y muerte relacionada con ETV |
68 (2.7) |
62 (2.4) |
|
Razón de riesgo vs. warfarina |
1.09 |
|
|
IC 95% |
(0.77, 1.54) |
|
|
Criterios de valoración de eficacia secundarios |
||
|
ETV sintomático recurrente y muertes por cualquier causa |
109 (4.3) |
104 (4.1) |
|
IC 95% |
3.52, 5.13 |
3.34, 4.91 |
|
TVP sintomático |
45 (1.8) |
39 (1.5) |
|
IC 95% |
1.29, 2.35 |
1.09, 2.08 |
|
EP sintomático |
27 (1.1) |
26 (1.0) |
|
IC 95% |
0.70, 1.54 |
0.67, 1.49 |
|
Muertes relacionadas con ETV |
4 (0.2) |
3 (0.1) |
|
IC 95% |
0.04, 0.40 |
0.02, 0.34 |
|
Muertes por cualquier causa |
51 (2.0) |
52 (2.0) |
|
IC 95% |
1.49, 2.62 |
1.52, 2.66 |
Otras medidas evaluadas:
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: El infarto al miocardio ocurrió en una frecuencia baja en los cuatro estudios de ETV para todos los grupos de tratamiento. Ocurrió muerte cardiaca en un paciente del grupo de tratamiento con warfarina.En los tres estudios controlados activos se reportó un desbalance numérico de infarto al miocardio en pacientes tratados con etexilato de dabigatrán (20; 0.5%) en comparación con los pacientes tratados con warfarina (5; 0.1%). En el estudio RE-SONATE, el cual comparó el etexilato de dabigatrán con el placebo, hubo 1 evento de IM en cada grupo de tratamiento, resultando en frecuencias de IM con etexilato de dabigatrán iguales a las frecuencias con el placebo.
Pruebas de función hepática:
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: En el estudio controlado activo RE-COVER, RE-COVER II y RE-MEDY, ocurrieron anormalidades potenciales de las pruebas de función hepática (LFT) con una incidencia comparable o menor en los pacientes tratados con etexilato de dabigatrán vs. los pacientes tratados con warfarina. En el estudio RE-SONATE, no hubo una diferencia notable entre los grupos tratados con etexilato de dabigatrán y placebo con respecto a posibles valores LFT anormales clínicamente significativos.
Estudios clínicos de prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada: La evidencia clínica ha demostrado que el etexilato de dabigatrán es un tratamiento efectivo y seguro para la TVP recurrente y/o EP. Dos estudios aleatorizados, de grupos paralelos, doble ciego fueron realizados en pacientes previamente tratados con anticoagulantes. En el estudio controlado con warfarina RE-MEDY, se incluyeron pacientes tratados durante 3 a 12 meses con la necesidad de continuar el tratamiento anticoagulante y el estudio controlado con placebo RE-SONATE, incluyó pacientes tratados durante 6 a 18 meses con inhibidores de la Vitamina K.
El objetivo del estudio RE-MEDY era comparar la seguridad y eficacia del etexilato de dabigatrán por vía oral (150 mg dos veces al día) con warfarina (INR 2.0-3.0) para el tratamiento a largo plazo y prevención de TVP sintomática y recurrente y/o EP. Un total de 2,866 pacientes fueron aleatorizados y 2,856 pacientes fueron tratados. El índice de eventos basales fue: TVP-65.1%, EP-23.1%, EP y TVP-11.7%. Las características basales de los pacientes fueron: edad promedio 54.6 años, varones 61.0%, caucásicos 90.1%, asiáticos 7.9%, raza negra 2.0%. La co-morbilidad incluyó hipertensión 38.6%, diabetes mellitus 9.0%, EAC 7.2% y úlcera gástrica o duodenal 3.8%. Medicamentos concomitantes: agentes que actúan sobre el sistema renina-angiotensina 27.9%, vasodilatadores 26.7%, agentes que disminuyen los lípidos 20.6%, AINEs 18,3%, beta bloqueadores 16.3%, bloqueadores de los canales de calcio 11.1%, aspirina 7.7%, inhibidores de la P-gp 2.7% (verapamilo 1.2% y amiodarona 0.7%), antiplaquetarios 0.9%. La duración del tratamiento con etexilato de dabigatrán osciló desde 6 hasta 36 meses (media de 534.0 días). El tiempo medio en el rango terapéutico (INR 2.0-3.0) para los pacientes aleatorizados a warfarina fue de 64.9%.
El estudio RE-MEDY demostró que el tratamiento con etexilato de dabigatrán con una dosis de 150 mg dos veces al día fue no-inferior a la warfarina (p = 0.0135 para la no-inferioridad). Los eventos de sangrado (ESMs/ESCRs; cualquier sangrado) fueron significativamente menores en pacientes que recibieron etexilato de dabigatrán en comparación con los pacientes que recibieron warfarina.
Así como en los estudios agrupados RE-COVER/RE-COVER II, en el estudio RE-MEDY pocos pacientes reportaron el uso concomitante de inhibidores de P-gp (2.7%); mientras que verapamilo (1.2%) y amiodarona (0.7%) fueron los más frecuentes. En los estudios agrupados que analizaron el tratamiento de ETV agudos, pocos pacientes reportaron el uso concomitante de inhibidores de P-gp (2.0%); mientras que verapamilo (1.2% general) y amiodarona (0.4% general) fueron los más frecuentes.
Figura 3. Estimado de tiempo para el primer ETV y muerte relacionada a ETV hasta el final del periodo planeado de tratamiento para el estudio RE-MEDY
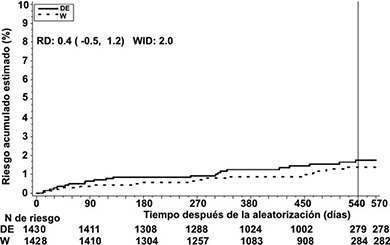
La tabla 8 incluye detalles de los resultados clave en el estudio RE-MEDY.
Tabla 8. Análisis de los criterios de valoración de eficacia primario y secundario (ETV es un compuesto de TVP y/o EP) hasta el final del periodo después del tratamiento para el estudio RE-MEDY
|
Etexilato de dabigatrán 150 mg |
Warfarina |
|
|
RE-MEDY |
||
|
Pacientes, n (%) |
1,430 (100.0) |
1,426 (100.0) |
|
ETV sintomático recurrente y muerte relacionada con ETV |
26 (1.8) |
18 (1.3) |
|
Razón de riesgo vs. warfarina |
1.44 |
|
|
IC 95% |
0.78, 2.64 |
|
|
Valor-p (no-inferioridad) |
0.0135 |
|
|
Pacientes con evento a 18 meses |
22 |
17 |
|
Riesgo acumulado a 18 meses (%) |
1.7 |
1.4 |
|
Diferencia de riesgo vs. warfarina (%) |
0.4 |
|
|
IC 95% |
-0.5, 1.2 |
|
|
Valor-p (no-inferioridad) |
< 0.0001 |
|
|
Criterios de valoración de eficacia secundarios |
||
|
ETV sintomático recurrente y muerte relacionado con todas las causas |
42 (2.9) |
36 (2.5) |
|
IC 95% |
2.12, 3.95 |
1.77, 3.48 |
|
TVP sintomático |
17 (1.2) |
13 (0.9) |
|
IC 95% |
0.69, 1.90 |
0.49, 1.55 |
|
EP sintomático |
10 (0.7) |
5 (0.4) |
|
IC 95% |
0.34, 1.28 |
0.11, 0.82 |
|
Muertes relacionadas con ETV |
1 (0.1) |
1 (0.1) |
|
IC 95% |
0.00, 0.39 |
0.00, 0.39 |
|
Muertes por cualquier causa |
17 (1.2) |
19 (1.3) |
|
IC 95% |
0.69, 1.90 |
0.80, 2.07 |
El objetivo del estudio RE-SONATE era evaluar la superioridad del etexilato de dabigatrán versus el placebo para la prevención de TVP sintomática recurrente y/o EP en pacientes que ya habían completado de 6 a 18 meses de tratamiento con antagonistas de la Vitamina K. La terapia prevista fue 6 meses de etexilato de dabigatrán 150 mg dos veces al día sin necesidad de monitoreo.
El índice de eventos basales fue: TVP-64.5%, EP-27.8%, EP y TVP-7.7%. Un total de 1,353 pacientes fueron aleatorizados y 1,343 pacientes fueron tratados. Las características basales de los pacientes fueron: edad promedio 55.8 años, varones 55.5%, caucásicos 89.0%, asiáticos 9.3%, raza negra 1.7%. La co-morbilidad incluyó hipertensión 38.8%, diabetes mellitus 8.0%, EAC 6.0% y úlcera gástrica o duodenal 4.5%. Medicamentos concomitantes: agentes que actúan sobre el sistema renina-angiotensina 28.7%, vasodilatadores 19.4%, agentes que disminuyen los lípidos 17.9%, beta bloqueadores 18.5%, bloqueadores de los canales de calcio 8.9%, AINEs 12,1%, aspirina 8.3%, antiplaquetarios 0.7% e inhibidores de la P-gp 1.7% (verapamilo 1.0% y amiodarona 0.3%).
El estudio RE-SONATE demostró que el etexilato de dabigatrán fue superior al placebo para la prevención de eventos de TVP sintomática recurrente/EP incluyendo muertes sin explicación, con una reducción del riesgo de 92% durante el periodo de tratamiento (p < 0.0001). Todos los análisis secundarios y de sensibilidad del criterio primario de valoración y todos los criterios secundarios de valoración mostraron superioridad del etexilato de dabigatrán sobre el placebo. Las tasas de ESM y la combinación de ESM/ESCR fueron significativamente mayores en pacientes recibiendo etexilato de dabigatrán en comparación con los pacientes que recibieron placebo.
El estudio incluyó un seguimiento de observación de 12 meses después de la conclusión del tratamiento. Después de la descontinuación del medicamento de estudio el efecto fue mantenido hasta el final del seguimiento, indicando que el efecto del tratamiento inicial del etexilato de dabigatrán fue sostenido. No se observó efecto de rebote. Al final del seguimiento los eventos de ETV en pacientes tratados con etexilato de dabigatrán fue de 6.9% vs 10.7% dentro del grupo tratado con placebo (razón de riesgo 0.61 (0.42, 0.88), p = 0.0082).
Figura 4. Estimado de tiempo para el primer ETV y muerte relacionada con ETV hasta el final del periodo planeado de tratamiento para el estudio RE-SONATE
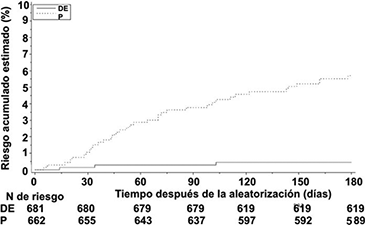
La tabla 9 muestra los detalles de los resultados clave del estudio RE-SONATE.
Tabla 9. Análisis de los criterios de valoración de eficacia primario y secundario (ETV es un compuesto de TVP y/o EP) hasta el final del periodo pos-tratamiento para el estudio RE-SONATE
|
Etexilato de dabigatrán 150 mg |
Placebo |
|
|
RE-SONATE |
||
|
Pacientes, n (%) |
681 (100.0) |
662 (100.0) |
|
ETV sintomático recurrente y muertes relacionadas |
3 (0.4) |
37 (5.6) |
|
Razón de riesgo |
0.08 |
|
|
IC 95% |
0.02, 0.25 |
|
|
Valor-p |
< 0.0001 |
|
|
Criterios de valoración de eficacia secundarios |
||
|
ETV sintomático recurrente y muerte por cualquier causa |
3 (0.4) |
37 (5.6) |
|
IC 95% |
0.09, 1.28 |
3.97, 7.62 |
|
TVP sintomático |
2 (0.3) |
23 (3.5) |
|
IC 95% |
0.04, 1.06 |
2.21, 5.17 |
|
EP sintomático |
1 (0.1) |
14 (2.1) |
|
IC 95% |
0.00, 0.82 |
1.16, 3.52 |
|
Muertes relacionadas con ETV |
0 (0) |
0 (0) |
|
IC 95% |
0.00, 0.54 |
0.00, 0.56 |
|
Muertes sin explicación |
0 (0) |
2 (0.3) |
|
IC 95% |
0.00, 0.54 |
0.04, 1.09 |
|
Muertes por cualquier causa |
0 (0) |
2 (0.3) |
|
IC 95% |
0.00, 0.54 |
0.04, 1.09 |
Otras medidas evaluadas:
Prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada: En los cuatro estudios que analizaron los ETV, el IM ocurrió a una frecuencia baja para todos los grupos de tratamiento. En el grupo de tratamiento con warfarina ocurrió muerte cardiaca en un paciente.
En tres estudios controlados activos se reportó un desbalance numérico de IM en pacientes que recibieron etexilato de dabigatrán (20; 0.5%) que en aquellos que recibieron warfarina (5; 0.1%).
En el estudio RE-SONATE, el cual comparó el etexilato de dabigatrán con el placebo, hubo 1 evento de IM en cada grupo de tratamiento, resultando en tasas de IM con dabigatrán igual a las tasas de IM con placebo.
Prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada:
Pruebas de función hepática: En los estudios con control activo RE-COVER, RE-COVER II y RE-MEDY se produjeron potenciales anormalidades de las pruebas de función hepática, con una incidencia comparable o más baja en los pacientes tratados con dabigatrán etexilato que en los pacientes tratados con warfarina. En el estudio RE-SONATE, no hubo ninguna diferencia marcada entre los grupos de dabigatrán y de placebo en lo que respecta a valores de pruebas de función hepática anormales posiblemente significativos desde el punto de vista clínico.
Farmacocinética: Después de la administración oral de etexilato de dabigatrán en voluntarios sanos, el perfil farmacocinético de dabigatrán en plasma está caracterizado por un incremento rápido en las concentraciones en plasma, alcanzando las concentraciones pico (Cmáx) en un lapso de 0.5 y 2.0 horas después de la administración. La Cmáx y el área bajo la curva tiempo-concentración plasmática (AUC) fueron proporcionales a la dosis. Después de la Cmáx, las concentraciones en plasma de dabigatrán mostraron un descenso biexponencial con una vida media terminal promedio de aproximadamente 11 horas en sujetos ancianos sanos. Después de dosis múltiples, se observó una vida media terminal de aproximadamente 12-14 horas. La vida media fue independiente de la dosis. Sin embargo, la vida media se prolonga si la función renal está dañada, como se muestra a continuación, en la Tabla 10.
Tabla 10. Vida media de dabigatrán total en sujetos sanos y sujetos con deterioro en la función renal
|
Tasa de filtración glomerular (depuración de creatinina) |
gMedia (gCV%; intervalo) vida media |
|
[mL/min] |
[h] |
|
> 80 |
13.4 (25.7%; 11.0-21.6) |
|
> 50-≤ 80 |
15.3 (42.7%; 11.7-34.1) |
|
> 30-≤ 50 |
18.4 (18.5%; 13.3-23.0) |
|
≤ 30 |
27.2 (15.3%; 21.6-35.0) |
La biodisponibilidad absoluta del dabigatrán después de la administración oral de etexilato de dabigatrán como cápsulas de HMPC fue aproximadamente 6.5%.
Los alimentos no afectan la biodisponibilidad del etexilato de dabigatrán, aunque retarda en 2 horas el tiempo necesario para alcanzar las concentraciones pico en plasma.
La biodisponibilidad oral puede aumentar en alrededor de 1.4 veces (+37%) en comparación con la formulación de la cápsula de referencia cuando se toman los gránulos sin la cubierta de la cápsula de HPMC. Por lo cual, siempre se deberá preservar la integridad de las cápsulas de HPMC en el uso clínico para evitar una biodisponibilidad aumentada involuntariamente del etexilato de dabigatrán. Por lo tanto, se debe aconsejar a los pacientes que no abran las cápsulas y tomen los gránulos solos (por ejemplo, espolvoreados sobre los alimentos o en las bebidas). (Ver Dosis y vía de administración).
Un estudio que evaluaba la absorción postoperatoria de etexilato de dabigatrán, que se realizó 1-3 horas después de la cirugía, demostró una absorción relativamente lenta en comparación con la de voluntarios sanos, mostrando un perfil uniforme de concentración plasmática/tiempo sin concentraciones plasmáticas pico elevadas. Las concentraciones pico en plasma se alcanzan a las 6 horas después de la administración, o entre 7 y 9 horas después de la cirugía (BISTRO 1b). Se observó, sin embargo, que factores influyentes, tales como la anestesia, la paresia gastrointestinal y los efectos quirúrgicos significarán que una proporción de pacientes experimentará en retraso en la absorción independientemente de la formulación del fármaco oral. Aunque este estudio no predijo si el deterioro en la absorción persiste con dosis posteriores, se demostró en otro estudio, que la absorción lenta y retrasada sólo suele observarse el día de la cirugía. En los días posteriores, la absorción del dabigatrán es rápida y las concentraciones plasmáticas pico se alcanzan 2 horas después de la administración del fármaco.
El metabolismo y la excreción de dabigatrán se estudiaron después de administrar una dosis única intravenosa de dabigatrán marcado radiactivamente en varones sanos. Después de una dosis intravenosa, la radioactividad derivada del dabigatrán fue eliminada principalmente en la orina (85%). La eliminación vía fecal representó el 6% de la dosis administrada. La recuperación de la radiactividad total osciló entre el 88-94% de la dosis administrada a las 168 horas después de la administración.
Después de la administración oral, el etexilato de dabigatrán se convierte rápida y completamente a dabigatrán, que es la forma activa en plasma. La escisión del profármaco etexilato de dabigatrán por hidrólisis catalizada por esterasas al principio activo dabigatrán es la reacción metabólica predominante. El dabigatrán se conjuga y forma acilglucurónidos farmacológicamente activos. Existen cuatro isómeros posicionales, 1-O, 2-O, 3-O, 4-O-acilglucurónido, y cada uno representa menos del 10% del dabigatrán total en plasma. Sólo pudieron detectarse trazas de otros metabolitos usando métodos analíticos de alta sensibilidad. El dabigatrán se elimina principalmente en forma intacta a través de la orina, a una tasa de aproximadamente 100 mL/min que corresponde con la tasa de filtración glomerular.
Se observó una baja unión de dabigatrán a las proteínas plasmáticas humanas independiente de la concentración (34-35%). El volumen de distribución de dabigatrán de 60-0-70 L superó el volumen de agua total de cuerpo, indicando una distribución tisular moderada del dabigatrán.
Farmacocinética en poblaciones especiales:
Insuficiencia renal: La exposición (AUC) al dabigatrán después de la administración oral del etexilato de dabigatrán en un estudio de fase I fue aproximadamente 3 veces mayor en voluntarios con insuficiencia renal moderada (depuración de la creatinina entre 30-50 mL/min) que en aquellos que no padecen insuficiencia renal.
En un pequeño número de voluntarios con insuficiencia renal severa (depuración de creatinina 10-30 mL/min), la exposición (AUC) al dabigatrán fue aproximadamente 6 veces mayor y la semivida aproximadamente 2 veces más prolongada que la observada en una población sin insuficiencia renal (ver secciones Dosis y vía de administración y Contraindicaciones).
Se investigó la depuración de dabigatrán con hemodiálisis en pacientes con insuficiencia renal terminal sin fibrilación auricular. La diálisis se realizó con una velocidad de flujo del dializado de 700 mL/min, durante cuatro horas, y un flujo sanguíneo de 200 mL/min o 350-390 mL/min, lo cual llevó a la eliminación del 50% o del 60% de las concentraciones de dabigatrán libre o total, respectivamente. La cantidad del medicamento depurada con la diálisis es proporcional al flujo sanguíneo. La actividad anticoagulante del dabigatrán disminuyó al disminuir las concentraciones plasmáticas y la relación PK/PD no se vio afectada por el procedimiento.
Prevención del accidente cerebrovascular y la embolia sistémica y reducción de la mortalidad vascular en pacientes con fibrilación auricular: La depuración de creatinina media en el RE-LY fue de 68,4 mL/min. Casi la mitad (45.8%) de los pacientes del RE-LY tenían una depuración de creatinina > 50-< 80 mL/min. Los pacientes con insuficiencia renal moderada (depuración de creatinina entre 30-50 mL/min), tuvieron concentraciones plasmáticas, respectivamente en promedio, 2.29 veces y 1.81 veces más altos antes y después de la dosis de dabigatrán, en comparación con los pacientes sin insuficiencia renal (depuración de creatinina ≥ 80 mL/min).
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: La mediana de CICr en el estudio RE-COVER fue 100.4 mL/min. El 21.7% de los pacientes tenía insuficiencia renal leve (CICr > 50-< 80 mL/min) y el 4.5% de los pacientes tenía insuficiencia renal moderada (CICr entre 30 y 50 mL/min). Los pacientes con insuficiencia renal leve y moderada tuvieron concentraciones valle en estado de equilibrio de dabigatrán que fueron en promedio 1.8 y 3.6 veces más altas en comparación con los pacientes con CICr > 80 mL/min. Se observaron valores similares para la CICr en el estudio RE-COVER II.
Prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada: La depuración de creatinina media en los estudios RE-MEDY y RE-SONATE fue de 99.0 mL/min y 99.7 mL/min, respectivamente. 22.9% y 22.5% de los pacientes tenían una depuración de creatinina > 50-< 80 mL/min y 4.1% y 4.8% tenían una depuración de creatinina entre 30-50 mL/min en los estudios RE-MEDY y RE-SONATE.
Ancianos: Los estudios de farmacocinética específicos con ancianos en estudios de fase I mostraron un incremento de 1.4 a 1.6 veces (+ 40 a 60%) en el AUC de no más de 1.25 veces (+25%) en Cmáx en comparación con sujetos jóvenes.
El AUCt,ss y Cmáx,ss en sujetos ancianos hombres y mujeres (> 65 años) fueron aproximadamente 1.9 veces y 1.6 veces mayores para mujeres ancianas en comparación con mujeres jóvenes y 2.2 y 2.0 veces mayores para varones ancianos que en sujetos varones de 18-40 años de edad.
El incremento observado de exposición al dabigatrán guardó relación con la reducción de la depuración de la creatinina relacionada con la edad.
El efecto de la edad en la exposición al dabigatrán fue confirmado en el estudio RE-LY con una concentración mínima alrededor de 1.3 veces más alta (+ 31%) para sujetos ≥ 75 años de edad y un nivel mínimo más bajo de aproximadamente 22% para sujetos de < 65 años en comparación con sujetos cuyas edades estaban entre 65 y 75 años.
Insuficiencia hepática: No se apreció cambio alguno en la exposición a dabigatrán en 12 sujetos en un estudio de fase I con insuficiencia hepática moderada (Child-Pugh B) en comparación con 12 controles.
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a cirugía ortopédica mayor: Pacientes con insuficiencia hepática moderada a severa (clasificación B y C de Child-Pugh) o enfermedad hepática que se espera tenga algún impacto en la supervivencia o con enzimas hepáticas elevadas ≥ 2 al límite superior normal (ULN, por sus siglas en inglés Upper Level of Normal) fueron excluidos en los ensayos clínicos.
Prevención de eventos vasculares cerebrales, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: Los pacientes con enfermedad hepática activa, incluyendo aunque no limitado a la elevación persistente de las enzimas hepáticas ≥ 2 ULN o hepatitis A, B o C, fueron excluidos en los ensayos clínicos.
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: Los pacientes con insuficiencia hepática moderada y grave (clasificación Child-Pugh B o C) o enfermedades hepáticas que se espera tengan algún impacto sobre la supervivencia o con enzimas hepáticas elevadas ≥ 2 veces el Límite Superior Normal (ULN) fueron excluidos en los ensayos clínicos.
Prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada: Los pacientes con insuficiencia hepática moderada y grave (clasificación Child-Pugh B o C) o enfermedades hepáticas que se espera tengan algún impacto sobre la supervivencia o con enzimas hepáticas elevadas ≥ 2 veces el Límite Superior Normal (ULN) fueron excluidos en los ensayos clínicos.
Peso corporal: Las concentraciones mínimas de dabigatrán fueron aproximadamente 20% más bajas en pacientes con un peso corporal > 100 kg en comparación con 50-100 kg. La mayoría (80.8%) de los sujetos estaban en la categoría de ≥ 50 kg y < 100 kg sin detectar alguna diferencia evidente. Se dispone de información limitada para pacientes ≤ 50 kg.
Género:
Prevención de los eventos de tromboembolia venosa en pacientes sometidos a una cirugía ortopédica mayor: La exposición al fármaco en los estudios de prevención primaria de TEV fue alrededor de 1.4 a 1.5 veces (+40% a 50%) mayor en pacientes femeninos. Este hallazgo no tuvo relevancia clínica.
Prevención de eventos vasculares cerebrales, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: En la fibrilación auricular, los pacientes femeninos tuvieron un promedio de concentraciones mínimas y post-dosis 1.3 veces (+30%) más elevadas. Este hallazgo no tuvo relevancia clínica.
Origen étnico: La farmacocinética del dabigatrán fue investigada en voluntarios caucásicos y japoneses después de dosis únicas y múltiples. El origen étnico no afecta a la farmacocinética del dabigatrán de un modo clínicamente relevante.
Existe información farmacocinética limitada en pacientes negros, la cual sugiere diferencias no relevantes.
Interacciones farmacocinéticas: Los estudios de interacción in vitro no mostraron alguna inhibición o inducción del citocromo P450.
Esto ha sido confirmado por estudios in vivo en voluntarios sanos, quienes no mostraron interacción alguna entre el tratamiento con etexilato de dabigatrán y los siguientes fármacos: atorvastatina (CYP3A4) y diclofenaco (CYP2C9).
Atorvastatina: Cuando el etexilato de dabigatrán fue coadministrado con atorvastatina, un sustrato de CYP3A4, no hubo interacción, ya que la exposición de atorvastatina, metabolitos de atorvastatina y de dabigatrán permanecieron sin cambios.
Diclofenaco: Cuando el etexilato de dabigatrán fue coadministrado con diclofenaco, un sustrato de CYP2C9, no hubo interacción, ya que la farmacocinética de ambos fármacos permaneció sin cambios.
Interacciones el inhibidor/inductor de la glicoproteína-P: El profármaco etexilato de dabigatrán, aunque no el dabigatrán, es un sustrato del transportador del flujo, la glicoproteína-P (gp-P). Por lo tanto, co-medicación con inhibidores e inductores del transportador de la gp-P han sido investigados.
Medicación conjunta con inhibidores de la gp-P:
Amiodarona: Cuando el etexilato de dabigatrán fue administrado junto con una dosis oral única de 600 mg de amiodarona, el alcance e índice de absorción de amiodarona y su metabolito activo DEA permanecieron básicamente intactos. El AUC y Cmáx de dabigatrán se incrementaron en aproximadamente 1.6 y 1.5 veces (+60% y 50%), respectivamente.
Prevención de eventos vasculares cerebrales, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: En la población del RE-LY, en los estudios farmacocinéticos no se observaron cambios importantes en los niveles de dabigatrán en los pacientes que recibieron amiodarona (véase la sección Interacciones medicamentosas y de otro género).
Dronedarona: Cuando dabigatrán etexilato y dronedarona se administraron en forma simultánea, los valores de AUC0-∞ y Cmáx de dabigatrán total se incrementaron a razón de aproximadamente 2.4 veces y 2.3 veces (+136% y 125%), respectivamente, tras la administración de dosis múltiples de 400 mg de dronedarona b.i.d., y por un factor de aproximadamente 2.1 y 1.9 (+114% y 87%), respectivamente, tras la administración de una dosis única de 400 mg. La semivida terminal y la depuración renal de dabigatrán no se vieron afectadas por la dronedarona. Cuando se administraron dosis únicas y dosis múltiples de dronedarona 2 horas después de la administración de dabigatrán etexilato, los valores de AUC0-∞ del dabigatrán se incrementaron por un factor de 1.3 y de 1.6, respectivamente.
Verapamilo: Cuando el etexilato de dabigatrán fue administrado conjuntamente con verapamilo oral, la Cmáx y el AUC del dabigatrán se incrementaron dependiendo del horario de la administración y de la formulación de verapamilo.
La mayor elevación de la exposición al dabigatrán se observó con la primera dosis de una formulación de liberación inmediata de verapamilo administrado una hora antes de la ingesta del etexilato de dabigatrán (incremento de la Cmáx) en aproximadamente 2.8 veces (+180%) y del AUC alrededor de 2.5 veces (+ 150%). El efecto se redujo progresivamente con la administración de una formulación de liberación prolongada (aumento de la Cmáx) en aproximadamente 1.9 veces (+90%) y del AUC en aproximadamente 1.7 veces (+70%), o administración de múltiples dosis de verapamilo (aumento de la Cmáx) en aproximadamente 1.6 veces (+60%) y del AUC en aproximadamente 1.5 veces (+50%). Esto puede explicarse por la inducción de la gp-P en el intestino por el tratamiento crónico con verapamilo.
No se observó interacción significativa cuando el verapamilo fue administrado 2 horas después del etexilato de dabigatrán (aumento de la Cmáx en aproximadamente un 10% y del AUC en alrededor de 20%). Esto se explica por la absorción completa del dabigatrán después de 2 horas. (Ver dosificación y formas de administración).
No se dispone de información para la aplicación parenteral del verapamilo; con base en el mecanismo de la interacción, no es de esperarse que haya interacción significativa.
Prevención de eventos vasculares cerebrales, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: En la población del RE-LY, en los estudios farmacocinéticos no se observaron cambios importantes en los niveles de dabigatrán en los pacientes que recibieron verapamilo (véase la sección Interacciones medicamentosas y de otro género).
Ketoconazol: El ketoconazol sistémico aumentó los valores totales de AUC0-∞ y Cmáx de dabigatrán en 2.4 veces (+138% y 135%), respectivamente, después de una dosis única de 400 mg, y alrededor de 2.5 veces (+153% y 149%), respectivamente, después de dosis múltiples de 400 mg de ketoconazol una vez al día. El tiempo para alcanzar el pico, vida media terminal y tiempo de residencia media no fueron afectados por el ketoconazol.
Claritromicina: Al administrar claritromicina, 500 mg dos veces al día, junto con etexilato de dabigatrán no se observaron interacciones farmacocinéticas relevantes clínicamente (aumento de la Cmáx en aproximadamente un 15% y del AUC en alrededor del 19%).
Quinidina: La quinidina fue administrada como dosis de 200 mg cada 2 horas hasta una dosis total de 1000 mg. El etexilato de dabigatrán se administró dos veces al día durante 3 días consecutivos, al tercer día con o sin quinidina. El AUCt,ss y Cmáx,ss de dabigatrán aumentaron en promedio de 1.5 veces (+53 y 56%), respectivamente con quinidina concomitante.
Ticagrelor: Cuando una dosis única de 75 mg de dabigatrán etexilato se coadministró en forma simultánea con una dosis de carga de 180 mg de ticagrelor, los valores de AUC y Cmax de dabigatrán se incrementaron por un factor de 1.73 y de 1.95 (+73% y 95%), respectivamente. Tras la administración de dosis múltiples de ticagrelor de 90 mg dos veces al día, el aumento en la exposición a dabigatrán se redujo a un factor de 1.56 y 1.46 (+56% y 46%) para Cmáx y AUC, respectivamente.
La administración concomitante de una dosis de carga de 180 mg de ticagrelor y 110 mg de dabigatrán etexilato (en estado estable) incrementó el ABCt,ss y Cmáx,ss 1.49-veces y 1.65-veces (+49% y 65%), respectivamente, comparando con etexilato de dabigatrán administrado solo. Cuando una dosis de carga de 180 mg de ticagrelor se administró 2 horas después de 110 mg de etexilato de dabigatrán (en estado estable), el aumento de dabigatrán ABCt,ss y Cmáx,ss se redujo 1.27 veces y 1.23 veces (+27% y 23%), respectivamente, comparando con etexilato dabigatrán administrado solo. La administración concomitante de 90 mg de ticagrelor BID (dosis de mantenimiento) con 110 mg de etexilato dabigatrán incrementó el ajuste del ABCt,ss y Cmáx,ss de dabigatrán 1.26 veces y 1.29 veces, respectivamente, comparando con etexilato de dabigatrán administrado solo.
Medicación conjunta con sustratos de la glicoproteína-P:
Digoxina: Cuando se administró conjuntamente etexilato y digoxina, un sustrato de la gp-P, no se observó interacción farmacocinética. Ni el dabigatrán, ni el profármaco etexilato de dabigatrán son inhibidores de la gp-P relevantes clínicamente.
Medicación conjunta con los inductores de la glicoproteína-P:
Rifampicina: La dosificación previa de la rifampicina, un inductor, en una dosis de 600 mg una vez al día durante 7 días disminuyó el pico de dabigatrán total y la exposición total en un 65.5 y 67%, respectivamente. Se redujo el efecto inductor, lo que dio como resultado una exposición al dabigatrán cercana a la referencia para el día 7 después de suspender el tratamiento con rifampicina. No se observó un incremento adicional en la biodisponibilidad después de otros 7 días.
Medicación conjunta con inhibidores plaquetarios:
Ácido acetilsalicílico (AAS): El efecto de la administración concomitante del etexilato de dabigatrán y el ácido acetilsalicílico (AAS) respecto al riesgo de sangrados fue estudiado en pacientes con fibrilación auricular en un estudio de fase II, en el cual se administró concomitantemente y aleatoriamente AAS. Con base en el análisis de regresión logística, la administración conjunta de AAS y 150 mg de etexilato de dabigatrán dos veces al día, puede incrementar el riesgo de cualquier sangrado de 12% a 18% y 24% con 81 mg y 325 mg de AAS, respectivamente.
A partir de los datos recopilados en el estudio de fase III, RE-LY, se observó que la medicación conjunta de AAS o clopidogrel con etexilato de dabigatrán en dosis de 110 o 150 mg dos veces al día puede incrementar el riesgo de sangrado mayor. La tasa más elevada de eventos de sangrado por medicaciones conjuntas con AAS o clopidogrel, sin embargo, también fue observada para warfarina.
AINEs administrados para analgesia perioperatoria de corto plazo han demostrado no estar asociados con incrementos en el riesgo de sangrado cuando se suministraron junto con etexilato de dabigatrán. Existe evidencia limitada respecto al uso de medicación regular con AINEs con vidas medias menores de 12 horas durante el tratamiento con etexilato de dabigatrán y esto no ha sugerido un riesgo de sangrado adicional.
Prevención de eventos vasculares cerebrales, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: AINEs aumenta el riesgo de sangrado en todos los grupos de tratamiento en el estudio RE-LY.
Clopidogrel: En un estudio de fase I en voluntarios varones sanos jóvenes, la administración concomitante de etexilato de dabigatrán y clopidogrel no resultó en una prolongación adicional de los tiempos de hemorragia capilar (CBT, por sus siglas en inglés) comparada con la monoterapia con clopidogrel. Además, el AUCt,ss y Cmáx,ss de dabigatrán y las mediciones de coagulación para el efecto del dabigatrán, TTPa, ECT o TT (anti FIIa), o la inhibición de la agregación plaquetaria (IAP) como medición del efecto del clopidogrel permanecieron esencialmente inalteradas en comparación con el tratamiento combinado y las respectivas monoterapias. Con una dosis de carga de 300 o 600 mg de clopidogrel, el AUCt,ss y Cmáx,ss de dabigatrán se incrementaron en aproximadamente 1.3 a 1.4 veces (+30 a 40%). (Ver en la subsección AAS).
Antiagregantes plaquetarios u otros anticoagulantes: El uso concomitante de dabigatrán y antiagregantes plaquetarios u otros anticoagulantes puede aumentar el riesgo de sangrado (ver Advertencias y precauciones especiales).
Co-medicación con inhibidores selectivos de la recaptura de serotonina (ISRS): ISRS en el estudio RELY-ABLE aumentan el riesgo de sangrado en todos los grupos de tratamiento.
Medicación conjunta con agentes que elevan el pH gástrico: Los cambios en la exposición al dabigatrán determinados por el análisis farmacocinético de la población causados por los inhibidores de la bomba de protones (IBP) y los antiácidos no fueron considerados clínicamente relevantes puesto que la magnitud del efecto fue menor (disminución fraccionaria en la biodisponibilidad no significativa para los antiácidos y 14.6% para los IBPs.
Pantoprazol: Cuando el etexilato de dabigatrán se administró conjuntamente con pantoprazol, se observó una reducción del área bajo la curva de tiempo-concentración plasmática de dabigatrán de aproximadamente el 30%. El pantoprazol y otros inhibidores de la bomba de protones fueron administrados con etexilato de dabigatrán en ensayos clínicos y no se observaron efectos sobre el sangrado o la eficacia.
Prevención de eventos vasculares cerebrales, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: En el estudio de fase III, RE-LY, la medicación conjunta con IBP no resultó en niveles mínimos y, en promedio, sólo concentraciones post-dosis ligeramente reducidas (-11%). En consecuencia, la medicación conjunta de IBP no pareció estar asociada con una mayor incidencia de evento vascular cerebral o EES, especialmente en comparación con la warfarina, y por ende, la reducida biodisponibilidad por la administración conjunta de pantoprazol pareció no tener relevancia clínica.
Ranitidina: La administración de ranitidina junto con el etexilato de dabigatrán no tuvo efecto significativo en el grado de absorción del dabigatrán.
CONTRAINDICACIONES:
• Hipersensibilidad conocida al dabigatrán o al etexilato de dabigatrán o a alguno de los excipientes de la fórmula.
• Insuficiencia renal severa (depuración de la creatinina < 30 mL/min).
• Pacientes con manifestaciones hemorrágicas, con diátesis hemorrágica o con alteración espontánea o farmacológica de la hemostasia.
• Lesiones orgánicas con riesgo de sangrado clínicamente significativo, incluyendo EVC hemorrágico dentro de los últimos 6 meses.
• Tratamiento concomitante con ketoconazol sistémico (ver Interacciones medicamentosas y de otro género).
• Pacientes con válvulas cardiacas protésicas.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No se dispone de información clínica de exposición durante el embarazo. Se desconoce el riesgo potencial para humanos.
Las mujeres en edad fértil deben evitar embarazarse durante el tratamiento con PRADAXAR® y si están embarazadas, no deben ser tratadas con PRADAXAR®, a menos que el beneficio esperado sea superior al riesgo.
Lactancia: No se cuenta con datos clínicos. Como precaución, se debe suspender la lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS: PRADAXAR® ha sido evaluado en pruebas clínicas en más de 64,000 pacientes, de los cuales más 35,000 fueron tratados con PRADAXAR®.
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a una cirugía ortopédica mayor: En los estudios de prevención primaria de TEV después de una cirugía ortopédica mayor, un total de 10,795 pacientes fueron tratados en 6 estudios controlados con al menos una dosis de etexilato de dabigatrán (150 mg una vez al día, 220 mg una vez al día, enoxaparin). De los 10,795 pacientes, 6,684 fueron tratados con 150 mg o 220 mg de etexilato de dabigatrán, una vez al día.
Prevención del accidente cerebrovascular y la embolia sistémica y reducción de la mortalidad vascular en pacientes con fibrilación auricular: En el estudio RE-LY que investigó la prevención de EVC y embolismo sistémico en pacientes con fibrilación auricular, un total de 12,042 pacientes fueron tratados con etexilato de dabigatrán. De ellos, 6,059 fueron tratados con etexilato de dabigatrán 150 mg, dos veces al día, mientras que 5,983 recibieron dosis de 110 mg dos veces al día.
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: En los estudios de tratamiento de TVP aguda/EP (RE-COVER, RE-COVER II), un total de 2,553 pacientes fueron incluidos en el análisis de seguridad para el etexilato de dabigatrán. Todos los pacientes fueron tratados con etexilato de dabigatrán 150 mg dos veces al día.
Prevención de la trombosis venosa profunda (TVP) y/o la embolia pulmonar (EP) recurrente y la muerte relacionada: En los estudios de prevención de la TVP recurrente/EP (RE-MEDY, RE-SONATE) un total de 2,114 pacientes fueron tratados con dabigatrán etexilato; 552 de dichos 2,114 pacientes eran pacientes que habían pasado del estudio RE-COVER (tratamiento de la TVP aguda/EP) al estudio RE-MEDY y están contabilizados en los totales de pacientes de los dos tipos de eventos de TVP/EP, agudos y recurrentes. Todos los pacientes fueron tratados con un dabigatrán etexilato 150 mg dos veces al día.
En total, alrededor de 9% de los pacientes tratados por cirugía electiva de cadera o rodilla (tratamiento a corto plazo hasta por 42 días). El 22% de los pacientes con fibrilación auricular tratados para la prevención de EVC y embolismo sistémico (tratamiento a largo plazo, hasta por 3 años), 14% de los pacientes tratados por TVP aguda/EP (tratamiento a largo plazo, hasta por 6 meses) y 15% de los pacientes tratados por prevención de TVP recurrente/EP (tratamiento a largo plazo hasta por 36 meses) experimentaron eventos adversos.
Sangrado: El sangrado es la reacción adversa más relevante de PRADAXAR®; dependiendo de la indicación, el sangrado de cualquier tipo o severidad se presentó en aproximadamente el 14% de los pacientes tratados a corto plazo, por cirugía de reemplazo electivo de cadera o rodilla y en el tratamiento a largo plazo anualmente en 16.6% de los pacientes con fibrilación auricular tratados para la prevención de EVC y el embolismo sistémico y en el 14.4% de los pacientes adultos con TVP aguda/EP. En el estudio de RE-MEDY, de TVP recurrente/EP, el 19.4% de los pacientes adultos tuvieron algún sangrado, mientras que en el estudio RE-SONATE el porcentaje de pacientes adultos con algún sangrado fue del 10.5%.
El sangrado mayor o severo puede ocurrir, aunque es rara su frecuencia en estudios clínicos, e independientemente de su localización, puede producir discapacidad, poner en riesgo la vida o incluso producir desenlaces fatales.
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a una cirugía ortopédica mayor:
En general, las tasas de sangrado fueron similares entre los grupos de tratamiento y no fueron diferentes significativamente.
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular:
El sangrado mayor cumplió uno o más de los siguientes criterios:
• Sangrado asociado con una reducción en la hemoglobina de al menos 20 gramos por litro o conducente a una transfusión de cuando menos 2 unidades de sangre o concentrado celular.
• Sangrado sintomático en un área u órgano crítico; intraocular, intracraneal, intraespinal o intramuscular con síndrome compartamental, sangrado retroperitoneal, sangrado intra-articular o sangrado pericárdico.
Los sangrados mayores fueron clasificados como riesgosos para la vida si cumplían uno o más de los siguientes criterios:
• Sangrado fatal; sangrado intracraneal sintomático; reducción en la hemoglobina de al menos 50 gramos por litro; transfusión de al menos 4 unidades de sangre o concentrado celular; un sangrado asociado con hipotensión que requiere el uso de agentes inotrópicos intravenosos; un sangrado que requirió intervención quirúrgica.
Los sujetos que fueron aleatorizados a etexilato de dabigatrán 110 mg dos veces al día y 150 mg dos veces al día tuvieron un riesgo significativamente menor de sangrados riesgosos para la vida, EVC hemorrágico y sangrado intracraneal en comparación con warfarina [p < 0.05]. Ambas concentraciones de dosis de etexilato de dabigatrán también tuvieron una tasa de sangrado total más baja estadísticamente significativa. Los sujetos aleatorizados a etexilato de dabigatrán 110 mg dos veces al día tuvieron un riesgo significativamente menor de sangrados mayores comparados con warfarina (razón de riesgo 0.81, p = 0.0027).
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: La definición de los eventos de sangrado grave (ESG) se generó de acuerdo con las recomendaciones de la Sociedad Internacional de Trombosis y Hemostasis (International Society on Thrombosis and Haemostasis). Un evento de sangrado se calificaba como un ESG si cumplía al menos uno de los siguientes criterios:
Sangrado fatal: Sangrado sintomático en una zona o un órgano críticos, como ser sangrado intracraneano, sangrado intraespinal, sangrado intraocular, sangrado retroperitoneal, sangrado intraarticular o sangrado pericárdico, o sangrado intramuscular con síndrome compartimental. Para que un sangrado en una zona o un órgano críticos se clasificara como un ESG, el mismo debía estar asociado con una presentación clínica sintomática.
Sangrado que provoca un descenso en los niveles de hemoglobina de 20 g/L (1.24 mmol/L) o más, o que conduce a una transfusión de 2 o más unidades de sangre completa o de glóbulos rojos.
En un análisis de datos combinados de los dos estudios pivote (RE-COVER y RE-COVER II) en el tratamiento de la TVP aguda/EP, los sujetos aleatorizados a dabigatrán etexilato tuvieron tasas más bajas de los siguientes eventos de sangrado, las cuales fueron estadísticamente significativas:
Eventos de sangrado graves (razón de riesgo 0.60 [0.36, 0.99]).
Eventos de sangrado graves o clínicamente relevantes (ESCR) (razón de riesgo 0.56 [0.45, 0.71]).
Cualquier evento de sangrado (razón de riesgo 0.67 [0.59, 0.77]).
Todas estas tasas fueron superiores en comparación con la warfarina.
Los eventos de sangrado de ambos tratamientos se contabilizan a partir de la primera toma de dabigatrán etexilato o warfarina tras la discontinuación de la terapia parenteral (periodo de tratamiento oral solamente). Esto incluye todos los eventos de sangrado que se produjeron durante el tratamiento con dabigatrán. Todos los eventos de sangrado que se produjeron durante el tratamiento con warfarina están incluidos, excepto los que se produjeron durante el lapso de superposición entre la warfarina y la terapia parenteral.
Prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada: La definición de los ESG se generó de acuerdo con las recomendaciones de la Sociedad Internacional de Trombosis y Hemostasis (International Society on Thrombosis and Haemostasis). En el estudio RE-MEDY, un evento de sangrado se calificaba como un ESG si cumplía al menos uno de los siguientes criterios:
Sangrado fatal: Sangrado sintomático en una zona o un órgano críticos, como ser sangrado intracraneano, sangrado intraespinal, sangrado intraocular, sangrado retroperitoneal, sangrado intraarticular o sangrado pericárdico, o sangrado intramuscular con síndrome compartimental. Para que un sangrado en una zona o un órgano críticos se clasificara como un ESG, el mismo debía estar asociado con una presentación clínica sintomática.
Sangrado que provoca un descenso en los niveles de hemoglobina de 20 g/L (1.24 mmol/L) o más, o que conduce a una transfusión de 2 o más unidades de sangre completa o de glóbulos rojos.
En el estudio RE-MEDY, los pacientes aleatorizados a dabigatrán etexilato tuvieron un número significativamente menor de eventos de sangrado en comparación con la warfarina en lo que respecta a las siguientes categorías: Eventos de sangrado graves o eventos de sangrado clínicamente relevantes (razón de riesgo 0.55 [0.41, 0.72], p < 0.0001) y cualquier evento de sangrado (razón de riesgo 0.71 [0.61, 0.83], p < 0.0001).
En el estudio RE-SONATE, un evento de sangrado se calificaba como un ESG si cumplía al menos uno de los siguientes criterios:
Sangrado fatal: Asociado con un descenso en el nivel de hemoglobina de 2 g/dL o más.
Condujo a transfusión de ≥ 2 unidades de concentrado de células sanguíneas o sangre completa.
Se produce en una zona crítica: sangrado intracraneano, sangrado intraespinal, sangrado intraocular, sangrado pericárdico, sangrado intraarticular, sangrado intramuscular con síndrome compartimental, sangrado retroperitoneal.
En el estudio RE-SONATE, las tasas de ESG fueron bajas (2 pacientes con ESG [0,3%] para dabigatrán etexilato versus 0 pacientes con ESG [0%] para placebo. Las tasas de eventos de sangrado graves o eventos de sangrado clínicamente relevantes fueron más altas con dabigatrán etexilato que con el placebo (5.3% versus 2.0%).
Efectos secundarios: Las reacciones adversas clasificadas por los términos preferidos según SOC y MedDRA, reportados de cualquier grupo de tratamiento por población de todos los estudios controlados, se presentan en los listados siguientes. La Tabla 8 enumera las reacciones adversas identificadas aplicables a todas las indicaciones. La Tabla 9 enumera las reacciones adversas específicas para la indicación que fueron identificados.
Los efectos secundarios están asociados generalmente con el mecanismo de acción farmacológico del etexilato de dabigatrán y representan los eventos asociados de sangrado que pueden presentarse en las diferentes regiones anatómicas y órganos.
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a una cirugía ortopédica mayor: En pacientes tratados para la prevención de TEV después de una cirugía de reemplazo de cadera o rodilla, las incidencias observadas de reacciones adversas del etexilato de dabigatrán estuvieron dentro del rango de la enoxaparina.
Prevención del accidente cerebrovascular y la embolia sistémica y reducción de la mortalidad vascular en pacientes con fibrilación auricular: Las incidencias observadas de reacciones adversas de etexilato de dabigatrán en pacientes tratados para la prevención de eventos vasculares cerebrales en pacientes con fibrilación auricular estuvieron dentro del rango de la warfarina, excepto los trastornos gastrointestinales que aparecieron en una tasa superior en los brazos de etexilato de dabigatrán.
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: La frecuencia general de reacciones adversas en pacientes recibiendo PRADAXAR® para el tratamiento de TVP aguda/EP fue menor para PRADAXAR® en comparación con la warfarina (14.2% vs. 18.9%).
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: La frecuencia general de reacciones adversas en los pacientes tratados para la prevención de la TVP/EP recurrente fue más baja para PRADAXAR® en comparación con la warfarina (14,6% versus 19,6%), en tanto que en comparación con el placebo la frecuencia fue más alta (14,6% versus 6,5%).
Tabla 12. Reacciones adversas identificadas a partir de estudios y datos posteriores a la comercialización en: prevención primaria de TEV después de un programa de cirugía ortopédica mayor, la Prevención de EVC tromboembólico y embolismo sistémico en pacientes con fibrilación auricular, el Tratamiento de TVP aguda y/o PE y muerte relacionada y la Prevención de TVP recurrente y/o PE y muerte relacionada
|
Trastornos de la sangre y del sistema linfático |
|
Anemia, trombocitopenia. Neutropenia, agranulocitosis |
|
Trastornos del sistema inmunológico |
|
Hipersensibilidad al fármaco, incluyendo urticaria, exantema y prurito, broncoespasmo, angioedema*, reacción anafiláctica* |
|
Trastornos del sistema nervioso |
|
Hemorragia intracraneal |
|
Trastornos vasculares |
|
Hematoma, hemorragia |
|
Trastornos respiratorios, torácicos y mediastinales |
|
Epistaxis, hemoptisis |
|
Trastornos gastrointestinales |
|
Hemorragia gastrointestinal, dolor abdominal, diarrea, dispepsia, náusea, úlcera gastrointestinal, incluyendo úlcera esofágica, gastroesofagitis, enfermedad por reflujo gastroesofágico, vómito, disfagia |
|
Trastornos hepatobiliares |
|
Función hepática anormal |
|
Trastornos de la piel y tejido subcutáneo |
|
Hemorragia cutánea, alopecia* |
|
Trastornos musculoesqueléticos, del tejido conectivo y óseo |
|
Hemartrosis |
|
Trastornos renales y urinarios |
|
Hemorragia urogenital |
|
Trastornos generales y alteraciones en el lugar de administración |
|
Hemorragia en el lugar de la inyección, hemorragia en el lugar del catéter |
|
Lesiones, intoxicaciones y complicación de los procedimientos |
|
Hemorragia traumática, hemorragia en el lugar de la incisión |
* Incluidos los datos posteriores a la comercialización.
Tabla 13. Reacciones adversas específicas adicionales identificados por indicación
|
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a cirugía ortopédica mayor |
|
Trastornos vasculares |
|
Hemorragia en la herida |
|
Trastornos generales y condiciones en el lugar de administración |
|
Flujo sangrante |
|
Lesiones, intoxicaciones y complicaciones de procedimientos |
|
Hematoma posterior al procedimiento, hemorragia posterior al procedimiento, anemia post-quirúrgica, flujo posterior al procedimiento, secreción de la herida |
|
Procedimientos quirúrgicos y médicos |
|
Drenaje de la herida, drenaje posterior al procedimiento |
|
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: Ninguno |
|
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: Ninguno |
|
Prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada: Ninguno |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Se llevaron a cabo estudios de teratología hasta con 200 mg/kg (equivalente a base libre) en ratas y conejos. Se observó un ligero efecto en la morfogénesis de los fetos en ratas a 200 mg/kg (equivalente a base libre). No se apreciaron efectos teratogénicos en conejos.
En el estudio de fertilidad en ratas, no se apreciaron hallazgos parenterales notables desde el punto de vista toxicológico. Con respecto a los parámetros de la camada, una ligera disminución en el cuerpo lúteo y un incremento en la pérdida de pre-implantación llevaron a una reducción en el número medio de implantaciones en el grupo con dosis de 200 mg/kg (equivalente a base libre).
Estudios exhaustivos in vitro e in vivo no revelaron evidencia de un potencial mutagénico.
Se realizaron estudios de toxicidad oral aguda en ratas y ratones. En ambas especies, la dosis letal aproximada después de una administración oral única fue superior a 2000 mg/kg. En perros y monos Rhesus, la administración oral de 600 mg/kg de etexilato de dabigatrán no indujo ningún cambio toxicológicamente significativo.
En estudios de toxicidad de dosis repetidas durante un máximo de 26 semanas en ratas y 52 semanas en monos Rhesus, se usaron dosificaciones hasta de 300 mg/kg (equivalente a base libre). Habitualmente, estas dosis fueron toleradas notablemente por ambos, ratas y monos Rhesus. Los problemas de sangrado se observaron en asociación con traumatismos (por ejemplo, toma de muestras de sangre) durante las primeras 4-6 horas después de la administración y están directamente relacionados con la actividad farmacodinámica del dabigatrán.
En los estudios de toxicología de por vida en ratas y ratones, no hubo evidencia de un potencial tumorígeno de etexilato de dabigatrán hasta dosis máximas de 200 mg/kg (equivalente a base libre).
En un estudio de toxicidad juvenil realizado en ratas Han Wistar, la mortalidad estuvo asociada con eventos de sangrado a exposiciones similares, en las cuales se observó sangrado en animales adultos. Tanto en ratas adultas como jóvenes, se considera que la mortalidad está relacionada con la actividad farmacológica excesiva del dabigatrán junto con el empleo de fuerzas mecánicas durante la dosificación y la manipulación. Los datos del estudio de toxicidad juvenil no indicaron un aumento de la sensibilidad a la toxicidad, ni toxicidad específica de animales jóvenes.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: El uso concomitante de PRADAXAR® con tratamientos que actúan sobre la hemostasia o la coagulación incluidos los antagonistas de la vitamina K pueden aumentar notablemente el riesgo de sangrado. (Ver Precauciones generales).
El etexilato de dabigatrán y el dabigatrán no son metabolizados por el sistema del citocromo P450 y no ejercen efectos in vitro sobre las enzimas del citocromo P450 en humanos. Por lo tanto, no se esperan interacciones medicamentosas relacionadas con el etexilato de dabigatrán ni con el dabigatrán (ver Farmacocinética en poblaciones especiales).
Interacciones con la glicoproteína-P:
Inhibidores de la glicoproteína-P: El etexilato de dabigatrán es un sustrato para el transportador de flujo de gp- P. La administración concomitante de inhibidores de la Gp-P, por ejemplo, amiodarona, verapamilo, quinidina, ketoconazol sistémico, dronedarona, ticagrelor, claritromicina y la combinación a dosis fija de glecaprevir/ pibrentasvir se espera que resulte en un aumento de las concentraciones plasmáticas de dabigatrán.
La administración concomitante de ketoconazol sistémico está contraindicada.
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: Para los otros inhibidores de la gp-P antes mencionados no se requieren ajustes de PRADAXAR® en esta indicación.
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a una cirugía ortopédica mayor: Para el uso concomitante de inhibidores de la gp-P y PRADAXAR® en esta indicación por favor consulte la sección Dosis y vía de administración y Farmacocinética en poblaciones especiales.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: Para los otros inhibidores de la gp-P antes mencionados no se requieren ajustes de PRADAXAR® en esta indicación.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: Para los otros inhibidores de la gp-P antes mencionados no se requieren ajustes de PRADAXAR® en esta indicación.
Amiodarona: La exposición al dabigatrán en sujetos sanos incrementó 1.6 veces (+60%) en presencia de la amiodarona (ver Farmacocinética en poblaciones especiales).
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: En los pacientes del estudio RE-LY las concentraciones no se incrementaron más de 14% y no se observó un incremento mayor del riesgo de hemorragia.
Verapamilo: Cuando PRADAXAR® (150 mg) fue coadministrada con verapamilo oral, la Cmáx y AUC de dabigatrán aumentaron, aunque la magnitud de este cambio difiere dependiendo del tiempo de administración y la formulación de verapamilo (ver Farmacocinética en poblaciones especiales).
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: En los pacientes del estudio RE-LY las concentraciones no se incrementaron más de 21% y no se observó un incremento mayor del riesgo de hemorragia.
Quinidina: La exposición al dabigatrán en sujetos sanos aumentó en 1.5 veces (+53%) en presencia de la quinidina (ver Farmacocinética en poblaciones especiales).
Claritromicina: La exposición al dabigatrán en sujetos sanos incrementó en aproximadamente un 19% en presencia de claritromicina, sin preocupación alguna respecto a la seguridad clínica (ver Farmacocinética en poblaciones especiales).
Ketoconazol: La exposición a dabigatrán se incrementó por un factor de 2.5 (+150%) tras la administración de dosis únicas y múltiples de ketoconazol por vía sistémica (ver Contraindicaciones y Farmacocinética en poblaciones especiales).
Dronedarona: La exposición al dabigatrán incrementó en 2.1 veces (+114%) después de una dosis única y 2.4 veces (+136%) después de dosis múltiples de dronedarona, respectivamente (ver Farmacocinética en poblaciones especiales).
Ticagrelor: La exposición al dabigatrán en sujetos sanos se incrementó en 1.46 veces (+46%) en presencia de ticagrelor en estado estacionario o 1.73 veces (+73%) cuando una dosis de carga de ticagrelor se administró simultáneamente con una dosis única de 75 mg de etexilato de dabigatrán. La exposición de estado estable de dabigatrán en sujetos sanos, se incrementó por un factor de 1.26 (+26%) en la presencia de ticagrelor en estado estable por un factor de 1.49 (+49%) cuando una dosis de carga de ticagrelor se administró simultáneamente con 110 mg de etexilato de dabigatrán. El aumento en exposición fue menos pronunciado cuando una dosis de carga de 180 mg de ticagrelor se administró dos horas posteriores a la ingesta de dabigatrán (+27%).
Sustrato de la glicoproteína-P:
Digoxina: En un estudio realizado con 24 sujetos sanos, cuando PRADAXAR® fue coadministrada con digoxina, no se observaron cambios en la digoxina, ni cambios relevantes clínicos en la exposición al dabigatrán (ver Farmacocinética en poblaciones especiales).
Inductores de glicoproteína-P: Después de 7 días de tratamiento con 600 mg de rifampicina una vez al día el AUC0-∞ y la Cmáx totales de dabigatrán fueron reducidos en un 67% y 66%, respectivamente, en comparación con el tratamiento de referencia.
El uso concomitante con inductores de la gp-P (por ejemplo, rifampicina) reduce la exposición a dabigatrán y debe ser evitado (ver Precauciones generales y Farmacocinética en poblaciones especiales).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Han sido observados algunos cambios no significativos en los niveles de enzimas hepáticas (ver Reacciones secundarias y adversas).
PRECAUCIONES GENERALES:
Riesgo hemorrágico: Al igual que con todos los anticoagulantes, PRADAXAR® deberá usarse con precaución en condiciones que aumenten el riesgo de sangrado. Puede ocurrir sangrado en cualquier lugar durante la terapia con PRADAXAR®. Una caída inexplicable en la hemoglobina y/o hematocrito o en la presión sanguínea pueden conducir a una búsqueda de un sitio de sangrado.
En situaciones que se encuentre en peligro la vida o en sangrado no controlado donde se requiera revertir inmediatamente el efecto anticoagulante del dabigatrán, se encuentra disponible el agente específico para revertir dichos efectos (Praxbind®, idarucizumab). (Ver "Cirugía e intervenciones", "Fase Preoperatoria" y "Sobredosis").
El tratamiento con PRADAXAR® no requiere monitoreo de la acción anticoagulante. La prueba INR no es confiable en los pacientes con PRADAXAR® se han reportado resultados falsos positivos (INR elevado). Por lo tanto, las pruebas de INR no se deben realizar.
Se dispone de pruebas de actividad anticoagulante, como el tiempo de trombina (TT), tiempo de coagulación de la ecarina (ECT) y tiempo de tromboplastina parcial activado (TTPa) para detectar la actividad excesiva de dabigatrán.
La anticoagulación relacionada con el uso de dabigatrán puede ser evaluada por la ECT o TT. Si no están disponibles el ECT, y el TT la prueba de TTPa proporciona una aproximación de la actividad anticoagulante de PRADAXAR®.
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: En pacientes con fibrilación auricular en el estudio RE-LY tratados con 150 mg 2 veces al día, un TTPa de más de 2.0 a 3.0 veces el límite normal se asoció con un mayor riesgo de hemorragia.
Estudios de farmacocinética demostraron un incremento en la exposición al fármaco en pacientes con función renal reducida, incluyendo deterioro de la función renal relacionado con la edad. PRADAXAR® está contraindicado en casos de insuficiencia renal severa (depuración de creatinina < 30 mL/min).
Los pacientes que desarrollen insuficiencia renal aguda deben suspender el uso de PRADAXAR®.
Factores como una disminución en la función renal (depuración de creatinina 30-50 mL/min), edad ≥ 75 años de edad o medicación conjunta con inhibidores potentes de la gp-P, están asociados con incremento en los niveles plasmáticos de dabigatrán. La presencia de uno o más de estos factores puede aumentar el riesgo de sangrado (ver Dosis y vía de administración).
El uso concomitante de PRADAXAR® con los siguientes tratamientos no ha sido estudiado y puede incrementar el riesgo de sangrado: heparinas no fraccionadas (excepto en dosis necesarias para mantener la permeabilidad del catéter arterial o venoso central o durante la ablación con catéter de la fibrilación auricular) y derivados de la heparina, heparinas de bajo peso molecular (HBPM), fondaparinux, desirudina, agentes trombolíticos, antagonistas de los receptores GPIIb/IIIa, ticlopidina, dextrano, sulfinpirazona, rivaroxaban, prasugrel, antagonistas de la vitamina K, inhibidores de la gp-P tales como, entre otros, itraconazol, tacrolimus, ciclosporina, ritonavir, tipranavir, nelfinavir y saquinavir.
Se ha demostrado que el uso concomitante de PRADAXAR® con la combinación a dosis fija de los inhibidores de la P-gp glecaprevir/pibrentasvir incrementa la exposición a dabigatrán y puede aumentar el riesgo de sangrado.
El uso concomitante de dronedarona aumenta la exposición a dabigatrán y no se recomienda (Ver Farmacocinética en poblaciones especiales).
El uso concomitante de ticagrelor incrementa la exposición del dabigatrán y puede presentar interacción farmacodinámica, la cual puede resultar en un aumento en el riesgo de sangrado.
El riesgo de sangrado puede ser mayor en pacientes tratados concomitantemente con inhibidores selectivos de la recaptura de la serotonina (ISRS) o inhibidores selectivos de la recaptura de serotonina/norepinefrina (ISRSN).
El uso de agentes fibrinolíticos para el tratamiento del EVC isquémico agudo: El uso de agentes fibrinolíticos para el tratamiento del EVC isquémico agudo se puede considerar si el paciente presenta un tiempo de trombina (TT), o tiempo de coagulación ecarina (ECT), o Tiempo de Tromboplastina Parcial Activado (TTPa) que no sobrepase el límite superior normal (ULN) en función del rango de referencia local.
En situaciones en las que existe un riesgo incrementado de hemorragia (p. ej., un traumatismo importante o una biopsia reciente, o endocarditis bacteriana) generalmente se requiere una observación estrecha del paciente (en pos de signos de sangrado o anemia).
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a cirugía ortopédica mayor: Los AINEs administrados para una analgesia perioperatoria a corto plazo han demostrado que no están asociados con un aumento en el riesgo de sangrado cuando se administran junto con PRADAXAR®. Existe evidencia limitada respecto al uso de medicación regular con AINEs con vidas medias de menos de 12 horas durante el tratamiento con PRADAXAR®, y dicha evidencia no ha sugerido riesgo de sangrado adicional.
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: La administración conjunta de antiplaquetarios (incluidos el AAS y el clopidrogel) y tratamientos con AINES aumenta el riesgo de sangrado. Específicamente, el riesgo de sangrado grave, incluido sangrado gastrointestinal, se incrementa con la toma concomitante de antiplaquetarios o inhibidores potentes de la P-gp en los pacientes de ≥75 años.
Ante una sospecha clínica de sangrado, se sugiere la implementación de las medidas apropiadas, como una prueba de detección de sangre oculta en heces, o una medición de los valores de hemoglobina para detectar un posible descenso de dicho parámetro.
Interacción con inductores de la gp-P: El uso concomitante de PRADAXAR® con el potente inductor de la gp-P como la rifampicina, reduce las concentraciones plasmáticas de dabigatrán. Otros inductores de la gp-P, como la Hierba de San Juan o carbamazepina también se espera que reduzcan las concentraciones plasmáticas de dabigatrán, y deben ser administrados conjuntamente con cuidado (ver Interacciones medicamentosas y de otro género y Farmacocinética en poblaciones especiales).
Pacientes con síndrome antifosfolipídico: Los pacientes con síndrome antifosfolipídico (especialmente aquellos con triple positividad de anticuerpos antifosfolipídicos) tienen un riesgo incrementado de eventos tromboembólicos.
Si bien la eficacia de PRADAXAR® está establecida para el tratamiento y la prevención del tromboembolismo venoso, no ha sido estudiada específicamente en la subpoblación de pacientes con síndrome antifosfolipídico.
Por consiguiente, se recomienda considerar cuidadosamente todas las opciones de tratamiento (incluido el tratamiento estándar tal como antagonistas de la vitamina K) antes del uso de PRADAXAR® en pacientes con síndrome antifosfolipídico.
Cirugía e intervenciones: Los pacientes que reciben PRADAXAR® que han sido sometidos a cirugía o a procedimientos invasivos tienen un mayor riesgo de sangrado. Por consiguiente, las intervenciones quirúrgicas pueden requerir de la suspensión temporal de PRADAXAR® (ver también Farmacocinética).
Los pacientes pueden continuar bajo tratamiento con PRADAXAR® durante la cardioversión. No es necesario interrumpir el tratamiento con PRADAXAR® (150 mg dos veces al día) en los pacientes sometidos a ablación con catéter de la fibrilación auricular (ver Dosis y vía de administración).
En casos de cirugías de emergencia o procedimientos urgentes cuando se requiera que se revierta el efecto anticoagulante, se encuentra disponible el agente específico (Praxibind®, idarucizumab) para revertir PRADAXAR®.
Revertir la terapia de dabigatrán expone al paciente a riesgo trombótico debido a su enfermedad subyacente. El tratamiento con PRADAXAR® puede reiniciarse 24 horas después de la administración de Praxibind® (idarucizumab), si el paciente se encuentra clínicamente estable y se ha logrado una hemostasis adecuada.
Fase preoperatoria: Debido a un mayor riesgo de hemorragia, PRADAXAR® puede ser suspendido temporalmente con antelación a un procedimiento invasivo o quirúrgico.
Cirugía de emergencia o procedimiento urgente: El agente específico para revertir PRADAXAR® se encuentra disponible (Praxibind®, idarucizumab) para revertir el efecto anticoagulante inmediatamente (Ver Cirugía e intervenciones).
Cirugía o Intervención aguda: PRADAXAR® debe ser suspendida temporalmente. Una cirugía o intervención de emergencia debe ser retrasada si es posible hasta al menos 12 horas después de la última dosis. Si la cirugía no puede ser retrasada, puede haber un riesgo mayor de sangrado.
Cirugía o intervención electiva: Si es posible, se debe suspender PRADAXAR® al menos 24 horas antes de un procedimiento invasivo o quirúrgico. En pacientes que presentan un riesgo de sangrado mayor o en una cirugía mayor en donde se requiera una hemostasia completa, puede considerar suspender PRADAXAR® de 2 a 4 días antes de la cirugía. La depuración de dabigatrán en pacientes con insuficiencia renal puede tomar más tiempo. Esto debe considerarse antes de cualquier procedimiento (tabla 11 y también Farmacocinética).
Tabla 11. Resumen de las reglas de interrupción antes de procedimientos invasivos o quirúrgicos
|
Función renal (CrCL mL/min) |
Vida media estimada (horas) |
Suspender dabigatrán antes de la cirugía electiva |
|
|
Riesgo elevado de sangrado o cirugía mayor |
Riesgo estándar |
||
|
≥ 80 |
~ 13* |
2 días antes |
24 h antes |
|
≥ 50-< 80 |
~ 15* |
2-3 días antes |
1-2 días antes |
|
≥ 30-< 50 |
~ 18* |
4 días antes |
2-3 días antes (> 48 horas) |
* Para más detalles, véase la tabla 10 Farmacocinética.
PRADAXAR® está contraindicado en pacientes con disfunción renal severa (depuración de creatinina < 30 mL/min) pero en caso de que esto ocurra, se debe suspender el uso de PRADAXAR® al menos 5 días antes de una cirugía mayor.
Si se requiere una intervención aguda, PRADAXAR® debe suspenderse temporalmente. Si es posible, la cirugía/intervención debe retrasarse al menos 12 horas después de la última dosis. Si no es posible retrasar la cirugía, entonces, el riesgo de sangrado puede aumentar. El riesgo de sangrado debe ser sopesado junto con la urgencia de la intervención. (Para cardioversión favor de revisar Dosis y vía de administración).
Anestesia espinal/anestesia epidural/punción lumbar: Los procedimientos como una anestesia espinal pueden requerir de una función hemostática completa.
El riesgo de hematoma espinal o epidural puede incrementarse en casos de punción traumática o repetida y por el uso prolongado de los catéteres epidurales. Después de remover un catéter, debe transcurrir un intervalo de cuando menos 1 hora antes de la administración de la primera dosis de PRADAXAR®. Estos pacientes requieren observación frecuente buscando signos y síntomas neurológicos de hematoma espinal o epidural.
Periodo posterior al procedimiento: Reanude/inicie el tratamiento con PRADAXAR® una vez que se haya logrado la hemostasia completa.
Efectos en la habilidad para manejar y operar maquinaria: No se han realizado estudios de los efectos sobre la capacidad para conducir, andar en bicicleta y usar maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Oral.
Cuadro de dosificación
|
Prevención de eventos Tromboembólicos Venosos (ETV) en pacientes que han sido sometidos a cirugía ortopédica mayor |
Prevención de evento vascular cerebral (EVC), embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular |
Prevención de trombosis venosa profunda (TVP) aguda y/o embolia pulmonar (EP) y prevención de muerte relacionada |
Tratamiento de trombosis venosa profunda (TVP) recurrente y/o embolia pulmonar (EP) y prevención de muerte relacionada |
|
La dosis recomendada de PRADAXAR® es 220 mg una vez al día administrada en 2 cápsulas de 110 mg. |
La dosis diaria recomendada de PRADAXAR® es de 300 mg vía oral, tomada como una cápsula de 150 mg dos veces al día. La terapia debe continuar de por vida. |
La dosis diaria recomendada de PRADAXAR® es de 300 mg tomada como una cápsula de 150 mg dos veces al día, luego del tratamiento con un anticoagulante parenteral durante un mínimo de 5 días. El tratamiento debe continuarse por un lapso de hasta 6 meses. |
La dosis diaria recomendada de PRADAXAR® es de 300 mg tomada como una cápsula de 150 mg dos veces al día. La terapia puede continuar de por vida dependiendo en el riesgo individual del paciente. |
Adultos:
Prevención de EVT en pacientes que han sido sometidos a cirugía ortopédica mayor: La dosis recomendada de PRADAXAR® es 220 mg una vez al día administrada en 2 cápsulas de 110 mg. Los pacientes con daño renal moderado presentan un aumento en el riesgo de sangrado para dichos pacientes, la dosis recomendada de PRADAXAR® es de 150 mg una vez al día, tomada como 2 cápsulas de 75 mg.
Prevención de EVT después de una cirugía de reemplazo de rodilla: El tratamiento con PRADAXAR® deberá iniciarse por vía oral dentro de las 1-4 horas posteriores a la finalización de la cirugía con una cápsula única (110 mg) y continuarse con 2 cápsulas de 110 mg una vez al día para un total de 10 días. Si no se puede garantizar la hemostasia, se deberá retrasar el inicio del tratamiento. Si no se comienza con el tratamiento al día de la cirugía, entonces éste deberá dar inicio con 2 cápsulas de 110 mg una vez al día.
Prevención de EVT después de una cirugía de reemplazo de cadera: El tratamiento con PRADAXAR® deberá iniciarse por vía oral dentro de las 1-4 horas posteriores a la finalización de la cirugía con una cápsula única (110 mg) y continuarse con 2 cápsulas de 110 mg una vez al día para un total de 28-35 días. Si no se puede garantizar la hemostasia, se deberá retrasar el inicio del tratamiento. Si no se comienza con el tratamiento al día de la cirugía, entonces éste deberá dar inicio con 2 cápsulas de 110 mg una vez al día.
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: La dosis diaria recomendada de PRADAXAR® es de 300 mg vía oral, tomada como una cápsula de 150 mg dos veces al día. La terapia debe continuar de por vida.
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: La dosis diaria recomendada de PRADAXAR® es de 300 mg tomada como una cápsula de 150 mg dos veces al día, luego del tratamiento con un anticoagulante parenteral durante un mínimo de 5 días. El tratamiento debe continuarse por un lapso de hasta 6 meses.
Prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada: La dosis diaria recomendada de PRADAXAR® es de 300 mg tomada como una cápsula de 150 mg dos veces al día. La terapia puede continuar de por vida dependiendo en el riesgo individual del paciente.
Población pediátrica:
Prevención de EVT en pacientes que han sido sometidos a cirugía ortopédica mayor: PRADAXAR® no ha sido investigado en pacientes menores de 18 años de edad en la indicación de prevención de eventos de tromboembolia venosa en pacientes que han sido sometidos a una cirugía ortopédica mayor. No se recomienda el tratamiento con PRADAXAR® en pacientes pediátricos en esta indicación.
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: PRADAXAR® no ha sido investigado en pacientes menores de 18 años de edad en la indicación prevención de eventos vascular cerebral, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular. No se recomienda el tratamiento con PRADAXAR® en pacientes pediátricos en esta indicación.
Tratamiento de la trombosis venosa profunda aguda (TVP) y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: No está registrado el tratamiento con PRADAXA® en pacientes pediátricos en la indicación de tratamiento de la TVP y/o la EP aguda y prevención de la muerte relacionada.
Prevención de la trombosis venosa profunda recurrente (TVP) y/o la embolia pulmonar (EP) y la muerte relacionada: No está registrado el tratamiento con PRADAXA® en pacientes pediátricos en la indicación de prevención de la TVP y/o la EP recurrente, y la muerte relacionada.
Insuficiencia renal: La función renal debe ser evaluada mediante el cálculo de la depuración de creatinina (CrCl) antes de iniciar el tratamiento con PRADAXAR® para excluir a los pacientes con insuficiencia renal grave (es decir, depuración de creatinina < 30 mL/min). No se cuenta con información para respaldar el uso de PRADAXAR® en pacientes con insuficiencia renal severa (depuración de creatinina < 30 mL/min). No se recomienda el tratamiento con PRADAXAR® para esta población (ver Contraindicaciones).
Mientras esté en tratamiento, la función renal debe ser evaluada en ciertas situaciones clínicas cuando se sospecha que la función renal podría disminuir o empeorar (como la hipovolemia, la deshidratación y con ciertas medicaciones concomitantes, etc.).
Dabigatrán puede ser dializado; existe experiencia clínica limitada en los estudios clínicos para demostrar la utilidad de esta medida en los estudios clínicos.
Prevención de TEV en pacientes que han sido sometidos a una cirugía ortopédica mayor: La dosis deberá reducirse a 150 mg de PRADAXAR® tomados una vez al día en forma de 2 cápsulas de 75 mg en pacientes con insuficiencia renal moderada (depuración de creatinina de 30-50 mL/min).
Prevención de EVC, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: No se requiere ajuste de dosis. Los pacientes deben ser tratados con una dosis diaria de 300 mg vía oral, administrada como una cápsula de 150 mg dos veces al día.
Prevención de los eventos de tromboembolia venosa luego de una cirugía de reemplazo de rodilla o de cadera: El tratamiento con PRADAXAR® debe iniciarse por vía oral dentro de un lapso de 1-4 horas de completada la cirugía, con una cápsula sola de 75 mg, y posteriormente debe continuarse con 2 cápsulas de 75 mg una vez al día durante un total de 10 días (luego de una cirugía de reemplazo de rodilla) o 28-35 días (luego de una cirugía de reemplazo de cadera). En ambas cirugías, si la hemostasia no está asegurada, el inicio del tratamiento debe postergarse. Si el tratamiento no se inicia en el día de la cirugía, en ese caso deberá iniciarse con 2 cápsulas administradas una vez al día.
En pacientes con insuficiencia renal moderada (depuración de creatinina 30-50 mL/min) la función renal debe ser evaluada por lo menos una vez al año.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: No se requiere ajuste de dosis en pacientes con una función renal por arriba de 30 mL/min. Los pacientes deben ser tratados con una dosis diaria de 300 mg vía oral, administrada como una cápsula de 150 mg dos veces al día.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: No se requiere ajuste de dosis en pacientes con una función renal por arriba de 30 mL/min. Los pacientes deben ser tratados con una dosis diaria de 300 mg vía oral, administrada como una cápsula de 150 mg dos veces al día.
En pacientes con insuficiencia renal moderada (depuración de creatinina 30-50 mL/min) la función renal debe ser evaluada por lo menos una vez al año.
Ancianos: Los estudios farmacocinéticos en sujetos de edad avanzada muestran un aumento en la exposición al fármaco relacionado con el deterioro de la función renal de estos pacientes. Véase también la dosis y la administración en pacientes con insuficiencia renal.
Prevención de TEV en pacientes que han sido sometidos a una cirugía ortopédica mayor: Debido a que la insuficiencia renal puede ocurrir frecuentemente en pacientes de edad avanzada (mayores de 75 años), la función renal debe ser evaluada a través de la depuración de creatinina (CrCl) antes de iniciar el tratamiento con PRADAXAR® para excluir del mismo a los pacientes con insuficiencia renal grave (depuración de creatinina menor a 30 mL/min). La función renal también debe ser evaluada en situaciones clínicas en la que se sospecha que la función renal podría disminuir o deteriorarse (como hipovolemia, deshidratación, y con la coadministración de ciertos medicamentos, etc.).
No se requiere ajustar la dosis. Los pacientes deberán ser tratados con 220 mg de PRADAXAR® una vez al día en forma de 2 cápsulas de 110 mg.
Prevención de EVC, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: Debido a que la insuficiencia renal puede ocurrir frecuentemente en pacientes de edad avanzada (mayores de 75 años), la función renal debe ser evaluada a través de la depuración de creatinina (CrCl) antes de iniciar el tratamiento con PRADAXAR® para excluir del mismo a los pacientes con insuficiencia renal grave (depuración de creatinina menor a 30 mL/min). La función renal también debe ser evaluada al menos una vez al año en los pacientes tratados con PRADAXAR® o con mayor frecuencia, según sea necesario, en situaciones clínicas en la que se sospecha que la función renal podría disminuir o deteriorarse (como hipovolemia, deshidratación, y con la coadministración de ciertos medicamentos, etc.).
Los pacientes de 80 años o mayores deben ser tratados con una dosis diaria de 220 mg por vía oral administrados como una cápsula de 110 mg dos veces al día.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: Debido a que la insuficiencia renal puede ocurrir frecuentemente en pacientes de edad avanzada (mayores de 75 años), la función renal debe ser evaluada a través de la depuración de creatinina (CrCl) antes de iniciar el tratamiento con PRADAXAR® para excluir del mismo a los pacientes con insuficiencia renal grave (depuración de creatinina menor a 30 mL/min). La función renal también debe ser evaluada al menos una vez al año en los pacientes tratados con PRADAXAR® o con mayor frecuencia, según sea necesario, en situaciones clínicas en la que se sospecha que la función renal podría disminuir o deteriorarse (como hipovolemia, deshidratación, y con la coadministración de ciertos medicamentos, etc.).
No se requiere ajustar la dosis. Los pacientes deben ser tratados con una dosis diaria de 300 mg administrada vía oral, como una cápsula de 150 mg dos veces al día.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: Debido a que la insuficiencia renal puede ocurrir frecuentemente en pacientes de edad avanzada (mayores de 75 años), la función renal debe ser evaluada a través de la depuración de creatinina (CrCl) antes de iniciar el tratamiento con PRADAXAR® para excluir del mismo a los pacientes con insuficiencia renal grave (depuración de creatinina menor a 30 ml/min). La función renal también debe ser evaluada al menos una vez al año en los pacientes tratados con PRADAXAR® o con mayor frecuencia, según sea necesario, en situaciones clínicas en la que se sospecha que la función renal podría disminuir o deteriorarse (como hipovolemia, deshidratación, y con la coadministración de ciertos medicamentos, etc.).
No se requiere ajustar la dosis. Los pacientes deben ser tratados con una dosis diaria de 300 mg administrada vía oral, como una cápsula de 150 mg dos veces al día.
Peso: No se requiere ajustar la dosis.
Uso concomitante de PRADAXAR® con inhibidores potentes de la glicoproteína-P, es decir, amiodarona, quinidina o verapamilo:
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a una cirugía ortopédica mayor: La dosis debe reducirse a 150 mg de PRADAXAR® tomados una vez al día en forma de 2 cápsulas de 75 mg en pacientes que reciben de manera simultánea PRADAXAR® y amiodarona, quinidina o verapamilo (ver Interacciones medicamentosas y de otro género).
Se debe evitar iniciar el tratamiento con verapamilo en pacientes que han sido sometidos a cirugía ortopédica mayor y que ya están siendo tratados con PRADAXAR®. También se debe evitar el inicio simultáneo de tratamiento con PRADAXAR® y verapamilo.
Prevención de los eventos de tromboembolia venosa luego de una cirugía de reemplazo de rodilla o de cadera: El tratamiento con PRADAXAR® debe iniciarse por vía oral dentro de un lapso de 1-4 horas de completada la cirugía, con una cápsula sola de 75 mg, y posteriormente debe continuarse con 2 cápsulas de 75 mg una vez al día durante un total de 10 días (luego de una cirugía de reemplazo de rodilla) o 28-35 días (luego de una cirugía de reemplazo de cadera). En ambas cirugías, si la hemostasia no está asegurada, el inicio del tratamiento debe postergarse. Si el tratamiento no se inicia en el día de la cirugía, en ese caso deberá iniciarse con 2 cápsulas administradas una vez al día.
Prevención de EVC, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: No se requiere ajuste de dosis. Los pacientes deben ser tratados con una dosis diaria de 300 mg vía oral, en cápsulas de 150 mg dos veces al día.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: No se requiere ajustar la dosis, los pacientes deben ser tratados con una dosis diaria de 300 mg administrada vía oral, como una cápsula de 150 mg dos veces al día.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: No se requiere ajustar la dosis. Los pacientes deben ser tratados con una dosis diaria de 300 mg administrada vía oral, como una cápsula de 150 mg dos veces al día.
Pacientes con riesgo de hemorragia:
Prevención de EVC, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: La presencia de los siguientes factores pueden aumentar el riesgo de sangrado: por ejemplo, edad ≥ 75 años, insuficiencia renal moderada (depuración de creatinina 30-50 mL/min), el tratamiento concomitante con fuertes inhibidores de la gp-P (ver Farmacocinética en poblaciones especiales), antiplaquetarios o sangrado gastrointestinal previo (Precauciones generales). Para los pacientes con uno o más de uno de estos factores de riesgo, reducir la dosis diaria a 220 mg, administrada como 110 mg dos veces al día de acuerdo con el criterio del médico tratante.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: La presencia de los siguientes factores pueden aumentar el riesgo de sangrado: por ejemplo, edad ≥75 años, insuficiencia renal moderada (depuración de creatinina 30-50 mL/min) o sangrado gastrointestinal previo (ver Precauciones generales). No es necesario un ajuste de dosis para pacientes que presenten sólo un factor de riesgo.
Existe evidencia clínica limitada para pacientes con factores de riesgo múltiples.
En estos pacientes, PRADAXAR® sólo debe ser administrado si los riesgos esperados sobrepasan los riesgos de sangrado.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: La presencia de los siguientes factores pueden aumentar el riesgo de sangrado: por ejemplo, edad ≥ 75 años, insuficiencia renal moderada (depuración de creatinina 30-50 mL/min) o sangrado gastrointestinal previo (Precauciones generales).
No es necesario un ajuste de dosis para pacientes que presenten sólo un factor de riesgo.
Existe evidencia clínica limitada para pacientes con factores de riesgo múltiples.
En estos pacientes, PRADAXAR® sólo debe ser administrado si los riesgos esperados sobrepasan los riesgos de sangrado.
Cambio del tratamiento con PRADAXAR® a un anticoagulante parenteral:
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a una cirugía ortopédica mayor: Espere 24 horas después de la última dosis antes de cambiar de PRADAXAR® a un anticoagulante parenteral.
Prevención de EVC, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: Espere 12 horas después de la última dosis, antes de cambiar de PRADAXAR® a un anticoagulante parenteral.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: Espere 12 horas después de la última dosis, antes de cambiar de PRADAXAR® a un anticoagulante parenteral.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: Espere 12 horas después de la última dosis, antes de cambiar de PRADAXAR® a un anticoagulante parenteral.
Cambio del tratamiento con anticoagulantes parenterales al tratamiento con PRADAXAR®:
PRADAXAR® deberá ser administrada de 0-2 horas antes del momento previsto para la administración de la siguiente dosis de la terapia alternativa, o al momento de la interrupción, en caso de tratamiento continuo (por ejemplo, HNF intravenosa).
Cambio de los antagonistas de la Vitamina K a PRADAXAR®:
Prevención de eventos vasculares cerebrales, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular: Se debe suspender el antagonista de la vitamina K. PRADAXAR® puede ser administrada tan pronto como el INR sea < 2.0.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: Se debe suspender el antagonista de la vitamina K. PRADAXAR® puede ser administrada tan pronto como el INR sea < 2.0.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: Se debe suspender el antagonista de la vitamina K. PRADAXAR® puede ser administrada tan pronto como el INR sea < 2.0.
Cambio de PRADAXAR® a antagonistas de Vit. K:
Prevención de eventos vasculares cerebrales, embolismo sistémico y reducción de mortalidad vascular en pacientes con fibrilación auricular:
El inicio de los AVK debe ajustarse de acuerdo con la depuración de creatinina del paciente de la siguiente manera:
• Depuración de creatinina ≥ 50 mL/min, iniciar AVK tres días antes de suspender dabigatrán.
• Depuración de creatinina ≥ 30-< 50 mL/min, iniciar AVK dos días antes de suspender dabigatrán.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: El inicio de los AVK debe ajustarse de acuerdo con la depuración de creatinina del paciente de la siguiente manera:
• Depuración de creatinina ≥ 50 mL/min, iniciar AVK tres días antes de suspender dabigatrán.
• Depuración de creatinina ≥ 30-< 50 mL/min, iniciar AVK dos días antes de suspender dabigatrán.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: El inicio de los AVK debe ajustarse de acuerdo con la depuración de creatinina del paciente de la siguiente manera:
• Depuración de creatinina ≥ 50 mL/min, iniciar AVK tres días antes de suspender dabigatrán.
• Depuración de creatinina ≥ 30-< 50 mL/min, iniciar AVK dos días antes de suspender dabigatrán.
Cardioversión:
Prevención de EVC, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: Los pacientes pueden permanecer con PRADAXAR® mientras se lleva a cabo la cardioversión.
Ablación con catéter de la fibrilación auricular:
Prevención de EVC, embolismo sistémico y reducción de la mortalidad vascular en pacientes con fibrilación auricular: La ablación con catéter puede realizarse en pacientes bajo tratamiento con 150 mg o 110 mg de PRADAXAR® dos veces al día. No es necesario interrumpir el tratamiento con PRADAXAR® (Farmacocinética y farmacodinamia).
Intervención coronaria percutánea (ICP) con colocación de stent:
Prevención del accidente cerebrovascular y la embolia sistémica, y reducción de la mortalidad vascular en pacientes con fibrilación auricular: Los pacientes con fibrilación auricular no valvular que sean sometidos a una ICP con colocación de stent pueden ser tratados con PRADAXAR® en combinación con antiplaquetarios después de haberse logrado la hemostasia (ver Propiedades farmacológicas”).
Si se salta una dosis:
Prevención de eventos tromboembólicos venosos en pacientes que han sido sometidos a una cirugía ortopédica mayor: Continúe con las dosis diarias restantes de PRADAXAR® a la misma hora, el día siguiente.
No tome una dosis doble para compensar las dosis individuales saltadas.
Una dosis de PRADAXAR® que se olvidó puede todavía tomarse hasta 6 horas antes de la siguiente dosis programada. A partir de las 6 horas previas a la siguiente dosis programada, se debe omitir la dosis saltada.
No tome una dosis doble para compensar las dosis individuales saltadas.
Tratamiento de la trombosis venosa profunda (TVP) aguda y/o la embolia pulmonar (EP) y prevención de la muerte relacionada: Una dosis de PRADAXAR® que se olvidó puede todavía tomarse hasta 6 horas antes de la siguiente dosis programada. A partir de las 6 horas previas a la siguiente dosis programada, se debe omitir la dosis saltada.
No tome una dosis doble para compensar las dosis individuales saltadas.
Prevención de la trombosis venosa profunda (TVP) recurrente y/o la embolia pulmonar (EP) y la muerte relacionada: Una dosis de PRADAXAR® que se olvidó puede todavía tomarse hasta 6 horas antes de la siguiente dosis programada. A partir de las 6 horas previas a la siguiente dosis programada, se debe omitir la dosis saltada.
No tome una dosis doble para compensar las dosis individuales saltadas.
Instrucciones de uso/manipulación:
Al retirar una cápsula dura del blíster, deben seguirse las instrucciones que se brindan a continuación: Corte el blíster por la línea punteada del troquelado para separar sólo una de las cápsulas del resto de la tira. Abra el blíster desprendiendo manualmente la lámina metalizada de la parte posterior y retire la cápsula.
No presione la cápsula contra la lámina metalizada para abrir el blíster.
Todo remanente del producto o material de desecho debe descartarse de acuerdo con los requerimientos locales.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: La sobredosis, después de la administración de PRADAXAR®, puede conducir a complicaciones hemorrágicas debido a sus propiedades farmacodinámicas. Las dosis de PRADAXAR® superiores a las recomendadas, exponen al paciente a un aumento en el riesgo de sangrado. La anticoagulación excesiva puede requerir la suspensión de PRADAXAR®. En el caso de complicaciones hemorrágicas, se debe suspender el tratamiento e investigar el origen del sangrado. Dado que el dabigatrán se excreta principalmente por vía renal, se debe mantener una diuresis adecuada.
Dependiendo de la situación clínica, se debe iniciar un tratamiento estándar adecuado, por ejemplo, hemostasia quirúrgica según se indique y reemplazo del volumen sanguíneo.
Para situaciones que requieran revertir inmediatamente el efecto anticoagulante, se encuentra disponible el agente reversor específico Praxbind® (idarucizumab), que antagoniza el efecto farmacodinámico de PRADAXAR®. (Ver sección Precauciones generales, Cirugía e intervenciones, Fase preoperatoria).
Además, se debe considerar el uso de sangre fresca o plasma fresco congelado. Puesto que la unión a proteínas es baja, el dabigatrán es dializable, sin embargo existe experiencia clínica limitada en el uso de diálisis en este entorno. (Ver Farmacocinética en poblaciones especiales).
Se pueden considerar los concentrados de factores (activados o no activados) o Factor VIIa recombinante concomitantemente con el uso de Praxbind®. Existe alguna evidencia experimental para respaldar el papel de estos agentes para revertir el efecto anticoagulante del dabigatrán, aunque todavía no se ha demostrado de manera sistemática su utilidad en entornos clínicos. Se debe considerar la administración de concentrados plaquetarios en los casos donde esté presente la trombocitopenia o se hayan usado fármacos antiplaquetarios de acción prolongada. Todos los tratamientos sintomáticos deben ser administrados a juicio del médico.
PRESENTACIONES: Caja de cartón con 10, 20, 30 o 60 cápsulas de 75 mg, 110 mg o 150 mg en envase de burbuja e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 25 °C.
El producto debe conservarse en el envase original para protegerlo de la humedad.
Las cápsulas no deben colocarse en pastilleros ni en organizadores de dosis, a menos que las cápsulas puedan mantenerse dentro del envase original.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Dosis, la que el médico señale. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni en la lactancia. No se administre en menores de 18 años.
Reporte las sospechas de reacción adversa a los correos:
farmacovigilancia@cofepris.gob.mx,
farmacovigilancia.mex@boehringer-ingelheim.com
BOEHRINGER INGELHEIM PROMECO, S.A. de C.V.
Calle del Maíz No. 49, Col. Barrio Xaltocan,
C.P. 16090, Xochimilco,
Ciudad de México, México
Reg. Núm. 358M2008 SSA IV
®Marca Registrada


