
PRAXBIND
IDARUCIZUMAB
Solución inyectable
1 Caja, 2 Frasco(s) ámpula de vidrio, 2.5/50 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula contiene:
Idarucizumab 2.5 g
Vehículo cbp 50 mL
Anticuerpo monoclonal humanizado (Fab) de origen ADN recombinante expresado en células de ovario de hámster chino (CHO)
INDICACIONES TERAPÉUTICAS:
PRAXBIND® es un agente específico de reversión para el dabigatrán y está indicado en pacientes tratados con PRADAXAR® cuando se requiere la reversión rápida de los efectos anticoagulantes de dabigatrán:
• Para cirugía de emergencia/procedimientos urgentes.
• En sangrado amenazante para la vida o no controlado.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacodinamia:
Código ATC: V03AB37.
Grupo farmacoterapéutico: Todos los demás productos terapéuticos; antídotos.
La farmacodinamia del idarucizumab después de la administración de etexilato de dabigatrán, se investigó en sujetos sanos de 45 a 64 años que recibieron una dosis de 5 g mediante infusión intravenosa. La mediana de la exposición máxima al dabigatrán en los sujetos sanos investigados estuvo en el rango de aquellos pacientes donde se administra 150 mg, dos veces al día de etexilato de dabigatrán.
Mecanismo de acción:
El idarucizumab es un agente específico de reversión para el dabigatrán. Es un fragmento humanizado de anticuerpo monoclonal (Fab) que se fija al dabigatrán con afinidad muy alta, aproximadamente 300 veces más potente que la afinidad de fijación del dabigatrán a la trombina. El complejo idarucizumab-dabigatrán se caracteriza por una activación rápida y una desactivación extremadamente lenta, de lo cual resulta un complejo muy estable. El idarucizumab se fija de manera potente y específica al dabigatrán y a sus metabolitos, neutralizando su efecto anticoagulante.
Efecto del idarucizumab en la exposición y la actividad anticoagulante del dabigatrán:
Inmediatamente después de administrar el idarucizumab, las concentraciones plasmáticas de dabigatrán no fijado se redujeron más de 99%, produciendo niveles sin actividad anticoagulante. La mayoría de los pacientes presentaron una reversión sostenida de hasta 12 horas (> 90%) de las concentraciones plasmáticas del dabigatrán.
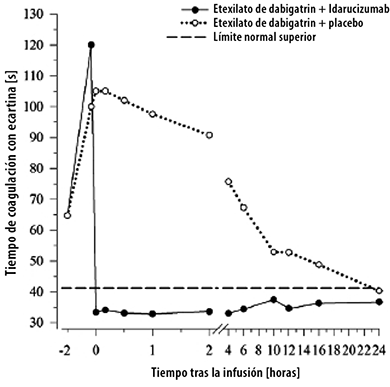
En un subgrupo de pacientes, se observó una reaparición de los niveles plasmáticos de dabigatrán libre y una prolongación concomitante de los tiempos de coagulación, quizás debida a la redistribución del dabigatrán procedente de la periferia. Esto ocurrió entre 2 y 24 horas tras la administración de idarucizumab, y sobre todo en los momentos de medición más allá de las 12 horas.
Figura 1. Niveles plasmáticos de dabigatrán libre (no unido) en el grupo representativo de sujetos sanos (administración de idarucizumab o placebo en el momento correspondiente a la hora 0 h).

El dabigatrán prolonga los tiempos de coagulación, medidos por tiempos de coagulación como el tiempo de trombina diluida (TTd), el tiempo de trombina (TT), el tiempo de tromboplastina parcial activado (aPTT) y el tiempo de coagulación de la ecarina (TCE), que proporcionan un indicio aproximado de la intensidad de la anticoagulación.
Un valor en el rango normal después de la administración del idarucizumab indica que un paciente ya no está bajo el efecto anticoagulante. Un valor superior al rango normal puede reflejar la presencia de dabigatrán activo residual u otras condiciones clínicas, p. ej., presencia de otros fármacos o coagulopatía por transfusión. Estas pruebas se usaron para evaluar el efecto anticoagulante del dabigatrán. Inmediatamente después de la infusión de idarucizumab, se observó una reversión completa y sostenida de la prolongación de los tiempos de coagulación inducida por el dabigatrán, que permaneció durante todo el periodo de observación de al menos 24 h.
Figura 2. Reversión de la anticoagulación inducida por el dabigatrán, determinada por TTd en el grupo representativo de sujetos sanos (administración de idarucizumab o placebo en el momento correspondiente a las 0 h).

Figura 3. Revisión de la anticoagulación inducida por dabigatrán en términos del TCE, en el grupo representativo de sujetos sanos (administración de idarucizumab o placebo en el momento correspondiente a las 0 h).
Parámetros de generación de trombina:
El dabigatrán ejerce efectos pronunciados sobre el potencial endógeno de la trombina (PTE). El tratamiento con idarucizumab normalizó la relación trombina-tiempo de retraso y la relación tiempo-máximo a los niveles basales, determinados 0.5 a 12 horas después de terminar la infusión de idarucizumab. El idarucizumab solo no ha mostrado efecto procoagulante medido como PTE. Esto sugiere que el idarucizumab no tiene efecto protrombótico.
Readministración de dabigatrán etexilato:
Veinticuatro horas después de la infusión de idarucizumab la readministración de dabigatrán etexilato produjo la actividad anticoagulante esperada.
Eficacia y seguridad clínica:
Estudios clínicos:
Se realizaron 3 estudios Fase I aleatorizados, doble ciego, controlados con placebo, en 283 sujetos (224 tratados con idarucizumab), para evaluar la seguridad, eficacia, tolerabilidad, farmacocinética y farmacodinamia del idarucizumab, administrado solo o después del etexilato del dabigatrán. La población de estudio consistió en sujetos sanos y en sujetos que mostraban características específicas de una población incluidas: edad, peso corporal, raza, sexo y disfunción renal. En estos estudios las dosis de idarucizumab variaron de 20 mg a 8 g, y los tiempos de infusión fluctuaron de 5 minutos a 1 hora.
Los valores representativos de los parámetros farmacocinéticos y farmacodinámicos se establecieron con base en sujetos sanos de 45 a 64 años que recibieron 5 g de idarucizumab.
Se llevó a cabo un estudio prospectivo, abierto, no aleatorizado, no pareado (RE-VERSE AD), para investigar el tratamiento en pacientes adultos que presentan un sangrado no controlado o con riesgo de muerte relacionado con el dabigatrán (Grupo B) o que necesitan una cirugía o procedimiento de urgencia (Grupo A). La variable primaria medida es la reversión porcentual máxima del efecto anticoagulante del dabigatrán en las 4 horas posteriores a la administración de idarucizumab, tomando como parámetros el tiempo de trombina diluida (TTd) o el tiempo de coagulación con ecarina (TCE) medidos en el laboratorio central. Una variable secundaria clave también analizada es la restauración de la hemostasia.
En el estudio RE-VERSE AD se incluyeron datos de 503 pacientes: 301 pacientes presentaron una hemorragia grave (Grupo A) y 202 pacientes requirieron un procedimiento/cirugía de urgencia (Grupo B). Aproximadamente la mitad de los pacientes de cada grupo eran varones. La mediana de la edad fue 78 años y la mediana de la depuración de creatinina fue 52.6 mL/min. El 61.5% de los pacientes del grupo A y el 62.4% de los pacientes del grupo B estaban tratados con dabigatrán 110 mg dos veces por día.
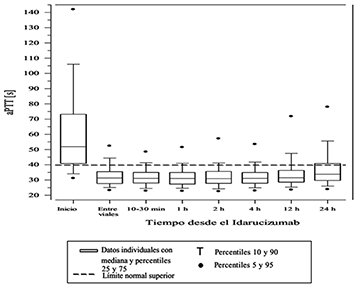
La reversión sólo pudo evaluarse en aquellos pacientes que presentaron tiempo de coagulación prolongada antes del tratamiento con idarucizumab. La mayoría de los pacientes, tanto en el Grupo A como el B, logró una reversión total del efecto anticoagulante del dabigatrán, (TTd 98.7%; TCE 82.2%; aPTT: 92.5% de los pacientes evaluables, respectivamente) en las primeras 4 horas posteriores a la administración de 5 g idarucizumab. La reversión se hizo evidente inmediatamente después de la administración.
Figura 4. Reversión de la anticoagulación inducida por el dabigatrán en términos del tiempo de trombina diluida (TTd) en pacientes del estudio RE-VERSE AD (N=487).

Figura 5. Reversión de la anticoagulación inducida por el dabigatrán en términos del tiempo de coagulación con ecarina (TCE) en pacientes del estudio RE-VERSE AD (N=487).
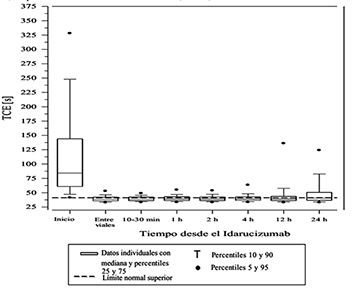
Figura 6. Reversión de la anticoagulación inducida por el dabigatrán en términos del tiempo de la tromboplastina activada (aPTT) en pacientes del estudio RE-VERSE AD (N=486).
La hemostasia se restauró en el 91% de los pacientes evaluables que habían sufrido hemorragias graves; y en el 93.4% de los pacientes que necesitaron procedimientos de urgencia se observó una hemostasia normal.
Del total de pacientes (503) murieron 101; cada una de estas muertes podría atribuirse tanto a una complicación del evento índice o a las comorbilidades asociadas. Se consignaron eventos trombóticos en 34 pacientes, (23 de los 34 pacientes no estaban bajo tratamiento antitrombótico cuando ocurrió el evento); en cada caso, el evento se podría atribuir a la condición médica subyacente. Se consignaron síntomas leves de una posible hipersensibilidad (pirexia, broncoespasmo, hiperventilación, exantema o prurito). No se pudo establecer ninguna relación causal con el idarucizumab.
Farmacocinética:
La farmacocinética del idarucizumab se investigó en sujetos sanos de 45 a 64 años que recibieron una dosis de 5 g mediante infusión intravenosa.
Distribución:
El idarucizumab mostró cinética de disposición multifásica y con distribución extravascular limitada. Después de la infusión intravenosa de una dosis de 5 g la media geométrica del volumen de distribución en estado estable (Vee) fue de 8.9 L (coeficiente de variación geométrico [CVg]: 24.8%). En la fase terminal el volumen de distribución (Vz) fue de 41.8 L (CVg: 22.3%).
Biotransformación:
Se han descrito varias rutas que pueden contribuir al metabolismo de anticuerpos. Todas estas rutas involucran la biodegradación del anticuerpo a moléculas más pequeñas, es decir, péptidos pequeños o aminoácidos que luego se reabsorben e incorporan en la síntesis general de proteínas.
Eliminación:
El idarucizumab es eliminado rápidamente, con aclaramiento total de 47.0 mL/min (CVg: 18.4%), vida media inicial de 47 minutos (CVg: 11.4%) y vida media terminal de 10.3 h (CVg: 18.9%). Después de la administración intravenosa de 5 g de idarucizumab, 32.1% (CVg: 60.0%) de la dosis se recuperó en orina dentro de un periodo de recolección de 6 horas y menos de 1% en las siguientes 18 horas. Se supone que la parte restante de la dosis se eliminó a través del catabolismo de proteínas, principalmente en los riñones.
Datos preclínicos:
Se utilizó un modelo en cerdos, en los cuales se realizó una lesión hepática en los que se dosificó dabigatrán hasta alcanzar concentraciones supra-terapéuticas de aproximadamente 10 veces los niveles plasmáticos en humanos. El idarucizumab revirtió eficaz y rápidamente el sangrado que amenazaba la vida en el curso de 15 minutos después de la inyección. Todos los cerdos sobrevivieron con dosis de idarucizumab de aproximadamente 2.5 y 5 g. Sin el idarucizumab, la mortalidad en el grupo que recibió anticoagulantes fue de 100%.
Investigaciones preclínicas con el idarucizumab han mostrado que no hay interacciones con:
• Expansores del volumen.
• Concentrados de factores de coagulación, como concentrados de complejo de protrombina (CCPs, p. ej., de 3 factores y 4 factores), CCPs activados (aPCCs) y factor recombinante VIIa.
• Otros anticoagulantes (p. ej., inhibidores de la trombina diferentes del dabigatrán, inhibidores del Factor Xa, incluyendo la heparina de bajo peso molecular, antagonistas de la vitamina K, heparina). Por tanto, el idarucizumab no revertirá los efectos de otros anticoagulantes.
CONTRAINDICACIONES:
Hipersensibilidad al principio activo o a cualquiera de los componentes de la fórmula.
Embarazo, lactancia y menores de 18 años.
Pacientes con intolerancia hereditaria a la fructosa.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
No hay datos del uso de PRADAXAR® en mujeres embarazadas. No se han realizado estudios de toxicidad reproductiva y del desarrollo, dada la naturaleza y el uso clínico previsto del producto medicinal. PRADAXAR® puede utilizarse durante el embarazo si el beneficio clínico esperado supera los riesgos potenciales.
Lactancia:
Se desconoce si idarucizumab se excreta en la leche humana.
REACCIONES SECUNDARIAS Y ADVERSAS:
En un estudio de Fase III, se ha evaluado la seguridad de PRADAXAR® en 503 pacientes que se presentaron con un sangrado no controlado o que requerían un procedimiento o cirugía de emergencia y estaban recibiendo tratamiento con PRADAXAR® así como en 224 voluntarios en un estudio de Fase I.
Se llevó a cabo un estudio prospectivo, abierto, no aleatorizado, no pareado (RE-VERSE AD), para investigar el tratamiento en pacientes adultos que presentan un sangrado no controlado o con riesgo de muerte relacionado con el dabigatrán (Grupo B) o que necesitan una cirugía o procedimiento de urgencia (Grupo A), en este estudio se encontraron 3 potenciales eventos de hipersensibilidad, cada uno ocurrió durante los primeros 5 días posteriores a la administración de idarucizumab y fueron reportados por los investigadores principales como relacionados al medicamento de estudio.
Rash (con uso concomitante de ondansetrón y tramadol).
Vómito y pérdida del estado de alerta (en paciente con una gran hemorragia intracraneal).
Hipotensión durante la infusión de idarucizumab que fue reportada como reacción anafiláctica.
Hubo un caso adicional de anafilaxia caracterizada por rash, vomito, alteraciones respiratorias y pérdida del estado de alerta en un paciente que tomaba amoxicilina de manera concomitante.
Se reportaron eventos adversos serios posteriores a la administración de idarucizumab en 117 pacientes (23.3%), 66 del grupo A y 51 en el grupo B.
Tabla 1. Eventos que ocurrieron con una frecuencia de al menos 1% en ambos grupos durante los primeros 5 días posteriores a la administración de idarucizumab (número total de casos y porcentaje en paréntesis).
|
Grupo A (N=301) |
Grupo B (N=202) |
Total (N=503) |
|
|
Total de eventos adversos serios |
66 (21.9) |
51 (25.2) |
117 (23.3) |
|
Delirum |
7 (2.3) |
4 (2.0) |
11 (2.2) |
|
Paro cardiaco |
1 (0.3) |
7 (3.5) |
8 (1.6) |
|
Sepsis |
3 (1.0) |
4 (2.0) |
7 (1.4) |
|
Choque séptico |
1 (0.3) |
6 (3.0) |
7 (1.4) |
|
Falla cardiaca |
4 (1.3) |
2 (1.0) |
6 (1.2) |
|
Edema pulmonar |
4 (1.3) |
2 (1.0) |
6 (1.2) |
|
Falla respiratoria |
3 (1.0) |
3 (1.5) |
6 (1.2) |
|
Neumonía |
0 (0.0) |
4 (2.0) |
4 (0.8) |
|
Falla renal aguda |
1 (0.3) |
2 (1.0) |
3 (0.6) |
|
Choque cardiogénico |
1 (0.3) |
2 (1.0) |
3 (0.6) |
|
Trombosis venosa profunda |
3 (1.0) |
0 (0.0) |
3 (0.6) |
|
Hemorragia intracraneal |
3 (1.0) |
0 (0.0) |
3 (0.6) |
|
Síndrome de disfunción orgánica múltiple |
1 (0.3) |
2 (1.0) |
3 (0.6) |
|
Hematoma subdural |
3 (1.0) |
0 (0.0) |
3 (0.6) |
|
Peritonitis |
0 (0.0) |
2 (1.0) |
2 (0.4) |
|
Evento vascular cerebral isquémico |
0 (0.0) |
2 (1.0) |
2 (0.4) |
|
Choque |
0 (0.0) |
2 (1.0) |
2 (0.4) |
Tabla 2. Frecuencia de las sospechas de reacciones adversas del estudio REVEARSE AD.
|
*Clase de órganos y sistemas (SOC) |
Frecuencia de la SOC N (%) |
TP basados en la incidencia N (%) |
|||||
|---|---|---|---|---|---|---|---|
|
Placebo N = 35 |
IDA solo N = 107 |
IDA o IDA + ED N = 224 |
Eventos adversos |
Placebo |
IDA solo |
IDA o IDA + ED |
|
|
Trastornos del sistema nervioso |
3 (8.6%) |
12 (11.2%) |
18 (8.0%) |
Cefalea Mareo Migraña Parestesia |
2 (5.7%) 1 (2.9%) 0 (0.0%) 0 (0.0%) |
12 (5.4%) 5 (2.2%) 2 (0.9%) 1 (0.4%) |
12 (5.4%) 5 (2.2%) 2 (0.9%) 1 (0.4%) |
|
Trastornos gastrointestinales |
1 (2.9%) |
5 (4.7%) |
9 (4.0%) |
Diarrea Náusea Dolor abdominal Estreñimiento Dispepsia |
0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) |
2 (1.9%) 1 (0.9%) 1 (0.9%) 1 (0.9%) 1 (0.9%) |
3 (1.3%) 1 (0.4%) 1 (0.4%) 1 (0.4%) 1 (0.4%) |
|
Trastornos generales y alteraciones en el lugar de la administración |
2 (5.7%) |
6 (5.6%) |
15 (6.7%) |
Dolor en el lugar del catéter Astenia Fatiga Hematoma en el lugar de la inyección Dermatitis en el lugar de la aplicación Irritación en el lugar de la aplicación Eritema en el lugar del catéter Sensación de calor Eritema en el lugar de la infusión Dolor en el lugar de la infusión Hinchazón en el lugar de la infusión |
1 (2.9%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) |
2 (1.9%) 0 (0.0%) 1 (0.9%) 1 (0.9%) 0 (0.0%) 1 (0.9%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 0 (0.0%) 1 (0.9%) |
3 (1.3%) 2 (0.9%) 2 (0.9%) 2 (0.9%) 1 (0.4%) 1 (0.4%) 1 (0.4%) 1 (0.4%) 1 (0.4%) 1 (0.4%) 1 (0.4%) |
|
Trastornos respiratorios, torácicos y mediastínicos |
1 (2.9%) |
1 (0.9%) |
2 (0.9%) |
Epistaxis Tos |
1 (2.9%) 0 (0.0) |
0 (0.0%) 1 (0.9%) |
1 (0.4%) 1 (0.4%) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
3 (8.6%) |
8 (7.5%) |
11 (4.9%) |
Dolor de espalda Espasmo muscular Dolor en las extremidades Rigidez musculoesquelética Mialgia |
1 (2.9%) 1 (2.9%) 1 (2.9%) 0 (0.0)% 0 (0.0%) |
4 (3.7%) 0 (0.0%) 1 (0.9%) 2 (1.9%) 1 (0.9%) |
4 (1.8%) 1 (0.4%) 2 (0.9%) 2 (0.9%) 2 (0.9%) |
|
Trastornos de la piel y deI tejido subcutáneo |
2 (5.7%) |
4 (3.7%) |
8 (3.6%) |
Irritación cutánea Eritema Reacción cutánea |
2 (5.7%) 0 (0.0%) 0 (0.0%) |
3 (2.8%) 1 (0.9%) 0 (0.0%) |
6 (2.7%) 1 (0.4%) 1 (0.4%) |
|
Trastornos renales y urinarios |
0 (0.0%) |
0 (0.0%) |
1 (0.4%) |
Glucosuria |
0 (0.0%) |
0 (0.0%) |
1 (0.4%) |
|
Trastornos del aparato reproductor y mamario |
0 (0.0%) |
0 (0.0%) |
1 (0.4%) |
Disfunción eréctil |
0 (0.0%) |
0 (0.0%) |
1 (0.4%) |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
0 (0.0%) |
0 (0.0%) |
1 (0.4%) |
Hematoma posterior al procedimiento |
0 (0.0%) |
0 (0.0%) |
1 (0.4%) |
|
*Clase de órganos y sistema |
TP basados en la incidencia ≥ 2% N (%) |
||||||
|---|---|---|---|---|---|---|---|
|
Grupo |
A Hemorragia |
B Cirugía |
Total |
||||
|
Número de pacientes (N/%) Pacientes con eventos adversos (N/%) |
66 53 |
100.0 80.3 |
57 38 |
100.0 66.7 |
123 91 |
100.0 74.4 |
|
|
Infecciones e infestaciones |
SOC Infección del tracto urinario Neumonía |
12 5 3 |
18.2 7.6 4.5 |
8 0 4 |
14.0 0.0 7.0 |
20 5 7 |
16.3 4.1 5.7 |
|
Neoplasias (incluyendo quistes y pólipos) benignas, malignas y no especificadas |
SOC |
4 |
6.1 |
0 |
0.0 |
4 |
3.3 |
|
Trastornos hemáticos y del sistema linfático |
SOC Trombocitopenia Anemia |
7 4 3 |
10.6 6.1 4.5 |
4 0 3 |
7.0 0.0 5.3 |
11 4 6 |
8.9 3.3 4.9 |
|
Metabolismo y nutrición |
SOC Hipopotasemia |
9 6 |
13.6 9.1 |
5 3 |
8.8 5.3 |
14 9 |
11.4 7.3 |
|
Trastornos psiquiátricos |
SOC Delirio Ansiedad Estado confusional Desorientación |
17 7 3 2 2 |
25.8 10.6 4.5 3.0 3.0 |
5 2 0 1 1 |
8.8 3.5 0.0 1.8 1.8 |
22 9 3 3 3 |
17.9 7.3 2.4 2.4 2.4 |
|
Trastornos del sistema nervioso |
SOC Cefalea |
13 5 |
19.7 7.6 |
3 1 |
5.3 1.8 |
16 6 |
13.0 4.9 |
|
Trastornos cardiacos |
SOC Bradicardia Fibrilación auricular |
8 3 1 |
12.1 4.5 1.5 |
7 1 2 |
12.3 1.8 3.5 |
15 4 3 |
12.2 3.3 2.4 |
|
Trastornos vasculares |
SOC Hipotensión Hematoma Hipertensión La trombosis venosa profunda |
10 3 0 2 2 |
15.2 4.5 0.0 3.0 3.0 |
9 1 2 2 1 |
15.8 1.8 3.5 3.5 1.8 |
19 4 2 4 3 |
15.4 3.3 1.6 3.3 2.4 |
|
Trastornos respiratorios, torácicos y mediastínicos |
SOC Neumonía por aspiración Hiperventilación Disnea Embolia pulmonar Edema pulmonar |
14 3 1 2 2 2 |
21.2 4.5 1.5 3.0 3.0 3.0 |
5 0 2 1 0 0 |
8.8 0.0 3.5 1.8 0.0 0.0 |
19 3 3 3 2 2 |
15.4 2.4 2.4 2.4 1.6 1.6 |
|
Trastornos gastrointestinales |
SOC Estreñimiento Diarrea Disfagia Náusea |
14 6 2 3 3 |
21.2 9.1 3.0 4.5 4.5 |
10 2 4 0 1 |
17.5 3.5 7.0 0.0 1.8 |
24 8 6 3 4 |
19.5 6.5 4.9 2.4 3.3 |
|
Trastornos hepatobiliares |
SOC |
0 |
0.0 |
2 |
3.5 |
2 |
1.6 |
|
Trastornos cutáneos y del tejido subcutáneo |
SOC Hiperhidrosis |
2 0 |
3.0 0.0 |
2 2 |
3.5 3.5 |
4 2 |
3.3 1.6 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
SOC Artralgia Dolor de cuello Dolor en las extremidades |
9 4 3 3 |
13.6 6.1 4.5 4.5 |
1 0 0 1 |
1.8 0.0 0.0 1.8 |
10 4 3 4 |
8.1 3.3 2.4 3.3 |
|
Trastornos renales y urinarios |
SOC Insuficiencia renal aguda |
5 0 |
7.6 0.0 |
3 2 |
5.3 3.5 |
8 2 |
6.5 1.6 |
|
Trastornos generales y alteraciones en el lugar de la administración |
SOC Pirexia Dolor de pecho El edema periférico Hinchazón periférica |
15 6 3 2 2 |
22.7 9.1 4.5 3.0 3.0 |
8 1 0 1 1 |
14.0 1.8 0.0 1.8 1.8 |
23 7 3 3 3 |
18.7 5.7 2.4 2.4 2.4 |
|
Investigaciones |
SOC Hemoglobina reducida Pérdida de peso |
10 3 2 |
15.2 4.5 3.0 |
4 1 0 |
/.0 1.8 0.0 |
14 4 2 |
11.4 3.3 1.6 |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
SOC |
5 |
7.6 |
6 |
10.5 |
11 |
8.9 |
* SOC = Clasificación por órganos y sistemas.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Datos preclínicos revelan que no hay riesgo especial para los humanos con base en estudios de toxicidad de dosis repetidas de hasta 4 semanas en ratas y 2 semanas en monos. Estudios de farmacología de la seguridad han demostrado que no hay efectos en el sistema respiratorio, el sistema nervioso central o el sistema cardiovascular.
No se han realizado estudios para evaluar el potencial mutagénico y carcinogénico del idarucizumab. Con base en su mecanismo de acción y las características de las proteínas, no se prevén efectos carcinogénicos ni genotóxicos.
No se han hecho estudios para evaluar los efectos reproductivos potenciales del idarucizumab. No se han identificado efectos relacionados con el tratamiento en los tejidos reproductores de ningún sexo durante estudios de toxicidad intravenosa con dosis repetidas de hasta 4 semanas en ratas y 2 semanas en monos. Además, no se observó fijación del idarucizumab a los tejidos reproductores humanos en un estudio de reactividad cruzada en tejidos. Por tanto, los resultados preclínicos no sugieren un riesgo para la fertilidad o el desarrollo embrio-fetal.
No se observaron irritaciones locales de los vasos sanguíneos después de la administración intravenosa o paravenosa de idarucizumab.
Fertilidad:
No hay datos sobre el efecto de PRAXBIND® en la fertilidad.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
No se han realizado estudios formales de interacciones con PRAXBIND® y otros fármacos. Con base a las propiedades farmacocinéticas y la alta especificidad de la fijación al dabigatrán, las interacciones clínicamente relevantes con otros fármacos se consideran improbables.
Investigaciones preclínicas han evidenciado que no hay interacciones con expansores de volumen, concentrados de factores de coagulación y otros anticoagulantes (véase la sección Farmacocinética y farmacodinamia).
El tratamiento con PRAXBIND® puede usarse junto con medidas de estándar de soporte, las cuales deben considerarse medicamente adecuadas.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
La formulación del idarucizumab no produjo hemólisis de la sangre entera humana in vitro.
Tras el tratamiento con idarucizumab, se observó proteinuria. La proteinuria transitoria es una reacción fisiológica a la sobrecarga de proteínas tras la aplicación de 5 g de idarucizumab intravenoso como un bolo o infusión rápida. La proteinuria transitoria en general alcanzó su máximo unas 4 h después de la administración de idarucizumab; los valores normales se recuperaron entre 12 y 24 horas después. En casos aislados, la proteinuria transitoria persistió más de 24 horas.
En un número limitado de pacientes, se observó un rebote en las concentraciones plasmáticas de dabigatrán libre y una prolongación concomitante de las pruebas de coagulación hasta 24 horas después de administrar idarucizumab (ver la sección Farmacocinética y farmacodinamia).
PRECAUCIONES GENERALES:
Idarucizumab se une específicamente al dabigatrán y revierte su efecto anticoagulante. Carece de todo efecto de reversión sobre los demás anticoagulantes (véase la sección Farmacocinética y farmacodinamia).
El tratamiento con PRADAXAR® puede usarse junto a las medidas de soporte estándar, las cuales deben considerarse en función de lo que sea médicamente apropiado.
Trazabilidad:
Para mejorar la trazabilidad de los medicamentos biotecnológicos, se deben registrar claramente el nombre y el número de lote del producto administrado en la historia clínica del paciente.
Hipersensibilidad:
El riesgo del uso en de PRADAXAR® en pacientes con hipersensibilidad conocida (p. ej., reacción anafiláctica) al idarucizumab o a cualquiera de los excipientes, debe ponderarse cuidadosamente contra el beneficio potencial de este tratamiento de emergencia. Si hay una reacción anafiláctica u otra reacción alérgica grave, la administración de PRADAXAR® debe interrumpirse inmediatamente e iniciarse la terapia adecuada.
Intolerancia hereditaria a la fructosa:
La dosis recomendada de PRADAXAR® contiene 4 g de sorbitol como excipiente. En pacientes con intolerancia hereditaria a la fructosa, la administración parenteral de sorbitol se ha asociado con reportes de hipoglucemia, hipofosfatemia, acidosis metabólica, elevación del ácido úrico, insuficiencia hepática aguda con colapso de la función excretora y sintética, y muerte. Por tanto, en pacientes con intolerancia hereditaria a la fructosa el riesgo del tratamiento con PRADAXAR® debe ponderarse contra el beneficio potencial de este tratamiento de emergencia.
Eventos tromboembólicos:
Los pacientes tratados con dabigatrán padecen enfermedades que los predisponen a padecer eventos tromboembólicos. Al revertir los efectos del dabigatrán, los pacientes vuelven a quedar expuestos al riesgo de trombosis propio de la enfermedad subyacente. Para reducir este riesgo, hay que considerar la posibilidad de reanudar el tratamiento anticoagulante lo más pronto posible, siempre que sea aconsejable desde un punto de vista médico (ver la sección Dosis y vía de administración).
Inmunogenicidad:
Las muestras de suero de 283 sujetos en estudio de Fase I (224 voluntarios tratados con idarucizumab) y de 501 pacientes fueron analizados para la detección anticuerpos anti-idarucizumab antes y después del tratamiento.
Se detectaron anticuerpos preexistentes con reactividad cruzada al idarucizumab se detectaron en aproximadamente 12% (33/283) de los sujetos de Fase I y el 3.8% (19/501) de los pacientes. No hubo impacto en la farmacocinética o el efecto de reversión del idarucizumab, ni reacciones de hipersensibilidad.
Se observó la presencia de anticuerpos asociados con el tratamiento potencialmente persistentes anti-idarucizumab del tratamiento con títulos bajos en 4% (10/224) de los sujetos de Fase I y el 1.6% (8/501) de los pacientes. Lo cual sugiere un potencial inmunogénico bajo del idarucizumab. En un subgrupo de 6 sujetos de Fase I, idarucizumab se administró una segunda vez, dos meses después de la primera administración. No se detectaron anticuerpos contra anti-idarucizumab en estos sujetos antes de la segunda administración. En un sujeto se detectaron anticuerpos contra idarucizumab asociados con el tratamiento luego de la segunda administración. A nueve pacientes se les readministró idarucizumab. Todos ellos recibieron una segunda dosis de idarucizumab dentro de los 6 días posteriores a la primera. Ninguno de los pacientes a los que se readministró idarucizumab dio positivo en los análisis para la detección de anticuerpos contra idarucizumab.
En un estudio fase III que estudió 503 pacientes que usaron Idarucizumab (REVERSE AD) se detectaron anticuerpos anti-idarucizumab en 28 de 501 pacientes evaluados (5.6%). De esos 28 pacientes, 19 pacientes tenían anticuerpos que generaban reacción cruzada contra idarucizumab previos a la administración del medicamento y sólo 9 desarrollaron nuevos anticuerpos durante el tratamiento.
Los anticuerpos encontrados previos a la administración no generaron ningún efecto detectable contra la actividad de idarucizumab.
Uso en poblaciones específicas:
Disfunción renal:
En estudios de Fase I se ha investigado PRADAXAR® en sujetos con un aclaramiento de creatinina que fluctúa entre 44 y 213 mL/min. No se han estudiado sujetos con aclaramiento de creatinina menor que 44 mL/min en la Fase I. Dependiendo del grado de disfunción renal, el aclaramiento total se redujo en comparación con sujetos sanos, causando una mayor exposición al idarucizumab.
Según datos farmacocinéticos de 347 pacientes con diferentes grados de funcionamiento renal (mediana de la depuración de creatinina 21-99 mL/min), se estima que la exposición media al idarucizumab (ABC0-24 h) aumenta 38% en pacientes con alteración leve de la función renal (depuración de creatinina: 50 < 80 mL/min), 90% en pacientes con alteración moderada (depuración de la creatinina: 30 < 50 mL/min) y 146% en pacientes con alteración grave (depuración de la creatinina: 0 < 30 mL/min). Dado que dabigatrán también se excreta principalmente por vía renal, con el empeoramiento de la función renal también se observa un aumento en la exposición a dabigatrán.
En base a esta información y al grado de reversión del efecto anticoagulante de dabigatrán observado en los pacientes, parecería que la alteración renal no influye en el efecto reversor del idarucizumab.
Disfunción hepática:
No se ha observado que la insuficiencia hepática, evaluada en función del daño hepático determinado por resultados elevados en las pruebas de función hepática, repercuta sobre la farmacocinética de idarucizumab. No se requieren ajustes de la dosis en los pacientes con daño hepático.
Idarucizumab se ha estudiado en 58 pacientes con diferentes grados de insuficiencia hepática. En comparación con 272 pacientes sin insuficiencia hepática, la mediana del AUC de idarucizumab se alteró en un -6%, un 37% y un 10% en pacientes con elevaciones de AST/ALT de 1 a < 2 x ULN (N=34), 2 a < 3 x ULN (N=3) y > 3 x ULN(N=21), respectivamente. Según datos farmacocinéticos de 12 pacientes con enfermedad hepática, el AUC de idarucizumab se incrementó en un 10% en comparación con pacientes sin enfermedad hepática.
Pacientes geriátricos/sexo/raza:
Conforme a análisis farmacocinéticos de la población, los factores como sexo, edad y raza no tienen un efecto clínicamente significativo en la farmacocinética del idarucizumab.
Pacientes pediátricos:
La seguridad y la eficacia de PRADAXAR® en la población pediátrica no se han establecido.
DOSIS Y VÍA DE ADMINISTRACIÓN:
La dosis recomendada de PRAXBIND® es de 5 g (2 frascos ámpula x 2.5 g/50 mL).
PRAXBIND® (2 frascos ámpula x 2.5 g/50 mL) se administra por vía intravenosa, en dos infusiones consecutivas durante 5 a 10 minutos cada una, o como una inyección de bolo.
La administración de una segunda dosis de 5 g de PRAXBIND® se podría considerar en las siguientes situaciones:
• Recurrencia de una hemorragia clínicamente relevantes asociada a tiempos de coagulación prolongados.
• Necesidad de una segunda cirugía o procedimiento urgente en pacientes que, además, tengan tiempos de coagulación prolongados.
Los parámetros de coagulación relevantes son el tiempo parcial de tromboplastina activada (aPTT), el tiempo de trombina diluida (TTd) y el tiempo de coagulación con ecarina (TCE) (ver la sección Farmacocinética y farmacodinamia).
No se requiere ajustar la dosis en pacientes con disfunción renal. Esta afección no tuvo impacto en el efecto de reversión que ejerce el idarucizumab.
Reanudación de la terapia antitrombótica:
El tratamiento con Pradaxar puede reanudarse 24 horas después de la administración de PRAXBIND® si el paciente está clínicamente estable y se ha alcanzado la hemostasis adecuada.
Después de administrar PRAXBIND® puede iniciarse otra terapia antitrombótica (p. ej., heparina de bajo peso molecular) en cualquier momento, si el paciente está clínicamente estable y se ha alcanzado la hemostasis adecuada.
La ausencia de terapia antitrombótica expone a los pacientes al riesgo trombótico de su enfermedad o condición subyacente.
PRAXBIND® no debe mezclarse con otros productos medicinales. Puede usarse una línea intravenosa preexistente para administrar PRAXBIND®. La línea debe enjuagarse con 9 mg/mL de solución estéril de cloruro de sodio (al 0.9%) antes y al finalizar la infusión. No debe administrarse otra infusión en paralelo usando el mismo acceso intravenoso.
No se han observado incompatibilidades entre PRAXBIND® y los equipos de infusión de cloruro de polivinilo, polietileno o poliuretano o las jeringas de polipropileno.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No hay experiencia clínica con sobredosis de PRAXBIND®.
La dosis más alta de PRAXBIND® estudiado en sujetos sanos fue de 8 g.
No se han identificado señales de seguridad en este grupo.
PRESENTACIÓN:
Caja de cartón con 2 frascos ámpula con 50 mL (2.5 g/50 mL) cada uno.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Vida útil: 48 meses.
Protéjase de la luz.
Manténganse en refrigeración de 2 a 8 °C, No se congele.
Antes de su uso, el frasco ámpula no abierto puede mantenerse a 25 °C hasta por 48 horas si se almacena en el empaque original para protegerlo de la luz, o hasta por 6 horas cuando está expuesto a la luz. Una vez que la solución se ha retirado del frasco ámpula, se ha demostrado la estabilidad química y física durante el uso de idarucizumab durante 6 horas a temperatura ambiente. La solución no debe estar expuesta a la luz durante más de 6 horas.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se use en menores de 18 años. Contiene 4 gramos de sorbitol por dosis, no se use en pacientes intolerantes a la fructosa. Sólo deberá ser administrado por médicos especialistas. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. Si no se administra todo el producto, deséchese el sobrante. No se administre si el cierre ha sido violado. PRAXBIND® es para un solo uso y no contiene conservadores. No se deje al alcance de los niños. No se use durante el embarazo y lactancia. 
Reporte las sospechas de reacción adversa a los correos:
farmacovigilancia@cofepris.gob.mx y
farmacovigilancia.mex@boehringer-ingelheim.com
Titular del registro:
BOEHRINGER INGELHEIM PROMECO, S.A. de C.V.
Calle del Maíz No. 49, Col. Barrio Xaltocan,
C.P. 16090, Xochimilco, Ciudad de México, México.
Reg. Núm. 013M2017 SSA IV
®Marca Registrada


