

PROGRAF XL
TACROLIMUS
Cápsulas de liberación prolongada
1 Caja, 30 Cápsulas de liberación prolongada, 0.5 mg
1 Caja, 30 Cápsulas de liberación prolongada, 1 mg
1 Caja, 30 Cápsulas de liberación prolongada, 5 mg
1 Caja, 50 Cápsulas de liberación prolongada, 0.5 mg
1 Caja, 50 Cápsulas de liberación prolongada, 1 mg
1 Caja, 50 Cápsulas de liberación prolongada, 5 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
PROGRAF® XL CÁPSULAS de liberación prolongada:
Cada CÁPSULA contiene:
Tacrolimus monohidratado equivalente a 0.5 mg, 1 mg, 5 mg de tacrolimus
Excipiente cbp
INDICACIONES TERAPÉUTICAS: PROGRAF® XL está indicado para la profilaxis del rechazo de órganos en pacientes que reciben trasplantes alogénicos de riñón o hígado. Se recomienda que PROGRAF® XL sea usado concomitantemente con corticosteroides adrenérgicos.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacocinéticas: La actividad de PROGRAF® XL se debe principalmente al fármaco precursor. Los parámetros farmacocinéticos (media ± DE) de tacrolimus se han determinado después de la administración intravenosa (IV) y/u oral (VO) a voluntarios sanos, y a pacientes con trasplante de riñón, trasplante de hígado y trasplante de corazón.
Parámetros farmacocinéticos de PROGRAF® XL
|
Población |
N |
Dosis |
Día |
Parámetros farmacocinéticos |
||
|
Cmáx (ng/mL) |
Tmáx (h) |
ABC0-24§ (ng• h/mL) |
||||
|
Voluntarios sanos |
24 |
4 mg |
Día 1, Día 10 |
6.2 11.6 |
2.0 2.0 |
74.2 155 |
|
Riñón adulto, de novo |
34 |
0.19 mg/kg, 0.20 mg/kg |
Día 1, Día 14 |
18.2 29.9 |
3.0 2.0 |
231.0 363.9 |
|
Riñón adulto, conversión |
66 |
6.1 mg |
Día 1, Día 14 |
14.8 14.2 |
2.0 2.0 |
204.6 197.6 |
|
Hígado adulto, de novo |
45 |
0.12 mg/kg, 0.22 mg/kg |
Día 1, Día 14 |
10.6 25.7 |
4.0 2.0 |
146.0 324.2 |
|
Hígado adulto, conversión |
62 |
5.2 mg |
Día 14 |
13.3 |
2.0 |
184.0 |
|
Hígado pediátrico, conversión |
18 |
5.4 mg |
Día 7 |
15.2 |
2.0 |
193.0 |
La dosis es el promedio para el grupo, administrada una vez al día (pacientes con trasplante).
La conversión se refiere a 1:1 (mg/mg) de PROGRAF® XL, basada en la dosis diaria total.
§ Días de tratamiento con PROGRAF® XL.
Hubo una marcada disminución de la variabilidad de la exposición (ABC0-24) entre sujetos en receptores de raza negra de trasplante de riñón, en estado estacionario posterior a la conversión de PROGRAF® (porcentaje de variación: %CV: 25.4%) a PROGRAF® XL (%CV: 12.2%). En receptores caucásicos de trasplante de riñón, la variabilidad de la exposición entre sujetos, en estado estacionario fue similar después de la conversión de PROGRAF® (%CV: 12.2%) a PROGRAF® XL (%CV: 14.1%).
Debido a la variabilidad entre sujetos en la farmacocinética de tacrolimus, es necesario individualizar el régimen de administración para que la terapia sea óptima (ver Dosis y vía de administración). Los datos farmacocinéticos indican que las concentraciones en la sangre, a diferencia de las concentraciones en el plasma, constituyen el compartimento de muestreo más adecuado para describir la farmacocinética de PROGRAF® XL.
Absorción: En humanos, se ha mostrado que tacrolimus se puede absorber a lo largo de todo el tracto gastrointestinal. Generalmente, el tacrolimus disponible se absorbe rápidamente. PROGRAF® XL es una formulación de tacrolimus de liberación prolongada que produce un perfil de absorción oral extenso, con un tiempo promedio hasta la concentración máxima (Cmáx) en sangre de aproximadamente 2 horas (Tmáx).
La absorción es variable, y la biodisponibilidad oral media de tacrolimus (investigada con la formulación de PROGRAF®) está en el intervalo de 20% a 25% (intervalo individual en pacientes adultos de 6%-43%). La biodisponibilidad oral de PROGRAF® XL se redujo cuando se administró después de una comida. Tanto la velocidad como el grado de absorción de PROGRAF® XL se redujeron cuando se administró con alimentos.
El flujo biliar no influye sobre la absorción de tacrolimus y, por lo tanto, el tratamiento con PROGRAF® XL puede iniciarse por vía oral.
Existe una importante correlación entre el ABC y la concentración sanguínea valle en estado estacionario para PROGRAF® XL. Por lo tanto, el monitoreo de la concentración sanguínea valle proporciona una buena estimación de la exposición sistémica.
Distribución: En humanos, la distribución de tacrolimus después de una infusión intravenosa se puede describir como bifásica.
En la circulación sistémica, tacrolimus se une fuertemente a los eritrocitos, resultando en una relación de distribución de concentraciones en sangre/plasma de aproximadamente 20:1. En el plasma, tacrolimus se une principalmente a las proteínas plasmáticas (> 98.8%), principalmente a la albúmina sérica y a la glicoproteína ácida α-1.
Tacrolimus se distribuye extensamente en el cuerpo. El volumen de distribución en estado estacionario, basado en las concentraciones plasmáticas, es de aproximadamente 1 300 L (en sujetos sanos). Los resultados correspondientes basados en determinaciones en sangre mostraron una media de 47.6 L.
Metabolismo: Tacrolimus se metaboliza ampliamente en el hígado, principalmente a través del citocromo P450-3A4. Tacrolimus también se metaboliza considerablemente en la pared intestinal. Se han identificado varios metabolitos. Se ha demostrado in vitro que solamente uno de estos metabolitos presenta actividad inmunosupresora in vitro similar a la de tacrolimus. Los otros metabolitos no tienen o tienen una leve actividad inmunosupresora. En la circulación sistémica, solamente uno de los metabolitos inactivos está presente a concentraciones bajas. Por lo tanto, los metabolitos no contribuyen a la actividad farmacológica de tacrolimus.
Eliminación: Tacrolimus es una sustancia de baja depuración. En sujetos sanos, la media de la depuración total del organismo, calculada a partir de las concentraciones en sangre, fue de 2.25 L/h. En pacientes adultos con trasplante de hígado, riñón y corazón, se ha observado una depuración total del organismo de 4.1 L/h, 6.7 L/h y 3.9 L/h, respectivamente. Algunos factores, como niveles bajos de hematócrito y proteínas, lo que resulta en un aumento de la fracción libre de tacrolimus, o el aumento del metabolismo inducido por el uso de corticosteroides, se consideran responsables de las tasas elevadas de depuración observadas después de un trasplante.
La vida media de tacrolimus es larga y variable. En sujetos sanos, la vida media de eliminación en sangre es de aproximadamente 43 horas.
Tras la administración intravenosa y oral de tacrolimus marcado con 14C, la mayor parte de la radiactividad se eliminó en las heces. Aproximadamente 2% de la radiactividad se eliminó en la orina. Menos del 1% de tacrolimus inalterado se detectó en la orina y en las heces, lo que indica que tacrolimus se metaboliza casi completamente antes de su eliminación; siendo la bilis la vía de eliminación principal.
Poblaciones especiales:
Pediátrica: En función de los datos de 18 receptores pediátricos de trasplante de hígado (5 a 13 años de edad), que cambiaron de PROGRAF® a PROGRAF® XL, los pacientes pediátricos con trasplante de hígado podrían cambiar de una dosis de PROGRAF® dos veces al día a una dosis diaria de PROGRAF® XL, basada en una dosis diaria total de 1:1 (mg:mg) para obtener la concentración sanguínea adecuada de tacrolimus (ver Dosis y vía de administración).
Después de la administración de PROGRAF®, las concentraciones valle sanguíneas de 31 pacientes menores de 12 años de edad mostraron que los pacientes pediátricos requieren mayores dosis que los adultos para alcanzar concentraciones valle similares de tacrolimus (ver Dosis y vía de administración).
Raza: Los datos de la administración de PROGRAF® XL a pacientes con trasplante de riñón de novo muestra que los pacientes afroamericanos requirieron una dosis mayor para obtener concentraciones valle comparables, en comparación con los pacientes caucásicos (ver Dosis y vía de administración).
Los receptores afroamericanos de un trasplante de riñón (n = 12) cambiaron de PROGRAF® a PROGRAF® XL con base en una dosis diaria total de 1:1 (mg:mg). La relación de las medias de mínimos cuadrados (PROGRAF® XL: PROGRAF®) para el ABC0-24 en estado estacionario fue de 109.8% [IC del 90%: 99.0%, 121.7%] para los pacientes afroamericanos disminuyó con PROGRAF® XL, en comparación con PROGRAF® (ver Propiedades farmacocinéticas).
Sexo: Los datos de los receptores de un trasplante de riñón, que cambiaron de PROGRAF® a PROGRAF® XL en un estudio abierto de fase 2, mostraron equivalencia en la exposición para los pacientes tanto hombres como mujeres; la relación de las medias de mínimos cuadrados (PROGRAF®:PROGRAF® XL) para el ABC0-24 en estado estacionario fue de 92.0% [IC del 90%: 86.1%, 98.3%] para las pacientes mujeres (n = 24), y de 96.7% [IC del 90%: 90.9%, 102.9%] para los pacientes hombres (n = 42). Se encontraron resultados similares en los datos de los estudios de conversión de fase 2 en pacientes con trasplante de hígado y de corazón; en los receptores de un trasplante de hígado, la relación de las medias de mínimos cuadrados (PROGRAF®: PROGRAF® XL) para el ABC0-24 en estado estacionario fue de 89.2% [IC del 90%: 82.7%, 96.1%] para las pacientes mujeres (n = 26), y de 88.5% [IC del 90%: 84.9%, 92.3%] para los pacientes hombres (n = 36).
Diabetes: Los receptores estables de trasplantes de riñón y de hígado que tenían diabetes o inicio de diabetes reciente después del trasplante, y que cambiaron a PROGRAF® XL, tuvieron relaciones de las medias de mínimos cuadrados (PROGRAF®:PROGRAF® XL) para el ABC0-24 de 92.0% [IC del 90%: 84.8%, 99.7%] en los trasplantes de riñón (n = 13), y de 85.4% [IC del 90%: 79.6%, 91.5%] en pacientes estables con trasplante de hígado (n = 23).
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Inmunosupresores, inhibidores de la calcineurina.
Código ATC: L04AD02.
PROGRAF® XL está disponible para administración oral como cápsulas (cápsulas de tacrolimus) que contienen el equivalente a 0.5 mg, 1 mg o 5 mg de tacrolimus anhidro de liberación prolongada.
Tacrolimus es el principio activo en PROGRAF® XL. Tacrolimus es un inmunosupresor macrólido producido por Streptomyces tsukubaensis.
El nombre químico de tacrolimus es [3S[3R*[E(1S*,3S*,4S*)],4S*,5R*,8S*,9E,12R*,14R*,15S*,16R*,18S*,19S*,26aR*]]-5,6,8,11,12,13,14,15,16,17,18,19,24,25,26,26a-hexadecahidro-5,19-dihidroxi-3-[2-(4-hidroxi-3-metoxiciclohexil)-1metiletenil]-14,16-dimetoxi-4,10,12,18-tetrametil-8-(2-propenil)-15,19-epoxi-3H-pirido[2,1-c][1,4]oxaazaciclotricosina-1,7,20,21(4H,23H)-tetrona, monohidratado.
La estructura química de tacrolimus es:
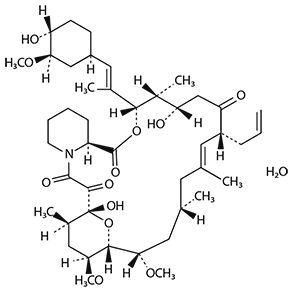
La fórmula empírica de tacrolimus es C44H69NO12• H2O, y su peso molecular es de 822.03. Tacrolimus tiene el aspecto de cristales blancos o polvo cristalino. Es prácticamente insoluble en agua, fácilmente soluble en etanol y muy soluble en metanol y cloroformo.
Mecanismo de acción: A nivel molecular, los efectos de tacrolimus parecen estar mediados por su unión a una proteína citosólica (FKBP12), que es la responsable de la acumulación intracelular del compuesto. El complejo FKBP12-tacrolimus se une específica y competitivamente a la calcineurina, inhibiéndola. Esto produce una inhibición dependiente del calcio de las vías de transducción de las señales de los linfocitos T, lo que impide la transcripción de un grupo concreto de genes de citocinas.
Tacrolimus es un potente agente inmunosupresor cuya actividad se ha demostrado en experimentos tanto in vitro como in vivo.
En particular, tacrolimus inhibe la formación de linfocitos citotóxicos, que son los principales responsables del rechazo de los trasplantes, también inhibe la activación de los linfocitos T y la proliferación de linfocitos B dependiente de linfocitos T auxiliares, así como la formación de linfocinas (como las interleucinas-2, -3 y el interferón-?), y la expresión del receptor de la interleucina-2).
Resultados de los estudios clínicos realizados con PROGRAF® XL, tacrolimus una vez al día:
Trasplante de hígado: La eficacia y la seguridad de PROGRAF® XL y PROGRAF®, ambos en combinación con corticosteroides, fue comparada en 471 receptores de trasplantes de hígado de novo. La tasa de episodios de rechazo agudo confirmados con biopsia dentro de las primeras 24 semanas después del trasplante fue de 32.6% en el grupo de PROGRAF® XL (N = 237), y de 29.3% en el grupo de PROGRAF® (N = 234). La diferencia entre tratamientos (PROGRAF® XL, PROGRAF®) fue de 3.3% (intervalo de confianza de 95% [-5.7%, 12.3%]). Las tasas de supervivencia de los pacientes después de 12 meses fueron de 89.2% para PROGRAF® XL y de 90.8% para PROGRAF®; en el grupo de PROGRAF® XL fallecieron 25 pacientes (14 mujeres, 11 hombres), y en el grupo de PROGRAF® fallecieron 24 pacientes (5 mujeres, 19 hombres). La supervivencia del trasplante después de 12 meses fue de 85.3% para PROGRAF® XL y de 85.6% para PROGRAF®.
Trasplante de riñón: La eficacia y seguridad de PROGRAF® XL y PROGRAF®, ambos en combinación con micofenolato de mofetilo (MMF) y corticosteroides, fue comparada en 667 receptores de trasplante de riñón de novo. La tasa de episodios de rechazo agudo confirmados con biopsia dentro de las primeras 24 semanas después del trasplante fue de 18.6% en el grupo de PROGRAF® XL (N = 331), y de 14.9% en el grupo de PROGRAF® (N = 336). La diferencia entre tratamientos (PROGRAF® XL-PROGRAF®) fue de 3.8% (intervalo de confianza de 95% [-2.1%, 9.6%]). Las tasas de supervivencia de los pacientes después de 12 meses fueron de 96.9% para PROGRAF® XL y de 97.5% para PROGRAF®; en el grupo de PROGRAF® XL fallecieron 10 pacientes (3 mujeres, 7 hombres), y en el grupo de PROGRAF® fallecieron 8 pacientes (3 mujeres, 5 hombres). La supervivencia del trasplante después de 12 meses fue de 91.5% para PROGRAF® XL y de 92.8% para PROGRAF®.
La eficacia y seguridad de PROGRAF®, ciclosporina y PROGRAF® XL, todos en combinación con inducción con anticuerpos a base de basiliximab, MMF y corticosteroides, fue comparada en 638 receptores de trasplantes de riñón de novo. La incidencia del fracaso de la eficacia a los 12 meses (definida como muerte, pérdida del trasplante, rechazo agudo confirmado con biopsia o pérdida de seguimiento) fue de 14.0% en el grupo de PROGRAF® XL (N = 214), 15.1% en el grupo de PROGRAF® (N = 212) y 17.0% en el grupo de ciclosporina (N = 212). La diferencia entre tratamientos fue -3.0% (PROGRAF® XL-ciclosporina) (intervalo de confianza de 95.2% [-9.9%, 4.0%]) para PROGRAF® XL frente a ciclosporina, y de -1.9% (PROGRAF®-ciclosporina) (intervalo de confianza de 95.2% [-8.9%, 5.2%]) para PROGRAF® frente a ciclosporina. Las tasas de supervivencia de los pacientes después de 12 meses fueron de 98.6% para PROGRAF® XL, 95.7% para PROGRAF® y 97.6% para ciclosporina; en el grupo de PROGRAF® XL, 3 pacientes murieron (todos hombres); en el grupo de PROGRAF® fallecieron 10 pacientes (3 mujeres y 7 hombres), y en el grupo de ciclosporina fallecieron 6 pacientes (3 mujeres y 3 hombres). La supervivencia del trasplante después de 12 meses fue de 96.7% para PROGRAF® XL, 92.9% para PROGRAF® y 95.7% para ciclosporina.
CONTRAINDICACIONES: Hipersensibilidad a tacrolimus o a cualquiera de los excipientes.
Hipersensibilidad a otros macrólidos.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: Los datos en humanos muestran que tacrolimus puede cruzar la placenta, y los fetos expuestos a tacrolimus in utero pueden estar en riesgo de nacer prematuramente, de tener defectos de nacimiento/anomalías congénitas, bajo peso al nacer y sufrimiento fetal.
El uso de tacrolimus durante el embarazo se ha asociado con parto prematuro, hiperpotasemia neonatal e insuficiencia renal.
Tacrolimus puede incrementar la hiperglucemia en mujeres embarazadas con diabetes (incluida la diabetes gestacional). Las concentraciones de glucosa en la sangre materna se deben monitorear con regularidad.
Tacrolimus puede exacerbar la hipertensión en mujeres embarazadas, y aumentar la preeclampsia.
La presión arterial se debe monitorear y controlar.
Las mujeres y los hombres con capacidad reproductiva deben considerar el uso de la anticoncepción adecuada antes de comenzar el tratamiento con tacrolimus.
Debido a la necesidad de tratamiento, tacrolimus se puede considerar en mujeres embarazadas cuando no hay ninguna alternativa más segura y cuando el beneficio previsto justifica el posible riesgo para el feto.
En ratas y conejos tacrolimus causó toxicidad embriofetal con dosis que demostraron toxicidad materna.
Lactancia: Los datos en humanos demuestran que tacrolimus se excreta en la leche materna. Como no se puede excluir que haya efectos dañinos para el recién nacido, las mujeres no deben amamantar mientras reciben PROGRAF® XL.
REACCIONES SECUNDARIAS Y ADVERSAS:
Receptores de trasplante de riñón: En un gran estudio (n = 668) comparativo, fase 3, aleatorizado, los receptores de novo de trasplante renal recibieron PROGRAF® XL más micofenolato de mofetilo (MMF) o PROGRAF® más MMF o neoral más MMF. Los tres regímenes incluyeron corticosteroides e inducción con basiliximab. La incidencia de eventos adversos que ocurrió en ≥ 15% de los receptores de novo de trasplante renal tratados con PROGRAF® XL se muestra en la siguiente tabla:
Trasplante renal de novo: eventos adversos que ocurrieron ≥ 15% de pacientes tratados con PROGRAF® XL + MMF
|
PROGRAF® XL + MMF % (n = 212) |
PROGRAF® XL + MMF % (n = 214) |
Neoral® + MMF % (n = 212) |
|
|
Trastornos gastrointestinales |
|||
|
Diarrea |
44.3 |
45.3 |
25.5 |
|
Náuseas |
38.7 |
42.1 |
46.7 |
|
Estreñimiento |
35.8 |
41.6 |
41.0 |
|
Vómito |
25.5 |
26.2 |
24.5 |
|
Dispepsia |
17.9 |
15.0 |
15.1 |
|
Daños, envenenamiento y complicaciones del procedimiento |
|||
|
Dolor post-procedimiento |
28.8 |
29.4 |
27.4 |
|
Complicación en el sitio de la incisión |
28.3 |
20.6 |
23.1 |
|
Trastornos del metabolismo y la nutrición |
|||
|
Hipomagnesemia |
28.3 |
25.7 |
22.2 |
|
Hipofosfatemia |
27.8 |
23.8 |
21.2 |
|
Hipercalemia |
25.5 |
22.0 |
19.3 |
|
Hiperglicemia |
21.2 |
19.2 |
15.1 |
|
Hiperlipidemia |
17.5 |
16.4 |
24.5 |
|
Hipocalemia |
16.0 |
15.9 |
17.5 |
|
Infecciones e infestaciones |
|||
|
Infección del tracto urinario |
25.5 |
15.9 |
22.2 |
|
Trastornos generales y condiciones del sitio de administración |
|||
|
Edema periférico |
34.9 |
35.5 |
45.8 |
|
Fatiga |
10.8 |
15.9 |
12.3 |
|
Trastornos del sistema nervioso |
|||
|
Tremor |
34.4 |
35.0 |
19.8 |
|
Cefalea |
24.1 |
21.5 |
24.5 |
|
Investigaciones |
|||
|
Creatinina en sangre aumentada |
23.1 |
18.7 |
22.6 |
|
Trastornos del sistema sanguíneo y linfático |
|||
|
Anemia |
30.2 |
33.6 |
27.8 |
|
Leucopenia |
15.6 |
16.4 |
11.8 |
|
Trastornos vasculares |
|||
|
Hipertensión |
32.1 |
29.9 |
34.9 |
|
Trastornos musculosqueléticos y del tejido conjuntivo |
|||
|
Dolor de espalda |
12.7 |
15.0 |
14.2 |
|
Trastornos psiquiátricos |
|||
|
Insomnio |
30.2 |
25.7 |
21.2 |
Las reacciones adversas observadas con menos frecuencia se describen en Reacciones adversas reportadas con menos frecuencia.
Receptores de trasplante de hígado: En un estudio fase 2, abierto, en receptores de novo de trasplante hepático, se reportaron los siguientes eventos adversos en < 15% de los pacientes tratados con PROGRAF® XL durante 1 año: hipertensión, diarrea, insuficiencia renal, hiperglicemia, anemia ascitis, insomnio, cefalea, tremores, efusión pleural, dolor de espalda, diabetes mellitus dependiente de insulina, leucopenia, dolor abdominal y náuseas.
Las reacciones adversas observadas con menos frecuencia con PROGRAF® XL se describen en Reacciones adversas reportadas con menos frecuencia.
Reacciones adversas reportadas con menos frecuencia (> 3 a < 15%): Las siguientes reacciones adversas también se reportaron en los estudios clínicos de receptores de trasplante de hígado, riñón y corazón que fueron tratados con PROGRAF® XL.
Infecciones e infestaciones: Infección con citomegalovirus, gastroenteritis, influenza, nasofaringitis, sinusitis, infección del tracto respiratorio superior.
Trastornos gastrointestinales: Dolor del abdomen superior, flatulencia.
Trastornos del metabolismo y la nutrición: Deshidratación, diabetes mellitus, hipocalcemia, acidosis metabólica.
Trastornos del sistema nervioso (véase Precauciones generales): Mareos.
Trastornos generales y condiciones del sitio de administración: Astenia, dolor de pecho, edema, pirexia, dolor.
Trastornos hepáticos: Aumento de enzimas hepáticas.
Trastornos musculosqueléticos y del tejido conjuntivo: Artralgia, calambres musculares, dolor en extremidades**.
Trastornos vasculares: Hipotensión.
Trastornos respiratorios, torácicos y mediastinales: Tos, disnea, dolor faringe-laríngeo.
Trastornos renales y urinarios (ver Precauciones generales): Hematuria, daño renal, insuficiencia renal.
Trastornos psiquiátricos: Ansiedad, depresión.
Trastornos de la piel y tejido subcutáneo: Acné, prurito.
Trastornos cardiacos: Taquicardia.
** En casos aislados, se ha reportado dolor en una extremidad como parte del síndrome de dolor inducido por inhibidor de la calcineurina (calcineurin-inhibitor induced pain syndrome, CIPS), que normalmente se presenta con dolor bilateral y simétrico intenso, ascendente, en las extremidades inferiores.
Reacciones adversas reportadas con menos frecuencia (> 1 a < 3%): Las siguientes reacciones adversas se reportaron en estudios clínicos de receptores de trasplante de hígado, riñón y corazón tratados con PROGRAF® XL y que tuvieron una tasa de frecuencia > 1 y < 3%.
Infecciones e infestaciones: Bronquitis, celulitis, infección con Escherichia del tracto urinario, infección con hongos, herpes simple, herpes zoster, infección con poliomavirus humano, candidiasis oral, faringitis, neumonía, pielonefritis, sepsis, infección de heridas.
Trastornos gastrointestinales: Malestar abdominal, distensión abdominal, dolor del abdomen bajo, ascitis, gastritis, reflujo gastroesofágico, hemorroides, deposiciones flojas, esofagitis, náuseas post-procedimiento, dolor de dientes.
Trastornos del metabolismo y la nutrición: Acidosis, anorexia, diabetes mellitus no dependiente de insulina, dislipidemia, sobrecarga de líquidos, gota, hipercalcemia, hipercolesterolemia, hiperhomocisteinemia, hiperfosfatemia, hiperuricemia, hipoalbuminemia, hipoglicemia, hiponatremia.
Trastornos del sistema nervioso: Hipoestesia, parestesia.
Trastornos generales y condiciones del sitio de administración: Anasarca.
Daños, envenenamiento y complicaciones del procedimiento: Complicaciones de la cirugía del trasplante, contusión, hernia incisional, conservación necrótica del daño al injerto, descarga post-procedimiento, toxicidad del agente terapéutico, desprendimiento de la herida.
Trastornos metabólicos: Aumento de glucosa en sangre, reducción de magnesio en sangre, reducción de fósforo en sangre, reducción de potasio en sangre, murmullo cardiaco, pruebas de función hepática anormales, reducción de la micción, reducción de peso, aumento de peso.
Trastornos musculoesqueléticos y del tejido conjuntivo: Mialgia, osteopenia, osteoporosis.
Trastornos vasculares: Hematoma, rubores, hipotensión ortostática.
Trastornos de la sangre y del sistema linfático: Leucocitosis, neutropenia, policitemia, trombocitopenia.
Trastornos respiratorios, torácicos y mediastinales: Disnea por esfuerzo, epistaxis, congestión nasal, tos productiva.
Trastornos renales y urinarios: Disuria, oliguria, proteinuria, daño renal agudo, dolor uretral.
Trastornos psiquiátricos: Agitación, estado de confusión.
Trastornos de la piel y del tejido subcutáneo: Alopecia, equimosis, sudores nocturnos, erupción, lesiones cutáneas.
Trastornos cardiacos: Fibrilación auricular.
Trastornos del sistema reproductivo y las mamas: Disfunción eréctil.
Trastornos hepatobiliares: Estenosis de conductos biliares, colestasis.
Trastornos de los ojos: Visión borrosa.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Los riñones y el páncreas fueron los órganos principales afectados en estudios de toxicidad realizados en ratas y monos. En ratas, tacrolimus produjo efectos tóxicos para el sistema nervioso y los ojos. En conejos, se observaron efectos cardiotóxicos reversibles después de la administración intravenosa de tacrolimus.
La administración subcutánea de tacrolimus a ratas macho en dosis de 2 o 3 mg/kg/día (1.6 a 6.4 veces el intervalo de la dosis clínica, en función del área de la superficie corporal) provocó una disminución relacionada con la dosis del recuento de espermatozoides.
Tacrolimus administrado por vía oral en dosis de 1.0 mg/kg (0.8 a 2.2 veces el intervalo de la dosis clínica, en función del área de la superficie corporal) a ratos macho y hembra, antes y durante el apareamiento, así como a hembras durante la gestación y la lactancia, se asoció con embrioletalidad y efectos adversos sobre la reproducción de la hembras, según lo indicaron una tasa alta de muerte fetal post-implantación y una disminución del número de crías no paridas y no viables. Cuando se administró en dosis de 3.2 mg/kg (2.6 a 6.9 veces el intervalo de la dosis clínica, en función del área de la superficie corporal, tacrolimus se asoció con toxicidad materna y paterna, así como toxicidad reproductiva, incluidos efectos adversos marcados sobre los ciclos estrales, el parto, la viabilidad de las crías y malformaciones en las crías.
Fertilidad: En ratas, se observó un efecto negativo de tacrolimus sobre la fertilidad masculina en la forma de una disminución del recuento y la motilidad de los espermatozoides.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones metabólicas: Tacrolimus, disponible sistémicamente, es metabolizado por el CYP3A4 hepático. También hay indicios de metabolismo gastrointestinal realizado por el CYP3A4 en la pared intestinal. El uso concomitante de medicamentos o remedios a herbales que se sabe que inhiben o que inducen al CYP3A4 puede afectar el metabolismo de tacrolimus y, por lo tanto, aumentar o disminuir sus concentraciones sanguíneas. Por lo tanto, se recomienda ampliamente monitorear estrechamente las concentraciones sanguíneas de tacrolimus, así como la prolongación del intervalo QT (con ECG), la función renal y otras reacciones adversas, siempre que se utilicen concomitantemente sustancias que tengan el potencial de alterar el metabolismo del CYP3A4 o de alterar de cualquier otra manera las concentraciones sanguíneas de tacrolimus, e interrumpir o ajustar la dosis de tacrolimus, según corresponda, para mantener una exposición similar a tacrolimus (ver Dosis y vía de administración y Precauciones generales).
lnhibidores del CYP3A4 que pueden causar un aumento de las concentraciones sanguíneas de tacrolimus: Clínicamente, las siguientes sustancias han mostrado incrementar las concentraciones sanguíneas de tacrolimus: se han observado interacciones fuertes con agentes antimicóticos, como ketoconazol, fluconazol, itraconazol y voriconazol, el antibiótico macrólido eritromicina, inhibidores de la proteasa del VIH (por ejemplo, ritonavir, nelfinavir, saquinavir) o inhibidores de la proteasa del VHC (por ejemplo, telaprevir, boceprevir). El uso concomitante de estas sustancias podría requerir la disminución de la dosis de tacrolimus en casi todos los pacientes. Los estudios farmacocinéticos han demostrado que el aumento de las concentraciones sanguíneas se debe principalmente al aumento de la biodisponibilidad oral de tacrolimus, debido a la inhibición del metabolismo gastrointestinal. El efecto sobre la depuración hepática es menor.
Se han observado interacciones más débiles con clotrimazol, claritromicina, josamicina, nifedipina, nicardipina, diltiazem, verapamilo, amiodarona, danazol, etinilestradiol, omeprazol, nefazodona y remedios herbales (chinos) que contienen extractos de Schisandra sphenanthera.
In vitro, las siguientes sustancias han demostrado ser posibles inhibidores del metabolismo de tacrolimus: bromocriptina, cortisona, dapsona, ergotamina, gestodeno, lidocaína, mefenitoína, miconazol, midazolam, nilvadipina, noretindrona, quinidina, tamoxifeno (triacetil) oleandomicina. Se ha reportado que el jugo de toronja aumenta la concentración sanguínea de tacrolimus y, por lo tanto, se debe evitar.
Lansoprazol y ciclosporina podrían potencialmente inhibir el metabolismo mediado por el CYP3A4 de tacrolimus y, por lo tanto, aumentar las concentraciones sanguíneas de tacrolimus.
Otras posibles interacciones que conducen a un aumento de las concentraciones sanguíneas de tacrolimus: Tacrolimus se une ampliamente a las proteínas plasmáticas. Se deben considerar las posibles interacciones con otros medicamentos que se sabe que tienen alta afinidad por las proteínas plasmáticas (por ejemplo, AINEs, anticoagulantes orales o antidiabéticos orales).
Entre otras posibles interacciones que podrían aumentar la exposición sistémica a tacrolimus se incluyen agentes procinéticos (como metoclopramida y cisaprida), cimetidina e hidróxido de magnesio-aluminio.
Inductores del CYP3A4 que pueden causar una disminución de las concentraciones sanguíneas de tacrolimus: Clínicamente, las siguientes sustancias disminuyen las concentraciones sanguíneas de tacrolimus: se han observado interacciones fuertes con rifampicina, fenitoína o hierba de San Juan (Hypericum perforatum), que podrían requerir un aumento de dosis de tacrolimus en casi todos los pacientes.
También se han observado interacciones clínicamente significativas con fenobarbital. Las dosis de mantenimiento de corticosteroides han mostrado reducir las concentraciones sanguíneas de tacrolimus.
Dosis elevadas de prednisolona o metilprednisolona, administradas para el tratamiento del rechazo agudo, tienen el potencial de aumentar o disminuir las concentraciones sanguíneas de tacrolimus.
Carbamazepina, metamizol e isoniazida tienen el potencial de disminuir las concentraciones de tacrolimus.
Efecto de tacrolimus sobre el metabolismo de otros medicamentos: Tacrolimus es un inhibidor conocido del CYP3A4; por lo tanto, su uso concomitante con medicamentos que se sabe que son metabolizados por el CYP3A4 puede afectar el metabolismo de dichos medicamentos.
La vida media de ciclosporina se prolonga cuando se administra concomitantemente con tacrolimus. Además, puede haber efectos nefrotóxicos sinérgicos/aditivos. Por este motivo, no se recomienda la administración concomitante de ciclosporina y tacrolimus, y se debe tener precaución al administrar tacrolimus a pacientes que han recibido ciclosporina previamente (ver Dosis y vía de administración y Precauciones generales).
Se ha demostrado que tacrolimus aumenta la concentración sanguínea de fenitoína. Debido a que tacrolimus puede disminuir la depuración de los anticonceptivos a base de esteroides, ocasionando un aumento de la exposición a hormonas, se debe tener particular cuidado al decidir qué métodos anticonceptivos utilizar.
El conocimiento sobre las interacciones entre tacrolimus y las estatinas es limitado. Los datos disponibles sugieren que la farmacocinética de las estatinas no se altera en gran medida por la administración concomitante de tacrolimus. Los datos en animales han mostrado que tacrolimus podría potencialmente disminuir la depuración y aumentar la semivida de pentobarbital y antipirina.
Otras interacciones que causan efectos clínicamente dañinos: El uso concomitante de tacrolimus con medicamentos que se sabe que tienen efectos nefrotóxicos o neurotóxicos puede incrementar el nivel de toxicidad (por ejemplo, antibióticos aminoglucósidos, inhibidores de la girasa, vancomicina, cotrimoxazol, AINEs, ganciclovir o aciclovir).
Se ha observado un aumento de la nefrotoxicidad después de la administración de anfotericina B e ibuprofeno junto con tacrolimus.
Debido a que el tratamiento con tacrolimus se puede asociar con hiperpotasemia, o puede aumentar la hiperpotasemia preexistente, se debe evitar la ingesta de potasio en dosis altas o de diuréticos ahorradores de potasio (por ejemplo, amilorida, triamtereno o espironolactona) (ver Precauciones generales).
Los inmunosupresores pueden afectar la respuesta a las vacunas y, por lo tanto, la vacunación durante el tratamiento con tacrolimus podría ser menos eficaz. Se debe evitar el uso de vacunas con organismos vivos atenuados (ver Precauciones generales).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Los niveles de creatinina sérica, potasio y glucosa en ayunas se deben evaluar con regularidad. Los sistemas metabólico y hematológico se deben monitorear de manera rutinaria, según se requiera clínicamente. No se sabe que tracolimus interfiera con las pruebas de laboratorio.
PRECAUCIONES GENERALES:
Errores en la medicación: Se han observado errores en la medicación, incluyendo cambio involuntario, no intencional o no supervisado entre las formulaciones de tacrolimus de liberación inmediata o de liberación prolongada. Esto ha ocasionado reacciones adversas graves, que incluyen el rechazo del trasplante, u otras reacciones adversas que podrían ser consecuencia de la subexposición o la sobrexposición a tacrolimus. Los pacientes se deben mantener con una sola formulación de tacrolimus con el correspondiente régimen de dosis diarias; las modificaciones de la formulación o de la dosis se deben hacer solamente bajo la estrecha supervisión de un especialista en trasplantes.
Para el tratamiento del rechazo de alotrasplantes resistentes al tratamiento con otros medicamentos inmunosupresores en pacientes adultos, no se dispone todavía de datos clínicos para la formulación de liberación prolongada PROGRAF® XL.
Para la profilaxis del rechazo de trasplantes en receptores adultos de alotrasplantes de corazón, los datos clínicos aún no están disponibles para PROGRAF® XL.
Durante el periodo post-trasplante inicial, se debe realizar un control de los siguientes parámetros de manera rutinaria: Presión arterial, ECG, estado neurológico y visual, concentración de glucosa sanguínea en ayunas, electrólitos (en particular, potasio), pruebas de la función hepática y renal, parámetros hematológicos, valores de la coagulación y determinaciones de las proteínas plasmáticas.
Si se observan cambios clínicamente significativos, se debe considerar un ajuste del régimen inmunosupresor.
Uso con inhibidores e inductores del CYP3A4: Las concentraciones de tacrolimus en sangre deben monitorearse cuando se combine con sustancias con potencial de interacción (ver Interacciones medicamentosas y de otro género), en particular los inhibidores potentes del CYP3A4 (como telaprevir, boceprevir, ritonavir, ketoconazol, voriconazol, itraconazol, telitromicina o claritromicina) o los inductores del CYP3A4 (como rifampicina, rifabutina), con el fin de ajustar la dosis de tacrolimus para mantener una exposición similar a tacrolimus.
Cuando se toma PROGRAF® XL se deben evitar las preparaciones herbales que contienen hierba de San Juan (Hypericum perforatum) u otras preparaciones herbales debido al riesgo de interacciones que conducen a una disminución de las concentraciones sanguíneas de tacrolimus y una reducción del efecto clínico de mismo, o bien, a un aumento de las concentraciones sanguíneas de tacrolimus y al riesgo de toxicidad de tacrolimus (ver Interacciones medicamentosas y de otro género).
Uso en pacientes que reciben ciclosporina: Se debe evitar la administración concomitante de ciclosporina y tacrolimus, y se debe tener precaución al administrar tacrolimus a pacientes que han recibido ciclosporina previamente (ver Dosis y vía de administración e Interacciones medicamentosas y de otro género).
Hiperpotasemia: Se debe evitar la ingesta de cantidades elevadas de potasio o el uso de diuréticos ahorradores de potasio (ver Interacciones medicamentosas y de otro género).
Nefrotoxicidad: Tacrolimus puede ocasionar el deterioro de la función renal en los pacientes después del trasplante debido a su efecto vasoconstrictor sobre la vasculatura renal. La insuficiencia renal aguda, que generalmente es reversible, puede ocasionar creatinina sérica alta, hiperpotasemia, disminución de la secreción de urea e hiperuricemia. Los pacientes con insuficiencia renal deben ser monitoreados estrechamente, ya que es posible que se requiera reducir o suspender temporalmente la dosis de tacrolimus. La insuficiencia renal aguda sin un manejo eficaz puede progresar a insuficiencia renal crónica, que se caracteriza por disfunción renal progresiva, aumento de la urea en sangre y proteinuria.
Se debe evitar el uso concomitante de tacrolimus con medicamentos con efectos nefrotóxicos conocidos.
Vacunas: Los inmunosupresores pueden afectar la respuesta a la vacunación, y por ello las vacunas pueden ser menos eficaces durante el tratamiento con tacrolimus. Se debe evitar el uso de vacunas con organismos vivos atenuados.
Trastornos gastrointestinales: Se ha reportado perforación gastrointestinal en pacientes tratados con tacrolimus. Debido a que la perforación gastrointestinal es un evento médicamente importante que puede causar un trastorno grave o potencialmente mortal, se deben considerar los tratamientos adecuados inmediatamente después de que ocurran sospechas de síntomas o signos.
Debido a que las concentraciones sanguíneas de tacrolimus pueden cambiar significativamente durante los episodios de diarrea, se recomienda el monitoreo adicional de las concentraciones de tacrolimus durante estos episodios.
Trastornos cardiacos: En raras ocasiones, se han observado casos de hipertrofia ventricular o hipertrofia septal, reportadas como cardiomiopatías, en pacientes tratados con PROGRAF®, y también podrían ocurrir con PROGRAF® XL. La mayoría de los casos han sido reversibles, y se han presentado principalmente con concentraciones sanguíneas valle de tacrolimus mucho mayores que las concentraciones máximas recomendadas. Otros factores que se ha observado que aumentan el riesgo de estos trastornos clínicos incluyen cardiopatía preexistente, uso de corticosteroides, hipertensión, insuficiencia renal o hepática, infecciones, sobrecarga de fluidos y edema. Por lo tanto, los pacientes de alto riesgo que reciben un tratamiento inmunosupresor importante, se deben monitorear utilizando procedimientos como ecocardiografía o ECG antes y después del trasplante (por ejemplo, inicialmente a los 3 meses y después a los 9-12 meses). Si se observan anomalías, se debe considerar la reducción de la dosis de PROGRAF® XL, o el cambio del tratamiento a otro agente inmunosupresor.
Tacrolimus puede prolongar el intervalo QT y causar Torsades de Pointes. Se debe tener precaución en pacientes con factores de riesgo de prolongación del intervalo QT, lo que incluye a pacientes con antecedentes personales o familiares de prolongación del intervalo QT, insuficiencia cardiaca congestiva, bradiarritmias y desequilibrios electrolíticos. Se debe tener precaución en pacientes con un diagnóstico o presunto síndrome de QT largo congénito o prolongación adquirida del intervalo QT, y en pacientes que reciben medicamentos concomitantes que se sabe que prolongan el intervalo QT, inducen anomalías de los electrólitos o se sabe que aumentan la exposición a tacrolimus (ver Interacciones medicamentosas y de otro género).
Trastornos linfoproliferativos y neoplasias malignas: Se ha reportado que los pacientes tratados con tacrolimus tienen un mayor riesgo de desarrollar trastornos linfoproliferativos asociados con el virus de Epstein-Barr (VEB) (ver Reacciones secundarias y adversas). Cuando se administra de forma concomitante una combinación de inmunosupresores como anticuerpos antilinfocitarios (por ejemplo basiliximab, daclizumab), aumenta el riesgo de trastornos linfoproliferativos asociados al VEB. Se ha reportado que los pacientes negativos al antígeno de la cápside viral (ACV) del VEB tienen un mayor riesgo de desarrollar trastornos linfoproliferativos. Por lo tanto, en este grupo de pacientes, se debe determinar la serología del ACV del VEB antes de iniciar el tratamiento con PROGRAF® XL. Durante el tratamiento, se recomienda el monitoreo cuidadoso con PCR del VEB. Un resultado positivo en la PCR del VEB puede persistir durante meses, y no es indicativo por sí mismo de enfermedad linfoproliferativa o linfoma.
Al igual que con otros agentes inmunosupresores, debido al posible riesgo de trastornos malignos en la piel, debe minimizarse la exposición a la luz solar y a la luz UV utilizando ropa protectora adecuada y protector solar con un factor de protección alto.
De la misma forma que con otros agentes inmunosupresores potentes, se desconoce el riesgo de cáncer secundario (ver Reacciones secundarias y adversas).
Neurotoxicidad:
Síndrome de encefalopatía posterior reversible (PRES): Se ha reportado el desarrollo de síndrome de encefalopatía posterior reversible (PRES, por sus siglas en inglés), en pacientes tratados con tacrolimus. Si los pacientes que están recibiendo tacrolimus presentan síntomas indicativos de PRES, como cefalea, alteración del estado mental, convulsiones o trastornos visuales, se debe llevar a cabo un procedimiento radiológico (por ejemplo, RMN). Si se diagnostica PRES, se aconseja el control adecuado de la presión arterial y de las convulsiones, y la suspensión inmediata de tacrolimus sistémico. La mayoría de los pacientes se recupera completamente después de que se han tomado las medidas adecuadas.
Infecciones oportunistas: Los pacientes tratados con inmunosupresores, incluido PROGRAF® XL, tienen un mayor riesgo de adquirir infecciones oportunistas (bacterianas, micóticas, virales y protozoarias). Entre estas afecciones se encuentran la nefropatía asociada con el virus BK y la leucoencefalopatía multifocal progresiva (LMP) asociada con el virus JC. Estas infecciones a menudo se relacionan con una carga inmunosupresora total alta, y pueden causar trastornos graves o potencialmente mortales, que los médicos deben considerar en el diagnóstico diferencial de pacientes inmunosuprimidos con deterioro de la función renal o síntomas neurológicos.
Aplasia eritrocitaria pura: Se han reportado casos de aplasia eritrocitaria pura (AEP) en pacientes tratados con tacrolimus. Todos los pacientes reportaron factores de riesgo para la AEP, como infección por parvovirus B19, enfermedad subyacente o medicamentos concomitantes asociados con la AEP.
Poblaciones especiales: La experiencia en pacientes no caucásicos y pacientes con un riesgo inmunológico alto (por ejemplo, retrasplante, indicios de anticuerpos reactivos contra el panel, ARP) es limitada.
En pacientes con insuficiencia hepática grave puede ser necesario reducir la dosis (ver Dosis yvía de administración).
No se recomienda su administración en menores de 18 años.
Excipientes: Las cápsulas de PROGRAF® XL contienen lactosa. Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa de los lapones o malabsorción de la glucosa-galactosa no deben recibir este medicamento.
La tinta de impresión utilizada en las cápsulas de PROGRAF® XL contiene lecitina de soya. En pacientes que son hipersensibles a los cacahuates o la soya, se debe ponderar el riesgo y la intensidad de la hipersensibilidad contra el beneficio de utilizar PROGRAF® XL.
Efectos sobre la habilidad de manejar y utilizar maquinaria: Tacrolimus puede causar trastornos visuales y neurológicos. Este efecto puede potenciarse si se administra junto con alcohol.
No se han realizado estudios de los efectos de tacrolimus (PROGRAF® XL) sobre la capacidad para conducir y utilizar máquinas.
DOSIS Y VÍA DE ADMINISTRACIÓN: La dosis inicial de PROGRAF® XL varía dependiendo del órgano trasplantado y de los otros agentes inmunosupresores.
En la siguiente tabla se muestra la dosis inicial y las típicas concentraciones valle en sangre completa; los detalles de la concentración en sangre se describen en Monitoreo de la concentración sanguínea.
Resumen de las recomendaciones para la dosis inicial y concentraciones típicas más bajas en sangre completa
|
Población de pacientes |
Dosis oral inicial recomendada diaria una vez al día |
Concentraciones típicas más bajas en sangre completa |
|
Pacientes adultos de trasplante de riñón |
0.15-0.2 mg/kg/día |
Mes 1-3: 7-16 ng/mL Mes 4-12: 5-15 ng/mL |
|
Pacientes adultos de trasplante hepático |
0.10-0.15 mg/kg/día |
Mes 1-2: 5-20 ng/mL |
|
Pacientes pediátricos de trasplante hepático |
0.15-0.20 mg/kg/día |
Mes 1-12: 5-20 ng/mL |
|
Pacientes adultos de trasplante de corazón |
0.075 mg/kg/día |
Mes 1-3: 10-20 ng/mL Mes ≥ 4: 5-15 ng/mL |
Para la conversión de los receptores estables de trasplante, se usa el mismo rango objetivo de baja concentración y de monitoreo de concentración en sangre que con PROGRAF®.
La dosis debe titularse para mantener el nivel de concentración más baja indicado antes; los detalles de la concentración en sangre se describen en Monitoreo de la concentración en sangre.
Trasplante renal: La dosis oral inicial recomendada del PROGRAF® XL es 0.15 a 0.2 mg/kg administrada una vez diaria en la mañana. La dosis inicial de PROGRAF® XL puede ser administrada dentro de 24 horas del trasplante, pero puede retrasarse hasta que se haya recuperado la función renal (por ejemplo, según lo indique una creatinina sérica > 4 mg/dL). La dosis debe titularse con base en las evaluaciones clínicas de rechazo y tolerancia, y para mantener las concentraciones valle como se indicó anteriormente.
A los pacientes que se convierten de PROGRAF® a PROGRAF® XL se les debe administrar una sola dosis diaria en la mañana de PROGRAF® XL, equivalente a la dosis diaria total estable anterior de los pacientes de PROGRAF®. Las dosis subsiguientes de PROGRAF® XL deben ajustarse con el fin de mantener las concentraciones valle similares a las de antes de la conversión.
Los datos de la administración de PROGRAF® XL en los pacientes de trasplante de riñón indican que los pacientes de raza negra requirieron una dosis mayor para alcanzar concentraciones valle comparables a las de los pacientes de raza blanca.
|
Tiempo después del trasplante |
Raza blanca n = 160 |
Raza negra n = 41 |
||
|
Dosis (mg/kg) |
Concentración valle media (ng/mL) |
Dosis (mg/kg) |
Concentración valle media (ng/mL) |
|
|
Día 7 |
0.14 |
10.79 |
0.14 |
7.85 |
|
Mes 1 |
0.14 |
11.11 |
0.18 |
10.83 |
|
Mes 6 |
0.10 |
7.96 |
0.13 |
8.50 |
|
Mes 12 |
0.09 |
7.54 |
0.12 |
7.52 |
Trasplante de hígado: La dosis inicial de PROGRAF® XL debe administrarse no antes de 6 horas después del trasplante. La dosis inicial recomendada de PROGRAF® XL es 0.10-0.15 mg/kg/día, administrada una vez al día en la mañana. La dosis debe titularse con base en las evaluaciones clínicas de rechazo y tolerancia, y para mantener las concentraciones valle como se indicó antes. Las dosis más bajas de PROGRAF® XL pueden ser suficientes como terapia de mantenimiento. Se recomienda la terapia de apoyo con corticosteroides adrenales al principio del postrasplante.
A los pacientes que se convierten de PROGRAF® a PROGRAF® XL se les debe administrar una sola dosis diaria en la mañana de PROGRAF® XL, equivalente a la dosis diaria total estable anterior de los pacientes de PROGRAF®. Las dosis subsiguientes de PROGRAF® XL deben ajustarse con el fin de mantener las concentraciones valle similares a las de antes de la conversión.
Pacientes pediátricos: Los receptores pediátricos de trasplante de hígado sin insuficiencia renal o hepática preexistente han requerido o tolerado dosis más elevadas de PROGRAF® que los adultos para alcanzar concentraciones similares en sangre. Por lo tanto, se recomienda que la terapia sea iniciada en pacientes pediátricos en una dosis inicial de 0.15-0.20 mg/kg/día en la mañana. Pueden requerirse ajustes de la dosis. La experiencia en receptores pediátricos de trasplante está más limitada que en los adultos.
A los pacientes que se convierten de PROGRAF® a PROGRAF® XL se les debe administrar una sola dosis diaria en la mañana de PROGRAF® XL, equivalente a la dosis diaria total estable anterior de los pacientes de PROGRAF®. Las dosis subsiguientes de PROGRAF® XL deben ajustarse con el fin de mantener las concentraciones valle similares a las de antes de la conversión.
Pacientes con daño hepático o renal: Debido a la depuración reducida y a la vida media prolongada, los pacientes con daño hepático severo (Child-Pugh > 10) pueden requerir dosis menores de PROGRAF® XL. Se justifica un monitoreo cercano de las concentraciones en sangre. Debido al potencial de nefrotoxicidad, los pacientes con daño renal o hepático deben recibir dosis en el valor más bajo del rango de dosis oral recomendada. Pueden requerirse reducciones adicionales de la dosis por debajo de estos rangos. La terapia con PROGRAF® XL generalmente debe retrasarse hasta 48 horas o más en pacientes con oliguria posoperatoria.
Conversión de un régimen inmunosupresor a otro: PROGRAF® XL no debe usarse simultáneamente con ciclosporina. Debe suspenderse PROGRAF® XL o la ciclosporina al menos 24 horas antes de iniciar el otro. En presencia de concentraciones elevadas de PROGRAF® o de ciclosporina, la administración con el otro fármaco debe retardarse más.
Los receptores de trasplante de riñón e hígado pueden convertirse de PROGRAF® a PROGRAF® XL una vez al día sobre una base de dosis diaria total 1:1 (mg:mg), para alcanzar las concentraciones apropiadas de tacrolimus en sangre.
Monitoreo de la concentración sanguínea: El monitoreo de las concentraciones sanguíneas de tacrolimus junto con otros parámetros clínicos y de laboratorio se considera una ayuda esencial para el manejo del paciente para la evaluación del rechazo, toxicidad, ajuste de dosis y cumplimiento. Los factores que influyen en la frecuencia del monitoreo incluyen, aunque no están limitados a la insuficiencia hepática o renal, la adición o suspensión de fármacos que potencialmente puedan interactuar y al tiempo postrasplante. El monitoreo de la concentración sanguínea no es un reemplazo para el monitoreo de la función del órgano y las biopsias de tejido.
Los métodos comúnmente usados para el ensayo de tacrolimus incluyen cromatografía de líquidos de alta resolución con detección acoplada de espectrometría de masas (CLAR/MS/MS), inmunoensayo enzimático (EIA), inmunoensayo enzimático de micropartículas (MEIA), y ensayo enzimático inmunoabsorbente (ELISA). La comparación de las concentraciones en la literatura publicada para las concentraciones de los pacientes, que usan los ensayos actuales, deben hacerse con un conocimiento detallado de los métodos de ensayo y de las matrices biológicas empleadas. La sangre completa es la matriz de elección y las muestras deben colectarse en tubos que contengan el anticoagulante ácido etilendiamin-tetraacético (EDTA). No se recomienda la anticoagulación con heparina debido a la tendencia a formar coágulos con el almacenamiento. Las muestras que no se analicen inmediatamente deben almacenarse a temperatura ambiente o en refrigeración y analizarse en un lapso de 7 días; si las muestras se van a mantener más tiempo, deben congelarse a -20 °C hasta por 12 meses.
Los datos de receptores de riñón e hígado que reciben tacrolimus administrado como PROGRAF® indican que las concentraciones valle de tacrolimus en sangre completa, medidas por IMx®† MEIA (riñón) y ELISA (hígado), fueron más variables durante la primera semana de administración, y el riesgo relativo de toxicidad se incrementa con las concentraciones valle más elevadas. Por lo tanto, se recomienda el monitoreo de las concentraciones valle en sangre completa para ayudar en la evaluación clínica de la toxicidad. Para receptores estables de trasplante convertidos de PROGRAF® a PROGRAF® XL, puede usarse el mismo tipo de monitoreo terapéutico.
El riesgo relativo de toxicidad se incrementa con las concentraciones valle más elevadas de tacrolimus. Por lo tanto, se recomienda el monitoreo de las concentraciones valle en sangre completa para ayudar en la evaluación clínica de la toxicidad. Los pacientes con postrasplante a largo plazo con frecuencia se mantienen en el extremo bajo del rango objetivo recomendado.
El Monitoreo terapéutico del fármaco, 1995, volumen 17, número 6, contiene un documento de acuerdo y varios artículos con respecto al monitoreo terapéutico de tacrolimus desde la Conferencia Internacional de Consenso sobre Fármacos Inmunosupresores de 1995. Referirse a estos manuscritos para discusiones adicionales del monitoreo de tacrolimus.
† IMx es una marca registrada de Abbott Laboratories, Inc.
Trasplante de riñon: Los datos del estudio fase 3 de PROGRAF® XL indican que las concentraciones valle de tacrolimus en sangre completa fueron más variables durante la primera semana de administración.
En el mes 2, 76% de los pacientes tuvo concentraciones valle entre 7-16 ng/mL, y más de 78% mantuvo concentraciones entre 5-15 ng/mL, desde el mes 4 hasta 1 año.
Trasplante de hígado: Los datos de un estudio fase 2 con PROGRAF® XL en receptores de novo de trasplante hepático indican que las concentraciones valle de tacrolimus en sangre completa fueron más variables durante la primera semana postrasplante. Las concentraciones valle medias de este estudio fueron 12.1 ng/mL para los meses 1-3 y 8.8 ng/mL para los meses 10-12 postrasplante.
Vía de administración: Oral.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: La experiencia con la sobredosificación es ilimitada. Se han reportado varios casos de sobredosificación accidental con tacrolimus; los síntomas han incluido temblor, cefalea, náuseas y vómito, infecciones, urticaria, letargo, y aumento del nitrógeno ureico en sangre, las concentraciones de creatinina sérica y los niveles de alanina aminotransferasa.
No existe un antídoto específico para la terapia con tacrolimus. Si ocurre una sobredosificación, se deben aplicar medidas de soporte generales y tratamiento sintomático.
En función de su alto peso molecular, limitada solubilidad en agua y amplia unión a las proteínas plasmáticas y a los eritrocitos, se prevé que tacrolimus no será dializable. En pacientes aislados con concentraciones plasmáticas muy altas, la hemofiltración o diafiltración han sido eficaces para disminuir las concentraciones tóxicas. En casos de intoxicación oral, el lavado gástrico y/o el uso de adsorbentes (por ejemplo, carbón activado) podrían ser útiles si se utilizan poco después de la ingesta.
PRESENTACIONES: Caja de cartón con 30 o 50 cápsulas de liberación prolongada de 0.5, 1 o 5 mg e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 30 °C y en lugar seco.
Consérvese la caja bien cerrada.
PROGRAF® XL Cápsulas debe usarse dentro de los 12 meses siguientes después de abrir el sobre de aluminio, sin exceder la fecha de caducidad impresa en la caja.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. No se deje al alcance de los niños. Su venta requiere receta médica. No se use durante el embarazo o la lactancia. Sólo pueden prescribir PROGRAF® XL médicos expertos en terapia de inmunosupresión y en manejo de pacientes con trasplante de órganos. Este medicamento contiene lactosa. Este medicamento puede producir somnolencia y afectar el estado de alerta, por lo que no deberá conducir vehículos automotores ni maquinaria pesada durante su uso.
Reporte las sospechas de reacciones adversas al correo:
farmacovigilancia@cofepris.gob.mx y
farmacovigilancia@asofarma.com.mx
Hecho en Irlanda por
Astellas lreland Co. Ltd.
Killorglin, Co., Kerry, Irlanda.
Representante legal e importador:
ASOFARMA DE MÉXICO, S.A. de C.V.
Calz. México-Xochimilco No. 43,
Col. San Lorenzo Huipulco,
C.P. 14370, Tlalpan, Ciudad de México, México.
Distribuido por:
New Transport Applications, S.A. de C.V.
Avenida Michoacán No. 20,
bodega 24-A y 24-B, Col. Renovación,
C.P. 09209 lztapalapa, Ciudad de México, México.
Reg. Núm. 055M2016 SSA IV
PROGRAF® es una marca comercial registrada de Astellas Pharma lnc. Todas las demás marcas comerciales o marcas comerciales registradas son propiedad de sus respectivos dueños.
248715-PXL-MEX


