RADIESSE
CALCIO
Implante inyectable
1 Implante inyectable
INDICACIONES DE USO: El implante inyectable RADIESSE® está indicado para el aumento de la mano para corregir la pérdida de volumen en el dorso de las manos.
El implante inyectable RADIESSE® está indicado para implantación sub-dérmica, extradérmica para la corrección de arrugas y pliegues faciales de moderados a severos, como los pliegues naso-labiales. También está indicado para la restauración y/o corrección de los signos de la pérdida de grasa facial (lipoatrofia) en las personas con el virus de inmunodeficiencia humana.
Nota: Estas instrucciones de uso son específicas para el tratamiento del dorso de la mano con RADIESSE®. Por favor, consulte las instrucciones de uso alternativas para el tratamiento de los pliegues nasolabiales y lipoatrofia-VIH con RADIESSE®.
CONTRAINDICACIONES:
• Está contraindicado para pacientes con alergias graves que se manifiestan por antecedentes de anafilaxia o antecedentes o presencia de múltiples alergias severas.
• No debe utilizarse en pacientes con hipersensibilidad conocida a cualquiera de los componentes.
• RADIESSE® implante inyectable está contraindicado para los pacientes con trastornos hemorrágicos.
Cuidados:
• La introducción de un producto en el sistema vascular puede conducir a la embolización, la oclusión de los vasos sanguíneos, isquemia o infarto. Tenga mucho cuidado cuando esté inyectando rellenos de tejidos blandos, por ejemplo inyectar el producto lentamente y aplicar la menor cantidad de presión necesaria. Se han reportado eventos adversos raros, pero graves asociados con la inyección intravascular de rellenos de tejidos blandos en la cara e incluyen la pérdida temporal o permanente de visión, ceguera, isquemia cerebral o hemorragia cerebral, lo que lleva a un accidente cerebrovascular, necrosis de la piel y daños a las estructuras faciales subyacentes. Detener inmediatamente la inyección si un paciente presenta alguno de los siguientes síntomas, como cambios en la visión, síntomas de un derrame cerebral, palidez de la piel o dolor inusual durante o poco después del procedimiento. Los pacientes deben recibir atención médica inmediata y, posiblemente, la evaluación por un profesional de la salud especialista competente si llegará a producirse una inyección intravascular.
• El uso del implante inyectable RADIESSE® en cualquier persona con inflamación activa de la piel o infección en o cerca de la zona de tratamiento debe aplazarse hasta que el proceso inflamatorio o infeccioso haya sido controlado.
• No sobrecorregir (sobrellenado) una deficiencia de contorno porque la depresión debe mejorar gradualmente dentro de varias semanas, mientras se produce el efecto del tratamiento con el implante inyectable RADIESSE®. Consulte la sección de individualización del tratamiento para obtener más detalle.
• Debe tenerse especial cuidado para evitar la inyección en las venas y tendones de la mano. La inyección en los tendones puede debilitar los tendones y provocar la rotura del tendón. La inyección en las venas puede causar embolización o trombosis.
• La inyección en la mano puede causar efectos adversos que duran más de 14 días. Consulte la sección de eventos adversos para los detalles.
• La inyección en el dorso de la mano puede resultar en dificultad temporal para la realización de las actividades cotidianas (48% de los pacientes del estudio informó de este evento adverso). Los tipos de piel Fitzpatrick IV-VI pueden tener un mayor riesgo en la dificultad para la realización de sus actividades (68% de los pacientes con tipos de piel Fitzpatrick IV-VI reportaron este evento).
• RADIESSE® puede causar nódulos, protuberancias o bultos en el dorso de la mano (12% informó de este evento) y pueden durar hasta 1 año.
• La inyección en los pacientes con pérdida muy grave de tejido graso con marcada visibilidad de venas y tendones no se ha estudiado. La seguridad y eficacia en esta población de pacientes no ha sido establecida.
• No se han estudiado volúmenes mayores a 3 cc de RADIESSE® por mano en una sesión de tratamiento. El aumento de los moretones se asocia con un mayor volumen de inyección. Re-tratamiento con RADIESSE® con volúmenes superiores a aproximadamente 1.6 cc por mano en una sesión de tratamiento puede dar lugar a un aumento de los eventos adversos (enrojecimiento, dolor, hinchazón y dificultad para realizar actividades).
PRECAUCIONES:
• Con el fin de minimizar los riesgos de complicaciones potenciales, este producto sólo debe ser utilizado únicamente por profesionales de la salud que tengan una adecuada formación, experiencia y que conozcan la anatomía en y alrededor del sitio de la inyección.
• Con el fin de minimizar los riesgos de complicaciones potenciales, los profesionales de la salud deben familiarizarse plenamente ellos mismos con el producto, los materiales educativos del producto y todo el con el instructivo completo.
• Las partículas de hidroxiapatita de calcio (CaHA) del implante inyectable RADIESSE® son radioopacas y son claramente visibles en la tomografía computarizada y pueden ser visibles en la radiografía simple estándar. En un estudio radiográfico de 58 caras, no hubo ninguna indicación de que el implante inyectable RADIESSE® posiblemente haya enmascarado tejidos anormales o fuera interpretado como tumores en la tomografía computarizada. Los pacientes deben ser informados de la naturaleza radioopaca del implante inyectable RADIESSE®, para que puedan informar a sus profesionales de la salud de atención primaria, así como a los radiólogos. No se han realizado estudios de imagen en la mano. Actualmente se desconoce si RADIESSE® podría enmascarar una lesión en la mano en los estudios de imagen.
• Se invita a los profesionales de la salud a discutir todos los riesgos potenciales de la inyección de tejido blando con sus pacientes antes del tratamiento y asegurarse de que los pacientes estén conscientes de los signos y síntomas de las complicaciones potenciales.
• Como con todos los procedimientos transcutáneos, la inyección del implante inyectable RADIESSE® conlleva un riesgo de infección. La infección puede requerir el intento de extirpación quirúrgica de RADIESSE®. Las precauciones estándar asociados con materiales inyectables deben ser seguidas.
• El uso de RADIESSE® en el dorso de la mano en pacientes con enfermedades, lesiones o discapacidades de la mano no ha sido estudiado. Se debe tener cuidado en el tratamiento de pacientes con enfermedades autoinmunes que afectan a la mano, implantes de mano, contractura de Dupuytren, antecedentes de tumor en la mano, malformaciones vasculares, enfermedad de Raynaud y en los pacientes en riesgo de ruptura del tendón.
• El uso de RADIESSE® en el dorso de la mano puede resultar en una hinchazón significativa del dorso de la mano. Los pacientes deben ser instruidos para quitarse las joyas (anillos) antes del tratamiento y hasta que la hinchazón haya desaparecido para evitar comprometer la circulación del dedo.
• Los efectos de la inyección de RADIESSE® en el funcionamiento de la mano son inciertos.
• Los pacientes que utilizan medicamentos que pueden prolongar el sangrado, como aspirina o warfarina pueden, como con cualquier inyección, aumentar la aparición de moretones o sangrado en el lugar de la inyección.
• Si un tratamiento con láser, peeling químico o cualquier otro procedimiento basado en la respuesta dérmica activa está considerado después del tratamiento con el implante inyectable RADIESSE®, hay un posible riesgo de provocar una reacción inflamatoria en el sitio del implante. Esto también aplica si el implante inyectable RADIESSE® es administrado antes de que la piel se haya recuperado por completo después de un procedimiento de este tipo.
• La seguridad del implante inyectable RADIESSE® por más de 3 años en la cara y 1 año en la mano no se ha investigado en ensayos clínicos.
• La seguridad del implante inyectable RADIESSE® para su uso durante el embarazo y en mujeres en período de lactancia no se ha establecido.
• La seguridad de RADIESSE® inyectado en el dorso de la mano en pacientes menores de 26 años y más de 79 años de edad no ha sido estudiada.
• La seguridad de RADIESSE® en pacientes con una susceptibilidad incrementada a la formación de queloides y cicatrices hipertróficas no ha sido estudiada.
• La seguridad del implante inyectable RADIESSE® con terapias dérmicas concomitantes como la depilación, la irradiación UV o láser, procedimientos de peeling mecánicos o químicos no ha sido evaluada en ensayos clínicos controlados.
• La inyección del implante inyectable RADIESSE® en pacientes con antecedentes de erupción herpética anterior puede estar asociada con la reactivación del herpes.
• No se han realizado estudios de interacciones del implante inyectable RADIESSE® con fármacos u otras sustancias o implantes.
• El paciente debe ser informado de que él o ella debe minimizar la actividad extenuante y la exposición prolongada de la zona tratada al sol o la exposición al calor durante aproximadamente 24 horas después del tratamiento y hasta que la hinchazón y el enrojecimiento inicial hayan desaparecido.
• Las precauciones universales deben ser observadas cuando existe un riesgo de contacto con los fluidos corporales de los pacientes. La sesión de inyección debe llevarse a cabo con una técnica aséptica.
• El implante inyectable RADIESSE® se envasa para ser usado por un solo paciente. No vuelva a esterilizar. No utilizar si el envase está abierto o dañado. No utilizar si la tapa del extremo de la jeringa o el émbolo de la jeringa no están en su lugar.
• Para ayudar a evitar la rotura de la aguja, no intente enderezar una aguja doblada. Deséchela y complete el procedimiento con una aguja de reemplazo.
• No vuelva a tapar las agujas usadas. Volver a tapar con la mano es una práctica peligrosa y debe ser evitada.
• Después de su uso, las jeringas y agujas utilizadas para el tratamiento pueden ser peligros biológicos potenciales. Por lo que se deben manejar en consecuencia y desechar de acuerdo con la práctica médica aceptada y los requerimientos locales, estatales y federales aplicables.
ENSAYO CLÍNICO DE PRE-COMERCIALIZACIÓN PARA EL AUMENTO DE LA MANO:
A. Eventos adversos:
La información aquí proporcionada contiene los eventos adversos para los 113 sujetos que completaron un estudio aleatorizado, enmascarado, controlado en seis centros de investigación de Estados Unidos de América (EUA). Un total de 78 sujetos fueron vueltos a tratar después de 6 meses posteriores al tratamiento inicial. Los eventos adversos fueron registrados en los diarios de los sujetos (30 días después del tratamiento), así como por las evaluaciones médicas.
RADIESSE® fue mezclado con lidocaína HCl y luego fue inyectado como pequeños bolos de hasta 0.5 cc en el dorso de la mano. A continuación, RADIESSE®/lidocaína fue masajeado en la mano hasta que se logró el efecto estético deseado.
Las tablas 1 y 2 resumen los eventos adversos reportados por todos los sujetos y los médicos, respectivamente, durante un período de 12 meses. Los eventos adversos se presentan por su grado de severidad (leve, moderada o grave).
Tabla 1. Eventos adversos reportados por los sujetos durante un periodo de 12 meses (n = 113 sujetos)
|
Tipo de evento adverso |
# de sujetos con evento (% total) |
Severidad máxima (N, % con evento) |
||
|
Leve |
Moderado |
Severo |
||
|
Moretones |
82 (72.6%) |
48 (58.5%) |
29 (35.4%) |
5 (6.1%) |
|
Hinchazón |
112 (99.1%) |
22 (19.6%) |
74 (66.1%) |
16 (14.3%) |
|
Enrojecimiento |
92 (81.4%) |
40 (43.5%) |
48 (52.2%) |
4 (4.3%) |
|
Comezón |
52 (46.0%) |
35 (67.3%) |
17 (32.7%) |
0 (0.0%) |
|
Dolor |
104 (92.0%) |
46 (44.2%) |
51 (49.0%) |
7 (6.7%) |
|
Hematoma |
1 (0.9%) |
1 (100.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, depresiones/protuberancias |
7 (6.2%) |
2 (28.6%) |
5 (71.4%) |
0 (0.0%) |
|
Dificultad para realizar actividades |
54 (47.8%) |
30 (55.6%) |
21 (38.9%) |
3 (5.6%) |
|
Pérdida de sensibilidad |
17 (15.0%) |
10 (58.8%) |
7 (41.2%) |
0 (0.0%) |
|
Otro |
10 (8.8%) |
4 (40.0%) |
5 (50.0%) |
1 (10.0%) |
|
Total |
113 (100.0%) |
14 (12.4%) |
78 (69.0%) |
21 (18.6%) |
* Otros eventos adversos reportados que estuvieron relacionados con el dispositivo incluyen respuesta vagal, sequedad de la piel, hipersensibilidad y pinchazos con la aguja.
Tabla 2. Eventos adversos reportados por el médico durante un periodo de 12 meses (n = 113 sujetos)
|
Tipo de evento adverso |
# de sujetos con evento (% Total) |
Severidad máxima (N, % con evento) |
||
|
Leve |
Moderado |
Severo |
||
|
Moretones |
21 (18.6%) |
13 (61.9%) |
6 (28.6%) |
2 (9.5%) |
|
Hinchazón |
23 (20.4%) |
7 (30.4%) |
14 (60.9%) |
2 (8.7%) |
|
Enrojecimiento |
9 (8.0%) |
5 (55.6%) |
4 (44.4%) |
0 (0.0%) |
|
Comezón |
4 (3.5%) |
3 (75.0%) |
1 (25.0%) |
0 (0.0%) |
|
Dolor |
7 (6.2%) |
4 (57.1%) |
2 (28.6%) |
1 (14.3%) |
|
Hematoma |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, irregularidades/ |
7 (6.2%) |
7 (100.0%) |
0 (0.0%) |
0 (0.0%) |
|
Dificultad para realizar actividades |
2 (1.8%) |
2 (100.0%) |
0 (0.0%) |
0 (0.0%) |
|
Pérdida de sensibilidad |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Otro |
13 (11.5%) |
7 (53.8%) |
5 (38.5%) |
1 (7.7%) |
|
Total |
50 (44.2%) |
24 (48.0%) |
21 (42.0%) |
5 (10.0%) |
* Otros eventos adversos reportados que estuvieron relacionados con el dispositivo incluyen respuesta vagal, sequedad de la piel, hipersensibilidad y pinchazos con la aguja.
La tabla 3 muestra la duración de los eventos adversos, reportados por los sujetos y/o médicos del estudio. Un total de 24 de los 113 sujetos (21%) experimentaron acontecimientos adversos descritos como “severos”. Todos los eventos se resolvieron sin secuelas.
Tabla 3. Duración de los eventos adversos severos durante un periodo de 12 meses
|
Tipo de evento adverso |
# de sujetos |
Duración promedio (días) |
Mediana duración (días) |
Rango de días |
Duración de reportados como “severo” en el diario (días) |
|
Hinchazón |
18 |
17.5 |
12 |
3-57 |
1-8 |
|
Moretones |
7 |
19.9 |
10.5 |
5-67 |
1-4 |
|
Dolor |
7 |
33.1 |
21.5 |
8-99 |
1-7 |
|
Dificultad para realizar actividades |
3 |
41.8 |
15 |
3-97 |
1-11 |
|
Enrojecimiento |
4 |
18.5 |
14.5 |
3-37 |
1-2 |
Eventos adversos con una duración mayor a 14 días:
Eventos reportados por los sujetos y/o médicos con una duración mayor a 14 días están enlistados abajo. Los porcentajes son el número de sujetos que experimentaron un evento adverso por más de 14 días de los 113 sujetos que fueron tratados en el estudio. Todos los eventos se resolvieron sin secuelas.
• 29% hinchazón.
• 25% dolor.
• 7% nódulos/irregularidades/protuberancia.
• 6% dificultad para realizar actividades.
• 6% enrojecimiento.
• 3% moretones.
• 1% hematomas.
Eventos adversos después del tratamiento inicial:
Las tablas 4 y 5 presentan los eventos adversos y la severidad máxima de aquellos eventos posteriores a los 6 meses después del tratamiento inicial, según lo reportado por los sujetos y por los médicos, respectivamente.
Tabla 4. Sujetos que experimentaron eventos adversos, durante los primeros seis meses del tratamiento inicial reportados en los diarios de los sujetos
n = 113 sujetos
|
Tipo de evento adverso |
# sujetos con evento |
Severidad máxima |
|||
|
N |
95% CI |
Leve |
Moderado |
Severo |
|
|
Moretones |
73 (64.6%) |
(55.0-73.4) |
48 (65.8%) |
22 (30.1%) |
3 (4.1%) |
|
Hinchazón |
110 (97.3%) |
(92.4-99.4) |
28 (25.5%) |
69 (62.7%) |
13 (11.8%) |
|
Enrojecimiento |
88 (77.9%) |
(69.1-85.1) |
46 (52.3%) |
39 (44.3%) |
3 (3.4%) |
|
Comezón |
49 (43.4%) |
(34.1-53.0) |
36 (73.5%) |
13 (26.5%) |
0 (0.0%) |
|
Dolor |
98 (86.7%) |
(79.1-92.4) |
48 (49.0%) |
45 (45.9%) |
5 (5.1%) |
|
Hematoma |
0 (0.0%) |
- |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, irregularidades/protuberancias |
4 (3.5%) |
(1.0-8.8) |
1 (25.0%) |
3 (75.0%) |
0 (0.0%) |
|
Dificultad para realizar actividades |
45 (39.8%) |
(30.7-49.5) |
26 (57.8%) |
17 (37.8%) |
2 (4.4%) |
|
Pérdida de sensibilidad |
11 (9.7%) |
(5.0-16.8) |
7 (63.6%) |
4 (36.4%) |
0 (0.0%) |
|
Otro |
9 (8.0%) |
(3.7-14.6) |
4 (44.4%) |
5 (55.6%) |
0 (0.0%) |
|
Total |
112 (99.1%) |
(95.2-100.0) |
21 (18.8%) |
75 (67.0%) |
16 (14.3%) |
Tabla 5. Sujetos que experimentaron eventos adversos, durante los primeros seis meses del tratamiento inicial reportados por la evaluación del médico
n = 113 sujetos
|
Tipo de evento adverso |
# sujetos con evento |
Severidad máxima |
|||
|
N |
95% CI |
N |
95% CI |
N |
|
|
Moretones |
20 (17.7%) |
(11.2-26.0) |
14 (70.0%) |
4 (20.0%) |
2 (10.0%) |
|
Hinchazón |
23 (20.4%) |
(13.4-29.0) |
7 (30.4%) |
14 (60.9%) |
2 (8.7%) |
|
Enrojecimiento |
9 (8.0%) |
(3.7-14.6) |
5 (55.6%) |
4 (44.4%) |
0 (0.0%) |
|
Comezón |
4 (3.5%) |
(1.0-8.8) |
3 (75.0%) |
1 (25.0%) |
0 (0.0%) |
|
Dolor |
7 (6.2%) |
(2.5-12.3) |
4 (57.1%) |
2 (28.6%) |
1 (14.3%) |
|
Hematoma |
0 (0.0%) |
- |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, irregularidades/ |
2 (1.8%) |
(0.2-6.2) |
2 (100.0%) |
0 (0.0%) |
0 (0.0%) |
|
Dificultad para realizar actividades |
2 (1.8%) |
(0.2-6.2) |
2 (100.0%) |
0 (0.0%) |
0 (0.0%) |
|
Pérdida de sensibilidad |
0 (0.0%) |
- |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Otro |
10 (8.8%) |
(4.3-15.7) |
6 (60.0%) |
3 (30%) |
1 (10%) |
|
Total |
44 (38.9%) |
(29.9-48.6) |
20 (45.5%) |
19 (43.2%) |
5 (11.4%) |
Las tablas 6 y 7 representan la aparición de eventos adversos después del tratamiento inicial, según lo reportado por los sujetos y los médicos, respectivamente.
Tabla 6. Aparición de eventos adversos reportados por los sujetos* después del tratamiento inicial (n = 914 eventos)
|
Tipo de evento adverso |
Toda primera aparición (N, % total) |
Eventos adversos reportados (n, % con evento) |
||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 y posteriores |
Semana 1 y 2 combinadas |
||
|
Moretones |
133 (14.6%) |
124 (93.2%) |
5 (3.8%) |
3 (2.3%) |
1 (0.8%) |
129 (97.0%) |
|
Hinchazón |
218 (23.9%) |
218 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
218 (100.0%) |
|
Enrojecimiento |
166 (18.2%) |
163 (98.2%) |
3 (1.8%) |
0 (0.0%) |
0 (0.0%) |
166 (100.0%) |
|
Comezón |
192 (21.0%) |
180 (93.8%) |
4 (2.1%) |
6 (3.1%) |
2 (1.0%) |
184 (95.8%) |
|
Dolor |
83 (9.1%) |
60 (72.3%) |
16 (19.3%) |
6 (7.2%) |
1 (1.2%) |
76 (91.6%) |
|
Hematoma |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, irregularidades/ |
7 (0.8%) |
2 (28.6%) |
4 (57.1%) |
0 (0.0%) |
1 (14.3%) |
6 (85.7%) |
|
Dificultad para realizar actividades |
82 (9.0%) |
71 (86.6%) |
7 (8.5%) |
4 (4.9%) |
0 (0.0%) |
78 (95.1%) |
|
Pérdida de sensibilidad |
16 (1.8%) |
8 (50.0%) |
5 (31.3%) |
3 (18.8%) |
0 (0.0%) |
13 (81.3%) |
|
Otro |
17 (1.9%) |
13 (76.5%) |
4 (23.5%) |
0 (0.0%) |
0 (0.0%) |
17 (100.0%) |
|
Total |
914 (100.0%) |
839 (91.8%) |
48 (5.3%) |
22 (2.4%) |
5 (0.5%) |
887 (97.0%) |
* Los sujetos registraron entradas en sus diarios para el periodo de 30 días después del tratamiento. Si un evento todavía estaba en curso en el momento de la recolección del diario a los 30 días, la fecha de la resolución fue registrada e informada por teléfono o en la próxima visita del estudio.
Tabla 7. Número total de aparición de eventos adversos después del tratamiento inicial reportados por el médico (n = 117 eventos)
|
Tipo de evento adverso |
Toda primera aparición (n, % total) |
Eventos adversos reportados (n, % con evento) |
||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 y posteriores |
Semana 1 Y 2 combinadas |
||
|
Moretones |
26 (22.2%) |
23 (88.5%) |
0 (0.0%) |
0 (0.0%) |
3 (11.5%) |
23 (88.5%) |
|
Hinchazón |
39 (33.3%) |
28 (71.8%) |
10 (25.6%) |
1 (2.6%) |
0 (0.0%) |
38 (97.4%) |
|
Enrojecimiento |
15 (12.8%) |
14 (93.3%) |
1 (6.7%) |
0 (0.0%) |
0 (0.0%) |
15 (100.0%) |
|
Comezón |
11 (9.4%) |
5 (45.5%) |
2 (18.2%) |
1 (9.1%) |
3 (27.3%) |
7 (63.6%) |
|
Dolor |
7 (6.0%) |
5 (71.4%) |
0 (0.0%) |
0 (0.0%) |
2 (28.6%) |
5 (71.4%) |
|
Hematoma |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, Irregularidades/Protuberancias |
3 (2.6%) |
2 (66.7%) |
0 (0.0%) |
0 (0.0%) |
1 (33.3%) |
2 (66.7%) |
|
Dificultad para realizar actividades |
4 (3.4%) |
2 (50.0%) |
2 (50.0%) |
0 (0.0%) |
0 (0.0%) |
4 (100.0%) |
|
Pérdida de sensibilidad |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Otro |
12 (10.3%) |
4 (33.3%) |
0 (0.0%) |
0 (0.0%) |
8 (66.7%) |
4 (33.3%) |
|
Total |
117 (100.0%) |
83 (70.9%) |
15 (12.8%) |
2 (1.7%) |
17 (14.5%) |
98 (83.8%) |
Eventos adversos recurrentes:
Un evento adverso fue considerado un evento adverso recurrente, si un evento adverso del mismo tipo fue reportado de nuevo después de más de 3 días. Un total de 58% de los sujetos (66 de 113) tuvieron un evento adverso recurrente después del tratamiento inicial. La tabla 8 proporciona el número de eventos adversos recurrentes reportados por los sujetos después del tratamiento inicial. Los médicos informaron como evento adverso recurrente la inflamación que se presentó a los 14 a 19 días (2 eventos) después del tratamiento inicial y a los 60 o más días (1 evento).
Tabla 8. Número total de EAs recurrentes después del tratamiento inicial reportados en los diarios de los sujetos* (n = 239 eventos)
|
Tipo de evento adverso |
Menos de 14 días |
14-19 días |
20-29 días |
30-59 días |
Total eventos adversos por tipo de evento |
|
Moretones |
4 (28.6%) |
4 (28.6%) |
6 (42.9%) |
0 (0.0%) |
14 (5.9%) |
|
Hinchazón |
44 (64.7%) |
17 (25.0%) |
6 (8.8%) |
1 (1.5%) |
68 (28.5%) |
|
Enrojecimiento |
16 (40.0%) |
10 (25.0%) |
11 (27.5%) |
3 (7.5%) |
40 (16.7%) |
|
Comezón |
43 (65.2%) |
6 (9.1%) |
14 (21.2%) |
3 (4.5%) |
66 (27.6%) |
|
Dolor |
17 (54.8%) |
7 (22.6%) |
7 (22.6%) |
0 (0.0%) |
31 (13.0%) |
|
Hematoma |
1 (33.3%) |
1 (33.3%) |
1 (33.3%) |
0 (0.0%) |
3 (1.3%) |
|
Nódulo, irregularidades/protuberancias |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Dificultad para realizar actividades |
11 (68.8%) |
1 (6.3%) |
3 (18.8%) |
1 (6.3) |
16 (6.7%) |
|
Pérdida de sensibilidad |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Otro |
1 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
1 (0.4%) |
|
Total |
137 (57.3%) |
46 (19.2%) |
48 (20.1%) |
8 (3.3%) |
239 (100.0%) |
* Los sujetos registraron entradas en sus diarios para el periodo de 30 días después del tratamiento. Si un evento todavía estaba en curso en el momento de la recolección del diario a los 30 días, la fecha de la resolución fue registrada e informada por teléfono o en la próxima visita del estudio.
Eventos adversos reportados después del retratamiento:
Las tablas 9 y 10 presentan los eventos adversos y el grado de severidad de aquellos eventos posteriores al tratamiento inicial y los posteriores al retratamiento, según lo reportado por los sujetos y los médicos, respectivamente.
Tabla 9. Eventos adversos reportados por los sujetos* posteriores al tratamiento inicial vs. retratamiento reportados en los diarios de los sujetos* (n = 78 sujetos retratados)
|
Tipo de evento adverso |
# Sujetos |
|||||||
|
Posterior al tratamiento inicial |
Posterior al retratamiento |
|||||||
|
N (%) |
Severidad máxima |
N (%) |
Severidad máxima |
|||||
|
Leve |
Moderado |
Severo |
Leve |
Moderado |
Severo |
|||
|
Moretones |
52 (66.7%) |
34 (65.4%) |
16 (30.8%) |
2 (3.8%) |
45 (57.7%) |
27 (60.0%) |
16 (35.6%) |
2 (4.4%) |
|
Hinchazón |
75 (96.2%) |
23 (30.7%) |
44 (58.7%) |
8 (10.7%) |
68 (87.2%) |
31 (45.6%) |
33 (48.5%) |
4 (5.9%) |
|
Enrojecimiento |
60 (76.9%) |
34 (56.7%) |
24 (40.0%) |
2 (3.3%) |
42 (53.8%) |
26 (61.9%) |
15 (35.7%) |
1 (2.4%) |
|
Comezón |
33 (42.3%) |
23 (69.7%) |
10 (30.3%) |
0 (0.0%) |
16 (20.5%) |
7 (43.8%) |
9 (56.3%) |
0 (0.0%) |
|
Dolor |
65 (83.3%) |
34 (52.3%) |
28 (43.1%) |
3 (4.6%) |
47 (60.3%) |
28 (59.6%) |
17 (36.2%) |
2 (4.3%) |
|
Hematoma |
2 (2.6%) |
0 (0.0%) |
2 (100.0%) |
0 (0.0%) |
3 (3.8%) |
1 (33.3%) |
2 (66.7%) |
0 (0.0%) |
|
Nódulo, irregularidades/protuberancias |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
1 (1.3%) |
1 (100.0%) |
0 (0.0%) |
0 (0.0%) |
|
Dificultad para realizar actividades |
26 (33.3%) |
15 (57.7%) |
10 (38.5%) |
1 (3.8%) |
21 (26.9%) |
15 (71.4%) |
5 (23.8%) |
1 (4.8%) |
|
Pérdida de sensibilidad |
8 (10.3%) |
6 (75.0%) |
2 (25.0%) |
0 (0.0%) |
6 (7.7%) |
3 (50.0%) |
3 (50.0%) |
0 (0.0%) |
|
Otro |
7 (9.0%) |
3 (42.9%) |
4 (57.1%) |
0 (0.0%) |
1 (1.3%) |
0 (0.0%) |
0 (0.0%) |
1 (100.0%) |
|
Total |
77 (98.7%) |
17 (22.1%) |
50 (64.9%) |
10 (13.0%) |
73 (93.6%) |
28 (38.4%) |
39 (53.4%) |
6 (8.2%) |
* Los sujetos registraron entradas en sus diarios para el periodo de 30 días después del tratamiento. Si un evento todavía estaba en curso en el momento de la recolección del diario a los 30 días, la fecha de la resolución fue registrada e informada por teléfono o en la próxima visita del estudio.
Tabla 10. Eventos adversos posteriores al tratamiento inicial reportados por el médico vs. retratamiento (n = 78 sujetos retratados)
|
Tipo de evento adverso |
# Sujetos |
|||||||
|
Posterior al tratamiento inicial |
Posterior al retratamiento |
|||||||
|
N (%) |
Severidad máxima |
N (%) |
Severidad máxima |
|||||
|
Leve |
Moderado |
Severo |
Leve |
Moderado |
Severo |
|||
|
Moretones |
11 (14.1%) |
9 (81.8%) |
1 (9.1%) |
1 (9.1%) |
5 (6.4%) |
3 (60.0%) |
2 (40.0%) |
0 (0.0%) |
|
Hinchazón |
12 (15.4%) |
5 (41.7%) |
6 (50.0%) |
1 (8.3%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Enrojecimiento |
6 (7.7%) |
3 (50.0%) |
3 (50.0%) |
0 (0.0%) |
1 (1.3%) |
1 (100.0%) |
0 (0.0%) |
0 (0.0%) |
|
Comezón |
2 (2.6%) |
1 (50.0%) |
1 (50.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Dolor |
4 (5.1%) |
3 (75.0%) |
1 (25.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Hematoma |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
5 (6.4%) |
5 (100.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, irregularidades/protuberancias |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Dificultad para realizar actividades |
1 (1.3%) |
1 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Pérdida de sensibilidad |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Otro |
4 (5.1%) |
3 (75.0%) |
1 (25.0%) |
0 (0.0%) |
3 (3.8%) |
1 (33.3%) |
2 (66.7%) |
0 (0.0%) |
|
Total |
24 (30.8%) |
14 (58.3%) |
9 (37.5%) |
1 (4.2%) |
14 (17.9%) |
10 (71.4%) |
4 (28.6%) |
0 (0.0%) |
Las tablas 11 y 12 representan la aparición de todos los eventos adversos después del Re-tratamiento según lo reportado por los sujetos y los médicos, respectivamente.
Tabla 11. Número total de aparición de eventos adversos después del Re-tratamiento reportados por los sujetos* (n = 473 eventos)
|
Tipo de evento adverso |
Primera aparición (n, % total) |
Eventos adversos reportados (n, % tipo de evento) |
||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 y |
Semana 1 y 2 combinadas |
||
|
Moretones |
82 (17.3%) |
82 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
82 (100.0%) |
|
Hinchazón |
133 (28.1%) |
133 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
133 (100.0%) |
|
Enrojecimiento |
83 (17.5%) |
82 (98.8%) |
1 (1.2%) |
0 (0.0%) |
0 (0.0%) |
83 (100.0%) |
|
Comezón |
91 (19.2%) |
91 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
91 (100.0%) |
|
Dolor |
30 (6.3%) |
30 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
30 (100.0%) |
|
Hematoma |
1 (0.2%) |
1 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
1 (100.0%) |
|
Nódulo, irregularidades/protuberancias |
5 (1.1%) |
0 (0.0%) |
2 (40.0%) |
0 (0.0%) |
3 (60.0%) |
2 (40.0%) |
|
Dificultad para realizar actividades |
36 (7.6%) |
32 (88.9%) |
2 (5.6%) |
1 (2.8%) |
1 (2.8%) |
34 (94.4%) |
|
Pérdida de sensibilidad |
11 (2.3%) |
9 (81.8%) |
0 (0.0%) |
2 (18.2%) |
0 (0.0%) |
9 (81.8%) |
|
Otro |
1 (0.2%) |
1 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
1 (100.0%) |
|
Total |
473 (100.0%) |
461 (97.5%) |
5 (1.1%) |
3 (0.6%) |
4 (0.8%) |
466 (98.5%) |
* Los sujetos registraron entradas en sus diarios para el periodo de 30 días después del tratamiento. Si un evento todavía estaba en curso en el momento de la recolección del diario a los 30 días, la fecha de la resolución fue registrada e informada por teléfono o en la próxima visita del estudio.
Tabla 12. Número total de aparición de eventos adversos después del Re-tratamiento reportados por el médico (n = 21 eventos)
|
Tipo de evento adverso |
Primera aparición (n, % total) |
Eventos adversos reportados (n, % tipo de evento) |
||||
|
Semana 1 |
Semana 2 |
Semana 3 |
Semana 4 y |
Semana 1 y 2 combinadas |
||
|
Moretones |
8 (38.1%) |
7 (87.5%) |
0 (0.0%) |
0 (0.0%) |
1 (12.5%) |
7 (87.5%) |
|
Hinchazón |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Enrojecimiento |
2 (9.5%) |
2 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
2 (100.0%) |
|
Comezón |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Dolor |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Hematoma |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, irregularidades/protuberancias |
7 (33.3%) |
1 (14.3%) |
0 (0.0%) |
0 (0.0%) |
6 (85.7%) |
1 (14.3%) |
|
Dificultad para realizar actividades |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Pérdida de sensibilidad |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Otro |
4 (19.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
4 (100.0%) |
0 (0.0%) |
|
Total |
21 (100.0%) |
10 (47.6%) |
0 (0.0%) |
0 (0.0%) |
11 (52.4%) |
10 (47.6%) |
La tabla 13 muestra el número total de eventos adversos recurrentes después del retratamiento, según lo reportado por los sujetos. Ningún evento adverso recurrente después del re-tratamiento fue reportado por los médicos.
Tabla 13. Número total de eventos adversos recurrentes después del Re-tratamiento reportados en los diarios de los sujetos* (n = 31 eventos)
|
Tipo de evento adverso |
Menos de 14 días |
14-19 días |
20-29 días |
30-59 días |
60 o más días |
Total eventos adversos por tipo de evento |
|
Moretones |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Hinchazón |
4 (33.3%) |
1 (8.3%) |
5 (41.7%) |
2 (16.7%) |
0 (0.0%) |
12 (38.7%) |
|
Enrojecimiento |
1 (20.0%) |
2 (40.0%) |
1 (20.0%) |
1 (20.0%) |
0 (0.0%) |
5 (16.1%) |
|
Comezón |
0 (0.0%) |
3 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
3 (9.7%) |
|
Dolor |
0 (0.0%) |
0 (0.0%) |
4 (66.7%) |
2 (33.3%) |
0 (0.0%) |
6 (19.4%) |
|
Hematoma |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Nódulo, irregularidades/protuberancias |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Dificultad para realizar actividades |
0 (0.0%) |
3 (100.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
3 (9.7%) |
|
Pérdida de sensibilidad |
0 (0.0%) |
0 (0.0%) |
2 (100.0%) |
0 (0.0%) |
0 (0.0%) |
2 (6.5%) |
|
Otro |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
0 (0.0%) |
|
Total |
5 (16.1%) |
9 (29.0%) |
12 (38.7%) |
5 (16.1%) |
0 (0.0%) |
31 (100.0%) |
* Los sujetos registraron entradas en sus diarios para el periodo de 30 días después del tratamiento. Si un evento todavía estaba en curso en el momento de la recolección del diario a los 30 días, la fecha de la resolución fue registrada e informada por teléfono o en la próxima visita del estudio.
B. Ensayo pivotal de la mano:
Diseño del estudio:
Un estudio prospectivo, aleatorizado, enmascarado, controlado en 114 sujetos en seis centros de investigación en los EUA se llevó a cabo para evaluar la seguridad y eficacia del implante inyectable RADIESSE® para el tratamiento de la pérdida de volumen en las manos. Ochenta y cinco (85) sujetos fueron asignados al azar a un grupo de tratamiento (tratamiento inmediato) y veintinueve (29) sujetos fueron asignados al azar a un grupo de control sin tratamiento (tratamiento diferido) a través de 3 meses a partir de la inscripción. Ciento trece (113) de los 114 sujetos (99%) completaron el estudio a través de 3 meses. Después de la recolección de los datos necesarios para el análisis entre estos dos grupos, el grupo de control fue cruzado y recibió tratamiento. Todos los sujetos fueron elegibles para retratamiento 6 meses después del tratamiento inicial. Setenta y ocho de los 113 sujetos (69%) recibieron retratamiento. A partir de la inscripción a los 12 meses, ciento once (111) de los 113 sujetos (98%) completaron el estudio de seguimiento.
Datos demográficos de los sujetos:
La tabla 14 resume los datos demográficos de los 114 sujetos que participaron en la investigación. El análisis estadístico para la comparación entre el grupo de tratamiento y el grupo control mostraron que no hubo diferencias estadísticas entre los grupos en ninguna de las categorías demográficas.
Tabla 14. Datos demográficos de los sujetos
n = 114 sujetos*
|
Grupo de tratamiento (inmediato) (n = 85) |
Grupo control (demorado) (n = 29) |
|
|
Edad (años) |
||
|
Media |
52.8 |
54.8 |
|
DE |
8.0 |
10.6 |
|
Mediana |
52.0 |
57.0 |
|
Rango |
(26-75) |
(34-79) |
|
Género-n (%) |
||
|
Femenino |
81 (95.3%) |
28 (96.6%) |
|
Masculino |
4 (4.7%) |
1 (3.4%) |
|
Raza-n (%) |
||
|
Caucásica |
66 (77.6%) |
21 (72.4%) |
|
Afro-americana |
3 (3.5%) |
3 (10.3%) |
|
Hispana |
12 (14.1%) |
3 (10.3%) |
|
Asiática |
3 (3.5%) |
1 (3.4%) |
|
Otra |
1 (1.2%) |
1 (3.4%) |
|
Tipo de piel Fitzpatrick-n (%) |
||
|
I |
3 (3.5%) |
0 (0.0%) |
|
II |
45 (52.9%) |
11 (37.9%) |
|
III |
19 (22.4%) |
11 (37.9%) |
|
IV |
13 (15.3%) |
4 (13.8%) |
|
V |
4 (4.7%) |
2 (6.9%) |
|
VI |
1 (1.2%) |
1 (3.4%) |
|
Mano dominante-n (%) |
||
|
Derecha |
79 (92.9%) |
26 (89.7%) |
|
Izquierda |
6 (7.1%) |
3 (10.3%) |
* Incluyendo a un sujeto que fue retirado antes del tratamiento.
Volumen de inyección:
Los sujetos recibieron el implante inyectable RADIESSE® mezclado con lidocaína HCL a 2% (concentración final 0.3% de acuerdo al protocolo de mezcla detallado en los Componentes del ensamble e instrucciones de mezclado) en el dorso de ambas manos (definido como el espacio unido lateralmente entre el primer y quinto metacarpianos, proximalmente al pliegue dorsal de la muñeca y distalmente por las articulaciones metacarpofalángicas) usando una aguja de calibre 27. El número de puntos de inyección fue variado y se dejó a la discreción del investigador tratante. Las alícuotas inyectadas tuvieron volúmenes de un máximo de 0.5 cc cada una.
Los volúmenes de RADIESSE® (incluyendo el volumen de lidocaína añadido) que fueron inyectados se detallan en la Tabla 15. Los datos se presentan para el tratamiento inicial, retratamiento, y para la cantidad combinada de ambos tratamientos.
Tabla 15. Volúmenes de inyección (cc)
n = 226 manos (todos los sujetos)
|
Tratamiento inicial n = 226 manos |
Retratamiento n = 156 manos |
Combinado n = 226 manos |
|||||||
|
Mano derecha |
Mano izquierda |
Total |
Mano derecha |
Mano izquierda |
Total |
Mano derecha |
Mano izquierda |
Total |
|
|
Media |
2.58 |
2.60 |
5.18 |
1.64 |
1.61 |
3.25 |
3.72 |
3.71 |
7.43 |
|
Desviación estándar |
0.68 |
0.69 |
1.37 |
0.52 |
0.61 |
1.08 |
1.16 |
1.15 |
2.29 |
|
Mediana |
2.64 |
2.64 |
5.28 |
1.76 |
1.76 |
3.52 |
3.52 |
3.54 |
7.20 |
|
Rango |
1.50-3.60 |
1.40-3.60 |
2.90-7.20 |
0.70-2.64 |
0.00-3.00 |
1.40-5.30 |
1.50-6.16 |
1.40-6.16 |
2.90-12.32 |
Criterios de valoración del estudio:
La eficacia primaria se evaluó mediante la escala de evaluación de la mano de Merz (MHGS, figura 1), que fue validada para las evaluaciones en vivo. La eficacia secundaria fue evaluada mediante la evaluación reportada por los sujetos en una escala de mejora estética global no validada (GAIS, tabla 16).
Figura 1. Escala de evaluación de la mano de Merz (MHGS)
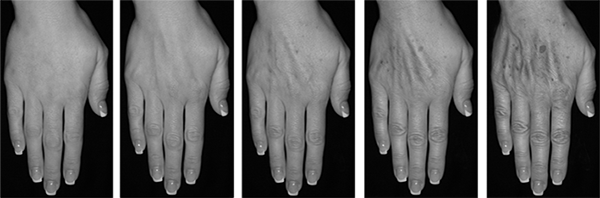
|
Sin pérdida de tejido graso |
Leve pérdida de tejido graso; ligera visibilidad de venas |
Moderada pérdida de tejido graso; leve visibilidad de venas y tendones |
Severa pérdida de tejido graso; moderada visibilidad de venas y tendones |
Muy severa pérdida de tejido graso; marcada visibilidad de venas y tendones |
|
|
|
|
|
|
Tabla 16. Escala de Mejora Estética Global (GAIS)
|
Clasificación |
Descripción |
|
Mucho muy mejorada |
Resultado cosmético óptimo para el implante en este sujeto. |
|
Muy mejorada |
Marcada mejoría en la apariencia a partir de la condición inicial, pero no completamente óptima en este sujeto. Un retoque podría mejorar ligeramente el resultado. |
|
Mejorada |
Mejoría obvia en la apariencia a partir de la condición inicial, pero un retoque o re-tratamiento es indicado. |
|
Sin cambio |
La apariencia es esencialmente la misma que la condición original. |
|
Peor |
La apariencia es peor que la condición original. |
La variable primaria de eficacia fue la mejora de ³ 1 punto en la MHGS entre el inicio y 3 meses en ambas manos para el grupo de tratamiento comparado contra el grupo de control. Las evaluaciones por MHGS en vivo fueron realizadas por un evaluador no médico enmascarado en cada uno de los sitios quien fue cegado para las asignaciones al azar de los sujetos. Las evaluaciones GAIS fueron realizadas por los sujetos, comparando la apariencia de su mano en vivo contra fotografías de su mano antes del tratamiento.
Evaluaciones de seguridad:
El criterio de valoración de la seguridad del estudio fue la evaluación de la incidencia, severidad, duración, su relación con el dispositivo y tratamiento en estudio, en su caso, de todos los eventos adversos observados por los sujetos y los investigadores tratantes. La seguridad también se evaluó mediante una serie de pruebas de función de la mano en tiempo real las cuales evaluaron el rango de movimiento, la sensibilidad, destreza y agarre y fuerza de pinza.
Resultados del criterio de valoración primario para la Eficacia:
La tabla 17 muestra que la mejoría en la apariencia de la mano en el grupo de tratamiento comparado contra el grupo de control a los 3 meses fue estadísticamente significativa y 75% de los sujetos tratados mostraban en ambas manos una mejoría ³ 1 punto en la MHGS.
Tabla 17. Mejoría de ³ 1 punto en MHGS en ambas manos a los 3 meses (n = 114 sujetos**)
|
n (%) |
Valor p* |
|
|
Grupo de tratamiento n = 85 |
Grupo de control n = 29 |
|
|
64 (75.3%) |
1 (3.4%) |
< 0.0001 |
* Prueba exacta de Fisher.
** Incluyendo un sujeto que fue retirado antes del tratamiento.
La tabla 18 muestra los resultados de MHGS, por mano, para ambos grupos, el de tratamiento y el de control a los 3 meses. En el grupo de tratamiento hubo una mejoría estadísticamente significativa a los 3 meses cuando es comparado contra el control. Además, el grupo de tratamiento mostró una mejoría estadísticamente significativa a partir de la condición inicial, mientras que el grupo de control no.
Tabla 18. MHGS por mano (n = 228 manos***)
|
Línea base |
3 meses |
Cambio |
||||
|
Grupo de tratamiento n = 170 |
Grupo de control n = 58 |
Grupo de tratamiento n = 170 |
Grupo de control n = 58 |
Grupo de tratamiento n = 170 |
Grupo de control n = 58 |
|
|
Media |
2.6 |
2.6 |
1.5 |
2.6 |
-1.1 |
-0.1 |
|
Mediana |
3.0 |
3.0 |
1.0 |
3.0 |
-1.0 |
0 |
|
Desviación estándar |
0.5 |
0.5 |
0.8 |
0.5 |
0.9 |
0.2 |
|
Rango |
2-3 |
2-3 |
0-3 |
2-3 |
-3, 1 |
-1, 0 |
|
Diferencia promedio |
0 |
-1.1 |
-1.0 |
|||
|
Valor p-tratamiento vs. control* |
0.56 |
<0.0001 |
<0.0001 |
|||
|
Valor p vs. línea base** |
<0.0001 |
0.25 |
||||
* Prueba de suma de rangos de Wilcoxon.
** Prueba de rango con signo de Wilcoxon.
*** Incluyendo un sujeto que fue retirado antes del tratamiento.
Se realizó un análisis de sensibilidad por sitio y se encontró que un sitio (sitio 7) tuvo las puntuaciones de eficacia significativamente más altas que los demás sitios. Cuando se evaluó la eficacia excluyendo al sitio 7, la mejoría promedio en MHGS fue de 0.7. Cuando se evaluó la eficacia excluyendo al sitio 7, 65.5% de los sujetos mostraron al menos una mejoría de 1 punto en la MHGS en ambas manos, contra 75.3% cuando el sitio 7 fue incluido. El porcentaje de sujetos que mostraron una mejoría ³ 1 punto en 3 meses por centro de investigación es proporcionado en la tabla 19.
Tabla 19. Mejoría de ³ 1 punto en MHGS a los 3 meses-por sitio de investigación n = 114 sujetos
|
Mejoría a partir de la línea base |
n (%) |
|||
|
Sitio 1, 2, 3 |
Sitio 4 |
Sitio 6 |
Sitio 7 |
|
|
n = 16 |
n = 44 |
n = 17 |
n = 37 |
|
|
³ 2 puntos |
0 (0.0%) |
1 (2%) |
5 (29%) |
25 (68%) |
|
1 punto |
8 (50%) |
27 (61%) |
10 (59%) |
11 (30%) |
|
0 puntos |
7 (44%) |
15 (34%) |
2 (12%) |
1 (3%) |
|
< 0 puntos |
1 (6%) |
1 (2%) |
0 (0.0%) |
0 (0.0%) |
Resultados del criterio de valoración secundario:
La tabla 20 describe los resultados de la escala de mejoría estética global (GAIS) para el grupo de tratamiento según la clasificación de los sujetos a los 3 meses. La evaluación de los resultados reportados por los sujetos demostró que 166/170 manos (97.6%) fueron mejoradas en comparación con la línea base. Sólo 4 manos (2%) fueron reportadas como sin cambios y ninguna mano fue clasificada como peor.
Tabla 20. GAIS para manos (n = 170 manos de 85 sujetos)
|
Clasificación |
n (%) |
|
Mucho muy mejorada |
54 (31.8%) |
|
Muy mejorada |
75 (44.1%) |
|
Mejorada |
37 (21.8%) |
|
Sin cambio |
4 (2.4%) |
|
Peor |
0 (0.0%) |
|
Total-al menos “mejorado” |
166 (97.6%) |
La tabla 21 proporciona los datos de eficacia a largo plazo de RADIESSE® inyectado en el dorso de la mano después del tratamiento inicial (tratamiento individual) y retratamiento de sujetos que tenían ³ 1 punto de mejoría en el MHGS a los 3, 6, 9 y 12 meses.
Tabla 21. Puntuaciones MHGS: ³ 1 Punto de Mejoría a los 3, 6, 9 y 12 meses después del Tratamiento Inicial y Después del Re-tratamiento (n = 113 Sujetos)
|
Número (n) o porcentaje (%) de sujetos |
|||||
|
Tiempo después del tratamiento inicial |
Tiempo después del retratamiento |
||||
|
3 meses n = 113 |
6 meses n = 113 |
9 meses n = 35 |
12 meses n = 22 |
3 meses n = 78 |
6 meses n = 61 |
|
87 (77%) |
82 (72.6%) |
25 (71.4%) |
15 (68.2%) |
64 (82.1%) |
54 (88.5%) |
Vigilancia Post-comercialización:
Se recibieron los siguientes eventos adversos de la vigilancia posterior a la comercialización para el implante inyectable RADIESSE®, independientemente de la indicación, en los EUA y fuera de los EUA y no fueron observados en los ensayos clínicos con el implante inyectable RADIESSE®: infección, sobre-inyección, inyección deficiente, pérdida del efecto, desplazamiento del producto, reacción alérgica, necrosis, granuloma, material expuesto, pérdida de cabello, hormigueo, ptosis, abscesos, parálisis, inyección superficial, infección herpética, escaldado, formación de ampollas, color azulado, ojeras, no le gustó los resultados, mareos, visión doble, festones, síntomas parecidos a la gripe, coloración gris, inflamación, reacción isquémica, hiperplasia linfoide, palidez de la piel, posible coágulo de sangre, cicatrices, sensibilidad al frío, cambios en la textura de la piel, masa de tejido desarrollada, embolia vascular que resulta en compromiso del tejido y la pérdida de visión o ceguera.
Los acontecimientos adversos graves más frecuentes (con una frecuencia mayor a 5 eventos reportados) fueron: necrosis, reacción alérgica, edema e infección. A continuación se describen estos eventos adversos graves:
• La necrosis fue precedida generalmente por dolor y palidez de la piel en el momento de la inyección acompañada de picazón u hormigueo y moretones, enrojecimiento e hinchazón. El inicio de la necrosis osciló entre inmediatamente en el momento de la inyección a 12 días después de la inyección. El tratamiento para la necrosis consistía en general de una combinación de ungüento de nitroglicerina/vasodilatación, ibuprofeno, paracetamol o aspirina, antibióticos, esteroides, ungüento no esteroideo para el tratamiento de heridas y compresas calientes. Para los casos en que se disponía de información, los pacientes se habían recuperado o se estaban recuperando con una mínima o ninguna cicatriz en el último contacto. Pocos casos requirieron la consulta con un cirujano plástico y una posible cirugía de escisión y la revisión quirúrgica para corregir el defecto resultante de la necrosis.
• La reacción alérgica fue identificada por una picazón e hinchazón severas, incluyendo hinchazón de la cara y la lengua. La aparición varió desde inmediatamente después de la inyección a 2 días después de la inyección. La reacción alérgica fue tratada generalmente con antihistamínicos y esteroides. Algunos casos requieren de hospitalización. Todos los pacientes se recuperaron de la reacción alérgica sin un resultado adverso permanente.
• Edema serio ha sido reportado con un inicio que va de 1 día a 3 semanas (inflamación relacionada con la formación de nódulos). El tratamiento generalmente consiste en la administración de antibióticos, antihistamínicos y esteroides. En algunos casos, los pacientes buscaron tratamiento en una sala de emergencias o fueron hospitalizados. Generalmente los eventos se resuelven dentro de 1 a 2 días, pero unos cuantos pacientes han reportado que presentan edema intermitente o edema persistente relacionado con una infección recurrente. Para los casos en que se disponía de información, la mayoría de los pacientes se han recuperado o están recuperándose.
• La infección, a menudo identificada como celulitis, fue acompañada por hinchazón, zonas endurecidas, enrojecimiento, pústulas y dolor. El inicio de la infección varió de 1 día a 2 meses y generalmente duró 2 días, pero en un caso, persistió durante 6 meses. Las infecciones fueron tratadas generalmente con antibióticos. Para los casos en que se disponía de información, los pacientes se habían recuperado o estaban en recuperación. Pocos pacientes experimentaron una cicatrización que pudiera requerir cirugía correctiva o decoloración en el sitio de la infección.
Individualización del tratamiento:
Antes del tratamiento, la idoneidad del paciente para el tratamiento y la necesidad del paciente para aliviar el dolor deben ser evaluadas. El resultado del tratamiento con el implante inyectable RADIESSE® variará entre los pacientes. En algunos casos, los tratamientos adicionales pueden ser necesarios dependiendo del tamaño del defecto y las necesidades del paciente.
MODO DE EMPLEO:
General:
Lo siguiente es necesario para el procedimiento de inyección percutánea:
• Jeringa(s) del implante inyectable RADIESSE®.
• Aguja(s) calibre 25 de diámetro externo (OD)-calibre 27 de diámetro interno (ID) con adaptador Luer lock.
1. Preparar al paciente para la inyección percutánea utilizando métodos estándar. El sitio de la inyección del tratamiento debe ser marcado y preparado con un antiséptico adecuado. La anestesia local o tópica en el sitio de la inyección se debe utilizar a criterio del médico. La joyería debe ser removida antes de la inyección, y hasta que la hinchazón después del procedimiento haya desaparecido.
2. Preparar las jeringas del implante inyectable RADIESSE® y la aguja(s) de inyección antes de la inyección percutánea. Una nueva aguja de inyección puede ser utilizada para cada jeringa, o la misma aguja de inyección puede ser conectada a cada jeringa nueva.
3. Retire la bolsa de aluminio de la caja. Abra la bolsa de aluminio rasgando en las muescas (marcadas como 1 y 2), y retire la jeringa de la bolsa de aluminio. Hay una pequeña cantidad de humedad normalmente presente en el interior de la bolsa de aluminio para fines de esterilización; esto no es una indicación de un producto defectuoso.
4. Retire o tuerza el empaque de la aguja para exponer el conector. Para el uso de agujas diferentes a la aguja(s) suministrada en este empaque, siga las instrucciones proporcionadas con la aguja(s).
5. Retirar la tapa del Luer de la jeringa desde el extremo distal de la jeringa antes de acoplar la aguja. La jeringa del implante inyectable RADIESSE® puede entonces ser girada sobre el adaptador de bloqueo Luer de la aguja teniendo cuidado de no contaminar la aguja. Deseche el empaque de la aguja. La aguja se debe apretar firmemente a la jeringa y ser purgada con el implante inyectable RADIESSE®. Si se encuentra un exceso de implante sobre la superficie de los dispositivos de cierre Luer, será necesario limpiar con una gasa estéril.
Empuje lentamente el émbolo de la jeringa hasta que el implante inyectable RADIESSE® sea expulsado por el extremo de la aguja. Si se observa una fuga por el conector Luer, puede ser necesario apretar la aguja, o extraer la aguja y limpiar las superficies del conector Luer o, en casos extremos, reemplazar tanto la jeringa como la aguja.
6. Localice el sitio inicial para el implante. El tejido cicatricial y el cartílago pueden ser difíciles o imposible de tratar. Evite en lo posible, pasar a través de este tipo de tejidos al introducir la aguja de inyección.
7. La cantidad inyectada puede variar dependiendo del sitio y el alcance de la restauración o aumento deseado. El implante inyectable RADIESSE® debe inyectarse por vía sub-dérmica.
8. Utilice un factor de corrección 1:1. No es necesaria una sobrecorrección.
9. Inserte la aguja con el bisel hacia abajo en un ángulo de aproximadamente 30° hacia la piel. La aguja debe deslizarse debajo de la dermis hasta el punto en que desea comenzar la inyección. Éste debe ser fácilmente palpable con la mano no dominante.
10. Si se encuentra una resistencia significativa al empujar el émbolo, la aguja de inyección se puede mover ligeramente para facilitar la colocación del material o puede ser necesario cambiar la aguja de inyección. Una obstrucción de la aguja se produjo en el estudio clínico de pliegues nasolabiales. La obstrucción de las agujas es más probable con el uso de agujas más pequeñas que el calibre 27 de diámetro interno (ID).
11. Avance la aguja en la subdermis a la ubicación inicial. Con cuidado, empujar el émbolo de la jeringa del implante inyectable RADIESSE® para iniciar la inyección e inyectar lentamente el material de implante en hilos lineales mientras se retira la aguja. Continuar la colocación de líneas adicionales de material hasta que se consiga el nivel deseado de corrección.
12. Aplicar lentamente una presión continua al émbolo de la jeringa para inyectar el implante a medida que se retira la aguja. El material de implante debe estar completamente rodeado por tejido blando sin dejar depósitos globulares. El área inyectada puede ser masajeada según sea necesario para lograr una distribución uniforme del implante.
13. Usar una sola vez y desechar de conformidad con las normas de seguridad locales.
Procedimiento de inyección para el aumento de la mano:
1. Preparar al paciente para la inyección percutánea utilizando métodos estándar. Haga que el paciente se lave las manos con agua jabonosa produciendo fricción durante 5-10 minutos y luego preparar las manos con un antiséptico adecuado. El sitio de inyección del tratamiento puede ser marcado para sitios de inyección planeados. La joyería debe ser removida antes de la inyección, y hasta después de que la hinchazón posterior al procedimiento haya desaparecido.
2. Usar la jeringa del implante inyectable RADIESSE® que se ha mezclado con lidocaína utilizando el procedimiento descrito más adelante en Componentes del ensamble e instrucciones de mezclado, y acoplada con la aguja de inyección, empuje lentamente el émbolo de la jeringa hasta que el implante inyectable RADIESSE® sea expulsado por el extremo final de la aguja, efectuar una aspiración antes de la inyección en bolo para evitar una inyección intravascular. Si se observa una fuga por el conector Luer, puede ser necesario apretar la aguja, o extraer la aguja y limpiar las superficies del conector Luer o, en casos extremos, reemplazar tanto la jeringa como la aguja. Una nueva aguja de inyección puede ser utilizado para cada jeringa, o la misma aguja de inyección puede ser conectada a cada jeringa nueva.
3. Localice el sitio inicial de la inyección. Los pacientes deben recibir las inyecciones en el dorso de las manos entre el primero y quinto metacarpianos. La inyección debe producirse inicialmente entre el segundo y cuarto metacarpianos, teniendo cuidado de no inyectar cerca de las articulaciones metacarpofalángicas. Si es necesario para lograr la corrección óptima, también se permite la inyección entre el primero y el segundo y cuarto y quinto metacarpianos.
4. Se debe realizar un abombamiento de la piel para separar la piel de las estructuras vasculares y tendinosas usando el pulgar y el dedo índice de la mano libre que no está inyectando para levantar la piel sobre la cara dorsal de la mano que está siendo tratada.
5. Avance la aguja entre la capa subcutánea y la fascia superficial con la jeringa paralela al dorso de la mano. Con cuidado, empujar el émbolo de la jeringa del implante inyectable RADIESSE® para iniciar la inyección e inyectar el material del implante inyectable RADIESSE® en pequeños bolos, 0.2-0.5 cc/bolo. No más de 0.5 cc debe inyectarse por bolo. El número de bolos variará dependiendo de la extensión del tratamiento deseado. No más de 3 cc del implante inyectable RADIESSE® (2 jeringas) deben ser inyectados por mano.
6. Si se encuentra una resistencia significativa al empujar el émbolo, la aguja de inyección se puede mover ligeramente para facilitar la colocación del material o puede ser necesario cambiar la aguja de inyección.
7. Inmediatamente después de la inyección, cubra el lugar de la inyección con una gasa estéril 4x4 y haga que el paciente se siente sobre esta mano mientras la mano contralateral está siendo inyectada. Esto calienta el implante inyectable RADIESSE® por lo que es más maleable para un masaje posterior.
8. Tratar la mano contralateral de la misma manera como se describe en los pasos 2 a 6 anteriores.
9. Inmediatamente después de la inyección de la mano contralateral, cubra el lugar de la inyección con una gasa estéril 4x4 y haga que el paciente se siente sobre esta mano.
10. Mientras que el lado contralateral se está calentando, quite la gasa de la mano que se inyectó en un principio, indíquele al paciente que cierre el puño con esta mano y de masaje suavemente en el dorso de la mano hasta que el implante inyectable RADIESSE® se haya extendido de forma homogénea en el dorso permaneciendo distal al pliegue de la muñeca y proximal a las articulaciones metacarpofalángicas.
11. Utilice un factor de corrección 1:1. No es necesaria una sobrecorrección.
Técnica para el mezclado del implante inyectable RADIESSE® y lidocaína HCL a 2%:
Precaución: No use la mezcla del implante inyectable RADIESSE® y lidocaína a 2% después de 2 horas de haber sido mezclados.
Precaución: Los componentes ensamblados están destinados para ser usados una sola vez. Dentro del estudio clínico, se utilizaron los siguientes componentes:
• Aguja estéril calibre 27, de pared regular de 0.5" con conector Luer-lock (no suministrada por Merz North America, Inc.).
• Jeringa de 3.0 cc estéril de polipropileno con Luer-lock (BD 309585).
• 0.2 cc de solución de lidocaína HCL a 2% para inyección USP, Hospira, Inc. (NDC 0409-4277-02) 2% (no suministrada por Merz North America, Inc.).
• Conector estéril Luer-lock hembra a hembra (Braun FDC1000 o Baxa 13901).
• Jeringa de 1.3 cc del implante inyectable RADIESSE®.
La jeringa de mezclado estéril de 3.0 cc de polipropileno (BD 309585) y el conector Luer-lock hembra a hembra (Baxa 13901) están disponibles por separado en el Kit de accesorios de Merz North America. Ni la lidocaína ni la aguja estéril calibre 27 de 0.5" son suministradas por Merz North America, Inc.
Componentes del ensamble e instrucciones de mezclado:
1. Ensamblar los componentes y realizar la mezcla utilizando una técnica estéril (figura 2).
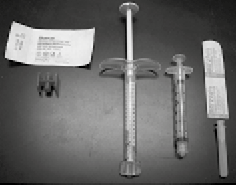
Figura 2. De izquierda a derecha: Conector Luer-lock hembra a hembra, jeringa de RADIESSE®, jeringa de mezclado de 3.0 cc, aguja estéril calibre 27 de 0.5"
2. Extraer la lidocaína dentro de la jeringa de mezclado estéril de polipropileno de 3.0 cc acoplada con una jeringa estéril calibre 27 de 0.5".
3. Golpee suavemente la jeringa de mezclado, conteniendo la lidocaína y bajar el embolo para eliminar todo el exceso de aire.
4. Remover la aguja estéril calibre 27 de 0.5".
5. Conectar firmemente la jeringa de mezclado a la jeringa de RADIESSE® usando el conector Luer-lock hembra a hembra (figuras 3 y 4).

Figura 3
Figura 4
6. Mezclar la lidocaína y el implante inyectable RADIESSE® pulsando alternativamente los émbolos, primero en la jeringa de mezcla y luego en la jeringa de RADIESSE® para diez golpes de mezcla (cada golpe de mezcla es una compresión completa del émbolo de la jeringa de mezcla seguido de una compresión completa del émbolo de la jeringa de RADIESSE®). Los émbolos deben ser comprimidos con firmeza y rapidez, a aproximadamente dos compresiones por segundo (figura 5).

Figura 5
7. Después del mezclado, remover la jeringa de mezclado y el conector Luer-lock hembra a hembra y desecharlos.
8. Acoplar la jeringa conteniendo la mezcla de lidocaína y RADIESSE® con una aguja de inyección.
9. Proceder con la inyección del implante inyectable RADIESSE®.
El estudio clínico se llevó a cabo mediante la mezcla de 0.2 cc de lidocaína a 2% con 1.3 cc del implante inyectable RADIESSE® en la jeringa de 3.0 cc BD. La tabla 22 proporciona la relación de lidocaína a 2% para ser mezclada con los diferentes volúmenes de las jeringas del implante inyectable RADIESSE®. Estas proporciones dan como resultado la misma concentración de lidocaína a 2% (p/v%) en el implante inyectable RADIESSE® que fue mezclado en el estudio clínico después de contar el espacio muerto en las jeringas de RADIESSE® y de mezclado de 3.0 cc BD (tabla 22).
Tabla 22. Concentración de lidocaína
|
RADIESSE® (cc) |
2% lidocaína (cc) |
Concentración resultante de lidocaína (p/v%) |
|
0.3 |
0.02 |
0.30-0.33% |
|
0.8 |
0.11 |
0.31-0.32% |
|
1.3 |
0.20 |
0.31-0.32% |
|
1.5 |
0.26 |
0.31-0.32% |
|
3.0 |
0.45 |
0.32-0.34% |
Información de orientación para el paciente: Consulte la guía de información del paciente del implante inyectable RADIESSE®.
DESCRIPCIÓN DEL DISPOSITIVO:
El implante inyectable RADIESSE® es un implante cohesivo opaco, estéril, apirógeno, semi-sólido, cuyo principal componente es la hidroxiapatita de calcio sintética suspendida en un gel acarreador de agua estéril para inyectables, glicerina y carboximetilcelulosa de sodio. El implante inyectable RADIESSE® (1.5 y 0.8 cc) tiene partículas de CaHA en un rango de tamaño de 25–45 micrones y debe ser inyectado con una aguja de calibre 25 de diámetro externo (OD) a calibre 27 de diámetro interno (ID).
ALMACENAMIENTO: El implante inyectable RADIESSE® se debe almacenar a temperatura ambiente controlada entre 15 y 32°C (59 y 90°F). La fecha de caducidad, si se almacena a estas temperaturas, es de dos años a partir de la fecha de fabricación para la jeringa con un volumen de 1.5 cc. La fecha de caducidad, si se almacena a estas temperaturas, es de dos años a partir de la fecha de fabricación para la jeringa con un volumen de 0.8 cc. No lo use si se ha excedido la fecha de caducidad.
Disposición: Las jeringas y agujas usadas y parcialmente usadas para la inyección podrían ser biológicamente peligrosos y deben ser manejados y eliminados de acuerdo con las prácticas médicas de la instalación y las regulaciones locales, estatales o federales.
GARANTÍA: Merz North America, Inc. garantiza que un cuidado razonable ha sido ejercitado en el diseño y la fabricación de este producto.
Esta garantía sustituye y excluye cualquier otra garantía no expresamente establecida en este documento, ya sea expresa o implícita por aplicación de la ley o de otra manera, incluyendo, pero no limitada a, las garantías implícitas de comercialización o idoneidad para su propósito particular.
La manipulación y almacenamiento de este producto, así como los factores relacionados con el paciente, diagnóstico, tratamiento, procedimientos quirúrgicos y otros asuntos más allá del control de Merz North America, Inc. afectan directamente al producto y a los resultados obtenidos de su uso. La obligación de Merz North America, Inc., bajo esta garantía se limita a la sustitución de este producto y Merz North America, Inc. no será responsable por cualquier pérdida incidental o consecuente, daños o gastos, originados directa o indirectamente a partir del uso de este producto. Merz North America, Inc. no asume ni autoriza a ninguna persona a asumir por Merz North America, Inc., cualquier otra obligación o responsabilidad adicional en relación con este producto.
Fabricado por:
Merz North America, Inc.
4133 Courtney Road, Suite #10
Franksville, WI 53126 U.S.A.
Fax: 262-835-3330
Teléfono: 262-835-3300
e-Mail: info@merz.com
Importado y distribuido por:
MERZ PHARMA, S.A. de C.V.
Parque Industrial Prologis
Av. Del Pozo S/N Nave 5C-2
Col. Recursos Hidráulicos
C.P. 54913, Tultitlán, México







