
REXULTI - Tabletas
Sustancia(s):
- Brexpiprazol
Presentaciones:
- 1 Caja, 7 Tabletas, 0.25 mg
- 1 Caja, 7 Tabletas, 0.5 mg
- 1 Caja, 7 Tabletas, 1 mg
- 1 Caja, 10 Tabletas, 1 mg
- 1 Caja, 14 Tabletas, 1 mg
- 1 Caja, 14 Tabletas, 2 mg
- 1 Caja, 14 Tabletas, 3 mg
- 1 Caja, 14 Tabletas, 4 mg
- 1 Caja, 28 Tabletas, 1 mg
- 1 Caja, 28 Tabletas, 2 mg
- 1 Caja, 28 Tabletas, 3 mg
- 1 Caja, 28 Tabletas, 4 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Brexpiprazol 0.25 mg, 0.5 mg, 1 mg, 2 mg, 3 mg y 4 mg
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
REXULTI® está indicado para:
• Tratamiento de la esquizofrenia en adultos y pacientes pediátricos de 13 años en adelante.
• Tratamiento adyuvante del trastorno depresivo mayor.
FARMACOCINÉTICA Y FARMACODINAMIA:
Mecanismo de acción: Se desconoce el mecanismo de acción de brexpiprazol en el tratamiento del trastorno depresivo mayor o de la esquizofrenia. Sin embargo, la eficacia de la brexpiprazol puede ser mediada a través de una combinación de actividad agonista parcial en receptores de serotonina 5-HT1A y de dopamina D2 y actividad antagonista en receptores 5-HT2A de serotonina.
Farmacodinamia: Brexpiprazol tiene afinidad (expresada como Ki) para múltiples receptores monoaminérgicos, incluyendo receptores de serotonina 5-HT1A (0.12 nM), 5-HT2A (0.47 nM), 5-HT2B (1.9 nM), 5-HT7 (3.7 nM), dopamina D2 (0.30 nM), D3 (1.1 nM) y noradrenérgicas α1A (3.8 nM), α1B (0.17 nM), α1D (2.6 nM) y α2C (0.59 nM). Brexpiprazol actúa como un agonista parcial en los receptores 5-HT1A, D2 y D3 y como antagonista en los receptores 5-HT2A, 5-HT2B, 5-HT7, α1A, α1B, α1D y α2C. Brexpiprazol también muestra afinidad por el receptor de histamina H1 (19 nM) y por el receptor muscarínico M1 (67% de inhibición a 10 μM).
Electrofisiología cardiaca: A una dosis de 3 veces la dosis humana máxima recomendada para el tratamiento de la esquizofrenia y de 4 veces la dosis humana máxima recomendada para el tratamiento adyuvante a los antidepresivos para el tratamiento del trastorno depresivo mayor, REXULTI® no prolonga el intervalo QTc a ningún grado clínicamente relevante.
Farmacocinética:
Absorción: Después de la administración de una dosis única de REXULTI® tabletas, las concentraciones plasmáticas máximas de brexpiprazol se presentaron a las 4 horas después de la administración; y la biodisponibilidad oral absoluta fue de 95%. Las concentraciones en estado estable de brexpiprazol se alcanzaron de los 10 a 12 días de dosificación.
REXULTI® se puede administrar con o sin alimentos. La administración de una tableta de REXULTI® de 4 mg con una comida alta en grasa estándar no afectó significativamente la Cmáx o el área bajo la curva de brexpiprazol. Después de la administración de una sola dosis y de dosis múltiples, una vez al día, la exposición de brexpiprazol (Cmáx y área bajo la curva) aumentó proporcionalmente a la dosis administrada. Los estudios in vitro de brexpiprazol no indicaron que brexpiprazol sea un sustrato de los transportadores de fluxión tales MDRI (P-gp) y BCRP.
Distribución: El volumen de distribución de brexpiprazol tras la administración intravenosa es alto (1.56 ± 0.42 L/kg), lo que indica distribución extravascular. Brexpiprazol se une fuertemente a las proteínas plasmáticas (más del 99%) a la albúmina sérica y a α-1-glicoproteína ácida y su unión a proteínas no se ve afectado por enfermedad renal o hepática. Con base en los resultados de los estudios in vitro, la unión de brexpiprazol a proteínas no se ve afectada por la warfarina, diazepam o digitoxina.
Eliminación:
Metabolismo: Con base en los estudios de metabolismo in vitro de brexpiprazol usando citocromo P450 recombinante humano (CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 y 3A4), se demostró que el metabolismo de brexpiprazol está mediado, principalmente, por CYP3A4 y CYP2D6.
Brexpirazol in vivo es metabolizado principalmente por enzimas CYP3A4 y CYP2D6. Después de la administración de una dosis única y de dosis múltiples, brexpiprazol y su principal metabolito, DM-3411, fueron las moléculas de fármaco predominantes en la circulación sistémica. En estado estable, DM-3411 representó del 23% al 48% de la exposición de brexpiprazol (área bajo la curva) en plasma. Se considera que DM-3411 no contribuye a los efectos terapéuticos de brexpiprazol.
Con base en los datos in vitro, brexpiprazol demostró poca o ninguna inhibición de las isoenzimas de CYP450.
Excreción: Después de una dosis oral única de brexpiprazol marcado en [14C], aproximadamente el 25% y el 46% de la radiactividad administrada se recuperó en la orina y heces, respectivamente. Menos del 1% de brexpiprazol, sin cambios, fue excretado en la orina y aproximadamente el 14% de la dosis oral se recuperó sin cambios en las heces. La depuración oral aparente de una tableta oral de brexpiprazol después de la administración una vez al día es 19.8 (± 11.4) mL/h/kg. Después de la administración de dosis múltiples, una vez al día, de REXULTI®, las vidas medias de eliminación terminal de brexpiprazol y su principal metabolito, DM-3411, fueron de 91 y 86 horas, respectivamente.
Estudios en poblaciones específicas: Las exposiciones de brexpiprazol en poblaciones específicas se resumen en la Figura 1. El análisis farmacocinético de población indicó la exposición de brexpiprazol, en pacientes con enfermedad renal moderada, fue mayor en comparación con pacientes con función renal normal.
Figura 1: Efectos de los factores intrínsecos en la farmacocinética de brexpiprazol
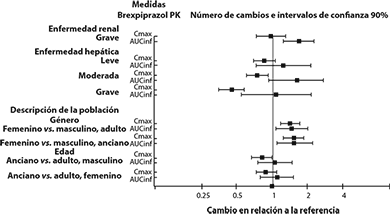
Pacientes pediátricos: Se realizó un estudio farmacocinético, de dosis múltiples (0.5, 1, 2, 3 o 4 mg/día) en 43 pacientes pediátricos, de 13 a 17 años de edad. El analisis farmacocinético poblacional indicó que la exposición sistémica (Cmáx y AUC) de brexpiprazol en pacientes pediátricos (de 13 a 17 años de edad) fue comparable con la de pacientes adultos en el intervalo de dosisde 0.5 a 4 mg.
Estudios de interacciones medicamentosas: El efecto de otros medicamentos sobre las exposiciones de brexpiprazol se resumen en la Figura 2. Con base en la simulación, se espera un incremento de 5.1 veces en los valores del área bajo la curva en estado estable, cuando se administran metabolizadores extensivos de CYP2D6 con inhibidores de CYP2D6 y CYP3A4 fuertes. Se espera un incremento de 4.8 veces en los valores medios de área bajo la curva en estado estable en metabolizadores pobres de CYP2D6 administrados con inhibidores fuertes de CYP3A4 [consultar Interacciones medicamentosas y de otro género].
Figura 2: El efecto de otros medicamentos sobre la farmacocinética de brexpiprazol
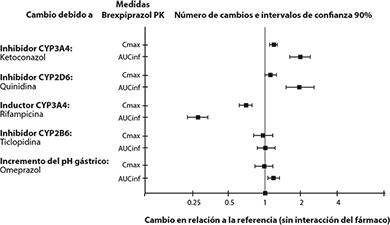
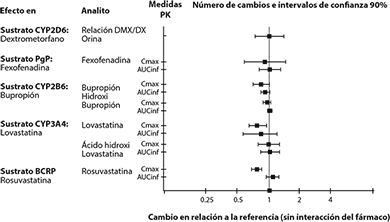
Figura 3: El efecto de REXULTI® sobre la farmacocinética de otros fármacos
Estudios clínicos:
Tratamiento adyuvante del trastorno depresivo mayor: La eficacia de REXULTI® en el tratamiento adyuvante del trastorno depresivo mayor se evaluó en dos estudios clínicos de 6 semanas, doble ciego, controlado con placebo, a dosis fijas de pacientes adultos que cumplían los criterios DSM-IV-TR para trastorno depresivo mayor, con o sin síntomas de ansiedad, que tuvieron una respuesta inadecuada a la terapia del antidepresivo previo (1 a 3 cursos) en el episodio actual y que también mostraron una respuesta inadecuada a lo largo de las 8 semanas de tratamiento antidepresivo prospectivo (con escitalopram, fluoxetina, paroxetina de liberación controlada, sertralina, duloxetina de liberación retardada o venlafaxina de liberación prolongada). La respuesta inadecuada durante la fase de tratamiento antidepresivo prospectivo se definió por tener síntomas persistentes sin una mejora sustancial en el transcurso del tratamiento.
Los pacientes en el estudio 228 (en adelante "estudio 1") fueron asignados, de manera aleatoria, a REXULTI® 2 mg una vez al día o al placebo. Los pacientes en el estudio 227 (en adelante "estudio 2") fueron asignados, de manera aleatoria, a REXULTI® 1 o 3 mg una vez al día o al placebo. Para los pacientes asignados de manera aleatoria a REXULTI®, todos los pacientes iniciaron el tratamiento a una dosis de 0.5 mg, una vez al día, durante 1 semana. En la semana 2, la dosis de REXULTI® se incrementó a 1 mg en todos los grupos de tratamiento y se mantuvo a 1 mg o se incrementó a 2 mg o 3 mg una vez al día, con base en la asignación de tratamiento, de la semana 3 en adelante. Las dosis se mantuvieron durante las 4 semanas restantes.
El criterio de valoración primario fue el cambio desde el estado basal hasta la Semana 6 en la Escala de Valoración de la Depresión de Montgomery-Asberg (MADRS), una escala relacionada con el médico, de 10-ítems, utilizada para evaluar el grado de sintomatología depresiva, en donde 0 representa sin síntomas y 60 representa los peores síntomas.
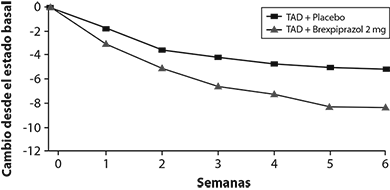
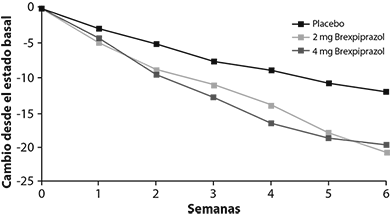
En la asignación aleatoria, la puntuación total de MADRS media fue 27. En los estudios 1 y 2, REXULTI® [+ antidepresivos (TAD)] 2 mg/día y 3 mg/día fue superior a placebo + TAD en la reducción de las medias de la puntuación total de MADRS. Los resultados de los parámetros de eficacia primaria para los estudios clínicos a dosis fijas se muestran en la Tabla 1. La Figura 4 a continuación muestra el curso del tiempo de respuesta con base en la medida de eficacia primaria (MADRS) en el Estudio 1.
Tabla 1: Resumen de resultados de eficacia para los estudios 1 y 2 para el tratamiento adyuvante del
trastorno depresivo mayor
|
Estudio |
Grupo de tratamiento |
N |
Medida de eficacia primaria: MADRS |
||
|
Puntuación basal media (SD) |
Media de mínimos cuadrados desde el estado basal (SE) |
Diferencia del placebo sustraídoa (IC 95%) |
|||
|
1 |
REXULTI® (2 mg/día) + TAD * |
175 |
26.9 (5.7) |
-8.4 (0.6) |
-3.2 (-4.9, -1.5) |
|
Placebo + TAD |
178 |
27.3 (5.6) |
-5.2 (0.6) |
-- |
|
|
2 |
REXULTI® (1 mg/día) + TAD |
211 |
26.5 (5.6) |
-7.6 (0.5) |
-1.3 (-2.7, 0.1) |
|
REXULTI® (3 mg/día) + TAD |
213 |
26.5 (5.3) |
-8.3 (0.5) |
-2.0 (-3.4, -0.5) |
|
|
Placebo + TAD |
203 |
26.5 (5.2) |
-6.3 (0.5) |
- |
|
SD: desviación estándar; SE: error estándar; CI: intervalo de confianza sin ajustar.
* Dosis estadísticamente significativas superiores al placebo.
a Diferencia (fármaco menos placebo) en cambio de la media de mínimos cuadrados desde el estado basal.
Un análisis de los subgrupos de población no sugirió respuesta diferencial con base en edad, género, raza o elección de antidepresivo prospectivo.
Figura 4: Cambio desde el estado basal en la puntuación total de MADRS por visita del estudio (semana) en los pacientes adultos con trastorno depresivo mayor en el estudio 1
Esquizofrenia: La eficacia de REXULTI® en el tratamiento de adultos con esquizofrenia se demostró en dos ensayos clínicos de 6 semanas, aleatorios, doble ciego, controlados con placebo, a dosis fijas en pacientes que cumplieron los criterios de DSM-IV-TR para esquizofrenia.
En ambos estudios, el estudio 231 (en adelante "estudio 3") y estudio 230 (en adelante "estudio 4"), los pacientes fueron asignados de manera aleatoria a REXULTI® 2 o 4 mg una vez al día o al placebo. Los pacientes en los grupos de REXULTI® iniciaron tratamiento a una dosis de 1 mg una vez al día, los días 1 a 4. La dosis de REXULTI® se aumentó a 2 mg los días 5 a 7. La dosis se mantuvo a 2 mg una vez al día o se aumentó a 4 mg una vez al día, dependiendo de la asignación del tratamiento, durante las 5 semanas restantes.
El criterio de valoración primario de eficacia de ambos estudios clínicos fue el cambio desde el estado basal hasta la Semana 6 en la puntuación total de la Escala del Síndrome Positivo y Negativo (PANSS). La PANSS es una escala de 30 ítems que mide síntomas positivos de la esquizofrenia (7 ítems), los síntomas negativos de la esquizofrenia (7 ítems) y psicopatología general (16 ítems), cada uno calificado en una escala de 1 (ausente) a 7 (extremo); el intervalo de puntuaciones PANSS totales de 30 (mejor) a 210 (peor).
En el estudio 3, REXULTI® 2 mg/día y 4 mg/día fue superior al placebo en la puntuación total de PANSS. En el estudio 4, REXULTI® 4 mg/día fue superior al placebo en la puntuación total de PANSS (tabla 2). La figura 5 muestra el curso del tiempo de respuesta con base en la medida de eficacia primaria (cambio desde el estado basal en la puntuación total de PANSS) en el estudio 3.
El análisis de subgrupos de población basado en edad, género y raza no sugiere sensibilidad diferencial.
Tabla 2: Resumen de resultados de eficacia para estudios en esquizofrenia en adultos (Estudios 3 y 4)
|
Estudio |
Grupo de tratamiento |
N |
Medida de eficacia primaria: PANSS |
||
|
Puntuación basal media (SD) |
Media de mínimos cuadrados desde el estado basal (SE) |
Diferencia del placebo sustraído a (IC 95%) |
|||
|
3 |
REXULTI® (2 mg/día)* |
180 |
95.9 (13.8) |
-20.7 (1.5) |
-8.7 (-13.1, -4.4) |
|
REXULTI® (4 mg/día)* |
178 |
94.7 (12.1) |
-19.7 (1.5) |
-7.6 (-12.0, -3.1) |
|
|
Placebo |
178 |
95.7 (11.5) |
-12.0 (1.6) |
--- |
|
|
4 |
REXULTI® (2 mg/día) |
179 |
96.3 (12.9) |
-16.6 (1.5) |
-3.1 (-7.2, 1.1) |
|
REXULTI® (4 mg/día)* |
181 |
95.0 (12.4) |
-20.0 (1.5) |
-6.5 (-10.6, -2.4) |
|
|
Placebo |
180 |
94.6 (12.8) |
-13.5 (1.5) |
--- |
|
SD: desviación estándar; SE: error estándar; CI: intervalo de confianza sin ajustar.
* Dosis estadísticamente significativas superiores al placebo.
a Diferencia (fármaco menos placebo) en cambio de la media de mínimos cuadrados desde el estado basal.
Figura 5: Cambio desde el estado basal en la puntuación total de PANSS por visita del estudio (semana) en pacientes adultos con esquizofrenia en estudio 3
La seguridad y la eficacia de REXULTI® como tratamiento de mantenimiento en adultos con esquizofrenia, de 18 a 65 años de edad, se demostraron en la fase de mantenimiento del estudio de suspensión aleatorio (estudio 331-10-232, en adelante "estudio 5"). Pacientes fueron estabilizados, durante al menos 12 semanas, en 1 a 4 mg/día de REXULTI® (N = 202). Después se asignaron de manera aleatoria en la fase de tratamiento doble ciego a REXULTI® continuo en su dosis estable alcanzada (N = 97) o se cambiaron al placebo (N = 105).
El criterio de valoración primario en el estudio 5 fue el tiempo desde la asignación aleatoria a la inminente recaída durante la fase doble ciego, definido como: 1) puntuación de mejora CGI ≥ 5 (mínimamente peor) y un aumento a una puntuación > 4 en la desorganización conceptual de PANSS, conducta alucinatoria, desconfianza o ítems de contenido inusual del pensamiento, con un aumento ≥ 2 en un ítem en específico artículo o un incremento ≥ 4 puntos en los cuatro ítems de PANSS combinados, 2) hospitalización por empeoramiento de los síntomas psicóticos, 3) comportamiento suicida actual, o 4) comportamiento violento/agresivo.
Un análisis provisional previamente especificado demostró, de manera estadísticamente significa, un mayor tiempo hasta la recaída en pacientes asignados al azar al grupo de REXULTI® en comparación con pacientes tratados con placebo. El estudio clínico terminó prematuramente debido a que el mantenimiento de la eficacia había sido demostrado. Las curvas de Kaplan-Meier de la proporción acumulada de pacientes con recaída durante la fase de tratamiento doble ciego para los grupos de REXULTI® y de placebo se muestran en la Figura 6. El criterio de valoración secundario clave, la proporción de pacientes que cumplieron los criterios de recaída inminente fue menor, de manera estadísticamente significativa, en los pacientes tratados con REXULTI® en comparación con el grupo placebo.
Figura 6: Estimación de Kaplan-Meier del porcentaje de recaída inminente en el estudio 5
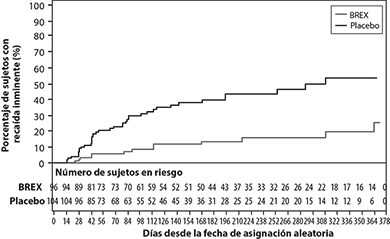
Nota: Un total de 202 pacientes fueron asignados de manera aleatoria. Entre ellos, un paciente del placebo no tomó un medicamento en investigación y un paciente de brexpiprazol no tuvo evaluaciones de eficacia posteriores a la asignación aleatoria. Estos dos pacientes fueron excluidos del análisis de eficacia.
CONTRAINDICACIONES:
REXULTI® está contraindicado en pacientes con hipersensibilidad conocida a brexpiprazol o a cualquiera de sus componentes. Las reacciones incluyen edema facial, erupción, urticaria y anafilaxia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Resumen de riesgos: No se han realizado estudios adecuados y bien controlados con REXULTI® en mujeres embarazadas para informar de los riesgos asociados al fármaco. Sin embargo, los neonatos cuyas madres se han expuesto a fármacos antipsicóticos, como REXULTI®, durante el tercer trimestre del embarazo, están en riesgo de experimentar síntomas extrapiramidales y/o síntomas de abstinencia. En los estudios de reproducción animal, no se observó teratogenicidad con la administración oral de brexpiprazol, en conejos y ratas preñadas, durante la organogénesis a dosis de hasta 73 y 146 veces, respectivamente, la dosis humana máxima recomendada de 4 mg/día sobre una base de un mg/m2. Sin embargo, cuando se administró brexpiprazol a las ratas preñadas, durante el periodo de organogénesis y hasta la lactancia, el número de muertes perinatales de crías aumentó a 73 veces la dosis humana máxima recomendada [consultar Datos]. Se desconoce el riesgo de antecedentes de defectos congénitos graves y de abortos espontáneos para la población indicada. En la población general de Estados Unidos, el riesgo de fondo estimado de defectos congénitos graves y de aborto involuntario en los embarazos, clínicamente reconocido, es de 2 a 4% y del 15 al 20%, respectivamente.
Consideraciones clínicas:
Reacciones adversas fetales/neonatales: Se han reportado síntomas extrapiramidales y/o de abstinencia, incluyendo agitación, hipertonía, hipotonía, temblor, somnolencia, dificultad respiratoria y trastornos de la alimentación en neonatos cuyas madres estuvieron expuestas a fármacos antipsicóticos durante el tercer trimestre del embarazo. Estos síntomas han variado en intensidad. Algunos neonatos se recuperaron dentro de horas o días sin un tratamiento específico; otros requirieron hospitalización prolongada. Vigilar a los neonatos en busca de aparición de síntomas extrapiramidal o síntomas de abstinencia y controlar adecuadamente los síntomas.
Datos:
Datos en animales: Las ratas preñadas fueron tratadas con dosis orales de 3, 10 y 30 mg/kg/día (7.3, 24 y 73 veces la dosis humana máxima recomendada, sobre una base mg/m2) de brexpiprazol durante el periodo de organogénesis. Brexpiprazol no fue teratogénico y no causó efectos adversos en el desarrollo con dosis de hasta 73 veces la dosis humana máxima recomendada.
Las hembras conejo preñadas fueron tratadas con dosis orales de 10, 30 y 150 mg/kg/día (49, 146 y 730 veces la dosis humana máxima recomendada) de brexpiprazol durante el periodo de organogénesis. Brexpiprazol no fue teratogénico y no causó efectos adversos en el desarrollo con dosis de hasta 146 veces la dosis humana máxima recomendada. Se observaron hallazgos de disminución de peso, osificación retardada y mayor incidencia de variaciones esqueléticas y viscerales, en fetos, a 730 veces la dosis humana máxima recomendada, una dosis que indujo toxicidad materna.
En un estudio en el que se administraron dosis orales de 3, 10 y 30 mg/kg/día (7.3, 24 y 73 veces la dosis humana máxima recomendada) a ratas preñadas, durante el periodo de la organogénesis y hasta la lactancia, el número de crías que nacieron vivas disminuyó y aumentó el número de muertes postnatales prematuras a una dosis de 73 veces la dosis humana máxima recomendada. La lactancia deteriorada por las madres, el bajo peso al nacer y la disminución del aumento de peso en las crías se observaron a 73 veces, pero no a 24 veces la dosis humana máxima recomendada.
Lactancia:
Resumen de riesgos: No se han realizado estudios de lactancia para evaluar la presencia de brexpiprazol en la leche humana, los efectos de brexpiprazol en el infante alimentado con leche materna o los efectos de brexpiprazol en la producción de leche. Brexpiprazol está presente en la leche de rata. Los beneficios, en el desarrollo y la salud, de la lactancia deben ser considerados junto con la necesidad clínica de la madre de tomar REXULTI®, así como los efectos adversos potenciales en el lactante de REXULTI® o de la condición materna subyacente.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las siguientes reacciones adversas se discuten con mayor detalle en otras secciones del prospecto:
• Aumento de la mortalidad en pacientes de edad avanzada con psicosis relacionada con demencia [consultar Precauciones generales].
• Pensamientos y comportamientos suicidas en adolescentes y adultos jóvenes [consultar Precauciones generales].
• Reacciones adversas cerebrovasculares, incluyendo accidente cerebrovascular en pacientes de edad avanzada con psicosis relacionada con demencia [consultar Precauciones generales].
• Síndrome Neuroléptico Maligno (SNM) [consultar Precauciones generales].
• Discinesia tardía [consultar Precauciones generales].
• Cambios metabólicos [consultar Precauciones generales].
• Leucopenia, neutropenia y agranulocitosis [consultar Precauciones generales].
• Hipotensión ortostática y síncope [consultar Precauciones generales].
• Caídas [consultar Precauciones generales].
• Convulsiones [consultar Precauciones generales].
• Desregulación de la temperatura corporal [consultar Precauciones generales].
• Disfagia [consultar Precauciones generales].
• Trastornos del control de impulsos/comportamientos compulsivos [consultar Precauciones generales].
• Potencial de deterioro cognitivo y motor [consultar Precauciones generales].
Experiencia de los estudios clínicos: Debido a que los estudios clínicos se llevan a cabo bajo condiciones muy variables, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden compararse directamente con las tasas de los estudios clínicos de otro fármaco, y podrían no reflejar las tasas observadas en la práctica.
Trastorno depresivo mayor: Se evaluó la seguridad de REXULTI® en 1,054 pacientes (18 a 65 años de edad) con diagnóstico de trastorno depresivo mayor que participaron en dos estudios clínicos de 6 semanas, controlados con placebo, a dosis fijas en pacientes con trastorno depresivo mayor, en los que REXULTI® se administró en dosis de 1 mg a 3 mg diarios como tratamiento adyuvante a la terapia antidepresiva continua; los pacientes en el grupo placebo continuaron para recibir terapia antidepresiva.
Reacciones adversas reportadas como razones para la suspensión del tratamiento: Un total de 3% (17/643) de los pacientes tratados con REXULTI® y 1% (3/411) de los pacientes tratados con placebo suspendieron el medicamento debido a reacciones adversas.
Reacciones adversas frecuentes: Las reacciones adversas asociadas con el uso adyuvante de REXULTI® (incidencia de 2% o mayor e incidencia de REXULTI® adyuvante mayor que la del placebo adyuvante) que ocurrieron durante la terapia aguda (hasta 6 semanas en pacientes con trastorno depresivo mayor) se muestran en la Tabla 8.
Tabla 8: Reacciones adversas en los estudios clínicos agrupados, en adultos,de 6 semanas, controlados con placebo, a dosis fijas de trastorno depresivo mayor (estudios 1 y 2)*
|
Placebo |
REXULTI® |
||||
|
1 mg/día |
2 mg/día |
3 mg/día (N = 229) |
REXULTI todas |
||
|
Trastornos gastrointestinales |
|||||
|
Estreñimiento |
1% |
3% |
2% |
1% |
2% |
|
Trastornos generales y condiciones del sitio de administración |
|||||
|
Fatiga |
2% |
3% |
2% |
5% |
3% |
|
Infecciones e infestaciones |
|||||
|
Nasofaringitis |
2% |
7% |
1% |
3% |
4% |
|
Investigaciones |
|||||
|
Aumento de peso |
2% |
7% |
8% |
6% |
7% |
|
Disminución de cortisol sanguíneo |
1% |
4% |
0% |
3% |
2% |
|
Metabolismo y nutrición |
|||||
|
Aumento del apetito |
2% |
3% |
3% |
2% |
3% |
|
Trastornos del sistema nervioso |
|||||
|
Acatisia |
2% |
4% |
7% |
14% |
9% |
|
Cefalea |
6% |
9% |
4% |
6% |
7% |
|
Somnolencia |
0.5% |
4% |
4% |
6% |
5% |
|
Temblores |
2% |
4% |
2% |
5% |
4% |
|
Mareo |
1% |
1% |
5% |
2% |
3% |
|
Trastornos psiquiátricos |
|||||
|
Ansiedad |
1% |
2% |
4% |
4% |
3% |
|
Inquietud |
0% |
2% |
3% |
4% |
3% |
* Reacciones adversas que ocurrieron en ≥ 2% de los pacientes tratados con REXULTI® y a una incidencia mayor que en pacientes tratados con placebo.
Reacciones adversas relacionadas con la dosis en los estudios clínicos del trastorno depresivo mayor:
En los estudios 1 y 2, entre las reacciones adversas que ocurrieron a una incidencia ≥ 2% en los pacientes tratados con REXULTI® + TAD, la incidencia de acatisia y de inquietud aumentaron con los incrementos en la dosis.
Esquizofrenia:
Adultos: Se evaluó la seguridad de REXULTI® en 852 pacientes adultos (de 18 a 65 años), diagnosticados con esquizofrenia, que participaron en dos estudios clínicos de 6 semanas, controlados con placebo, a dosis fijas en los que REXULTI® se administró en dosis diarias de 1 mg, 2 mg y 4 mg [consultar Farmacocinética y farmacodinamia (Estudios clínicos)].
Reacciones adversas frecuentes:
Las reacciones adversas asociadas con REXULTI® (incidencia de 2% o mayor e incidencia con REXULTI® mayor que la incidencia con placebo) durante los estudios clínicos a corto plazo (hasta 6 semanas) en pacientes adultos con esquizofrenia se muestran en la Tabla 9.
Tabla 9: Reacciones adversas en estudios clínicos de esquizofrenia, agrupados, de 6 semanas, controlados con placebo, a dosis fijas, en pacientes adultos (estudios 3 y 4)*
|
Placebo (N = 368) |
REXULTI® |
||||
|
1 mg/día (N = 120) |
2 mg/día (N = 368) |
4 mg/día (N = 364) |
REXULTI® todas (N = 852) |
||
|
Trastornos gastrointestinales |
|||||
|
Dispepsia |
2% |
6% |
2% |
3% |
3% |
|
Diarrea |
2% |
1% |
3% |
3% |
3% |
|
Investigaciones |
|||||
|
Aumento de peso |
2% |
3% |
4% |
4% |
4% |
|
Aumento de creatinfosfocinasa en sangre |
1% |
4% |
2% |
2% |
2% |
|
Trastornos del sistema nervioso |
|||||
|
Acatisia |
5% |
4% |
5% |
7% |
6% |
|
Temblores |
1% |
2% |
2% |
3% |
3% |
|
Sedación |
1% |
2% |
2% |
3% |
2% |
* Reacciones adversas que ocurrieron en ≥ 2% de los pacientes tratados con REXULTI® y a una incidencia mayor que en pacientes tratados con placebo.
Síntomas extrapiramidales:
Trastorno depresivo mayor: La incidencia de reacciones adversas relacionadas con síntomas extrapiramidales reportadas, excluyendo acatisia, fue de 6% para pacientes tratados con REXULTI® más TAD vs. el 3% para los pacientes tratados con placebo más TAD. La incidencia de eventos de acatisia para los pacientes tratados con REXULTI® más TAD fue de 9% vs. el 2% para los pacientes tratados con placebo más TAD.
En los estudios de trastorno depresivo mayor, de 6 semanas, controlados con placebo, los datos fueron recolectados, objetivamente en la Escala de Valoración de Simpson Angus (SAS) para síntomas extrapiramidales, la Escala de Valoración de Acatisia de Barnes (BARS) para la acatisia y la Puntuación de Movimiento Involuntarios Anormales (AIMS) para discinesia. El cambio medio desde el estado basal a la última visita para los pacientes tratados con REXULTI® más TAD para la Escala de Simpson-Angus, Escala de acatisia de Barnes y la Escala de Movimientos Involuntarios Anormales fue comparable al de los pacientes tratados con placebo. El porcentaje de pacientes que cambió de normal a anormal fue mayor en pacientes tratados con REXULTI® + TAD vs. pacientes tratados con placebo más TAD para la Escala de acatisia de Barnes (4% vs. 0.6%) y la Escala de Simpson Angus (4% versus 3%).
Esquizofrenia: La incidencia de reacciones adversas relacionadas con síntomas extrapiramidales reportadas, excluyendo acatisia, fue de 5% para pacientes tratados con REXULTI® vs. el 4% para los pacientes tratados con placebo. La incidencia de eventos de acatisia para los pacientes tratados con REXULTI® fue de 6% vs. el 5% para los pacientes tratados con placebo.
En los estudios de esquizofrenia, de 6 semanas, controlados con placebo, a dosis fijas, los datos fueron recolectados, objetivamente en la Escala de Valoración de Simpson Angus (SAS) para síntomas extrapiramidales, la Escala de Valoración de Acatisia de Barnes (BARS) para la acatisia y la Puntuación de Movimiento Involuntarios Anormales (AIMS) para discinesia. El cambio medio desde el estado basal a la última visita para los pacientes tratados con REXULTI® para la Escala de Simpson-Angus, Escala de acatisia de Barnes y la Escala de Movimientos Involuntarios Anormales fue comparable al de los pacientes tratados con placebo. El porcentaje de pacientes que cambió de normal a anormal fue mayor en los pacientes tratados con REXULTI® + vs. los pacientes tratados con placebo para la Escala de acatisia de Barnes (2% vs. 1%) y la Escala de Simpson Angus (7% vs. 5%).
Distonía: Los síntomas de distonía se pueden presentar en individuos susceptibles durante los primeros días de tratamiento. Los síntomas distónicos incluyen: espasmo de los músculos del cuello, progresando algunas veces a la opresión de la garganta, dificultad para deglutir, dificultad para respirar y/o protrusión de la lengua. Mientras que estos síntomas pueden ocurrir a dosis bajas, en general se presentan con más frecuencia y con mayor intensidad con alta potencia y a dosis más altas de los fármacos antipsicóticos de primera generación. Se observó un elevado riesgo de distonía aguda en grupos de hombres y jóvenes.
Otras reacciones adversas observadas durante la evaluación previa a la comercialización de REXULTI®:
Otras reacciones adversas (frecuencia ≥ 1% y mayor que la del placebo) dentro de los estudios clínicos a corto plazo, controlados con placebo en pacientes adultos con trastorno depresivo mayor y esquizofrenia se muestran a continuación. La siguiente lista no incluye las reacciones adversas: 1) que ya aparecen en las tablas anteriores o en otros lugares en el etiquetado, 2) para las que la causa del fármaco sea remoto, 3) las que fueron tan generales como poco informativas, 4) las que no se considera que tengan implicaciones clínicamente significativas o 5) las que ocurrieron a una tasa igual o menor que las del placebo.
Trastornos oculares: Visión borrosa.
Trastornos gastrointestinales: Náuseas, boca seca, hipersecreción salival, dolor abdominal, flatulencia.
Infecciones e infestaciones: Infección de vías urinarias.
Investigaciones: Aumento de la prolactina en sangre.
Trastornos musculoesqueléticos y del tejido conectivo: Mialgia.
Trastornos psiquiátricos: Sueños anormales, insomnio.
Trastornos de la piel y del tejido subcutáneo: Hiperhidrosis.
Pacientes pediátricos (13 a 17 años de edad): En un estudio abierto, en curso de 2 años en pacientes pediátricos, de 13 a 17 años con esquizofrenia, en el que se evaluó la seguridad en 194 pacientes, de los cuales 140 recibieron REXULTI® durante al menos 6 meses. Las reacciones adversas notificadas en Ios estudios clínicos, para este grupo de edad fueron generalmente similares a las observadas en pacientes adultos.
Experiencia posterior a la comercialización:
Trastornos del sistema nervioso: Síndrome neuroléptico maligno.
El síndrome neuroléptico maligno ha sido identificado durante el uso posterior a la aprobación de REXULTI®. Debido a que estas reacciones se reportan voluntariamente en una población de tamaño desconocido, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal a la exposición del fármaco.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogénesis, mutagénesis, deterioro de la fertilidad:
Carcinogénesis: Se realizaron estudios de carcinogenicidad de por vida en ratones ICR y ratas SD. Brexpiprazol se administró por vía oral, durante dos años, a ratones machos y hembras a dosis de 0.75, 2 y 5 mg/kg/día (0.9 a 6.1 veces la dosis humana máxima recomendada oral de 4 mg/día con base en mg/m2 área de superficie corporal) y a ratas, machos y hembras, a dosis de 1, 3 y 10 mg/kg y 3, 10 y 30 mg/kg/día, respectivamente (2.4 a 24 y 7.3 a 73 veces la dosis humana máxima recomendada oral, machos y hembras). En ratones hembra, se incrementó la incidencia de adenocarcinoma de la glándula mamaria en todas las dosis y la incidencia del carcinoma adenoescamoso se incrementó en 2.4 y 6.1 veces la dosis humana máxima recomendada. Se observó ningún aumento en la incidencia de tumores en ratones machos. En el estudio con ratas, brexpiprazol no fue carcinógeno en ambos géneros a dosis de hasta 73 veces la dosis humana máxima recomendada.
Se han observado cambios proliferativos y/o neoplásicos en las glándulas mamarias y pituitarias de roedores después de la administración crónica de fármacos antipsicóticos y se considera que están mediados por prolactina. Se mostró el potencial de brexpiprazol para aumentar el nivel de prolactina sérica, tanto en ratones como en ratas. Se desconoce la relevancia para el riesgo humano de los hallazgos de los tumores endocrinos mediada por prolactina en roedores.
Mutagénesis: Brexpiprazol no fue mutagénico cuando se evaluó en el ensayo de mutación reversa bacteriana in vitro (prueba de Ames). Brexpiprazol fue negativo para la actividad clastogénica en el ensayo de micronúcleos in vivo en ratas y no fue genotóxico en el ensayo de síntesis de ADN no programado in vivo/in vitro en ratas. Brexpiprazol fue clastogénico in vitro con células de mamífero, pero sólo a dosis que indujeron citotoxicidad. Con base en el peso de la evidencia, no se considera que brexpiprazol presente un riesgo genotóxico para los seres humanos.
Deterioro de la fertilidad: Se administraron dosis orales de 0.3, 3 o 30 mg/kg/día (0.7, 7.3 y 73 veces la dosis humana máxima recomendada oral sobre una base mg/m2) a ratas hembras antes de aparearse con machos no tratados y continuaron con el tratamiento durante la concepción y la implantación. Se observaron irregularidades en el ciclo estral y disminución de la fertilidad a dosis de 3 y 30 mg/kg/día. También se observó una duración prolongada del apareamiento y aumentó de las pérdidas preimplantación a dosis de 30 mg/kg/día.
Se trató a ratas macho con dosis orales de 3, 10 o 100 mg/kg/día (7.3, 24 y 240 veces la dosis humana máxima recomendada oral sobre una base mg/m2) durante 63 días antes del apareamiento con hembras no tratadas y durante los 14 días de apareamiento. No se observaron diferencias en la duración de los índices de apareamiento o fertilidad en los machos a cualquiera de las dosis de brexpiprazol.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Fármacos que tienen interacciones clínicamente importantes con REXULTI®:
Tabla 10: Interacciones medicamentosas clínicamente importantes con REXULTI®
|
Inhibidores de CYP3A4 fuertes |
|
|
Impacto clínico |
El uso concomitante de REXULTI® con inhibidores fuertes de CYP3A4 aumentó la exposición de brexpiprazol en comparación con el uso de REXULTI® solo [consultar Farmacocinética y farmacodinamia] |
|
Intervención: |
Con el uso concomitante de REXULTI® con un inhibidor fuerte de CYP3A4, reducir la dosis de REXULTI® [consultar Dosis y vía de administración] |
|
Inhibidores de CYP2D6 fuertes* |
|
|
Impacto clínico |
El uso concomitante de REXULTI® con inhibidores fuertes de CYP2D6 aumentó la exposición de brexpiprazol en comparación con el uso de REXULTI® solo [consultar Farmacocinética y farmacodinamia] |
|
Intervención: |
Con el uso concomitante de REXULTI® con un inhibidor fuerte de CYP2D6, reducir la dosis de REXULTI® [consultar Dosis y vía de administración] |
|
Tanto inhibidores de CYP3A4 como inhibidores de CYP2D6 |
|
|
Impacto clínico |
El uso concomitante de REXULTI® con 1) un inhibidor fuerte de CYP3A4 y un inhibidor fuerte de CYP2D6; o 2) un inhibidor moderado de CYP3A4 y un inhibidor fuerte de CYP2D6; o 3) un inhibidor fuerte de CYP3A4 y un inhibidor moderado de CYP2D6; o 4) un inhibidor moderado del CYP3A4 y un inhibidor moderado de CYP2D6, aumentó la exposición de brexpiprazol en comparación con el uso de REXULTI® solo [consultar Farmacocinética y farmacodinamia] |
|
Intervención: |
Con el uso concomitante de REXULTI® con 1) un inhibidor fuerte de CYP3A4 y un inhibidor fuerte de CYP2D6; o 2) un inhibidor moderado de CYP3A4 y un inhibidor fuerte de CYP2D6; o 3) un inhibidor fuerte de CYP3A4 y un inhibidor moderado de CYP2D6; o 4) un inhibidor moderado de CYP3A4 y un inhibidor moderado de CYP2D6, disminuyó la dosis de REXULTI® [consultar Dosis y vía de administración] |
|
Inductores de CYP3A4 fuertes |
|
|
Impacto clínico |
Con el uso concomitante de REXULTI® y un inductor de CYP3A4 fuerte disminuyó la exposición de brexpiprazol en comparación con el uso de REXULTI® solo [consultar Farmacocinética y farmacodinamia] |
|
Intervención: |
Con el uso concomitante de REXULTI® con un inductor de CYP3A4 fuerte aumentó la dosis de REXULTI® [consultar Dosis y vía de administración] |
* En los estudios clínicos que investigan el uso adyuvante de REXULTI® en el tratamiento del trastorno depresivo mayor, la dosis no se ajustó para inhibidores de CYP2D6 fuertes (por ejemplo, paroxetina, fluoxetina). Por lo tanto, los factores de CYP están considerados en las recomendaciones generales de dosificación y REXULTI® se puede administrar sin ajuste de dosis en los pacientes con trastorno depresivo mayor.
Fármacos que no tienen interacciones clínicamente importantes con REXULTI®: Con base en los estudios farmacocinéticos, no es necesario ajustar la dosis de REXULTI® cuando se administra, de manera concomitante, con inhibidores de CYP2B6 (por ejemplo, ticlopidina) o modificadores del pH gástrico (por ejemplo, omeprazol). Además, no es necesario ajustar la dosis para sustratos de CYP2D6 (por ejemplo, dextrometorfano), CYP3A4 (por ejemplo, lovastatina), CYP2B6 (por ejemplo, bupropión), BCRP (por ejemplo, rosuvastatina) o P-gp (por ejemplo, fexofenadina) cuando se administran de manera concomitante con REXULTI®.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No conocidas.
PRECAUCIONES GENERALES:
Aumento de la mortalidad en pacientes de edad avanzada con psicosis relacionada con demencia:
Los pacientes de edad avanzada con psicosis relacionada con demencia tratados con fármacos antipsicóticos tienen mayor riesgo de muerte. Los análisis de 17 estudios clínicos controlados con placebo (duración modal de 10 semanas), en su mayoría, en pacientes que tomaban fármacos antipsicóticos atípicos, reveló un riesgo de muerte, en pacientes tratados con fármacos, de 1.6 a 1.7 veces mayor que el riesgo de muerte en pacientes tratados con placebo. Durante el transcurso de un típico estudio clínico, controlado de 10 semanas, la tasa de mortalidad en pacientes tratados con fármacos fue de aproximadamente 4.5%, en comparación con una tasa de aproximadamente el 2.6% en el grupo placebo. Aunque las causas de muerte fueron variadas, parece que la mayoría de las muertes fueron de naturaleza cardiovascular (por ejemplo, insuficiencia cardiaca, muerte súbita) o infecciosa (por ejemplo, neumonía). REXULTI® no está aprobado para el tratamiento de pacientes con psicosis relacionada con demencia.
Pensamientos y comportamientos suicidas en niños, adolescentes y adultos jóvenes: En los análisis agrupados de los estudios clínicos, controlados con placebo, de fármacos antidepresivos (ISRS y otras clases de antidepresivos) que incluyeron aproximadamente 77,000 pacientes adultos y más de 4,400 pacientes pediátricos, la incidencia de pensamientos y comportamientos suicidas en pacientes menores de 24 años fue mayor en pacientes tratados con antidepresivos que en pacientes tratados con placebo. Las diferencias fármaco-placebo en el número de casos de pensamientos y comportamientos suicidas por 1000 pacientes tratados se presentan en la Tabla 3.
No hubo suicidios en los estudios pediátricos. Hubo suicidios en los estudios en adultos, pero el número no fue suficiente para llegar a alguna conclusión sobre el efecto del fármaco antidepresivo sobre el suicidio.
Tabla 3: Diferencias de riesgo del número de pacientes con pensamientos o comportamientos suicidas en los estudios clínicos controlados con placebo, agrupados, de antidepresivos en pacientes pediátricos* y adultos
|
Intervalo de edad (años) |
Diferencias fármaco-placebo en el número de pacientes con pensamientos o comportamientos suicidas por 1000 pacientes tratados |
|
Aumentos en comparación con el placebo |
|
|
< 18 |
14 pacientes adicionales |
|
18-24 |
5 pacientes adicionales |
|
Disminuciones en comparación con el placebo |
|
|
25-64 |
1 paciente menos |
|
≥ 65 |
6 pacientes menos |
* REXULTI® no está aprobado en pacientes pediátricos con TDM.
Se desconoce si el riesgo de pensamientos y comportamientos suicidas en niños, adolescentes y adultos jóvenes se extiende al uso a largo plazo, es decir, más allá de cuatro meses. Sin embargo, hay evidencia sustancial de los estudios de mantenimiento, controlados con placebo, en adultos con trastorno depresivo mayor, de que los antidepresivos retrasan la recurrencia de la depresión.
Vigilar el agravamiento clínico y la presentación de pensamientos y comportamientos suicidas en todos los pacientes tratados con antidepresivos, especialmente durante los primeros meses de tratamiento farmacológico y en los momentos de cambio de dosis. Aconsejar a los miembros de la familia o a los cuidadores del paciente que estén alertas ante cambios en el comportamiento y avisen de esto al médico. Considerar el cambio de régimen terapéutico, incluyendo la posible suspensión de REXULTI®, en pacientes que experimenten un empeoramiento persistente de la depresión o que presenten la aparición de pensamientos o comportamientos suicidas.
Reacciones adversas cerebrovasculares, incluyendo accidente cerebrovascular en pacientes de edad avanzada con psicosis relacionada con demencia:
En los estudios clínicos, controlados con placebo, en pacientes de edad avanzada con demencia, los pacientes asignados de manera aleatoria al grupo de risperidona, de aripiprazol y de olanzapina tuvieron una mayor incidencia de accidente cerebrovascular y accidente isquémico transitorio, incluyendo el accidente cerebrovascular fatal. REXULTI® no está aprobado para el tratamiento de pacientes con psicosis relacionada con demencia.
Síndrome neuroléptico maligno (SNM): Se ha informado de un complejo de síntomas potencialmente mortales denominado, algunas veces, Síndrome Neuroléptico Maligno (SMN) en asociación con la administración de fármacos antipsicóticos, incluyendo REXULTI®. Las manifestaciones clínicas del Síndrome Neuroléptico Maligno son hiperpirexia, rigidez muscular, alteración del estado mental y evidencia de inestabilidad autonómica. Los signos adicionales pueden incluir un incremento en la creatinfosfocinasa, mioglobinuria (rabdomiólisis) e insuficiencia renal aguda.
Si se sospecha síndrome neuroléptico maligno, suspender inmediatamente REXULTI® y proporcionar tratamiento sintomático intensivo y vigilancia.
Discinesia tardía: La discinesia tardía, un síndrome que consiste en movimientos potencialmente irreversibles, involuntarios y discinéticos, puede desarrollarse en pacientes tratados con fármacos antipsicóticos. El riesgo parece ser más alto entre las personas de edad avanzada, especialmente las mujeres de edad avanzada, pero no es posible predecir cuáles pacientes son más propensos a desarrollar el síndrome. Se desconoce si los fármacos antipsicóticos difieren en su potencial para causar discinesia tardía.
El riesgo de discinesia tardía y la probabilidad de que se vuelva irreversible aumentan con la duración del tratamiento y la dosis acumulada. El síndrome se puede desarrollar después de un periodo de tratamiento, relativamente breve, incluso a dosis bajas. También se puede presentar después de la suspensión del tratamiento.
La discinesia tardía puede remitir, parcial o completamente, si se suspende el tratamiento antipsicótico. El tratamiento antipsicótico, en sí mismo, sin embargo, puede suprimir (o suprimir parcialmente) los signos y síntomas del síndrome, posiblemente, enmascarando el proceso subyacente. Se desconoce el efecto que tiene la supresión sintomática sobre el curso, a largo plazo, de la discinesia tardía.
Dadas estas consideraciones, REXULTI® se debe prescribir de manera, que sea más probable, que se reduzca el riesgo de discinesia tardía. El tratamiento antipsicótico crónico, generalmente, se debe reservar para los pacientes: (1) que padecen una enfermedad crónica que responde a los antipsicóticos; y (2) para quienes los tratamientos alternativos, efectivos y potencialmente menos dañinos no están disponibles o no son apropiados. En pacientes que requieren tratamiento crónico, usar la dosis más baja y la duración más corta, del tratamiento, necesaria para producir una respuesta clínica satisfactoria. Reevaluar periódicamente la necesidad de tratamiento continuo.
Si se presentan signos y síntomas de discinesia tardía en un paciente que toma REXULTI®, se debe considerar la suspensión del fármaco. Sin embargo, algunos pacientes pueden requerir tratamiento con REXULTI® a pesar de la presencia del síndrome.
Cambios metabólicos: Los fármacos antipsicóticos atípicos, incluyendo REXULTI®, han causado cambios metabólicos, como hiperglucemia, diabetes mellitus, dislipidemia y aumento de peso. Aunque se ha demostrado que todos los medicamentos en la clase, hasta la fecha, producen algunos cambios metabólicos, cada medicamento tiene su propio perfil de riesgo específico.
Hiperglucemia y diabetes mellitus: La hiperglucemia, en algunos casos extremos, asociada con cetoacidosis, coma hiperosmolar o muerte, se ha descrito en pacientes tratados con antipsicóticos atípicos. Se han descrito casos de hiperglucemia en pacientes tratados con REXULTI® [consultar Reacciones secundarias y adversas]. Evaluar la glucosa plasmática en ayunas, antes o poco después del inicio de la medicación antipsicótica y supervisar periódicamente durante el tratamiento a largo plazo.
Trastorno depresivo mayor: En los estudios clínicos de dosis fijas, controlados con placebo, de 6 semanas de duración en pacientes adultos con trastorno depresivo mayor, las proporciones de pacientes con cambios en la glucosa en ayunas de normal (< 100 mg/dL) a alta (≥ 126 mg/dL) y de limítrofe (≥ 100 y < 126 mg/dL) a alta, fueron similares en pacientes tratados con REXULTI® y con placebo.
En los estudios abiertos de depresión, a largo plazo, el 5% de los pacientes adultos con glucosa en ayunas, basal normal, experimentaron un cambio a glucosa alta mientras tomaban REXULTI® más un antidepresivo (TAD). El 25% de los pacientes con glucosa en ayunas, limítrofe, experimentaron cambios a glucosa alta. En combinación, el 9% de los pacientes con glucosa en ayunas, normal o limítrofe, experimentaron cambios a glucosa en ayunas alta durante los estudios de depresión a largo plazo.
Esquizofrenia:
Adultos: En los estudios clínicos de dosis fijas, controlados con placebo, de 6 semanas de duración en pacientes adultos con esquizofrenia, las proporciones de pacientes con cambios en la glucosa en ayunas de normal (< 100 mg/dL) a alta (≥ 126 mg/dL) o de limítrofe (≥ 100 y < 126 mg/dL) a alta, fueron similares en pacientes tratados con REXULTI® y con placebo.
En los estudios abiertos de esquizofrenia, a largo plazo, el 8% de los pacientes adultos con glucosa en ayunas, basal normal, experimentó un cambio de normal a alta mientras tomaba REXULTI®, el 17% de los pacientes con glucosa en ayunas limítrofe experimentaron cambios de limítrofe a alta. En combinación, el 10% de los pacientes con glucosa en ayunas, normal o limítrofe, experimentaron cambios a glucosa en ayunas alta durante los estudios de esquizofrenia a largo plazo.
Pacientes pediátricos (13 a 17 años de edad): En el estudio abierto a largo plazo en pacientes pediátricos con esquizofrenia, el 2.7% de los pacientes pediátricos, con glucosa en ayunas basal normal, experimentaron un cambio de normal (< 100 mg/dL) a alto (≥ 126 mg/dL) mientras tomaban REXULTI®.
Dislipidemia: Los antipsicóticos atípicos causan alteraciones adversas en los lípidos. Antes o poco después del inicio de la medicación antipsicótica, obtener un perfil de lípidos en ayunas, basal y vigilar periódicamente durante el tratamiento.
Lípidos:
Trastorno depresivo mayor: En los estudios clínicos, de 6 semanas, controlados con placebo, de dosis fijas, en pacientes adultos con trastorno depresivo mayor, los cambios en colesterol total, colesterol LDL y colesterol HDL en ayunas fueron similares en pacientes tratados con REXULTI ® y con placebo. La tabla 4 muestra las proporciones de pacientes con cambios en triglicéridos, en ayunas.
Tabla 4: Cambios en los triglicéridos en ayunas en los estudios clínicos de 6 semanas, controlados con placebo,
de dosis fijas de trastorno depresivo mayor
|
Proporción de pacientes con cambios desde el estado basal al estado post-basal |
||||
|
|
Placebo |
1 mg/día |
2 mg/día |
3 mg/día |
|
Triglicéridos |
6% |
5% |
13% |
9% |
|
Normal a alto (< 150 mg/dL a ≥ 200 y < 500 mg/dL) |
(15/257)* |
(7/145)* |
(15/115)* |
(13/150)* |
|
Normal/limítrofe a muy alto |
0% |
0% |
0.7% |
0% |
|
(0/309)* |
(0/177)* |
(1/143)* |
(0/179)* |
|
* Denota n/N donde N = el número total de pacientes que tenían una medición basal y al menos un resultado post-basal.
n = número de pacientes con cambio.
En los estudios abiertos, de depresión, a largo plazo, los cambios en el colesterol basal, en ayunas, de normal a alto se reportaron en 9% (colesterol total), 3% (colesterol LDL) y cambios en el estado basal de normal a bajo se reportaron en el 14% (colesterol HDL) de los pacientes que tomaban REXULTI®. De pacientes con triglicéridos basales normales, 17% experimentaron cambios a alto y 0.2% experimentaron cambios a muy alto. En combinación, el 0.6% de los pacientes con triglicéridos en ayunas, normal o limítrofe, experimentaron cambios a triglicéridos en ayunas, muy alto, durante los estudios de depresión a largo plazo.
Esquizofrenia:
Adultos: En los estudios clínicos, de 6 semanas, controlados con placebo, de dosis fijas, en pacientes adultos con esquizofrenia, los cambios en colesterol total, colesterol LDL y colesterol HDL en ayunas fueron similares en pacientes tratados con REXULTI® y con placebo. La Tabla 5 muestra las proporciones de pacientes con cambios en triglicéridos, en ayunas.
Tabla 5: Cambios en los triglicéridos, en ayunas, en los estudios clínicos de 6 semanas,
controlados con placebo, de dosis fijas de esquizofrenia
|
Proporción de pacientes con cambios desde el estado basal al estado post-basal |
||||
|
|
Placebo |
1 mg/día |
2 mg/día |
3 mg/día |
|
Triglicéridos |
6% |
10% |
8% |
10% |
|
Normal a alto (< 150 mg/dL a ≥ 200 y < 500 mg/dL) |
(15/253)* |
(7/72)* |
(19/232)* |
(22/226)* |
|
Normal/limítrofe a muy alto (< 200 mg/dL a ≥ 500 mg/dL) |
0% |
0% |
0% |
0% |
|
(0/303)* |
(0/94)* |
(1/283)* |
(0/283)* |
|
* Denota n/N donde N = el número total de pacientes que tenían una medición basal y al menos un resultado post-basal.
n = número de pacientes con cambio.
En los estudios abiertos, de esquizofrenia, a largo plazo, los cambios en el colesterol basal, en ayunas, de normal a alto se reportaron en 6% (colesterol total), 2% (colesterol LDL) y cambios en el estado basal, de normal a bajo, se reportaron en el 17% (colesterol HDL) de los pacientes adultos que tomaban REXULTI®. De los pacientes con triglicéridos basales normales, 13% experimentaron cambios a alto, y 0.4% experimentaron cambios a triglicéridos muy altos. En combinación, el 0.6% de los pacientes con triglicéridos en ayunas, normal o limítrofe, experimentaron cambios a triglicéridos en ayunas, muy alto, durante los estudios de esquizofrenia a largo plazo.
Pacientes pediátricos (13 a 17 años de edad): En el estudio abierto, a largo plazo, en pacientes pediátricos con esquizofrenia, se informaron cambios en el nivel basal de colesterol total en ayunas de normal a alto (< 170 a ≥ 200 mg/dL) en el 7% de los pacientes que tomaban REXULTI® y se informaron cambios en eI nivel basal de colesterol HDL de normal a bajo (≥ 40 a < 40 mg/dL) en el 12.9% de los pacientes que tomaban REXULTI®. De los pacientes con triglicéridos basales normales, el 8.5% experimentó cambios de normal a alto (< 150 a ≥ 200 mg/dL).
Aumento de peso: Se observó aumento de peso en pacientes tratados con antipsicóticos atípicos, incluyendo REXULTI®. Vigilar el peso en el estado basal y en adelante, con frecuencia.
Trastorno depresivo mayor: La Tabla 6 muestra datos de ganancia de peso en la última visita y un porcentaje de pacientes adultos con ≥ 7% de aumento de peso al final de los estudios clínicos de 6 semanas, controlados con placebo, de dosis fijas en pacientes con trastorno depresivo mayor.
Tabla 6: Aumento de peso en los estudios clínicos, de 6 semanas, controlados con placebo, a dosis fijas de trastorno depresivo mayor
|
Placebo n = 407 |
1 mg/día n = 225 |
2 mg/día n = 187 |
3 mg/día n = 228 |
|
|
Cambio medio desde el estado basal (kg) a la última visita |
||||
|
Todos los pacientes |
+0.3 |
+1.3 |
+1.6 |
+1.6 |
|
Proporción de pacientes con ≥ 7% de aumento en el peso (kg) en cualquier visita (*n/N) |
||||
|
2% (8/407)* |
5% (11/225)* |
5% (9/187)* |
2% (5/228)* |
|
* N = el número total de pacientes que tenían una medición basal y al menos un resultado post-basal.
n = número de pacientes con un cambio ≥ 7%.
En los estudios abiertos de depresión, a largo plazo, 4% de los pacientes suspendieron el fármaco debido al aumento de peso. REXULTI® se asoció con un cambio medio desde el estado basal en el peso de 2.9 kg en la semana 26 y de 3.1 kg en la semana 52. En los estudios abiertos de depresión, a largo plazo, 30% de los pacientes presentaron un aumento ≥ 7% en el peso y 4% presentó una disminución de ≥ 7% en el peso.
Esquizofrenia: La Tabla 7 muestra datos de ganancia de peso en la última visita y un porcentaje de pacientes adultos con ≥ 7% de aumento de peso al final de los estudios clínicos de 6 semanas, controlados con placebo, de dosis fijas en pacientes con esquizofrenia.
Tabla 7: Aumento de peso en los estudios clínicos, de 6 semanas, controlados con placebo, a dosis fijas de esquizofrenia
|
Placebo n = 362 |
1 mg/día n = 120 |
2 mg/día n = 362 |
4 mg/día n = 362 |
|
|
Cambio medio desde el estado basal (kg) a la última visita |
||||
|
Todos los pacientes |
+0.2 |
+1.0 |
+1.2 |
+1.2 |
|
Proporción de pacientes con ≥ 7% de aumento en el peso (kg) en cualquier visita (*n/N) |
||||
|
4% (15/362)* |
10% (12/120)* |
11% (38/362)* |
10% (37/362)* |
|
* N = el número total de pacientes que tenían una medición basal y al menos un resultado post-basal.
n = número de pacientes con un cambio ≥ 7%.
En los estudios abiertos de esquizofrenia, a largo plazo, 0.6% de los pacientes suspendieron el fármaco debido al aumento de peso. REXULTI® se asoció con un cambio medio de línea de base en el peso de 2.9 kg en la semana 26 y 3.1 kg en la semana 52. En los estudios abiertos de esquizofrenia, a largo plazo, 20% de los pacientes presentaron un aumento ≥ 7% en el peso y 10% presentó una disminución ≥ 7% en el peso.
Pacientes pediátricos (13 a 17 años de edad): En el estudio abierto, a largo plazo, en pacientes pediátricos con esquizofrenia, el 0.5% de los pacientes interrumpieron el tratamiento debido al aumento de peso. El incremento medio de peso, desde el inicio del estudio abierto hasta la última visita fue de 3.8 kg. Para ajustar el crecimiento normal, se derivaron las puntuaciones z (medidas en desviaciones estándar [SD]), que se normalizan para el crecimiento natural de niños y adolescentes en comparación con estándares de población equiparados por edad y género. Un cambio en la puntuación z < 0.5 SD se considera no significativo desde el punto de vista clínico. En este estudio clínico, abierto, el cambio medio en la puntuación z, desde el valor basal hasta la última visita fue de 0.10 SD para el peso corporal, mientras que el 20% de los pacientes tuvo un aumento en la puntuación z del peso corporal, ajustado por edad y género de al menos 0.5 SD desde el valor basal. Cuando se trata a pacientes pediátricos, se debe controlar y evaluar el aumento de peso contra el esperado para un crecimiento normal.
Leucopenia, neutropenia y agranulocitosis: Durante el tratamiento con agentes antipsicóticos se han reportado leucopenia y neutropenia. Se ha reportado agranulocitosis (incluyendo casos fatales) con otros agentes en esta clase.
Posibles factores de riesgo de leucopenia y neutropenia son recuento de leucocitos o recuento absoluto de neutrófilos bajos preexistentes y antecedentes de leucopenia o neutropenia inducida por fármacos. En pacientes con un recuento de leucocitos o recuento absoluto de neutrófilos preexistente bajo o con antecedentes de leucopenia o neutropenia inducida por fármacos, realizar un recuento sanguíneo completo, frecuentemente, durante los primeros meses de terapia. En tales pacientes, considerar la suspensión del REXULTI® a la primera señal de una disminución, clínicamente significativa, de los leucocitos en ausencia de otros factores causales.
Vigilar la presentación de fiebre u otros síntomas o signos de infección en los pacientes con neutropenia, clínicamente significativa y tratarlos con prontitud. Suspender REXULTI® en pacientes con recuento absoluto de neutrófilos < 1000/mm3 y seguir sus leucocitos hasta la recuperación.
Hipotensión ortostática y síncope: Los antipsicóticos atípicos causan hipotensión ortostática y síncope. En general, el riesgo es mayor durante el inicio del tratamiento y al aumentar la dosis. En los estudios clínicos a corto plazo, controlados con placebo de REXULTI® más TAD en los pacientes adultos con trastorno depresivo mayor, la incidencia de reacciones adversas relacionadas con la hipotensión ortostáticos en pacientes tratados con REXULTI® más TAD en comparación con los pacientes que recibieron placebo más TAD incluyeron: mareos (2% vs. 2%) e hipotensión ortostática (0.1% vs. 0%). En los estudios clínicos a corto plazo, controlados con placebo de REXULTI® en los pacientes adultos con esquizofrenia, la incidencia de reacciones adversas relacionadas con la hipotensión ortostática en pacientes tratados con REXULTI® en comparación con los pacientes que recibieron placebo incluyeron: mareos (2% vs. 2%), hipotensión ortostática (0.4% vs. 0.2%) y síncope (0.1% versus 0%).
Los signos vitales ortostáticos deben ser vigilados en pacientes que son vulnerables a la hipotensión (por ejemplo, pacientes de edad avanzada, pacientes con deshidratación, hipovolemia, tratamiento concomitante con medicación antihipertensiva), pacientes con enfermedad cardiovascular conocida (historia de infarto de miocardio, cardiopatía isquémica, insuficiencia cardiaca o anormalidades de la conducción) y pacientes con enfermedad cerebrovascular. REXULTI® no se ha evaluado en pacientes con un historial reciente de infarto de miocardio o enfermedad cardiovascular inestable. Tales pacientes se excluyeron de los estudios clínicos previos a la comercialización.
Caídas: Los antipsicóticos, incluyendo REXULTI®, pueden causar somnolencia, hipotensión postural, inestabilidad motora y sensorial, lo que puede conducir a caídas y, en consecuencia, fracturas u otras lesiones. Para los pacientes con enfermedades, condiciones o medicamentos que podrían exacerbar estos efectos, completar las evaluaciones de riesgo de caída cuando se inicia el tratamiento con antipsicóticos y de manera recurrente para pacientes con tratamiento antipsicótico a largo plazo.
Convulsiones: Al igual que otros antipsicóticos, REXULTI® puede causar convulsiones. Este riesgo es mayor en pacientes con antecedentes de convulsiones o con condiciones que disminuyen el umbral de la convulsión. Las condiciones que disminuyen el umbral de la convulsión pueden ser más frecuentes en pacientes de edad avanzada.
Desregulación de la temperatura corporal: Los antipsicóticos atípicos pueden interrumpir la capacidad del cuerpo para reducir la temperatura central del cuerpo. El ejercicio extenuante, la exposición al calor extremo, la deshidratación y los medicamentos anticolinérgicos pueden contribuir a una elevación en la temperatura central del cuerpo; utilizar REXULTI® con precaución en pacientes que experimenten estas condiciones.
Disfagia: La aspiración y la dismotilidad esofágica se han asociado con el uso de fármacos antipsicóticos. Los fármacos antipsicóticos, incluyendo REXULTI®, deben usarse con precaución en pacientes con riesgo de aspiración.
Trastornos del control de impulsos/comportamientos compulsivos: Muy raramente se han notificado reportes posteriores a la comercialización de los trastornos de control de los impulsos, incluido el juego en pacientes tratados con brexpiprazol y otros antipsicóticos con actividad agonista parcial en los receptores de dopamina. Los pacientes con antecedentes de trastornos de control de los impulsos pueden estar en mayor riesgo y deben controlarse cuidadosamente. Debido a que los pacientes pueden no reconocer estas conductas como anormales, es importante que los prescriptores pregunten a los pacientes o sus cuidadores específicamente sobre el desarrollo de nuevos o aumento de trastornos de control de los impulsos u otras conductas compulsivas mientras reciben tratamiento con brexpiprazol. Cabe señalar que los síntomas de control de los impulsos pueden estar asociados con el trastorno subyacente. Los comportamientos compulsivos pueden provocar daños al paciente y a otros si no se los reconoce.
Potencial de deterioro cognitivo y motor: REXULTI®, al igual que otros antipsicóticos, tiene el potencial para deteriorar el juicio, el pensamiento o las habilidades motoras. En los estudios clínicos de 6 semanas, controlados con placebo, en pacientes con trastorno depresivo mayor, se reportó somnolencia (incluyendo sedación e hipersomnia) en el 4% de los pacientes tratados con REXULTI® más TAD en comparación con el 1% de pacientes con placebo + TAD.
En los estudios clínicos de 6 semanas, controlados con placebo, en pacientes adultos con esquizofrenia, se reportó somnolencia (incluyendo sedación e hipersomnia) en el 5% de los pacientes tratados con REXULTI® en comparación con el 3% de los pacientes con placebo.
Los pacientes deben ser advertidos acerca de operar maquinaria peligrosa, incluyendo vehículos de motor, hasta que tengan una certeza razonable de que la terapia con REXULTI® no les afecta de manera adversa.
Población especial:
Uso pediátrico: La seguridad y efectividad de REXULTI®, para el tratamiento de la esquizofrenia, se han establecido en pacientes pediátricos de 13 años de edad y mayores. El uso de REXULTI® en esta población está respaldado por evidencia de estudios, adecuados y bien controlados, en adultos con esquizofrenia, datos farmacocinéticos de pacientes adultos y pediátricos y datos de seguridad en pacientes pediátricos de 13 a 17 años de edad [consultar Precauciones generales, Reacciones secundarias y adversas, y Farmacocinética y farmacodinamia].
Trastorno depresivo mayor: No se ha establecido la seguridad y efectividad en pacientes pediátricos con trastorno depresivo mayor.
Los antidepresivos aumentaron el riesgo de pensamientos y comportamientos suicidas en pacientes pediátricos.
Uso geriátrico: Los estudios clínicos de eficacia de REXULTI® no incluyeron pacientes mayores de 65 años, para determinar si ellos responden de manera diferente a los pacientes más jóvenes. En general, la selección de dosis, para un paciente de edad avanzada, se debe elegir con precaución, de manera usual comenzando en el extremo inferior del intervalo de dosificación, reflejando la mayor frecuencia de disminución de la función hepática, renal y cardiaca, enfermedades concomitantes y otros tratamientos farmacológicos.
Con base en los resultados de los estudios clínicos de seguridad, tolerabilidad y farmacocinética, la farmacocinética de la administración oral una vez al día de brexpiprazol (hasta 3 mg/día durante 14 días) como terapia adyuvante en el tratamiento de los pacientes de edad avanzada (70 a 85 años, N = 11) con trastorno depresivo mayor fueron comparables a los observados en pacientes adultos con trastorno depresivo mayor.
Los fármacos antipsicóticos aumentan el riesgo de muerte en los pacientes de edad avanzada con psicosis relacionada con demencia. REXULTI® no está aprobado para el tratamiento de pacientes con psicosis relacionada con demencia.
Metabolizadores pobres de CYP2D6: Se recomienda un ajuste de dosis en metabolizadores pobres de CYP2D6 conocidos, ya que estos pacientes tienen concentraciones más altas de brexpiprazol que los metabolizadores normales de CYP2D6. Aproximadamente 8% de los caucásicos y 3 a 8% de los estadounidenses negro/africanos no pueden metabolizar los sustratos de CYP2D6 y se clasifican como metabolizadores pobres [consultar Dosis y vía de administración, Farmacocinética y farmacodinamia].
Enfermedad hepática: Reducir la dosis máxima recomendada en pacientes con enfermedad hepática de moderada a grave (puntuación Child-Pugh 27). Los pacientes con enfermedad hepática, de moderada a grave (puntuación de Child-Pugh 27) tuvieron generalmente una mayor exposición a brexpiprazol que los pacientes con función hepática normal [consultar Farmacocinética y farmacodinamia]. Una mayor exposición puede aumentar el riesgo de reacciones adversas asociadas con REXULTI® [consultar Dosis y vía de administración].
Enfermedad renal: Reducir la dosis máxima recomendada en pacientes con enfermedad renal, moderada, grave o en etapa terminal (CLcr < 60 mL/minuto). Los pacientes con función renal deteriorada (CLcr < 60 mL/minuto) tuvieron mayor exposición a brexpiprazol que los pacientes con función renal normal [consultar Farmacocinética y farmacodinamia]. Una mayor exposición puede aumentar el riesgo de reacciones adversas asociadas con REXULTI® [consultar Dosis y vía de administración].
Otras poblaciones específicas: No se requiere ajustar la dosis de REXULTI® con base en el género, raza o estatus de tabaquismo de un paciente [consultar Farmacocinética y farmacodinamia].
Abuso y dependencia del fármaco:
Sustancia controlada: REXULTI ® no es una sustancia controlada.
Abuso: Los animales que a los que se les permitió el acceso a REXULTI® no se autoadministraron el fármaco, lo que sugiere que REXULTI® no tiene propiedades adictivas.
Dependencia: Los seres humanos y los animales que recibieron la administración crónica de REXULTI® no mostraron signos de abstinencia después de suspender el fármaco. Esto sugiere que REXULTI® no produce dependencia física.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Tratamiento adyuvante del trastorno depresivo mayor (adultos): La dosis de inicio recomendada para REXULTI® como tratamiento adyuvante del trastorno depresivo mayor en adultos es 0.5 mg o 1 mg una vez al día, tomada por vía oral, con o sin alimentos [consultar Farmacocinética y farmacodinamia].
Ajustar la dosis, de 1 mg una vez al día, con incrementos posteriores hasta alcanzar la dosis objetivo de 2 mg una vez al día. Se debe incrementar la dosis en intervalos semanales, con base en la tolerabilidad y la respuesta clínica del paciente. La dosis diaria máxima recomendada es de 3 mg. Reevaluar periódicamente para determinar la necesidad continua y la dosis apropiada para el tratamiento.
Tratamiento de la esquizofrenia (adultos y pacientes pediátricos 13 a 17 años):
Adultos: La dosis de inicio recomendada de REXULTI® para el tratamiento de la esquizofrenia en adultos es 1 mg una vez al día, los días 1 a 4, tomada por vía oral, con o sin alimentos [consultar Farmacocinética y farmacodinamia].
Ajustar la dosis a 2 mg una vez al día, del día 5 hasta el día 7, e incrementar a 4 mg el día 8 con base en la tolerabilidad y la respuesta clínica del paciente. La dosis objetivo recomendada de REXULTI® es de 2 mg a 4 mg, una vez al día. La dosis diaria máxima recomendada es de 4 mg.
Pacientes pediátricos (13 a 17 años): La dosis de inicio recomendada de REXULTI® para el tratamiento de la esquizofrenia en pacientes pediátricos (13 a 17 años) es 0.5 mg una vez al día, los días 1 a 4, tomada por vía oral, con o sin alimentos [consultar Farmacocinética y farmacodinamia].
Ajustar la dosis a 1 mg una vez al día, del día 5 hasta el día 7, e incrementar a 2 mg el día 8 con base en la tolerabilidad y la respuesta clínica del paciente. Semanalmente se pueden realizar incrementos de 1 mg. La dosis objetivo recomendada de REXULTI® es de 2 mg a 4 mg, una vez al día. La dosis diaria máxima recomendada es de 4 mg.
Ajustes de la dosis para la enfermedad hepática:
Para pacientes con enfermedad hepática moderada a severa (puntuación Child-Pugh ≥ 7), la dosis máxima recomendada es de 2 mg, una vez al día, para pacientes con trastorno depresivo mayor y 3 mg, una vez al día, para pacientes con esquizofrenia [consultar Precauciones generales, Farmacocinética y farmacodinamia].
Ajustes de la dosis para la enfermedad renal: Para pacientes con enfermedad renal moderada, severa o en etapa terminal (depuración de creatinina Clcr < 60 mL/minuto), la dosis máxima recomendada es de 2 mg, una vez al día, para pacientes con trastorno depresivo mayor y 3 mg, una vez al día, para pacientes con esquizofrenia [consultar Precauciones generales, Farmacocinética y farmacodinamia].
Modificaciones de la dosis para metabolizadores pobres de CYP2D6 y para uso concomitante con inhibidores o inductores CYP: Se recomiendan ajustes de la dosis en pacientes metabolizadores pobres de citocromo P450 (CYP) 2D6 conocidos y en pacientes que toman inhibidores de CYP3A4 o inhibidores de CYP2D6 o inductores fuertes de CYP3A4 concomitantes (ver tabla 11). Si se suspende el fármaco coadministrado, ajustar la dosis de REXULTI® a su nivel original. Si se suspende el inductor de CYP3A4 coadministrado, reducir la dosis REXULTI® al nivel original en un periodo de 1 a 2 semanas [consultar Interacciones medicamentosas y de otro género, Farmacocinética y farmacodinamia].
Tabla 11: Ajustes de la dosis de REXULTI® para metabolizadores pobres de CYP2D6 y uso concomitante con inhibidores de CYP2D6 y CYP3A4 y/o inductores de CYP3A4
|
Factores |
Dosis ajustada de REXULTI® |
|
Metabolizadores pobres de CYP2D6 |
|
|
Metabolizadores pobres de CYP2D6 |
Administrar la mitad de la dosis usual |
|
Metabolizadores pobres de CYP2D6 conocidos que toman inhibidores de CYP3A4 fuertes/moderados |
Administrar un cuarto de la dosis usual |
|
Pacientes que toman inhibidores de CYP2D6 y/o inhibidores de CYP3A4 |
|
|
Inhibidores de CYP2D6 fuertes* |
Administrar la mitad de la dosis usual |
|
Inhibidores de CYP3A4 fuertes |
Administrar la mitad de la dosis usual |
|
Inhibidores de CYP2D6 fuertes/moderados con inhibidores CYP3A4 fuertes/moderados |
Administrar un cuarto de la dosis usual |
|
Pacientes que toman inductores de CYP3A4 |
|
|
Inductores de CYP3A4 fuertes |
Doble de la dosis usual durante 1 a 2 semanas |
* En los estudios clínicos que investigan el uso adyuvante de REXULTI® en el tratamiento del trastorno depresivo mayor, la dosis no se ajustó para inhibidores de CYP2D6 fuertes (por ejemplo, paroxetina, fluoxetina). Por lo tanto, los factores de CYP están considerados en las recomendaciones generales de dosificación y REXULTI® se puede administrar sin ajuste de dosis en los pacientes con trastorno depresivo mayor.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
La experiencia de los estudios clínicos acerca de sobredosis en humanos con REXULTI® es limitada.
El tratamiento de la sobredosis debe concentrarse en una terapia de soporte, manteniendo una adecuada aireación, oxigenación y ventilación y tratamiento sintomático. Es necesario que el paciente continúe con una estrecha supervisión y vigilancia clínica hasta su recuperación.
Carbón: El carbón activado y el sorbitol (50 g/240 mL) orales, administrados una hora después de ingerir brexpiprazol oral, disminuyeron la Cmáx y el área bajo la curva (AUC) de brexpiprazol aproximadamente de 5% a 23% y de 31% a 39%, respectivamente; sin embargo, la información disponible sobre el potencial terapéutico del carbón activado en el tratamiento de una sobredosis con REXULTI® es insuficiente.
Hemodiálisis: No hay información sobre el efecto de la hemodiálisis en el tratamiento de una sobredosis con REXULTI®; no es probable que la hemodiálisis sea útil porque el brexpiprazol se une fuertemente a las proteínas plasmáticas.
El manejo de la sobredosis debe consistir en tratar los síntomas clínicos y la monitorización relevante. Se recomienda el seguimiento médico en un entorno especializado.
PRESENTACIONES:
REXULTI® 0.25 mg y 0.5 mg o 1 mg: Caja de cartón con 7 tabletas en envase de burbuja e instructivo anexo. REXULTI® 1 mg: Caja de cartón con 10 tabletas en envase de burbuja e instructivo anexo.
REXULTI® 1 mg, 2 mg, 3 mg y 4 mg: Caja de cartón con 14 o 28 tabletas en envase de burbuja e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 30 °C. Consérvese la caja bien cerrada.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje al alcance de los niños. No se administre en menores de 13 años. No se use durante el embarazo o lactancia. Literatura exclusiva para médicos. Este medicamento puede producir somnolencia y afectar el estado de alerta, deberá tener precaución al conducir vehículos u operar maquinaria pesada durante su uso.
Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y SafetyLuMexico@lundbeck.com
Representante Legal en México:
LUNDBECK MÉXICO, S.A. de C.V.
Avenida Insurgentes Sur 1457, Piso 14 de Torre Manacar, Col. Insurgentes Mixcoac, C.P. 03920,
Benito Juárez, Ciudad de México, México.
Reg. Núm. 210M2018 SSA IV