
SOMATULINE AUTOGEL
LANREÓTIDA
Solución inyectable
1 Caja, 1 Jeringa(s) prellenada(s), 1 Dosis, 60 Miligramos
1 Caja, 1 Jeringa(s) prellenada(s), 1 Dosis, 90 Miligramos
1 Caja, 1 Jeringa(s) prellenada(s), 1 Dosis, 120 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada JERINGA PRELLENADA contiene:
|
Acetato de lanreótida equivalente a de lanreótida |
60 mg |
90 mg |
120 mg |
|
Vehículo c.b.p. |
0.5 mL* |
0.5 mL* |
0.5 mL* |
* Cada jeringa prellenada contiene una solución sobresaturada de acetato de lanreótida que corresponde a 0.246 mg de lanreótida base/mg de solución, que equivale a una dosis de 60 mg, 90 mg y 120 mg de lanreótida respectivamente.
INDICACIONES TERAPÉUTICAS: SOMATULINE® AUTOGEL® está indicado en el tratamiento a largo plazo de pacientes con acromegalia, cuando los niveles de la hormona de crecimiento (GH) y/o de factor de crecimiento-1 similar a la insulina (IGF-1) se mantienen anormales después de la cirugía y/o la radioterapia y en los pacientes que por su condición clínica requieren de tratamiento farmacológico. El objetivo del manejo de la acromegalia con este medicamento es reducir los niveles de GH y de IGF-1 y, en la medida de lo posible, normalizar sus valores.
SOMATULINE® AUTOGEL® mejora el cuadro sintomatológico asociado a la acromegalia.
SOMATULINE® AUTOGEL® también está indicado para el tratamiento de los tumores neuroendocrinos para el manejo de síntomas asociados.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Hormonas anticrecimiento, código ATC: H01C B03.
La Lanreótida Autogel, es un octapéptido análogo de la somatostatina humana. Al igual que la somatostatina natural, la Lanreótida Autogel es un inhibidor de diversas funciones endocrinas, neuroendocrinas, exocrinas y paracrinas. Presenta una elevada afinidad de fijación a los receptores de la somatostatina humana (SSTR) 2, 3 y 5; y menos afinidad por los SSTR 1 y 4. Se considera que la actividad en los SSTR 2 y 5 es el principal mecanismo responsable de la inhibición de la GH. La lanreótida es mucho más activo que la somatostatina natural y presenta un efecto de acción mucho más prolongado.
Al igual que la somatostatina, la lanreótida presenta una acción antisecretora exocrina general inhibiendo la secreción basal de la motilina, péptido inhibidor gástrico y polipéptido pancreático; sin embargo, no tiene efectos significativos sobre la secretina producida durante el ayuno ni sobre la secreción de gastrina. La lanreótida inhibe notablemente los aumentos de flujo sanguíneo de la arteria mesentérica superior y el flujo sanguíneo venoso portal inducidos por las comidas. La lanreótida disminuye de manera significativa la secreción del yeyuno estimulada tanto por la prostaglandina E-1, como por agua, sodio, potasio y cloro. Además, en los pacientes acromegálicos con tratamiento prolongado reduce los niveles de prolactina.
Propiedades farmacocinéticas: Los parámetros farmacocinéticos de la lanreótida, después de su administración intravenosa a voluntarios sanos indicaron una distribución vascular limitada, con un volumen de distribución en estado de equilibrio de 13 L. La depuración total se alcanza a 20 L/h, la vida media terminal fue de 2,5 horas, y el tiempo medio de residencia fue de 0,68 horas.
Después de una inyección subcutánea única de SOMATULINE® AUTOGEL® 60 mg en voluntarios sanos, se alcanzó una concentración máxima en suero (Cmáx) de 5,8 ± 4 ng/mL a las 6 horas; seguida por una disminución lenta (tiempo medio de residencia: 30 ± 6 días, vida media aparente: 33 ± 14 días). La biodisponibilidad absoluta fue de 63 ± 10%.
Después de una inyección intramuscular única de SOMATULINE® AUTOGEL® 60 mg en voluntarios sanos, se alcanzó una concentración máxima en suero (Cmáx) de 6,8 ± 3 ng/mL a las 15 horas; seguida por una disminución lenta (tiempo medio de residencia: 23 ± 11 días, vida media aparente: 23 ± 9 días). La biodisponibilidad absoluta fue de 79 ± 10%.
Por lo tanto, la vía de administración (subcutánea o intramuscular) no demuestra tener una influencia notable sobre el perfil farmacocinético de la lanreótida.
Después de una inyección intramuscular única de SOMATULINE® AUTOGEL® 90 mg en voluntarios sanos, se alcanzó una concentración máxima en suero (Cmáx) de 9,8 ± 5 ng/mL a las 10 horas; seguida por una disminución lenta (tiempo medio de residencia: 26 ± 4 días, vida media aparente: 31 ± 16 días). La biodisponibilidad absoluta fue de 58 ± 10%.
Después de una inyección intramuscular única de SOMATULINE® AUTOGEL® 120 mg en voluntarios sanos, se alcanzó una concentración máxima en suero (Cmáx) de 12,8 ± 7 ng/mL a las 16 horas; seguida por una disminución lenta (tiempo medio de residencia: 29 ± 3 días, vida media aparente: 28 ± 6 días). La biodisponibilidad absoluta fue de 55 ± 10%.
Por lo tanto, la concentración sérica de Lanreótida después de la administración intramuscular de SOMATULINE® AUTOGEL® 60, 90 y 120 mg muestra en representación semilogarítmica, un perfil de liberación de lanreótida de primer orden.
Los niveles séricos mínimos de Lanreótida obtenidos después de tres inyecciones subcutáneas profundas de SOMATULINE® AUTOGEL® 60, 90 y 120 mg aplicadas cada 28 días son similares a los niveles séricos mínimos en estado de equilibrio de Lanreótida hallados en los pacientes acromegálicos tratados previamente con administraciones intramusculares de micropartículas de liberación prolongada de lanreótida 30 mg (Somatuline L.P.) cada 7, 10 ó 14 días.
CONTRAINDICACIONES: Hipersensibilidad a la somatostatina u otros péptidos relacionados o a cualquiera de los excipientes.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: Estudios en animales no muestran evidencia de efectos teratógenos durante la organogénesis asociados a la lanreótida.
El número de exposiciones durante la gestación es muy limitado. Aunque lanreótida puede ser administrada en mujeres embarazadas, sólo si es claramente necesario de acuerdo con el criterio y evaluación riesgo-beneficio del médico tratante.
Lactancia: No es conocido si la lanreótida es excretada por la leche materna.
Como existen muchos fármacos que se excretan por la leche materna, deben tomarse precauciones durante la administración de lanreótida durante la lactancia.
Fertilidad: Se ha observado disminución de la fertilidad en ratas hembra durante la inhibición de la secreción de la hormona de crecimiento en dosis superiores a las alcanzadas en humanos a dosis terapéuticas.
REACCIONES SECUNDARIAS Y ADVERSAS: Los efectos secundarios reportados por pacientes que sufren acromegalia tratados con lanreótida en estudios clínicos se enlistan a continuación con la siguiente clasificación: muy común (≥ 1/10); común (≥ 1/100 a < 1/10); no común (≥1/1,000 a < 1/100).
Las reacciones esperadas como reacción adversa al tratamiento con lanreótida son:
Reacciones en el sitio de la inyección (dolor, nódulos e induraciones) ALAT aumentada, ASAT anormal, ALAT anormal, bilirrubinas séricas aumentadas, glucosa sérica aumentada, hemoglobina glucosilada aumentada, disminución de peso:
No común: ASAT aumentada, fosfatasa alcalina aumentada, bilirrubinas séricas anormales, sodio sérico disminuido.
Trastornos gastrointestinales:
Común: diarrea y dolor abdominal (usualmente transitorios, de leve a moderada intensidad), colelitiasis (comúnmente asintomática).
Trastornos cardiacos:
Común: bradicardia sinusal.
Trastornos del sistema nervioso:
Común: vértigo, cefalea.
Trastornos gastrointestinales:
Muy común: diarrea, heces blandas, dolor abdominal.
Común: náuseas, vómito, dispepsia, flatulencias, malestar abdominal, distensión.
No Común: acolia.
Trastornos del apéndice y la piel:
No común: alopecia, hipotricosis.
Trastornos del metabolismo y nutrición:
Común: hipoglucemia.
Poco común: diabetes mellitus, hipercalcemia.
Trastornos vasculares:
No común: sofocos.
Trastornos generales y reacciones del sitio de aplicación:
Común: fatiga, reacciones en el sitio de inyección (dolor, induración, nódulos, prurito).
No Común: astenia.
Trastornos hepatobiliares:
Muy común: colelitiasis.
Común: dilatación biliar.
Trastornos psiquiátricos:
No Común: insomnio.
Experiencia Post-comercialización: No se ha identificado otra información relevante además de reportes ocasionales de pancreatitis.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: En estudios de bioensayos carcinogénicos en ratas y ratones, no se observaron cambios neoplásicos sistémicos a dosis superiores a las alcanzadas en humanos a dosis terapéuticas.
Se observó aumento de la incidencia de tumores subcutáneos en los sitios de inyección probablemente debido a la mayor frecuencia en la dosis de los animales (diaria) en comparación con la dosificación mensual en los seres humanos y por lo tanto no pueden ser clínicamente relevantes.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Los efectos gastrointestinales de SOMATULINE® AUTOGEL® pueden reducir la absorción intestinal de medicamentos administrados concomitantemente incluyendo la ciclosporina A.
No es probable la interacción con fármacos de elevada unión a proteínas plasmáticas, dada la moderada unión a las proteínas séricas de Lanreótida Autogel® (fijación sérica promedio: 78%).
Efectos sobre la capacidad de conducir y utilizar maquinaria: No se han descrito efectos de SOMATULINE® AUTOGEL® sobre las habilidades para manejar o el uso de máquinas.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Los pacientes diabéticos tratados con SOMATULINE® AUTOGEL® pueden experimentar hipoglucemia o hiperglucemia
Los niveles de glucosa en la sangre deben ser monitoreados al inicio del tratamiento con SOMATULINE® AUTOGEL®.
En los pacientes diabéticos el tratamiento debe ser ajustado de acuerdo a las necesidades individuales. En pacientes insulinodependientes, los requerimientos de insulina pueden ser reducidos.
PRECAUCIONES GENERALES: Los estudios farmacológicos en animales y en humanos muestran que la lanreótida, al igual que la somatostatina y sus análogos, produce la inhibición de la secreción de insulina y de glucagón. Por ello, los pacientes tratados con SOMATULINE® AUTOGEL® pueden experimentar hipoglucemia o hiperglucemia. Los niveles de glucosa en la sangre deben ser monitoreados al inicio del tratamiento con lanreótida. En los pacientes diabéticos el tratamiento debe ser ajustado de acuerdo a las necesidades individuales. En pacientes insulinodependientes, los requerimientos de insulina pueden ser reducidos.
La lanreótida reduce la motilidad de la vesícula biliar y aumenta el riesgo de formación de cálculos biliares. Por ello, se aconseja realizar de manera sistemática una ecografía de la vesícula biliar al inicio del tratamiento y posteriormente de manera periódica durante el tratamiento, en los pacientes que no hayan sido sometidos a una colecistectomía.
En el caso de insuficiencia renal grave, se ha observado una disminución de aproximadamente la mitad en la depuración total de lanreótida, lo cual tiene como consecuencia un aumento en la vida media y en el Área Bajo la Curva (ABC).
En caso de insuficiencia hepática se ha observado un aumento en el volumen de distribución y en el tiempo medio de retención; sin que existan modificaciones en la depuración o en el ABC.
En las personas de edad avanzada se ha observado un aumento en la vida media y en el tiempo medio de retención en comparación con personas jóvenes y sanas. Tomando en cuenta el margen de seguridad hepática, no es necesario modificar la dosis.
En pacientes con tumores neuroendocrinos, la lanreótida no se debe prescribir antes de excluir la presencia de la obstrucción ocasionada por un tumor intestinal.
La lanreótida puede llegar a disminuir el latido del corazón, sin alcanzar necesariamente el umbral de bradicardia en pacientes sin un problema cardiaco fundamental. En pacientes que sufren de problemas cardiacos antes de iniciar el tratamiento con SOMATULINE® AUTOGEL®, puede iniciar la bradicardia, por eso las pulsaciones del corazón deben ser monitoreadas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis:
Acromegalia:
Inicio del tratamiento: La posología recomendada varía de 60 a 120 mg cada 28 días.
Disminuir la dosis cuando las concentraciones lleguen a niveles normales (niveles de GH < 1 ng/mL y niveles de IGF-1 normalizado y/o desaparición de los síntomas clínicos).
Mantener la dosis cuando las concentraciones de la GH están entre 2.5 ng/mL y 1 ng/mL.
Para aumentar la dosis cuando las concentraciones de la GH son superiores a 2.5 ng/mL, el médico determinará la frecuencia de la monitorización de los niveles de GH y de IGF 1; así como de los síntomas, en función de la respuesta clínica del paciente.
Tratamiento Extendido: En los pacientes bien controlados con análogos de la somatostatina, puede administrarse SOMATULlNE® AUTOGEL® 120 mg cada 42 ó 56 días.
En los pacientes tratados previamente con Somatuline® L.P. 30 mg con una dosis cada 14 días, la dosis inicial de SOMATULINE® AUTOGEL® deberá ser de 60 mg cada 28 días.
En los pacientes tratados previamente con Somatuline® L.P. 30 mg, con una dosis cada 10 días, la dosis inicial de SOMATULINE® AUTOGEL® deberá ser de 90 mg cada 28 días.
En los pacientes tratados previamente con Somatuline® L.P. 30 mg, con una dosis cada 7 días, la dosis inicial de SOMATULINE® AUTOGEL® deberá ser de 120 mg cada 28 días.
Adaptación al tratamiento: El tratamiento debe ser adaptado para cada paciente, por un médico especializado.
La dosis debe individualizarse y adaptarse en función de la respuesta del paciente (valorada por la mejoría de los síntomas y/o disminución de los niveles de GH y/o IGF 1).
Tumores neuroendocrinos: La dosis inicial recomendada es de 90 mg cada 28 días (4 semanas) durante 2 meses.
En caso de que la respuesta sea insuficiente, a juzgar por los síntomas clínicos (episodios de eritemas y evacuaciones diarreicas), la dosis puede aumentarse a 120 mg cada 28 días (4 semanas).
En caso de que la respuesta sea buena, a juzgar por los síntomas clínicos (eritema y deposiciones diarreicas), la posología puede disminuirse a 60 mg cada 28 días (4 semanas).
Insuficiencia hepática y/o renal: En pacientes con insuficiencia renal o hepática no es necesario ajustar dosis.
Pacientes jóvenes: En pacientes jóvenes, no es necesario ajustar dosis.
Población pediátrica: SOMATULINE® AUTOGEL® no es recomendado para el uso en niños y adolescentes debido a la falta de datos sobre seguridad y eficacia.
Vía de administración: Parenteral (Subcutánea).
SOMATULINE® AUTOGEL® se presenta en una jeringa para un solo uso, lista para usarse.
La solución inyectable contenida en la jeringa prellenada es una solución sobresaturada de lanreótida lista para usarse formando un autogel de aspecto blanquizco a amarillo pálido translúcido.
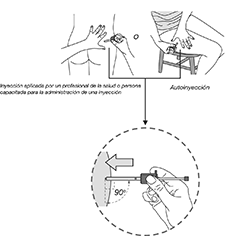
Forma de administración: La inyección debe ser aplicada por un profesional de la salud.
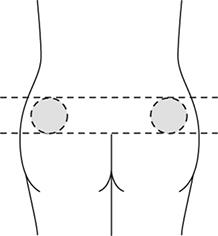
La solución debe ser inyectada vía subcutánea profunda en el cuadrante superior externo de la nalga.
Autoaplicación: Sin embargo, para los pacientes que reciban una dosis estable continua de SOMATULINE® AUTOGEL®, el producto puede ser administrado por el paciente o por una persona de confianza con previa capacitación por parte de un profesional de la salud. En caso de autoinyección, ésta deberá ser realizada en la región superior externa del muslo.
La decisión de administración por el paciente y/otra persona capacitada deberá ser tomada por el profesional de la salud.
Independientemente del lugar de la inyección, la piel no debe ser pellizcada y la aguja debe insertarse rápidamente en toda su longitud, perpendicularmente a la piel.
Instrucciones para el uso del producto:
1. Extraer SOMATULINE® AUTOGEL® del refrigerador 30 minutos antes de la inyección.
2. Lavarse las manos y asegurarse de la limpieza de la superficie de preparación.
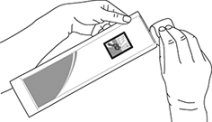
3. Antes de abrir el sobre laminado comprobar que está intacto y verificar la fecha de caducidad impresa. No usar si el medicamento ha caducado o si el sobre laminado está dañado de alguna manera.
4. Abra el sobre y saque la jeringa prellenada.
5. Seleccione el sitio de inyección:
• Inyección aplicada por un profesional de la salud o persona que conozca el procedimiento para administrar una inyección (cuadrante superior-externo de la nalga).
• Autoinyección (parte superior de la cara externa del muslo).
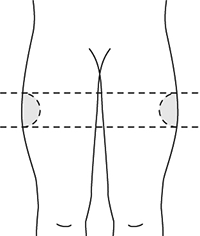
6. Alternar el sitio de la inyección entre el lado derecho e izquierdo para cada aplicación.
7. Desinfectar el sitio de la inyección sin frotar la piel.
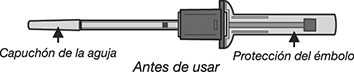
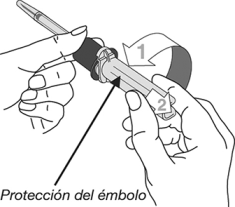
8. Girar (1) y sacar (2) la protección del émbolo como se muestra a continuación y desechar.
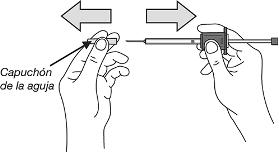
9. Retirar el capuchón de la aguja.
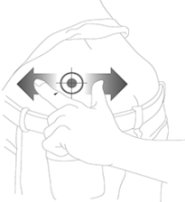
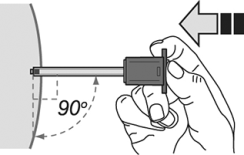
10. Mantener la piel alrededor del sitio de la inyección plana con el dedo pulgar y el dedo índice sin pellizcar o presionar la piel en el sitio de inyección. La aguja debe ser introducida en toda su longitud (inyección subcutánea profunda), es muy importante que la aguja se introduzca perpendicularmente en ángulo recto (90°) a la piel.
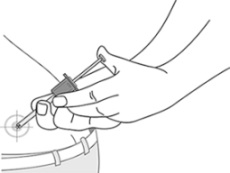
11. Inyectar lentamente y regularmente todo el producto, sin mover la aguja hasta que el émbolo no avance más. En este punto, usted escuchará un "clic".
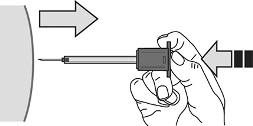
12. Sin dejar de ejercer presión sobre el émbolo, retirar la aguja del lugar de inyección.
13. A continuación, liberar la presión en el émbolo. La aguja se retraerá automáticamente en el escudo de la aguja, donde será bloqueada de forma permanente.
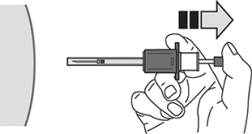
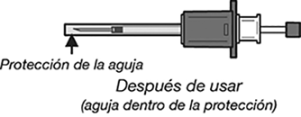
14. Aplique una leve presión sobre la zona de inyección con un algodón o gasa seca estéril para evitar cualquier sangrado. No frotar ni masajear el punto de inyección después de la administración.
15. Desechar la jeringa de manera apropiada. Su médico o enfermera le explicará las precauciones particulares de eliminación de material de inyección usado.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: En caso de sobredosis, está indicado un tratamiento sintomático.
PRESENTACIONES:
Caja con un sobre laminado conteniendo una dosis individual de 60 mg en una jeringa prellenada de 0.5 mL con aguja e instructivo anexo.
Caja con un sobre laminado conteniendo una dosis individual de 90 mg en una jeringa prellenada de 0.5 mL con aguja e instructivo anexo.
Caja con un sobre laminado conteniendo una dosis individual de 120 mg en una jeringa prellenada de 0.5 mL con aguja e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese en refrigeración entre 2°C y 8°C. No se congele.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx
Hecho en Francia por:
Ipsen Pharma Biotech
Parc d"Activités du Plateau de Signes
Chemin départemental N° 402
83870 Signes, Francia
Importado y Distribuido por:
IPSEN MÉXICO, S. de R. L. de C. V.
CPA Logistic Center Tlalnepantla, Edificio 1, Bodega 7,
Almacén No. 11, Km. 12.5 De la Vía Gustavo Baz Prada
Col. San Pedro Barrientos
Tlalnepantla de Baz, C.P. 54010, Estado de México.
Reg. Núm. 540M2004, SSA IV


