
SPEVIGO
ESPESOLIMAB
Solución inyectable
1 Caja, 2 Frasco de vidrio, 450/7.5 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula contiene:
Espesolimab 450 mg/7.5 mL
Vehículo cbp
La jeringa prellenada contiene:
Espesolimab 150 mg/mL
Anticuerpo monoclonal humanizado de tipo inmunoglobulina G1 (IgG1) antagonista, que bloquea la señalización del IL-36R humano expresado en células de ovario de hámster chino (CHO)
INDICACIONES TERAPÉUTICAS:
SPEVIGO® está indicado para el tratamiento de la psoriasis pustulosa generalizada (PPG), lo que incluye el tratamiento y la prevención de brotes agudos en adultos y adolescentes desde los 12 años de edad.
FARMACOCINÉTICA Y FARMACODINAMIA:
Mecanismo de acción:
Espesolimab es un anticuerpo monoclonal humanizado de tipo inmunoglobulina G1 (IgG1) antagonista, que bloquea la señalización del receptor de la interleucina 36 IL-36R humano. La unión de espesolimab al IL-36R evita la posterior activación del IL-36R por medio de sus ligandos relacionados (IL-36 α, β y ?) y la activación de las vías proinflamatorias y profibróticas. Estudios genéticos en humanos han establecido un vínculo sólido entre la señalización del IL-36R y la inflamación de la piel.
Farmacodinamia:
Tras el tratamiento con SPEVIGO® intravenoso en pacientes con psoriasis pustulosa generalizada (PPG), se observó una disminución en los niveles de proteína-C reactiva (PCR), interleucina 6 (IL-6), citocinas mediadas por linfocitos T colaboradores (Th1/Th17), marcadores de inflamación mediada por queratinocitos, mediadores neutrófilos y citocinas proinflamatorias en suero y piel en la Semana 1 en comparación con los valores iniciales, y se asociaron con una disminución en la severidad clínica. Estas disminuciones en los biomarcadores fueron más pronunciadas en la última medición en la semana 8 en el estudio Effisayil 1.
Estudios clínicos:
Effisayil 1 (1368.13)
Se llevó a cabo un estudio aleatorizado, doble ciego, comparado con placebo (Effisayil 1) para evaluar la eficacia y la seguridad clínicas de SPEVIGO® en pacientes adultos con brotes agudos de psoriasis pustulosa generalizada (PPG), diagnosticada conforme a los criterios de la Red Europea de Asociaciones de Psoriasis Raras y Graves (European Rare and Severe Psoriasis Expert Network [ERASPEN]), independientemente del estado de mutación del IL36RN. Los pacientes se aleatorizaban si tenían un brote agudo de PPG de intensidad entre moderada y grave, de acuerdo con lo definido por una puntuación total (en el intervalo de 0 [aclarada] a 4 [grave]) de un mínimo de 3 (moderada) de la Evaluación Global del Médico de la PPG (GPPGA), presencia de pústulas nuevas (nueva aparición o agravamiento de las pústulas existentes), subpuntuación de pustulación GPPGA de un mínimo de 2 (leve), y eritema y presencia de pústulas en, al menos, el 5% de la superficie corporal. Los pacientes debían interrumpir el tratamiento sistémico y tópico de PPG antes de recibir el medicamento en investigación.
El criterio de valoración principal del estudio fue el porcentaje de pacientes con subpuntuación de pustulación GPPGA de 0 (sin pústulas visibles) en la semana 1 después del tratamiento. El criterio de valoración secundario clave del estudio fue el porcentaje de pacientes con una puntuación total GPPGA de 0 o 1 (piel aclarada o casi aclarada) en la semana 1. Los otros criterios secundarios de valoración en la semana 4 fueron el porcentaje de pacientes con una reducción del 75% en el Índice de la Severidad del Área de PPG (GPPASI 75) y los desenlaces informados por los pacientes, que incluían el cambio desde el inicio en la puntuación de la Escala Visual Analógica (EVA) de dolor, el cambio desde el inicio en la puntuación de la Escala de Síntomas de la Psoriasis (PSS), y el cambio desde el inicio en la puntuación de la Evaluación Funcional del Tratamiento de Enfermedades Crónicas (FACIT) de fatiga.
Se aleatorizó un total de 53 pacientes (2:1) a recibir una dosis única intravenosa de 900 mg de SPEVIGO® (n=35) o placebo (n=18). Los pacientes en ambos grupos de tratamiento que todavía experimentaban síntomas de brote agudo en la semana 1 eran elegibles para recibir una dosis única intravenosa de 900 mg de SPEVIGO® con rótulos a la vista, lo que dio como resultado que 12 pacientes (34%) en el grupo SPEVIGO® recibieran una segunda dosis de SPEVIGO® y 15 pacientes (83%) del grupo placebo recibieran una dosis de SPEVIGO® el día 8. Además, 6 pacientes (4 en el grupo SPEVIGO® y 2 en el grupo placebo) recibieron tratamiento para los brotes agudos con una dosis única intravenosa de 900 mg de SPEVIGO® por reaparición de un brote agudo después del día 8.
La población del estudio estaba compuesta por 32% de hombres y 68% de mujeres. La media de edad de los pacientes era 43 (intervalo 21-69) años; el 55% eran asiáticos y el 45% eran caucásicos. La mayoría de los participantes del estudio tenía una subpuntuación de pustulación GPPGA de 3 (43%) o 4 (36%), y los pacientes tenían una puntuación total GPPGA de 3 (81%) o 4 (19%). El 24,5% de los pacientes había recibido anteriormente tratamiento con biológicos para PPG.
En la semana 1, se observó una diferencia estadísticamente significativa en el porcentaje de pacientes que alcanzaron una subpuntuación de pustulación GPPGA de 0 (sin pústulas visibles) y una puntuación total GPPGA de 0 o 1 (piel aclarada o casi aclarada) en el grupo SPEVIGO® en comparación con placebo (véase la Tabla 1).
Tabla 1. Subpuntuación de pustulación GPPGA y puntuación total GPPGA en la semana 1
|
Placebo |
SPEVIGO® 900 mg IV |
|
|
Cantidad de pacientes analizados |
18 |
35 |
|
Pacientes que alcanzaron una subpuntuación de pustulación GPPGA de 0, n (%) |
1 (5.6) |
19 (54.3) |
|
Diferencia de riesgo vs. placebo,% (IC del 95%) |
48.7 (21.5; 67.2) |
|
|
Valor p* |
0.0004 |
|
|
Pacientes que alcanzaron una puntuación total GPPGA de 0 o 1, n (%) |
2 (11.1) |
15 (42.9) |
|
Diferencia de riesgo vs. placebo,% (IC del 95%) |
31.7 (2.2; 52.7) |
|
|
Valor p* |
0.0118 |
|
GPPGA = Evaluación Global del Médico de la PPG; IV = intravenoso.
* Valor p unilateral.
En los pacientes aleatorizados a recibir SPEVIGO®, el aclaramiento pustular (subpuntuación de pustulación GPPGA de 0) se alcanzó tempranamente un día después del tratamiento en el 11,4% (4/35) de los pacientes. El efecto de un máximo de dos dosis de SPEVIGO® en la subpuntuación de pustulación GPPGA y la puntuación total GPPGA fue sostenido hasta la semana 12 (véanse las Figuras 1 y 2).
Figura 1. Porcentaje de pacientes con una subpuntuación de pustulación GPPGA de 0 en el tiempo (Effisayil 1)
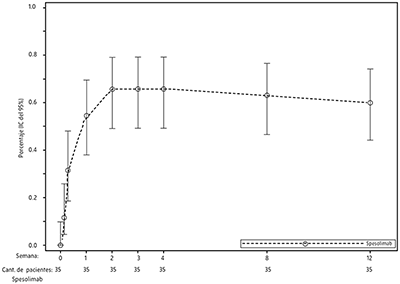
GPPGA = Evaluación Global del Médico de la PPG.
Figura 2. Porcentaje de pacientes con una puntuación total GPPGA de 0 o 1 en el tiempo (Effisayil 1)
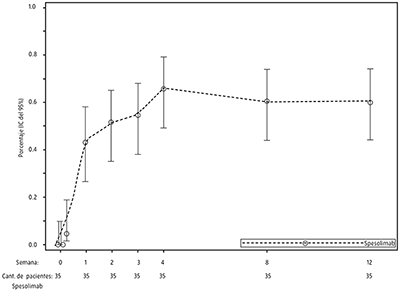
GPPGA = Evaluación Global del Médico de la PPG.
Los resultados del criterio de valoración principal y de los criterios secundarios clave fueron uniformes entre los subgrupos, incluidos sexo, edad, raza, subpuntuación de pustulación GPPGA al inicio, puntuación total GPPGA al inicio, estado de mutación de IL-36RN, e independientemente de cualquier tratamiento por PPG antes de la aleatorización.
En la semana 4, 16 pacientes (46%) aleatorizados a recibir SPEVIGO® alcanzaron un GPPASI 75.
En los pacientes aleatorizados a recibir SPEVIGO®, se observaron mejorías en la puntuación EVA de dolor, la puntuación PSS (medición de síntomas de dolor, enrojecimiento, picazón y ardor), la puntuación FACIT de fatiga en la Semana 4 (mediana de cambio desde el inicio: -22,45; -2,00 y 3,00 para la puntuación EVA de dolor, la puntuación PSS y la puntuación FACIT de fatiga, respectivamente).
Effisayil 2 (1368.27): Se llevó a cabo un estudio aleatorizado, doble ciego, comparado con placebo de fase II b (Effisayil 2) para evaluar la eficacia y la seguridad de SPEVIGO® en administración subcutánea en pacientes adultos y adolescentes con antecedentes de PPG, diagnosticada conforme a los criterios de la ERASPEN, independientemente del estado de mutación del IL-36RN y con al menos dos brotes agudos de PPG de intensidad entre moderada y severa en el pasado. Los pacientes se aleatorizaban si tenían una puntuación total de 0 o 1 de la GPPGA, en la selección y aleatorización. Los pacientes debían interrumpir el tratamiento sistémico y tópico de PPG antes o en el momento de la aleatorización. Estos pacientes debían haber tenido antecedentes de brotes agudos mientras recibían tratamiento concomitante para la PPG o antecedentes de brotes agudos luego de reducir la dosis o de discontinuar estos medicamentos concomitantes.
El criterio de valoración principal del estudio fue el tiempo hasta el primer brote agudo de PPG hasta la semana 48 (definido por una subpuntuación de pustulación GPPGA ≥ 2 y un aumento en la puntuación total GPPGA ≥ 2 desde el inicio). El criterio de valoración secundario clave del estudio fue la aparición de al menos un brote agudo de PPG hasta la semana 48. Otros criterios de valoración secundarios a la semana 48 fueron el tiempo hasta el primer empeoramiento en la PSS y en el Índice de Calidad de Vida Dermatológica (DLQI) definido como un aumento de 4 puntos en la puntuación total desde el inicio.
Se aleatorizó un total de 123 pacientes (1:1:1:1) a recibir uno de los cuatro tratamientos (véase la Tabla 2).
Tabla 2. Grupos de tratamiento en Effisayil 2:
|
Dosis de carga |
Dosis subsiguientes |
|
|
SPEVIGO® |
600 mg por vía subcutánea |
300 mg por vía subcutánea cada 4 semanas |
|
SPEVIGO® |
600 mg por vía subcutánea |
300 mg por vía subcutánea cada 12 semanas |
|
SPEVIGO® |
300 mg por vía subcutánea |
150 mg por vía subcutánea cada 12 semanas |
|
Placebo |
Tratamiento subcutáneo |
Tratamiento subcutáneo cada 4 semanas |
La población del estudio estaba compuesta por 38.2% de hombres y 61.8% de mujeres. La media de edad de los pacientes era 40.4 (intervalo 14-75) años, de los cuales 8 (6.5%) eran adolescentes (2 por grupo de tratamiento); el 64.2% de los pacientes eran asiáticos y el 35.8% eran caucásicos. Los pacientes incluidos en el estudio tenían una subpuntuación de pustulación GPPGA de 1 (28.5%) o 0 (71.5%), y los pacientes tenían una puntuación total GPPGA de 1 (86.2%) o 0 (13.8%). Al momento de la aleatorización, el 74.8% de los pacientes fueron tratados con tratamiento sistémico para la PPG, que se discontinuó al comienzo del tratamiento del estudio aleatorizado.
Mientras que en Effisayil 2 se estudiaron 3 pautas posológicas, la pauta posológica recomendada para la prevención de brotes agudos de PPG es una dosis de carga subcutánea de 600 mg de SPEVIGO® seguida de un tratamiento subcutáneo de 300 mg administrado cada 4 semanas (véase la sección Posología y administración). Los resultados resumidos a continuación corresponden a la pauta posológica recomendada.
Los pacientes que habían experimentado un brote agudo eran elegibles para recibir hasta dos dosis intravenosas de 900 mg de SPEVIGO® con rótulos a la vista (véase la sección Dosis y vía de administración). Dos pacientes (6.7%) en el grupo SPEVIGO® con la dosis recomendada y 15 (48.4%) en el grupo placebo recibieron tratamiento intravenoso para brotes agudos. El tratamiento con la dosis recomendada de SPEVIGO® en comparación con placebo llevó a una mejoría estadísticamente significativa con base en criterio de valoración principal y el criterio de valoración secundario clave (véase la Tabla 3).
Tabla 3. Tiempo hasta el primer brote agudo de PPG y la aparición de al menos un brote agudo de PPG hasta la Semana 48 (Effisayil 2)
|
Placebo |
Dosis recomendada de SPEVIGO® |
|
|
Cantidad de pacientes analizados, N |
31 |
30 |
|
Pacientes con brotes agudos de PPG, N (%)* |
16 (51.6) |
3 (10.0) |
|
Razón de riesgos instantáneos (HR)** para el tiempo hasta el primer brote agudo vs. placebo (IC del 95%) |
0.16 (0.05; 0.54) |
|
|
Valor p*** |
0.0005 |
|
|
Diferencia de riesgo para la aparición de brotes agudos de PPG vs. placebo (IC del 95%) |
-39.0% (-62.1; 15.9) |
|
|
Valor p**** |
0.0013 |
|
*El uso del tratamiento intravenoso SPEVIGO® o el tratamiento habitual indicado por el investigador para tratar el empeoramiento de la PPG se consideró al inicio del brote agudo de PPG.
**Modelo de regresión de Cox estratificado por el uso de medicamentos sistémicos para la PPG al momento de la aleatorización.
***Prueba de Log-rank estratificada por el uso de medicamentos sistémicos para la PPG al momento de la aleatorización, valor p unilateral.
****Prueba de Cochran-Mantel-Haenszel luego de imputación múltiple, estratificada por el uso de medicamentos sistémicos para la PPG al momento de la aleatorización, valor p unilateral.
La eficacia de la dosis subcutánea recomendada de SPEVIGO® en comparación con placebo se observó prontamente luego de la aleatorización y se mantuvo hasta la semana 48 (véase la Figura 3).
Figura 3. Tiempo hasta el primer brote agudo de PPG hasta la Semana 48 (Effisayil 2)
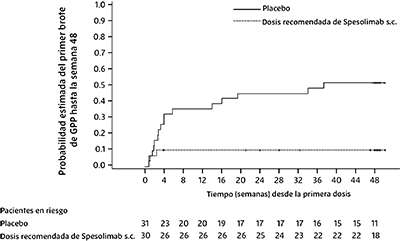
Los resultados del criterio de valoración principal y de los criterios secundarios clave fueron uniformes entre los subgrupos, incluidos sexo, edad, raza, IMC, peso corporal, estado de mutación de IL-36RN, psoriasis de placa concurrente, puntuación total GPPGA al inicio e independientes de cualquier tratamiento sistémico para la PPG al momento de la aleatorización.
Un paciente adolescente en el grupo placebo recibió tratamiento habitual indicado por el investigador para tratar el empeoramiento de la PPG y se consideró que presentaba un brote agudo de PPG. Ningún paciente adolescente en el grupo de dosis recomendada de SPEVIGO® experimentó brotes agudos de PPG.
El tratamiento con la dosis subcutánea recomendada de SPEVIGO® en comparación con el placebo redujo el riesgo de empeoramiento en la PSS (HR 0.42; IC del 95% 0.20 a 0.91; valor p nominal 0.0134) y empeoramiento en la DLQI (HR 0.26; IC del 95% 0.11 a 0.62; valor p nominal 0.0010) hasta las 48 semanas.
Inmunogenicidad:
Como en el caso de todas las proteínas terapéuticas, existe el potencial de inmunogenicidad. La detección de la formación de anticuerpos depende en gran manera de la sensibilidad y la especificidad del ensayo.
En los pacientes con PPG tratados con espesolimab en el estudio Effisayil 1, los anticuerpos antifármaco (ADA) se formaron con una mediana de inicio de 2,3 semanas. Después de la administración de 900 mg IV de espesolimab, intravenoso el 24% de los pacientes tenía un título de ADA máximo superior a 4000 y un resultado positivo para anticuerpos neutralizantes (Nab) al final del estudio (Semanas 12 a 17). En Effisayil 2, los ADA se formaron con una mediana de inicio de 8 semanas. Después de la administración de una dosis de carga de 600 mg subcutánea de espesolimab seguida de 300 mg de espesolimab por vía subcutánea cada 4 semanas durante un total de 48 semanas, el 24% de los pacientes tenía un título de ADA máximo superior a 4000 y un resultado positivo para NAb.
En Effisayil 1, luego de la administración de espesolimab intravenoso,las mujeres parecieron tener una respuesta de inmunogenicidad más alta; el porcentaje de pacientes con título de ADA superior a 4000 fue del 30% en las mujeres y del 12% en los hombres. En Effisayil 2, luego de la administración de espesolimab subcutáneo, los datos sobre respuesta de inmunogenicidad en hombres vs. mujeres fueron inconclusos.
En algunos pacientes con valores de título de ADA superiores a 4000, las concentraciones plasmáticas de espesolimab disminuyeron, sin un impacto aparente en la farmacocinética de los títulos de ADA inferiores a 4000. En presencia de ADA, se observó eficacia en el retratamiento de brotes agudos subsiguientes con SPEVIGO® en un estudio de extensión con rótulos a la vista. En los pacientes que recibieron la dosis recomendada de SPEVIGO® en Effisayil 2 (véase la sección Estudios clínicos), la media de las concentraciones valle en los pacientes ADA positivos con títulos superiores a 4000 fue aproximadamente del 77 al 107% de las concentraciones valle en pacientes ADA negativos o ADA positivos con valores de títulos ≤ 4000. Luego de la administración subcutánea de espesolimab, no se evidenció impacto de la presencia de ADA en la eficacia.
No se evidenció correlación entre la presencia de ADA a espesolimab y las reacciones de hipersensibilidad.
Farmacocinética:
Se desarrolló un modelo farmacocinético poblacional a partir de los datos provenientes de individuos sanos, pacientes con PPG y pacientes con otras enfermedades. Después de una dosis única intravenosa de 900 mg, los valores del AUC0-∞ (IC del 95%) y de la Cmáx (IC del 95%) estimados con el modelo farmacocinético poblacional en un paciente ADA negativo con PPG típico fueron 4750 (4510; 4970) μg·día/mL y 238 (218; 256) μg/mL, respectivamente. Después de la administración de una dosis de carga subcutánea de 600 mg de espesolimab seguida de 300 mg de espesolimab por vía subcutánea cada 4 semanas, la media de concentración valle en estado estacionario osciló entre 33.4 μg/mL y 42.3 μg/mL.
Absorción: Luego de la administración de la dosis única subcutánea de espesolimab en voluntarios sanos, se alcanzaron las concentraciones plasmáticas pico entre los 5.5 y los 7.0 días. Luego de la administración subcutánea en el abdomen, la biodisponibilidad absoluta fue levemente superior con las dosis mayores con valores estimados del 58, 65 y 72% al administrar 150 mg, 300 mg y 600 mg, respectivamente. Con base en los datos limitados, la biodisponibilidad absoluta en el muslo fue de aproximadamente el 85% tras una dosis subcutánea de 300 mg de espesolimab.
Distribución:
Sobre la base del análisis farmacocinético poblacional, el volumen de distribución típico en estado estacionario fue 6,4 L.
Biotransformación:
La vía metabólica de espesolimab no ha sido caracterizada. Como anticuerpo monoclonal humanizado IgG1, es esperable que espesolimab se degrade en pequeños péptidos y aminoácidos por las vías catabólicas de manera similar a la IgG endógena.
Eliminación:
En el intervalo lineal de dosis (0,3-20 mg/kg), sobre la base del modelo farmacocinético poblacional, la depuración de espesolimab (IC del 95%) en un paciente con PPG típico sin ADA de 70 kg de peso fue 0,184 (0,175; 0,194) L/día. La semivida terminal fue de 25,5 (24,4; 26,3) días.
Linealidad y no linealidad:
Al administrarse por vía intravenosa, espesolimab mostró una farmacocinética lineal con aumento proporcional a la dosis en exposiciones entre los intervalos de dosis de 0.3 a 20 mg/kg. Tanto la depuración (CL) como la vida media terminal fueron independientes de la dosis. Tras la administración de la dosis única subcutánea, la exposición a espesolimab aumentó levemente más que en forma proporcional a la dosis en el intervalo de 150 mg a 600 mg debido al leve aumento en la biodisponibilidad en dosis mayores.
Farmacocinética en poblaciones específicas:
Edad avanzada, género y raza:
Sobre la base de los análisis farmacocinéticos poblacionales, la edad, el género y la raza no tienen efecto sobre la farmacocinética de espesolimab.
Deterioro hepático y renal:
Como anticuerpo monoclonal, no es esperable la eliminación de espesolimab por la vía hepática o renal. No se realizaron estudios formales del efecto del deterioro hepático o renal sobre la farmacocinética de espesolimab.
El análisis farmacocinético poblacional no identificó una influencia del deterioro hepático leve o del deterioro renal leve o moderado sobre la exposición sistémica a espesolimab.
Peso corporal:
Las concentraciones de espesolimab fueron incrementadas en pacientes con peso corporal más bajo y reducidas en pacientes con peso corporal más alto. El impacto clínico del peso corporal sobre las concentraciones plasmáticas de espesolimab es desconocido.
Población pediátrica:
La farmacocinética de espesolimab en los pacientes pediátricos por debajo de los 12 años de edad no ha sido aún investigada.
La farmacocinética plasmática de espesolimab observada en adolescentes fue uniforme con la observada en adultos. No hubo evidencia de una diferencia en la relación exposición-respuesta en adolescentes en comparación con adultos.
CONTRAINDICACIONES:
Hipersensibilidad grave o potencialmente mortal a SPEVIGO® o a cualquiera de los excipientes (véase la sección Composición y Advertencias y precauciones especiales).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Existen datos limitados sobre el uso de espesolimab en embarazadas. Los estudios preclínicos con un anticuerpo monoclonal anti-IL-36R específico sustituto de ratón no indican efectos nocivos directos o indirectos respecto de la toxicidad reproductiva (véase la sección Toxicología). Como medida precautoria, se recomienda evitar el uso de SPEVIGO® en el embarazo, a menos que el beneficio clínico previsto supere claramente los riesgos potenciales.
Lactancia:
Se desconoce si espesolimab se excreta en la leche humana. No existen datos acerca de los efectos en el lactante o en la producción de leche. Espesolimab es un anticuerpo monoclonal y su presencia en la leche humana es esperable. No puede descartarse el riesgo para los recién nacidos y los lactantes. Se debe decidir si interrumpir la lactancia o abstenerse del tratamiento con SPEVIGO® tomando en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer.
Fertilidad:
No hay datos disponibles acerca del efecto de espesolimab sobre la fertilidad en humanos. Los estudios preclínicos en ratones con un anticuerpo monoclonal anti-IL-36R específico sustituto de ratón no indican efectos nocivos directos o indirectos con respecto a la fertilidad a partir del antagonismo de IL-36R.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad:
Se ha estudiado SPEVIGO® en estudios clínicos que incluyeron 183 pacientes con PPG.
Los datos de seguridad presentados a continuación provienen de dos estudios aleatorizados, doble ciego, comparados con placebo que compararon el tratamiento con SPEVIGO® respecto del placebo (Effisayil 1 y Effisayil 2, véase la sección Estudios clínicos) en pacientes con psoriasis pustulosa generalizada y siete estudios de SPEVIGO® doble ciego, comparados con placebo para otras enfermedades.
En los estudios clínicos, las reacciones adversas más frecuentes informadas asociadas a SPEVIGO® fueron las infecciones.
Resumen tabulado de reacciones adversas:
Categorías de frecuencia:
Muy frecuente: ≥ 1/10.
Frecuente: ≥ 1/100 - < 1/10.
Poco frecuente: ≥ 1/1000 - < 1/100.
Rara: ≥ 1/10000 - < 1/1000.
Muy rara: < 1/10000.
Frecuencia desconocida: no se puede estimar a partir de los datos disponibles.
Nota: las categorías de frecuencia descriptas se basan en la Guía del Resumen de las Características del Producto (SmPC) de la Unión Europea (UE) (de septiembre de 2009); por ende, en países que no pertenecen a la UE es posible que sean apropiadas otras definiciones.
Tabla 4. Resumen de reacciones adversas y las frecuencias correspondientes según la Guía del SmPC de la UE
|
Terminología de la Clasificación por órganos y sistemas |
Reacciones adversas a SPEVIGO® |
Frecuencias conforme a la Guía del SmPC de la UE |
|
Infecciones e infestaciones |
Infección del tracto urinario. Infección del tracto respiratorio alto |
Frecuente Frecuente |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
Frecuente |
|
Trastornos generales y alteraciones en el lugar de administración |
Reacciones en la zona de inyección. Fatiga |
Frecuencia desconocida* Frecuente |
* No reportado en el estudio Effisayil 1.
Descripción de las reacciones adversas seleccionadas:
Infecciones:
Durante el periodo de 1 semana comparado con placebo en el estudio Effisayil 1, se informaron infecciones en el 17,1% de los pacientes tratados con SPEVIGO® en comparación con el 5,6% de los pacientes tratados con placebo. En Effisayil 1,se informó infección seria (infección del tracto urinario) en 1 paciente (2,9%) en el grupo SPEVIGO® y en ningún paciente en el grupo placebo. Durante el periodo comparado con placebo de hasta 48 semanas en Effisayil 2, se informaron infecciones en el 33.3% de los pacientes tratados con SPEVIGO® y en el 33.3% de los pacientes tratados con placebo. En Effisayil 2, se informaron infecciones serias en 3 pacientes (3.2%) en el grupo SPEVIGO® y en ningún paciente del grupo placebo.
Las infecciones observadas en los estudios clínicos con espesolimab fueron generalmente de intensidad entre leve y moderada sin un patrón distintivo en cuanto al agente patógeno o al tipo de infección.
Reacciones en la zona de inyección: Las reacciones en la zona de inyección incluyen eritema, hinchazón, dolor, induración, calor, exfoliación, pápula, prurito, exantema y urticaria en el lugar de inyección. La gravedad de las reacciones en el lugar de inyección fue generalmente entre leve y moderada.
Población pediátrica: Los datos disponibles sobre adolescentes son limitados (véase la sección Estudios clínicos). No se han detectado nuevos problemas de seguridad en base a la cantidad limitada de pacientes adolescentes tratados.2
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Los datos preclínicos revelan que no existe riesgo especial para los seres humanos.
Se llevaron a cabo estudios de toxicología con dosis repetidas en ratones con un anticuerpo monoclonal anti-IL-36R específico sustituto de ratón, administrado por la vía intravenosa dos veces por semana durante 26 semanas en una dosis (50 mg/kg) que fue 5 veces más alta que la dosis protectora en un modelo experimental de inflamación de colon en ratones. No se evidenciaron cambios adversos en el peso corporal, la ingesta de alimentos o las observaciones clínicas con esta dosis.
La especificidad de unión de espesolimab a los tejidos humanos se evaluó en un estudio de reactividad cruzada en tejidos. No se observó una unión a tejidos inesperada.
Toxicidad reproductiva y del desarrollo:
Los estudios preclínicos realizados en ratones con un anticuerpo anti-IL-36R sustituto de ratón no indican efectos nocivos directos o indirectos con respecto al embarazo, al desarrollo embrionario y fetal o a la fertilidad en dosis intravenosas de hasta 50 mg/kg dos veces por semana.
Genotoxicidad:
No se realizaron estudios de genotoxicidad con espesolimab.
Carcinogenia:
No se realizaron estudios de carcinogénesis ni mutagénesis con espesolimab.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
No se han realizado estudios formales de interacciones con SPEVIGO® y otros fármacos. En pacientes con PPG, no se espera que espesolimab cause interacciones CYP mediadas por citoquinas como perpetrador.
Las vacunas con organismos vivos no deben administrarse simultáneamente con SPEVIGO® (véase la sección Precauciones generales).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
No se hallaron efectos adversos en los parámetros clínico-patológicos de laboratorio, incluidos hematológicos, inmunofenotipificación, clínico-bioquímicos e histopatológicos, que incluyó tejido linfático.
PRECAUCIONES GENERALES:
Trazabilidad:
A fin de mejorar la trazabilidad de los medicamentos biológicos, el nombre SPEVIGO® y el número de lote del producto administrado deben estar claramente indicados en el registro del paciente.
Infecciones: SPEVIGO® puede aumentar el riesgo de infecciones. (Véase la sección Reacciones adversas).
En pacientes con infección crónica o antecedentes de infección recurrente, es necesario considerar los riesgos potenciales y los beneficios clínicos previstos del tratamiento antes de prescribir SPEVIGO®. En pacientes con cualquier infección activa clínicamente importante, el tratamiento con SPEVIGO® no debe iniciarse hasta que la infección se resuelva o se trate adecuadamente. Es necesario indicar a los pacientes que deben consultar al médico si aparecen signos o síntomas de infección clínicamente importante durante o luego del tratamiento con SPEVIGO®.
Solución inyectable en jeringa prellenada SPEVIGO®:
Si un paciente está recibiendo tratamiento con inyección subcutánea SPEVIGO® para la prevención de brotes agudos de PPG y sufre una infección activa clínicamente importante, se debe interrumpir el tratamiento con SPEVIGO®. Se puede considerar reiniciar el tratamiento una vez que la infección se haya resuelto o se haya tratado adecuadamente.
Evaluación de tuberculosis antes del tratamiento:
Debe realizarse una evaluación de infección por tuberculosis (TB) en los pacientes antes de iniciar el tratamiento con SPEVIGO®. SPEVIGO® no debe administrarse en pacientes con infección TB activa.
Es necesario considerar el tratamiento para TB antes de iniciar SPEVIGO® en pacientes con TB latente o antecedentes de TB en los que no es posible confirmar un ciclo de tratamiento adecuado. Durante y tras el tratamiento con SPEVIGO®, debe realizarse el monitoreo de signos y síntomas de TB activa en los pacientes.
Hipersensibilidad y reacciones relacionadas con la infusión:
La aparición de hipersensibilidad y reacciones relacionadas con la infusión puede ocurrir con el uso de anticuerpos monoclonales como SPEVIGO®. La hipersensibilidad puede incluir reacciones inmediatas como anafilaxis y reacciones retardadas como reacciones al fármaco con eosinofilia y síntomas sistémicos (DRESS).
Si un paciente presenta signos de anafilaxis u otra hipersensibilidad seria, SPEVIGO® debe interrumpirse de inmediato, y debe iniciarse un tratamiento adecuado (véase la sección Contraindicaciones).
Solución para infusión SPEVIGO®:
Si un paciente presenta hipersensibilidad leve o moderada durante una infusión intravenosa u otras una reacciones relacionada con el medicamento leve o moderada, SPEVIGO® debe interrumpirse, y debe considerarse una terapéutica adecuada (p. ej., antihistamínicos y/o corticosteroides sistémicos). Una vez resuelta la reacción, la infusión puede retomarse a una velocidad de infusión más lenta, con aumento gradual hasta completar la infusión (véase Posología y administración).
Inmunogenicidad:
No se han realizado estudios específicos en pacientes que han recibido vacunas elaboradas con virus vivos o bacterias vivas recientemente. El intervalo entre la vacunación con organismos vivos y el inicio del tratamiento con SPEVIGO® debe ser de un mínimo de 4 semanas. Las vacunas con organismos vivos no deben administrarse durante y por un periodo mínimo de 16 semanas tras el tratamiento con SPEVIGO®.
Solución inyectable en jeringa prellenada SPEVIGO®:
Antes de iniciar SPEVIGO® para la prevención de brotes agudos de PPG, se debe considerar completar todas las inmunizaciones correspondientes según los lineamientos de inmunización vigentes.
Neuropatía periférica:
Se desconoce el potencial de ocurrencia de neuropatía periférica con SPEVIGO®. Los casos de neuropatía periférica se informaron en estudios clínicos con espesolimab. Los médicos deben estar atentos a la detección de síntomas que potencialmente indiquen la aparición de neuropatía periférica.
Excipientes:
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, es decir, es esencialmente “libre de sodio”.
Uso en poblaciones específicas:
Conducción y uso de maquinarias:
La influencia de SPEVIGO® sobre la capacidad de conducir y usar maquinarias es inexistente o insignificante.
Pacientes pediátricos:
La seguridad y la eficacia de SPEVIGO® se han establecido en adolescentes con PPG de 12 años de edad o más (véase la sección Estudios clínicos).
No se cuenta con datos clínicos sobre niños menores de 12 años. No es relevante el uso de espesolimab en niños menores de 12 años de edad.
Pacientes de edad avanzada:
No se requiere ajuste de la dosis.
Existe información limitada en pacientes a partir de los 65 años.
Pacientes con deterioro renal y/o hepático:
SPEVIGO® no se ha investigado formalmente en estas poblaciones de pacientes. Por lo general, no es esperable que estas afecciones tengan un impacto clínicamente de interés sobre la farmacocinética de los anticuerpos monoclonales, y los ajustes de dosis no se consideran necesarios.
DOSIS Y VÍA DE ADMINISTRACIÓN:
El tratamiento con SPEVIGO® debe ser administrado y supervisado por médicos con experiencia en el manejo de pacientes con enfermedades inflamatorias de la piel.
El tratamiento con SPEVIGO®puede iniciarse con la jeringa prellenada SPEVIGO® como inyección subcutánea para prevenir los brotes agudos de PPG o con una dosis intravenosa de SPEVIGO® para tratar un brote agudo de PPG.
La solución inyectable en jeringa prellenada SPEVIGO®está prevista para uso subcutáneo en la prevención de brotes agudos de PPG solamente. El concentrado para solución para infusión SPEVIGO® está previsto para uso intravenoso en el tratamiento de brotes agudos de PPG solamente.
Dosis:
Dosis recomendada para la prevención de brotes agudos de PPG: La dosis recomendada de SPEVIGO® para la prevención de brotes agudos de PPG en adultos y adolescentes desde los 12 años de edad es una dosis de carga subcutánea de 600 mg (cuatro inyecciones de 150 mg), seguida de 300 mg (dos inyecciones de 150 mg) administrados por vía subcutánea cada 4 semanas.
No se ha estudiado SPEVIGO® en pacientes con un peso corporal inferior a los 40 kg. No se pueden realizar recomendaciones de dosis (véase la sección Farmacocinética).
Tratamiento de brotes agudos de PPG durante el tratamiento preventivo subcutáneo de PPG: Si un paciente experimenta un brote agudo de PPG mientras recibe SPEVIGO® subcutáneo, el brote agudo de PPG puede tratarse con SPEVIGO® intravenoso (véase la sección Dosis recomendada para el tratamiento de brotes agudos de PPG).
Inicio o reinicio del tratamiento preventivo subcutáneo de PPG luego del tratamiento intravenoso de brotes agudos de PPG:
Cuatro semanas luego del tratamiento intravenoso con SPEVIGO®, se puede iniciar o reiniciar SPEVIGO® subcutáneo con una dosis de 300 mg (dos inyecciones de 150 mg) administrada cada 4 semanas. No se requiere dosis de carga subcutánea.
Dosis recomendada para el tratamiento de brotes agudos de PPG: La dosis recomendada de SPEVIGO® solución para infusión para tratar un brote agudo de PPG es una dosis única de 900 mg (dos frascos ámpula de 450 mg/7.5 mL) administradas como infusión intravenosa.
Si los síntomas de brote agudo persisten, puede administrarse otra dosis de 900 mg (dos frascos ámpula de 450 mg/7.5 mL) 1 semana después de la dosis inicial.
Dosis omitida:
Prevención de brotes agudos de PPG con solución inyectable en jeringa prellenada SPEVIGO®:
Si se omite una dosis, esta debe administrarse lo antes posible. De allí en adelante, la administración debe reanudarse en el momento programado habitual.
Vía de administración:
Solución inyectable en jeringa prellenada SPEVIGO®:
La inyección debe administrarse por vía subcutánea en la parte superior de los muslos o en el abdomen (véase la sección Instrucciones de uso). La jeringa prellenada SPEVIGO® no debe inyectarse en áreas donde la piel esté sensible, con hematomas, eritematosa, indurada o con cicatrices.
De requerirse una dosis de carga subcutánea de 600 mg de SPEVIGO® (véase la sección Dosis y vía de administración), la dosis de carga debe ser administrada por un profesional sanitario. Se deberá elegir un lugar de inyección diferente para cada inyección, al menos a 2 cm de distancia de los otros lugares de inyección.
Para las dosis subcutáneas subsiguientes de 300 mg de SPEVIGO®, si el profesional sanitario lo determina correcto, los pacientes podrán autoadministrarse o sus cuidadores administrarles la jeringa prellenada SPEVIGO® luego de la correspondiente capacitación sobre la técnica de inyección subcutánea.
Para una dosis completa de 300 mg, se deben inyectar dos jeringas prellenadas de 150 mg/mL, una seguida inmediatamente de la otra. Se deberá elegir un lugar de inyección diferente para cada una de las dos inyecciones, al menos a 2 cm de distancia del otro lugar de inyección.
Solución para infusión SPEVIGO®:
El concentrado para solución para infusión SPEVIGO® debe diluirse antes de usar (véase la sección Instrucciones de uso).
SPEVIGO® se administra como infusión intravenosa continua a través de una vía intravenosa que contiene un filtro estéril, apirogénico y de baja unión a proteínas (tamaño de poro de 0.2 micrones) durante 90 minutos.
En caso de que la infusión se reduzca o se interrumpa temporariamente, el tiempo total de infusión (incluido el tiempo de detención) no debe superar los 180 minutos (véase la sección Precauciones generales).
Instrucciones de uso:
Condiciones de almacenamiento:
Solución inyectable en jeringa prellenada SPEVIGO®:
Antes de usar, la jeringa prellenada SPEVIGO® puede conservarse a temperaturas de hasta 25 ºC durante un lapso de hasta 14 días si se mantiene en el envase original para resguardarla de la luz. La jeringa prellenada SPEVIGO® debe descartarse si se ha conservado a temperaturas de hasta 25 ºC durante un lapso mayor a 14 días.
La jeringa prellenada SPEVIGO® no debe usarse si se ha congelado, ni siquiera si se ha descongelado.
Solución para infusión SPEVIGO®:
Antes de usar, el frasco ámpula sin abrir puede conservarse a temperatura ambiente (hasta 30 ºC) durante un lapso de hasta 24 horas en el envase original para resguardarla de la luz.
Instrucciones de manejo y uso:
Solución inyectable en jeringa prellenada SPEVIGO®:
Estas instrucciones de manejo y uso de la jeringa prellenada SPEVIGO® contienen información sobre cómo inyectar SPEVIGO®, si la dosis indicada a usted o a su hijo requiere de 2 jeringas prellenadas SPEVIGO®.
Lea estas instrucciones de manejo y uso antes de usar SPEVIGO® por primera vez y cada vez que obtenga reposición. Puede haber información nueva.
Esta información no reemplaza la consulta con su profesional sanitario sobre la enfermedad o sobre el tratamiento. Un profesional sanitario deberá enseñarle la forma correcta de inyectar SPEVIGO® antes de que usted intente autoinyectarse o inyectar a otra persona por primera vez.
Su médico le ha indicado una dosis de SPEVIGO® que requiere de dos inyecciones para obtener la dosis completa. Debe inyectar el contenido de dos jeringas prellenadas de SPEVIGO® provistas en el envase para obtener la dosis completa.
SPEVIGO® sirve para un único uso. No reutilice la jeringa prellenada.
Conozca SPEVIGO®:
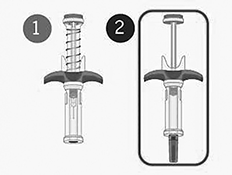
SPEVIGO® se presenta en una jeringa prellenada con una tapa de seguridad. La jeringa retrocede hacia adentro del protector de seguridad luego de la inyección.
Guía de piezas:
En la Figura 1 se muestra la jeringa prellenada SPEVIGO® antes de su uso y luego de su uso con el protector de seguridad activado.
Figura 1:
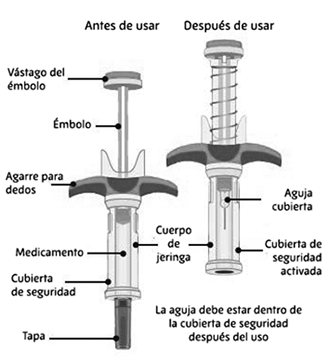
Información importante que debe conocer antes de inyectar SPEVIGO®:
• Debe inyectar el contenido de ambas jeringas prellenadas SPEVIGO®para obtener una dosis completa.
• Revise el envase del producto para asegurarse de tener el medicamento correcto, la cantidad apropiada de jeringas prellenadas para la dosis indicada a usted o a su hijo, descartar daños y verificar la fecha de vencimiento.
• No use SPEVIGO® si el líquido tiene aspecto turbio o contiene escamas o partículas grandes.
• No use SPEVIGO® si la fecha de vencimiento (VTO) ya ha pasado.
• No use SPEVIGO® si la jeringa prellenada se ha caído.
• No remueva la tapa hasta que esté listo para inyectar.
• Inyecte SPEVIGO® debajo de la piel (inyección subcutánea) en la parte superior de los muslos o en el área del estómago (abdomen). No inyecte SPEVIGO®en ninguna otra área del cuerpo.
• Mantenga SPEVIGO® y todo medicamento fuera del alcance de los niños.
Primer paso:
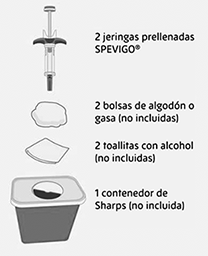
Reunir suministros:
• Saque el envase de SPEVIGO® del refrigerador y retire las jeringas prellenadas del envase.
• Reúna los suministros enumerados anteriormente y colóquelos sobre una superficie de trabajo limpia y plana en una zona bien iluminada.
• Si no dispone de todos los suministros mencionados, póngase en contacto con su farmacéutico.
• Consulte el paso 10: "Eliminación de las jeringas prellenadas y tapones SPEVIGO® usados".
Paso 2:
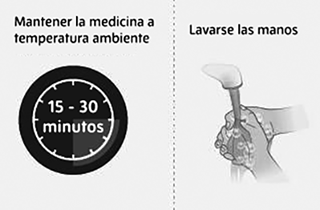
Preparación de la inyección de SPEVIGO®:
• Espere de 15 a 30 minutos para que el medicamento alcance la temperatura ambiente, a fin de evitar molestias durante la inyección. No acelere el proceso de calentamiento de ninguna manera, como utilizando el microondas o colocando la jeringa en agua caliente.
• No deje las jeringas prellenadas a la luz directa del sol.
• No retire la tapa de la aguja hasta que esté listo para inyectarse.
• Lavar bien las manos con agua y jabón y secarlas.
Paso 3:
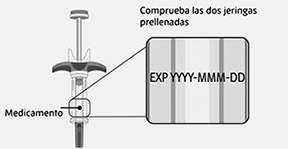
Inspección de las jeringas prellenadas:
Comprueba que ambas jeringas estén prellenadas:
• Compruebe que el nombre del medicamento SPEVIGO® y la dosis en sus jeringas prellenadas coinciden con la dosis prescrita a usted o a su hijo.
• Compruebe la fecha de caducidad en ambas jeringas prellenadas. No las utilice si la fecha de caducidad ha pasado.
• Compruebe que ambas jeringas prellenadas no estén dañadas, agrietadas o presenten fugas.
No utilizar si alguna parte de las jeringas prellenadas aparece agrietada, rota o tiene fugas.
• Asegúrese de que el medicamento de ambas jeringas prellenadas es transparente e incoloro o ligeramente amarillo parduzco. Puede contener pequeñas partículas blancas o transparentes. No lo utilice si el medicamento está turbio o contiene copos o partículas grandes o de color.
• Es normal ver burbujas de aire, no es necesario eliminarlas.
• No utilizar si las jeringas prellenadas SPEVIGO® se han caído.
Preparación para la primera inyección:
Prepárese para la primera de las 2 inyecciones.
Repetirá los siguientes pasos con la segunda jeringa inmediatamente después de la primera inyección.
Se necesitan 2 inyecciones para una dosis completa.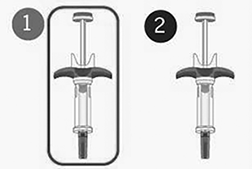
Paso 4:
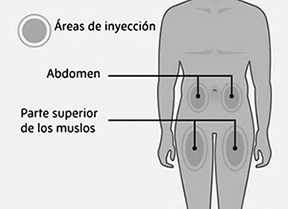
Elección del lugar de inyección:
Elija un lugar de inyección.
• Puede utilizar una zona de su:
• parte superior de los muslos o
• la zona del estómago (abdomen), excepto una zona de 5 cm alrededor del ombligo.
• Elija un lugar de inyección diferente cada vez que se inyecte, a una distancia mínima de 2.5 cm del último lugar de inyección. Alterne entre la parte superior del muslo o la zona del estómago para cada dosis completa.
• No se inyecte cerca de la cintura o el ombligo.
• No inyecte en zonas sensibles, con hematomas, enrojecidas, duras o con cicatrices.
• No inyectar a través de la ropa.
Paso 5:
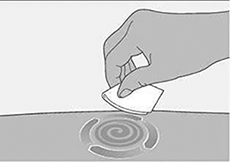
Limpiar el punto de inyección:
• Limpiar el lugar de la inyección con una toallita con alcohol y dejar secar al aire.
• No vuelva a tocar esta zona antes de la inyección.
• No abanique ni sople sobre la zona limpia.
Paso 6:
Retirar el tapón de la jeringa prellenada:
• Sujete la jeringa prellenada por el agarre para dedos con una mano. Con la otra mano, tire de la tapa hacia fuera.
• No tire del émbolo ni lo sujete.
• No gire la tapa. Si lo hace, podría dañar la aguja.
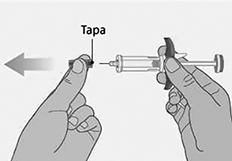
• No utilice la jeringa prellenada si la aguja está doblada o dañada. Si dobla accidentalmente la aguja, no intente enderezarla.
• Tire la tapa.
• Inyecte SPEVIGO® inmediatamente después de retirar la tapa.
• No intente volver a tapar la aguja. Volver a tapar puede provocar lesiones por pinchazo de aguja.
• No toque la aguja ni deje que toque nada antes de inyectarse.
Paso 7:
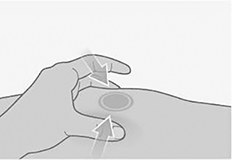
Pellizque la piel:
• Pellizque suavemente la zona de piel limpia alrededor del lugar de la inyección y sujétela con firmeza.
• Mantenga la piel pellizcada durante toda la inyección. Se inyectará en la piel pellizcada.
• No la suelte hasta que haya retirado la aguja de la piel al final de la inyección.
Paso 8:
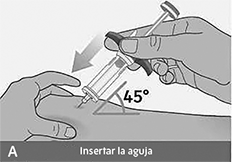
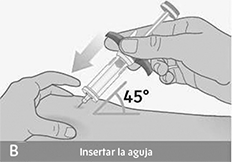
Antes de inyectar, revise los pasos A,B y C para aprender la forma correcta de inyectar.
Importante: No mueva la jeringa prellenada cuando esté introduciendo la aguja en su piel, mientras esté inyectando o al retirar la aguja de su piel.
Inserción de la aguja:
• Sujete la jeringa prellenada por el agarre para dedos azul. Evite tocar el vástago del émbolo azul.
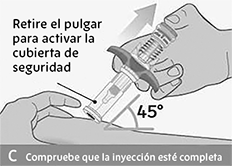
• Con un rápido movimiento en forma de dardo, inserte la aguja en la piel pellizcada en un ángulo de unos 45 grados.
• No mueva la aguja mientras se inserta o durante la inyección.
Inyectar el medicamento:
• Presione lentamente con el pulgar sobre el vástago del émbolo azul para empujar el émbolo hacia el interior del cuerpo de la jeringa.
• Continúe presionando el vástago del émbolo azul hasta que el émbolo se haya movido completamente hacia abajo.
• Asegúrese de que el vástago del émbolo azul no se pueda seguir pulsando para que se active la cubierta de seguridad incorporada.
Comprobación de inyección completada:
• Retire lentamente el pulgar del vástago del émbolo para sacar la aguja de la piel y colocarla en la cubierta de seguridad.
• Compruebe que el vástago del émbolo salta hacia atrás y que la aguja está dentro de la cubierta de seguridad.
• Si la aguja no está dentro de la cubierta de seguridad, llame a su médico. Es posible que no haya recibido una dosis completa.
• Si hay hemorragia, presione un algodón o una gasa sobre la zona durante unos segundos.
• No frote el lugar de la inyección.
• Aplique un vendaje adhesivo si es necesario.
Importante: No mueva la jeringa prellenada al insertar la aguja en su piel, durante la inyección o al retirar la aguja de su piel.
Paso 9:
Segunda inyección:
• Elija otro lugar de inyección.
El nuevo punto de inyección debe estar al menos a 2.5 cm del último punto de inyección. Alterne entre la parte superior del muslo o la zona del estómago para cada dosis completa.
• Consiga una segunda jeringa prellenada.
• Repita inmediatamente los pasos 4 a 8.
• Continúe con el paso 10.
Importante: Debe inyectar el contenido de ambas jeringas prellenadas de SPEVIGO® para administrar una dosis completa.
Paso 10:
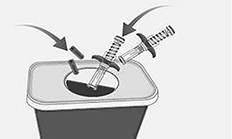
Eliminación de las jeringas prellenadas y las tapas de SPEVIGO® usadas:
• Deposite las jeringas prellenadas y los tapones usados en un contenedor de desecho de objetos punzantes inmediatamente después de su uso.
• No tire (deseche) las jeringas prellenadas y las tapas en la basura doméstica.
• Si no dispone de un contenedor para desechar objetos punzantes, puede utilizar un contenedor doméstico:
• Fabricado en plástico resistente,
• Puede cerrarse con una tapa hermética y resistente a los pinchazos, sin que puedan salir objetos punzantes,
• Erguido y estable durante el uso,
• Resistente a las fugas, y
• Debidamente etiquetado para advertir de la presencia de residuos peligrosos en el interior del contenedor.
Cuando su contenedor de residuos punzantes esté casi lleno, deberá seguir las directrices de su comunidad sobre la forma correcta de deshacerse del contenedor de residuos punzantes.
• No reutilice las jeringas prellenadas.
• No tire (deseche) el contenedor de objetos punzantes usado en la basura doméstica, a menos que las normas de su comunidad lo permitan.
• No recicle su contenedor de residuos punzantes usado.
Importante: Mantenga siempre el contenedor de residuos punzantes fuera del alcance de los niños.
Si tiene algún problema con su inyección, no utilice otra jeringa prellenada de SPEVIGO®. Pida ayuda a su médico.
 Solución para infusión SPEVIGO®:
Solución para infusión SPEVIGO®:
El frasco ámpula debe someterse a inspección visual antes de usar. SPEVIGO® es una solución incolora a levemente amarillo parduzca, transparente a ligeramente opalescente. Si la solución presenta aspecto turbio, decoloración o partículas grandes o coloreadas, se debe descartar el frasco ámpula.
La solución para infusión debe prepararse con técnicas asépticas. Extraer y descartar 15 mL de un envase de 100 mL de solución estéril de cloruro de sodio al 0.9% y reemplazar lentamente por 15 mL de SPEVIGO® (dos frascos ámpula de 450 mg/7.5 mL). Mezclar despacio antes de usar. La solución para infusión SPEVIGO®: diluida debe usarse de inmediato.
SPEVIGO® no debe mezclarse con otros medicamentos. SPEVIGO® puede administrarse con una vía intravenosa preexistente. La vía debe lavarse con una solución estéril de cloruro de sodio al 0.9% antes y después de la infusión. No debe administrarse otra infusión simultáneamente por el mismo acceso intravenoso.
SPEVIGO® es solo de uso único y no contiene conservadores.
La estabilidad físicoquímica durante el uso de la solución diluida se ha demostrado para un lapso de 24 horas entre 2 y 30 ºC, seguido de un tiempo de infusión de 3 horas.
Desde la perspectiva microbiológica, la solución para infusión diluida debe usarse de inmediato. De no ser así, las condiciones de almacenamiento durante el uso son responsabilidad del usuario y, por lo general, no deberían exceder las 24 horas a 2 a 8 ºC, a menos que la dilución se haya realizado en condiciones de asepsia validadas y controladas. Durante el periodo transcurrido entre la preparación y el inicio de la administración, la solución para infusión debe conservarse a resguardo de la luz conforme a los procedimientos estándares locales.
No se han observado incompatibilidades entre SPEVIGO® y los conjuntos para infusión compuestos de policloruro de vinilo (PVC), polietileno (PE), polipropileno (PP), polibutadieno y poliuretano (PUR), y los filtros de membrana en la vía compuestos de polietersulfona (PES, neutra y con carga positiva) y poliamida (PA) con carga positiva.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No existe experiencia clínica con sobredosis de SPEVIGO®.
La dosis máxima de SPEVIGO® administrada en los estudios clínicos fue de 1200 mg intravenosa o subcutánea. Los eventos adversos observados en los individuos que recibieron dosis únicas o repetidas de hasta 1200 mg concordaron con el perfil de seguridad conocido de SPEVIGO®.
En caso de sobredosis, se recomienda monitorear al paciente para detectar cualquier signo o síntoma de reacciones adversas y administrar un tratamiento sintomático según corresponda.
PRESENTACIONES:
Caja con 2 jeringas prellenadas de 150 mg/mL
Caja con 2 frascos ámpula de vidrio de 450 mg/7.5 mL cada uno (60 mg/mL).
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Solución inyectable en jeringa prellenada SPEVIGO®:
Manténgase en el envase original para protegerlo de la luz.
Manténganse en refrigeración de 2 a 8 °C, no se congele.
No utilizar si se ha congelado, aunque se haya descongelado.
Solución para infusión SPEVIGO®:
Manténgase en el envase original para protegerlo de la luz.
Manténganse en refrigeración de 2 a 8 °C, No se congele.
Antes de su uso, el frasco ámpula no abierto puede mantenerse a 30 °C durante un lapso de 24 horas si se almacena en el empaque original para protegerlo de la luz.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. Mantenga el recipiente para descartar agujas fuera del alcance de los niños. No se recomienda su uso en el embarazo y lactancia. No se recomienda su uso en niños menores de 12 años. El frasco ámpula deberá ser administrado por médicos especialistas. Si no se administra todo el producto, deséchese el sobrante. SPEVIGO® es para un solo uso y no contiene conservadores.
No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos.
No se administre si el cierre ha sido violado.
Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y
farmacovigilancia.mex@boehringer-ingelheim.com
BOEHRINGER INGELHEIM PROMECO, S.A. de C.V.
Calle del Maíz No. 49, Col. Barrio Xaltocan,
C.P. 16090, Xochimilco, Ciudad de México, México
®Marca Registrada


