SPIRIVA
BROMURO DE TIOTROPIO
Cápsulas
1 Caja, 1 Dispositivo dosificador (HandíHaler), 10 Cápsulas,
1 Caja, 20 Cápsulas,
1 Caja, 30 Cápsulas,
1 Caja, 1 Dispositivo dosificador (HandíHaler), 30 Cápsulas,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada cápsula dura con polvo para inhalación contiene:
Bromuro de tiotropio monohidratado equivalente a 18 μg tiotropio
Excipiente monohidrato de lactosa (que contiene proteínas de la leche) 1 cápsula
INDICACIONES TERAPÉUTICAS: SPIRIVA® está indicado en el tratamiento de mantenimiento de la enfermedad pulmonar obstructiva crónica (incluyendo bronquitis crónica y enfisema), de la disnea asociada y en la prevención de las exacerbaciones.
FARMACOCINÉTICA Y FARMACODINAMIA: El tiotropio es un compuesto de amonio cuaternario no quiral y es escasamente soluble en agua. El tiotropio se administra a través de la inhalación de polvo seco. Generalmente cuando se inhala, gran proporción de la dosis administrada se deposita en el tracto gastrointestinal y una porción mucho menor se aloja en el pulmón. Muchos de los datos farmacocinéticos que se describen a continuación, fueron obtenidos con dosificaciones mayores a las recomendadas en la terapéutica:
Absorción: Tras la inhalación de polvo seco por jóvenes voluntarios sanos, la biodisponibilidad absoluta del 19.5%, sugiere que la fracción que alcanza a los pulmones es altamente biodisponible. Dada la estructura química del compuesto (componente amonio cuaternario), se espera que el tiotropio sea escasamente absorbido en el tracto gastrointestinal. Por la misma razón, no se ha considerado que la ingesta de alimento influya en su absorción. Las soluciones orales de Tiotropio tienen una biodisponibilidad absoluta de 2-3%. Las concentraciones plasmáticas máximas fueron observadas 5 minutos después de la inhalación.
Distribución: El fármaco se une a las proteínas plasmáticas en un 72% y muestra un volumen de distribución de 32 L/Kg. En estado estable, los niveles plasmáticos máximos del tiotropio en los pacientes con EPOC fueron de 17-19 pg/mL a los 5 minutos de haberse efectuado la inhalación de 18 μg en polvo seco y descendieron rápidamente en forma multicompartamental. En estado estable, las concentraciones plasmáticas fueron de 3-4 pg/mL. Las concentraciones locales en el pulmón no son conocidas, pero la forma de administración sugiere que existen altas concentraciones en el tejido pulmonar. Los estudios en ratas han mostrado que el tiotropio no penetra la barrera hematoencefálica en forma relevante.
Biotransformación: El alcance de la biotransformación es pequeño. Lo anterior se evidencia por la excreción urinaria del 74% en forma inmodificada tras su administración intravenosa en voluntarios sanos. Tiotropio es un éster el cual no es enzimáticamente separable en el alcohol N-metilescopina y el ácido ditienilglicólico, ninguno de los cuales se une a receptores muscarínicos.
Los experimentos in vitro llevados a cabo en microsomas hepáticos y en hepatocitos humanos sugieren que cierta proporción del fármaco (< 20% de la dosis administrada por vía intravenosa) se metaboliza mediante oxidación dependiente del citocromo P450 y su subsiguiente conjugación con glutatión a una variedad de metabolitos fase II. Esta vía enzimática, puede ser bloqueada por los inhibidores del citocromo CYP450 2D6 (y 3A4), la quinidina, el ketoconazol y el gestodeno. Por tanto, los citocromos CYP450 2D6 y 3A4 se encuentran involucrados en la vía metabólica responsable en la eliminación de una más pequeña porción de la dosis administrada. El tiotropio, aún a concentraciones de dosis supraterapéuticas, no inhibe al citocromo P450 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 ó 3A, dentro de los microsomas hepáticos humanos.
Eliminación: Tras su inhalación, la vida media de eliminación terminal del tiotropio está entre 5 a 6 días. La depuración total es de 880 mL/min. tras una dosis intravenosa administrada a voluntarios jóvenes sanos, con una variabilidad interindividual del 22%. Cuando el tiotropio se administra por vía intravenosa, el fármaco inmodificado se excreta por la orina (74%). Tras la administración inhalada en polvo seco, la excreción urinaria corresponde al 14% y resto del fármaco no absorbido se elimina a través de las heces. La depuración renal de tiotropio excede a la depuración de la creatinina, lo que es indicativo de su excreción por vía urinaria. Tras la administración diaria prolongada en pacientes con EPOC, el estado estable farmacocinético se alcanzó después de 2 a 3 semanas sin evidencia de acumulación.
Linealidad/no linealidad. A rangos terapéuticos, el tiotropio presenta una farmacocinética lineal tras su administración tanto intravenosa como por inhalación en forma de polvo.
Pacientes de edad avanzada: Al igual que sucede con todos los fármacos que se excretan por vía renal, se ha asociado a la edad avanzada con el decremento de la depuración renal del tiotropio (326 mL/min. en pacientes con EPOC menores de 58 años y de 163 mL/min. en pacientes con EPOC mayores de 70 años) que puede explicarse por la disminución de la función renal en dichos grupos. La excreción urinaria de tiotropio tras su inhalación, desciende del 14% (voluntarios jóvenes sanos) al 7% (pacientes con EPOC), sin embargo, las concentraciones plasmáticas no cambian en forma significativa en pacientes de edad avanzada con EPOC si se comparan con la variabilidad intra- e inter-individual (43% de incremento en el área bajo la curva [ABC]0-4 h tras la administración del tiotropio en polvo).
Pacientes con insuficiencia renal: Acorde con otros fármacos, cuya vía de eliminación es predominantemente renal, la insuficiencia renal ha sido asociada con el incremento de las concentraciones plasmáticas del fármaco y con una reducción de la depuración del fármaco tras la administración intravenosa o por inhalación del tiotropio en polvo. La insuficiencia renal leve (depuración de la creatinina de 50-80 mL/min.), que es común en pacientes de edad avanzada, sólo incrementa ligeramente las concentraciones plasmáticas del fármaco (39% de incremento en el ABC0-4h tras su administración intravenosa). En pacientes con EPOC con insuficiencia renal moderada a severa (depuración de la creatinina de < 50 mL/min.), la administración del tiotropio por vía intravenosa, dio como resultado la duplicación de las concentraciones plasmáticas (82% de incremento del ABC0-4h, lo cual fue confirmado mediante las concentraciones plasmáticas después de la inhalación de tiotropio en polvo.
Pacientes con insuficiencia hepática: No se espera que ésta tenga influencia relevante en la farmacocinética del tiotropio. El fármaco es predominantemente depurado por vía renal (74% en jóvenes voluntarios sanos) y por clivaje éster no enzimático simple a productos que no se unen a los receptores muscarínicos.
El tiotropio es un agente antimuscarínico específico de acción prolongada, en medicina clínica es llamado frecuentemente anticolinérgico. Presenta afinidad similar por los subtipos de los receptores muscarínicos M1 a M5. En las vías aéreas, la inhibición de los receptores M3 de la musculatura lisa bronquial da como resultado su relajación. La naturaleza competitiva y reversible del antagonismo se demostró con receptores de origen humano y animal y en aislados de preparaciones orgánicas. En estudios no clínicos in vitro e in vivo, los efectos broncoprotectores fueron dosis dependientes y duraron más de 24 horas. La prolongada duración del efecto es probable que se deba a su muy lenta disociación desde los receptores muscarínicos M3, mostrando una vida media de disociación significativamente prolongada comparada con la del ipratropio. El tiotropio es una amina cuaternaria anticolinérgica tópicamente (bronquios) selectiva cuando se administra mediante inhalación, y posee un rango terapéutico aceptable antes de dar paso a efectos anticolinérgicos sistémicos. La disociación desde los receptores M2 es más rápida que desde los M3, lo cual en los estudios funcionales in vitro (cinéticamente controlados) se obtuvo alta afinidad de los receptores M3 sobre los M2. La alta potencia del fármaco y la lenta disociación desde los receptores, encontraron su correlación clínica en la significativa y prolongada broncodilatación en pacientes con EPOC.
La acción broncodilatadora del tiotropio es resultante de un efecto local en un sitio específico (en las vías aéreas) y no producto de un efecto sistémico.
El programa del desarrollo clínico incluyó cuatro estudios al azar de un año de duración y dos de 6 meses en estudios doble ciego que involucró a 2663 pacientes con EPOC (1308 administrados con SPIRIVA®). El programa de un año de duración consistió de dos estudios controlados con placebo y dos controlados con ipratropio. Los estudios a 6 meses de duración fueron controlados ambos con salmeterol y placebo. Estos estudios incluyeron una evaluación de la función pulmonar, disnea, exacerbaciones de la EPOC y evaluaciones de pacientes de su calidad de vida relacionada con la salud.
En los estudios mencionados, SPIRIVA® se administró una vez al día, generando una mejoría significativa en la función pulmonar (volumen de espiración forzada en un segundo, VEF1 y capacidad vital forzada, CVF) dentro de los 30 minutos tras la administración de la primera dosis y se mantuvo durante 24 horas. La farmacodinamia en estado estable se alcanzó durante la primera semana, observándose la mayor broncodilatación al tercer día. SPIRIVA® mejoró las tasas de flujo espiratorio pico tanto matutinos como vespertinos según los registros diarios de los pacientes.
La mejoría de la función respiratoria con SPIRIVA® se demostró a través del periodo de administración en seis estudios a largo plazo (figuras 1-3). Estas mejorías se mantuvieron sin evidencia de tolerancia.
Figura 1. VEF1 promedio (antes y después de la administración del medicamento en estudio) en los días 1 y 344 en dos estudios de 1 año de duración controlados contra placeboƒ
Estudio A
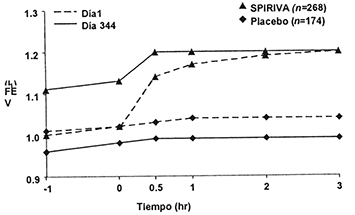
Estudio B
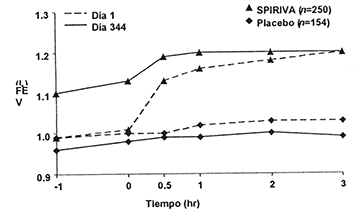
ƒ Los promedios son ajustados por centro y efectos basales.
Figura 2. VEF1 promedio (antes y después de la administración del medicamento en estudio) en los días 1 y 364 en dos estudios de un año de duración controlados con Ipratropioƒ
Estudio A
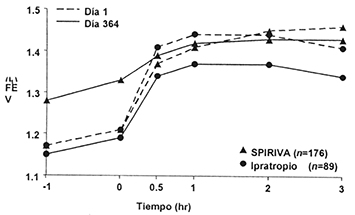
Estudio B
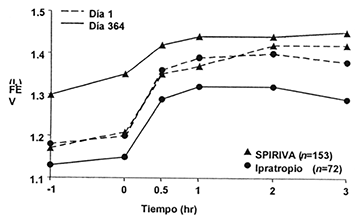
ƒ Los promedios son ajustados por centro y efectos basales.
Figura 3. VEF1 promedio en el tiempo (antes y después de la administración del medicamento en estudio) durante los días 1 y 169 en dos estudios de seis meses controlado con placebo y salmeterolƒ
Estudio A
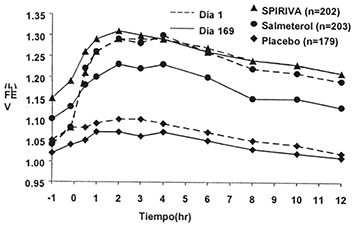
Estudio B
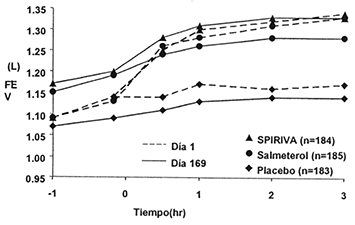
ƒ Los promedios son ajustados por centro y efectos basales.
Un estudio clínico con distribución al azar controlado con placebo, el cual incluyó a 105 pacientes con EPOC, demostró que la broncodilatación fue mantenida durante un intervalo de dosificación de 24 horas en comparación con el placebo sin considerar si SPIRIVA® era administrado por la mañana o por la noche.
Los siguientes resultados sobre la salud fueron demostrados en los estudios clínicos a largo plazo (mayor de un año):
• SPIRIVA® mejoró significativamente la disnea (evaluada utilizando el índice de disnea de transición). Esta mejoría fue mantenida a través de todo el periodo de tratamiento.
• SPIRIVA® redujo significativamente el número de exacerbaciones de la EPOC y retrasó el tiempo a la primera exacerbación en comparación con el placebo.
• SPIRIVA® mejoró significativamente la calidad de vida de los pacientes como se demostró mediante el "St. George"s Respiratory Questionnaire". Esta mejoría se mantuvo a través de todo el periodo de tratamiento.
Adicionalmente, en los estudios clínicos controlados con placebo con duración de un año, SPIRIVA® redujo significativamente el número de hospitalizaciones asociadas con exacerbaciones de EPOC y retrasó el tiempo a la primera exacerbación.
Se investigó mediante dos estudios controlados al azar doble ciego contra placebo en pacientes con EPOC, el impacto de la mejoría de la disnea sobre las actividades funcionales. En estos estudios SPIRIVA® mejoró significativamente los síntomas de la tolerancia limitada al ejercicio en 19.7% y 28.3% comparadas contra placebo.
Se realizó un estudio para el análisis del intervalo QT en donde intervinieron 53 pacientes sanos, se administró SPIRIVA® (p. ej. dosis terapéuticas tres veces al día) de 18 μg y 54 μg durante 12 días. En el electrocardiograma se observó que no ocasiona una prolongación del intervalo QT.
En un estudio de 4 años de 5,993 pacientes, SPIRIVA® sostuvo mejorías en el VEF1 y en la CVF a lo largo de los 4 años. Mejoró la tasa anualizada de declinación del VEF1 sin diferencia estadística significativa.

Durante el tratamiento, se redujo en un 16% el riesgo de muerte. La tasa de incidencia de la mortalidad fue de 4.79 por 100 pacientes/año en el grupo placebo vs. 4.10 por 100 pacientes año en el grupo tiotropio (razón de riesgo (tiotropio placebo) = 0.84, 95% IC = 0.73, 0.97). El tratamiento con tiotropio redujo el riesgo de insuficiencia respiratoria en 19% (2.09 vs 1.68 casos por 100 pacientes/año, riesgo relativo (tiotropio/placebo) = 0.81,95% IC = 0.65,1.00).
Un estudio de grupo paralelo aleatorizado a un año, doble ciego, doble simulación compare el efecto del tratamiento con 18 mcg de SPIRIVA® una vez al día con el de 50 mcg de salmeterol HFA pMDI dos veces al día con la incidencia de exacerbaciones moderadas y severas en 7,376 pacientes con EPOC y una historia de exacerbaciones en el año anterior. [64, 65, 66]
Figura 5. Estimaciones de Kaplan-Meier del tiempo hacia la primera exacerbación de EPOC/Grupo Tratado

Figura 6. Estimaciones de Kaplan-Meier del tiempo hasta la primera exacerbación de EPOC hospitalizado/Grupo Tratado

Tabla 1: Resumen de criterios de valoración de exacerbación
|
Criterio de valoración |
SPIRIVA® 18 microgramos (HandiHaler) N = 3,707 |
Salmeterol 50 microgramos (HFA pMDI) N = 3,669 |
Proporción (95% CI) |
valor-p |
|
Tiempo [días] para la primera exacerbación† |
187 |
145 |
0.83 (0.77-0.90) |
< 0.001 |
|
Tiempo para la primera (hospitalizada) exacerbación§ severa |
- |
- |
0.72 (0.61-0.85) |
< 0.001 |
|
Pacientes con ≥ 1 exacerbación, n (%)* |
1,277 (34.4) |
1,414 (38.5) |
0.90 (0.85-0.95) |
< 0.001 |
|
Pacientes con ≥ 1 exacerbación severa (hospitalizada), n (%)* |
262 (7.1) |
336 (9.2) |
0.77 (0.66-0.89) |
< 0.001 |
|
Tasa de incidencia de exacerbación media por paciente año# |
0.64 |
0.72 |
0.89 (0.83-0.96) |
= 0.002 |
|
Tasa de incidencia de exacerbación severa por paciente año# (hospitalizado) |
0.09 |
0.13 |
0.73 (0.66-0.82) |
< 0.001 |
† Tiempo [días] se refiere al primer cuartil de pacientes. El tiempo para el análisis del evento se hizo usando el modelo de regresión de riesgos proporcionales de Cox con (agrupamiento) centro y tratamiento como covariante; la proporción se refiere a la proporción de riesgo.
§ El tiempo para el análisis del evento se hizo usando el modelo de regresión de Cox de los riesgos proporcionales con (agrupamiento) centro y tratamiento como covariante; la proprción se refiere a la proporción de riesgo. El tiempo [días] para el primer cuartil de pacientes no puede calcularse, porque la proporción de pacientes con exacerbación severa es demasiado baja.
* El número de pacientes con evento se analizó usando la prueba de Cochran-Mantel-Haenszel estratificada con agrupamiento de centro; la proporción se refiere a la proporción de riesgo.
# El número de análisis de evento se hizo usando la regresión de Poisson conectando la sobredispersión y ajustando la exposición del tratamiento; la proporción se refiere a la proporción de riesgo.
En comparación con el salmeterol, SPIRIVA® incrementa el tiempo hacia la primera exacerbación (187 días vs. 145 días), con un 17% de reducción en riesgo (proporción de riesgo, 0.83; 95% intervalo de confianza [CI por sus siglas en inglés], 0.77 a 0.90; P < 0.001). SPIRIVA® también incrementó el tiempo hasta la primera exacerbación severa (hospitalizado) (proporción de riesgo, 0.72; 95% CI, 0.61 a 0.85; P < 0.001), redujo el número anual de exacerbaciones moderadas a severas (hospitalizado) (0.64 vs. 0.72; tasa de proporción, 0.89; 95% CI, 0.83 a 0.96; P = 0.002), y redujo el número anual de exacerbaciones severas (hospitalizado) (0.09 vs. 0.13; tasa de proporción, 0.73; 95% CI, 0.66 a 0.82; P < 0.001).
CONTRAINDICACIONES: SPIRIVA® está contraindicado en pacientes con antecedentes de hipersensibilidad a los componentes de la fórmula o a la atropina y sus derivados, como por ejemplo, ipratropio u oxitropio o a cualquier componente de este producto (ver lista de excipientes).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: No hay información clínica disponible sobre el empleo de SPIRIVA® durante el embarazo. Los estudios preclínicos no indican efectos dañinos directos o indirectos sobre el embarazo, desarrollo embrionario/fetal, nacimiento o desarrollo postnatal. No hay información clínica disponible sobre mujeres lactando expuestas al tiotropio. En base a estudios realizados en roedores lactando, una escasa cantidad de tiotropio se excreta a través de la leche materna, por lo que SPIRIVA® no debe ser administrada durante el embarazo y la lactancia, a menos que los beneficios para la madre superen los riesgos para el producto.
Los datos clínicos sobre fertilidad no están disponibles para el tiotropio. Un estudio no clínico llevado a cabo con tiotropio no mostró indicación alguna de ningún efecto adverso sobre la fertilidad (favor de consultar la sección Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
REACCIONES SECUNDARIAS Y ADVERSAS: Muchos de los efectos indeseables enlistados abajo pueden ser atribuidos a las propiedades anticolinérgicas de SPIRIVA®.
Las reacciones adversas al medicamento se obtuvieron de la información recabada de los estudios clínicos y de los reportes espontáneos durante el uso posterior a su aprobación. La base de datos de los estudios clínicos incluyó 9,647 pacientes que usaron tiotropio en 28 estudios clínicos controlados con placebo con periodos de tratamiento que oscilaron entre cuatro semanas y cuatro años, contribuyendo a un total de 12,469 personas-año de exposición al tiotropio.
Trastornos del metabolismo y de la nutrición:
Deshidratación.
Trastornos del sistema nervioso:
Vértigo.
Insomnio.
Trastornos oculares:
Visión borrosa.
Glaucoma.
Aumento de la presión intraocular
Trastornos cardiacos:
Fibrilación auricular.
Taquicardia supraventricular.
Taquicardia.
Palpitaciones.
Trastornos del aparato respiratorio, región torácica y Trastornos del mediastino:
Tos.
Disfonía.
Faringitis.
Broncoespasmo.
Epistaxis.
Laringitis.
Sinusitis.
Trastornos gastrointestinales:
Obstrucción intestinal incluyendo al íleo paralítico.
Estomatitis.
Gingivitis.
Glositis.
Candidiasis orofaríngea.
Disfagia.
Constipación.
Enfermedad por reflujo gastroesofágico.
Xerostomía, usualmente leve.
Trastornos del tejido subcutáneo y de la piel, Trastornos del sistema inmune:
Angioedema
Angioneurótico.
Hipersensibilidad (incluyendo reacciones inmediatas).
Infección cutánea y úlcera cutánea.
Urticaria.
Prurito.
Xerodermia.
Exantema.
Trastornos músculo-esqueléticas y del tejido conectivo:
Edema articular.
Trastornos del aparato urinario y renal:
Retención urinaria (usualmente en hombres con factores predisponentes) .
Infección del tracto urinario.
Disuria.
Efectos sobre la capacidad de conducir y usar maquinaria: No se han realizado estudios sobre la capacidad de conducir y usar maquinaria. La presencia de mareos o visión borrosa pueden influir sobre la capacidad para conducir o utilizar maquinaria.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: La inhalación aguda y la toxicidad oral en ratones, ratas y perros fue baja; por lo tanto los efectos tóxicos en humanos por sobredosis aguda del medicamento son poco probables. Los estudios farmacológicos de seguridad de dosis única demostraron que los efectos esperados de un medicamento anticolinérgico incluyeron midriasis, aumento de la frecuencia cardiaca y tiempo prolongado del tránsito gastro-intestinal.
Los efectos secundarios de los estudios de dosis repetidas en ratas, ratones y perros estuvieron relacionados a las propiedades anticolinérgicas del tiotropio e incluyeron midriasis, aumento de la frecuencia cardiaca, constipación, disminución en la ganancia de peso corporal y reducción en la secreción de las glándulas salivales y lacrimales. Otros cambios relevantes observados fueron: Leve irritación del tracto respiratorio superior en ratas evidenciado por rinitis y cambios epiteliales de la cavidad nasal y laringe, y prostatitis junto con depósitos proteináceos y litiasis en la vejiga de las ratas macho, aumento del peso pulmonar en ratas y disminución del peso cardiaco en perros.
Se pudieron demostrar efectos peligrosos en los estudios de reproducción en conejos y ratas con respecto al embarazo, desarrollo embrio/fetal, parto o desarrollo post-natal a niveles de dosis maternas tóxicas. En un estudio general de reproducción y fertilidad en ratas, no hubo indicación alguna de ningún efecto adverso sobre la fertilidad o el desempeño del apareamiento de ninguno de los padres tratados o de sus crías a ninguna dosis.
En una serie de estudios de mutagenicidad tanto in vivo como in vitro, el bromuro de tiotropio monohidratado no ocasionó mutación genética en procariontes ni en eucariontes, ni tampoco generó condiciones para el daño cromosómico o daños al ADN primario.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Aunque no se han llevado a cabo estudios formales acerca de las posibles interacciones farmacológicas, se ha administrado SPIRIVA® en forma concomitante con otros fármacos utilizados comúnmente en el control y manejo del EPOC, incluidos los broncodilatadores simpaticomiméticos, metilxantinas y esteroides orales e inhalados, sin presentarse evidencia clínica de interacciones farmacológicas.
Se dispone de información limitada, generada a partir de dos estudios clínicos acerca de la administración concomitante de SPIRIVA® en forma concomitante con otros fármacos anticolinérgicos. En un estudio se administró una dosis aguda de bromuro de ipratropio administrada crónicamente con SPIRIVA® en pacientes con EPOC (n = 64) y voluntarios sanos (n = 35), no encontrándose una asociación que incrementara los eventos adversos, cambios en los signos vitales o hallazgos en el electrocardiograma. Sin embargo, la administración concomitante y crónica de SPIRIVA® con fármacos anticolinérgicos no ha sido estudiada y, por lo tanto, no se recomienda.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No han sido descritas hasta el momento.
PRECAUCIONES GENERALES: SPIRIVA® es un broncodilatador de mantenimiento de una administración al día y no debe ser empleado para el tratamiento de episodios agudos de broncoespasmo ni como terapia de rescate.
Las reacciones de hipersensibilidad inmediata pueden suceder tras la inhalación del polvo de SPIRIVA®.
Al igual que con otros anticolinérgicos, SPIRIVA® debe ser utilizado con precaución en pacientes con glaucoma del ángulo estrecho, hiperplasia prostática o con obstrucción vesical.
En general, todos los medicamentos inhalados pueden dar lugar a broncoespasmo inducido.
Al igual que con otros fármacos que se excretan predominantemente por vía renal, los pacientes con insuficiencia renal moderada o grave (depuración de creatinina ≤ 50 mL/min.) deberán ser vigilados estrechamente durante el tratamiento con SPIRIVA®.
Los pacientes deben ser instruidos en relación a la administración correcta de SPIRIVA®. Se debe de evitar que el polvo penetre en los ojos. El dolor ocular o la molestia, visión borrosa, halos visuales o imágenes coloreadas asociadas con ojo rojo por congestión conjuntival y edema corneal, pueden ser signos de glaucoma del ángulo estrecho. Cuando se encuentren asociados dichos síntomas, deberá acudirse inmediatamente al especialista. Los colirios que producen miosis no son considerados como tratamiento efectivo.
SPIRIVA® no debe ser empleado más de una vez al día.
SPIRIVA® solamente debe ser usado a través del inhalador HandiHaler®.
Este producto contiene 5 mg de monohidrato de lactosa por cápsula.
DOSIS Y VÍA DE ADMINISTRACIÓN: La dosis recomendada de SPIRIVA® es la inhalación del contenido de una cápsula al día con el aparato de inhalación HandiHaler® a la misma hora del día (ver Instrucciones para su uso).
Las cápsulas de SPIRIVA® no deben deglutirse.
Los pacientes ancianos pueden usar SPIRIVA® a la dosis recomendada.
Los pacientes con insuficiencia renal pueden usar SPIRIVA® a la dosis recomendada. Sin embargo, como todos los medicamentos con excreción predominantemente renal, debe vigilarse estrechamente el uso de SPIRIVA® en pacientes con insuficiencia renal moderada a severa.
Los pacientes con insuficiencia hepática pueden usar SPIRIVA® a la dosis recomendada.
No hay experiencia con SPIRIVA® en lactantes y niños por lo tanto no debe usarse en estos grupos de edad.
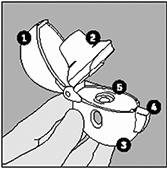
HandiHaler®
Instrucciones de uso: Siga cuidadosamente las instrucciones que su médico le ha dado para la correcta administración del medicamento.
El HandiHaler® ha sido diseñado especialmente para facilitar la inhalación del medicamento contenido en la cápsula de SPIRIVA®. No deberá ser usado con otro medicamento.
El HandiHaler® podrá ser utilizado hasta por un año, para administrar SPIRIVA®.
|
|
|
|
1 Tapa. 2 Boquilla. 3 Base. 4 Botón perforador de la cápsula. 5 Cámara central. |
|
|
1. Para abrir la tapa presione el botón perforador completamente y después suéltelo. |
|
|
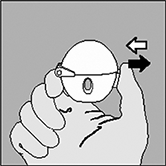
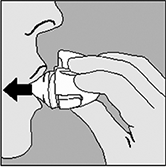
2. Abra la tapa del HandiHaler® jalándola hacia arriba. Luego jale la boquilla hacia arriba. |
|
|
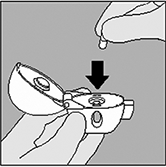
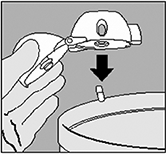
3. Tome la cápsula de SPIRIVA® del envase de burbuja (sólo inmediatamente antes del uso) y colóquela en la cámara central del HandiHaler®, tal y como se muestra en la figura. No tiene importancia en qué dirección se coloque la cápsula dentro de la cámara. |
|
|
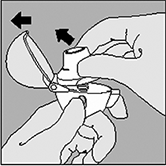
4. Cierre la boquilla firmemente hasta que se escuche un clic, manteniendo abierta la tapa del HandiHaler®. |
|
|
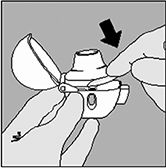
5. Mantenga el HandiHaler® con la boquilla hacia arriba, a continuación, presione completamente el botón perforador una vez, y luego suéltelo. Esto permite liberar el medicamento contenido en la cápsula. |
|
|
6. Antes de colocar el HandiHaler® en la boca, exhale completamente. Importante: Por favor evite respirar dentro de la boquilla en cualquier momento. |
|
|
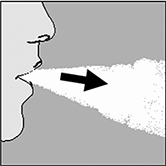
7. Coloque el HandiHaler® en la boca y cierre los labios estrechamente alrededor de la boquilla. Mantenga la cabeza en posición hacia arriba e inhale lenta y profundamente, pero a una velocidad suficiente para escuchar la cápsula vibrar. Inhale hasta que sus pulmones estén llenos; a continuación, contenga la respiración tanto como sea posible y, simultáneamente, retire el HandiHaler® de la boca. Reanude la respiración normal y repita los pasos 6 y 7. Esto vaciará la cápsula completamente. |
|
|
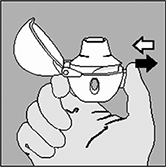
8. Abra la boquilla de nuevo. Voltee el HandiHaler® como se indica en la figura y deseche la cápsula utilizada. Cierre la boquilla y la tapa para el almacenamiento del HandiHaler®. |
|
|
Limpieza del HandiHaler®: |
|
|
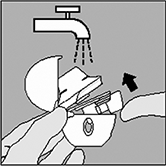
Limpie el HandiHaler® una vez al mes. Abra la tapa y la boquilla. Luego abra la base levantando el botón perforador. Enjuague completamente el HandiHaler® con agua tibia para remover cualquier residuo de medicamento. Vacíe completamente el exceso de agua sobre una toalla de papel y deje secar bajo la acción del aire, manteniendo abiertas la tapa, boquilla y la base. La limpieza mensual del HandiHaler® debe realizarse justo después de haber sido usado y deberá quedar listo para la administración de la próxima dosis dado que el secado tarda aproximadamente 24 horas. De ser necesario, el lado exterior de la boquilla se puede limpiar con un paño suave, húmedo, pero no empapado. |
|
|
Manejo del envase de burbuja: |
|
|
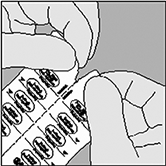
A. Separe las tiras del envase de burbuja siguiendo la línea de perforación. |
|
|
B. Remueva la capa de aluminio (sólo inmediatamente antes del uso) por medio de la pestaña como se indica en la figura, hasta que la cápsula sea totalmente visible. En caso de que inadvertidamente se exponga al aire una segunda cápsula, ésta debe ser descartada. |
|
|
C. Extraiga la cápsula. |
|
Las cápsulas no deben ser expuestas a temperaturas extremas.
La cápsula de SPIRIVA® contiene sólo una pequeña cantidad de polvo, así que sólo está parcialmente llena.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Altas dosis de SPIRIVA® pueden provocar signos y síntomas de tipo anticolinérgico; sin embargo, no se produjeron eventos de este tipo en voluntarios sanos tras la administración inhalada de hasta de 282 microgramos de tiotropio como dosis única.
Se observaron conjuntivitis bilateral y xerostomía en voluntarios sanos a quienes se les administraron dosis repetidas de tiotropio hasta completar 141 μg al día resolviéndose esto aún estando bajo tratamiento. En un estudio de dosis múltiples en pacientes con EPOC, con una dosis máxima diaria de 36 microgramos de tiotropio durante cuatro semanas, se observó como única reacción adversa xerostomía, atribuible al tiotropio.
La intoxicación aguda por la ingestión oral de cápsulas de tiotropio es muy poco probable debido a la baja biodisponibilidad del fármaco por vía oral.
PRESENTACIONES:
Caja de cartón con 10 ó 30 cápsulas con polvo, dispositivo inhalador (HandiHaler®) e instructivo anexo.
Caja de cartón con 20 ó 30 cápsulas con polvo (como repuesto) e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 25°C y en lugar seco. Después de abrir el envase de burbuja, úsese dentro de los 9 días.
LEYENDAS DE PROTECCIÓN:
Léase instructivo anexo. Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo ni en la lactancia. No ingerible.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx
Almacenado y distribuido por:
BOEHRINGER INGELHEIM PROMECO, S.A. de C.V.
Calle del Maíz No. 49, Col. Barrio Xaltocán,
C.P. 16090, Xochimilco, Ciudad de México.
Reg. Núm. 039M2002, SSA IV
®Marca Registrada