
STAQUIS - Ungüento
Sustancia(s):
- Crisaborol
Presentaciones:
- 1 Caja, 1 Tubo, 30 g, 2 %
- 1 Caja, 1 Tubo, 60 g, 2 %
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada 100 g contienen:
Crisaborol 2.0 g
Excipiente csp 100 g
INDICACIONES TERAPÉUTICAS:
STAQUIS® se indica para el tratamiento cutáneo de la dermatitis atópica leve a moderada en pacientes a partir de los 3 meses de edad.
FARMACOCINÉTICA Y FARMACODINÁMICA:
Propiedades farmacocinéticas:
Absorción:
La farmacocinética (PK) de STAQUIS® se investigó en 33 sujetos pediátricos de 2 a 17 años que tenían dermatitis atópica de leve a moderada y una media ± desviación estándar (DE) de área de superficie corporal (ASC) afectada del 49% ± 20% (rango de 27% a 92%). En este estudio, los sujetos aplicaron aproximadamente 3 mg/cm2 de ungüento STAQUIS® (el rango de dosis aproximado fue de 6 g a 30 g por aplicación) dos veces al día durante 8 días. Se midieron las concentraciones plasmáticas en todos los sujetos. La concentración plasmática máxima (Cmáx) media ± DE y el área bajo la curva de tiempo de la concentración de 0 a 12 horas posteriores a la dosis (ABC0-12) para crisaborol el Día 8 fueron 127 ± 196 ng/mL y 949 ± 1240 ng*h/mL, respectivamente. Las concentraciones sistémicas de crisaborol se presentaron en estado de equilibrio antes del Día 8. Con base en las proporciones de ABC0-12 entre el Día 8 y el Día 1, el factor de acumulación medio de crisaborol fue de 1.9. Los niveles sistémicos de crisaborol y sus metabolitos principales fueron similares entre las cohortes de edad de 2 a 5 años, de 6 a 11 años y de 12 a 17 años.
Para sujetos de 3 meses y mayores, las exposiciones sistémicas (ABC0-12 y Cmáx) al crisaborol son comparables y no presentan diferencias clínicamente significativas en ASC tratadas similarmente.
Distribución:
Según un estudio in vitro, crisaborol se une a las proteínas plasmáticas humanas en un 97%.
Biotransformación y eliminación:
Crisaborol se metaboliza considerablemente en metabolitos inactivos. El metabolito principal 5- (4-cianofenoxi)-2-hidroxil alcohol bencílico (metabolito 1), se forma mediante hidrólisis; este metabolito se metaboliza en metabolitos posteriores, entre los cuales está el 5-(4-cianofenoxi)-2-hidroxil ácido benzoico (metabolito 2) como metabolito principal que se forma mediante oxidación. La farmacocinética de los metabolitos 1 y 2 se evaluó en el estudio farmacocinético descrito anteriormente, y las concentraciones sistémicas se encontraron en el estado de equilibrio, o cerca de éste, antes del Día 8. Con base en las proporciones de ABC0-12 entre el Día 8 y el Día 1, los factores de acumulación media de los metabolitos 1 y 2 fueron 1.7 y 6.3, respectivamente. La excreción renal de los metabolitos es la vía de eliminación principal.
Interacciones medicamentosas:
Potencial de crisaborol de influir en la farmacocinética de otros medicamentos:
Los estudios in vitro en los cuales se utilizaron microsomas de hígado humano indicaron que, en condiciones de uso clínico y según se prevé, crisaborol y el metabolito 1 no inhiben CYP1A2, 2B6, 2C8, 2C9, 2C19 y 3A4.
Los estudios in vitro de microsomas de hígado humano para el metabolito 2 mostraron que este no inhibió las actividades de CYP2C19, 2D6 y 3A4, fue un inhibidor débil de CYP1A2 y 2B6, y un inhibidor moderado de CYP2C8 y 2C9. La enzima más sensible, CYP2C9, se investigó posteriormente en un ensayo clínico con warfarina como sustrato de CYP2C9. Los resultados de este estudio no mostraron posibles interacciones medicamentosas.
Los estudios in vitro indican que, en condiciones de uso clínico y según se prevé, crisaborol y los metabolitos 1 y 2 no inducen enzimas CYP.
Los estudios in vitro demostraron que crisaborol y el metabolito 1 no inhibieron las actividades de UGT1A1, 1A4, 1A6, 1A9, 2B7 y 2B15. El metabolito 2 no inhibió UGT1A4, 1A6, 2B7 ni 2B15. El metabolito 2 mostró una inhibición débil de UGT1A1; sin embargo, no se prevén interacciones medicamentosas clínicamente significativas entre crisaborol (y sus metabolitos) y los sustratos de UGT1A1 en concentraciones terapéuticas. El metabolito 2 mostró una inhibición moderada de UGT1A9 y puede causar un aumento moderado de las concentraciones de sustratos de UGT1A9 sensibles.
Los estudios in vitro indican que, en condiciones de uso clínico, no se prevé que crisaborol y los metabolitos 1 y 2 causen interacciones clínicamente significativas con los sustratos de transportadores como los transportadores de glicoproteína P, de proteína de resistencia al cáncer de mama (BCRP) y transportadores aniónicos o catiónicos orgánicos.
Propiedades farmacodinámicas:
Mecanismo de acción:
Crisaborol es un inhibidor antiinflamatorio de benzoxaborol fosfodiesterasa-4 (PDE4) que suprime la secreción de ciertas citocinas, como el factor de necrosis tumoral-α (TNF-α), las interleucinas (IL-2, IL-4, IL-5) y el interferón gamma (IFNℽ), y mejora la función de la barrera de la piel medida por la pérdida de agua transepidérmica (TEWL, por sus siglas en inglés). Crisaborol aplicado en pacientes con lesiones de dermatitis atópica, reduce la expresión de quimiocinas asociadas a la inflamación atópica, incluidas CCL17, CCL18 y CCL22.
Eficacia clínica:
Dos ensayos multicéntricos, controlados por excipiente, con grupos en paralelo, doble ciego, aleatorizados (Ensayos 1 y 2) e idénticos en cuanto a su diseño incluyeron un total de 1522 sujetos de 2 a 79 años (86.3% de los sujetos tuvieron de 2 a 17 años) con un área de superficie corporal (ASC) tratable de 5% a 95%. En el periodo inicial (datos combinados del estudio), en una evaluación global estadística del investigador (ISGA, por sus siglas en inglés), el 38.5% de los sujetos tuvieron una ISGA de leve (2) y el 61.5% tuvieron una ISGA de moderada (3) en la evaluación general de dermatitis atópica (eritema, induración/pápulas y supuración/formación de costras) en una escala de severidad de 0 a 4.
En ambos ensayos, los sujetos fueron aleatorizados 2:1 para recibir STAQUIS® o el excipiente aplicado dos veces al día durante 28 días. El criterio primario de valoración de la eficacia fue la proporción de los sujetos en el Día 29 que alcanzaron una ISGA de limpio (0) o casi limpio (1) con una mejora mínima de 2 grados desde el valor inicial, al comparar sujetos tratados con STAQUIS® dos veces al día con sujetos tratados con el excipiente. En ambos ensayos, un porcentaje estadísticamente significativamente mayor de sujetos logró alcanzar este criterio de valoración en el grupo tratado con STAQUIS® en comparación con el grupo tratado con el excipiente.
Los criterios de valoración de la eficacia secundarios fueron la proporción de sujetos en el Día 29, con una ISGA de limpio o casi limpio, y el tiempo para obtener un grado de ISGA de limpio o casi limpio con una mejora mínima de 2 grados desde el valor inicial. La proporción de sujetos que obtuvieron un puntaje de ISGA de limpio o casi limpio en el Día 29 del grupo tratado con STAQUIS® dos veces al día en comparación con los del grupo tratado con el excipiente dos veces al día fue estadísticamente significativamente mayor. Los sujetos tratados con STAQUIS® dos veces al día mostraron un estadísticamente significativamente menor tiempo para obtener una ISGA de limpio o casi limpio con una mejora mínima de 2 grados desde el valor inicial en comparación con los sujetos tratados con el excipiente mediante la prueba de rango logarítmico de cada estudio.
Los resultados de eficacia de los dos ensayos se resumen en la Tabla 1.
Tabla 1: Resultados de eficacia en sujetos con dermatitis atópica de leve a moderada en el Día 29
|
Ensayo 1 |
Ensayo 2 |
|||
|
STAQUIS® Dos veces al día (N = 503) |
Excipiente dos veces al día (N = 256) |
STAQUIS® dos veces al día (N = 513) |
Excipiente dos veces al día (N = 250) |
|
|
ISGAa |
32.8% |
25.4% |
31.4% |
18.0% |
|
Valor p |
0.038b |
< 0.001b |
||
|
ISGA limpio o casi limpioc |
51.7% |
40.6% |
48.5% |
29.7% |
|
Valor p |
0.005d |
< 0.001d |
||
|
Tiempo hasta ISGAa, e |
NCf |
NCf |
NCf |
NCf |
|
Valor p |
< 0.001g |
< 0.001g |
||
a Definido como una ISGA de limpio o casi limpio con una mejora con al menos una mínima de 2 grados desde el valor inicial.
b El valor p desde una prueba de regresión logística (con opción de Firth) con factores del grupo de tratamiento y del centro de análisis. A partir de la regresión logística, los cálculos estimados del Ensayo 1 son de 29.1% y 22.0% para STAQUIS® y el excipiente respectivamente. A partir de la regresión logística, los cálculos estimados del Ensayo 2 son de 26.5% y 14.2% para STAQUIS® y el excipiente respectivamente. Los valores se ajustaron para la imputación múltiple.
c No se requirió una mejora mínima de 2 grados desde el valor inicial.
d El valor p desde una prueba de regresión logística (con opción de Firth) con factores del grupo de tratamiento y del centro de análisis. A partir de la regresión logística, los cálculos estimados del Ensayo 1 son de 49.0% y 37.7% para STAQUIS® y el excipiente, respectivamente. A partir de la regresión logística, los cálculos del Ensayo 2 son de 45.2% y 25.5% para STAQUIS® y el excipiente, respectivamente. Los valores se ajustaron para la imputación múltiple.
e Medianas calculadas mediante el método de Kaplan-Meier.
f No fue posible calcular (NC) el tiempo mediano para obtener una ISGA de limpio o casi limpio con una mejora al menos de 2 grados desde el valor inicial, ya que menos del 50% de los sujetos obtuvo una ISGA de limpio o casi limpio con una mejora al menos de 2 grados desde el valor inicial.
g Valor p de la prueba de rango logarítmico.
En ambos ensayos clínicos (Ensayo 1 y 2), se evaluaron los signos (eritema, induración/pápulas, exudación, excoriación y liquenificación) y un síntoma (prurito) de dermatitis atópica.
La proporción de sujetos con mejoras en los signos de dermatitis atópica (definidos como Ninguna [0] o Leve [1] con una mejora al menos de 1 grado desde el valor inicial en una escala de 4 puntos) en el Día 29 fue mayor en los sujetos tratados con STAQUIS® dos veces al día que en los sujetos tratados con el excipiente dos veces al día para los 5 signos clínicos de dermatitis atópica. En el Ensayo 2, los 5 signos clínicos de dermatitis atópica mejoraron de forma estadísticamente significativamente. En el Ensayo 1, se alcanzó la importancia estadística para eritema, exudación y excoriación. Los resultados de cada ensayo se resumen en la Tabla 2.
Tabla 2: Proporción de sujetos que alcanzaron mejoríaa en los signos de dermatitis atópica en el día 29
|
Ensayo 1 |
Ensayo 2 |
|||||
|
STAQUIS® Dos veces al día (N = 503) |
Excipiente dos veces al día (N = 256) |
Valor pb |
STAQUIS® Dos veces al día (N = 513) |
Excipiente dos veces al día (N = 250) |
Valor pb |
|
|
Eritema |
62.8% |
46.1% |
< 0.001 |
54.9% |
33.9% |
< 0.001 |
|
Induración/pápulas |
57.7% |
54.8% |
0.375 |
51.9% |
40.2% |
0.005 |
|
Exudación |
41.0% |
33.3% |
0.027 |
38.1% |
27.2% |
0.004 |
|
Excoriación |
63.0% |
51.8% |
0.004 |
57.2% |
44.2% |
0.001 |
|
Liquenificación |
51.7% |
46.5% |
0.128 |
51.4% |
35.3% |
< 0.001 |
a Definida como Ninguna o Leve con una mejora mínima de 1 grado desde el valor inicial.
b Valor p obtenido de la prueba de Cochran-Mantel-Haenszel (CMH) estratificado por centro de análisis.
La proporción de sujetos que alcanzaron una mejora en el prurito, definida como un puntaje medio semanal en la escala de severidad de prurito (SPS, por sus siglas en inglés) de ≤ 1 con una mejora mínima de 1 punto en comparación con el valor inicial en una escala de severidad de 0 a 3, se evaluó en cada visita del estudio programada. Se observó una proporción estadísticamente significativamente mayor de sujetos que alcanzaron una mejoría en el prurito con STAQUIS® dos veces al día en comparación con los sujetos tratados con el excipiente dos veces al día en la Semana 4, tal como se resume en la Tabla 3.
Tabla 3: Mejora en la Escala de Severidad de Prurito (SPS) en sujetos con dermatitis atópica de leve a moderada en la semana 4
|
Ensayo 1 |
Ensayo 2 |
|||||
|
STAQUIS® Dos veces al día (N = 363) |
Excipiente dos veces al día (N = 170) |
Valor p |
STAQUIS® Dos veces al día (N = 363) |
Excipiente dos veces al día (N = 165) |
Valor p |
|
|
Mejora en SPS |
37.2% |
21.2% |
p < 0.001 |
34.4% |
20.6% |
p < 0.001 |
La mejora en la SPS se define como la obtención de un puntaje medio semanal en la escala de severidad de prurito (SPS) de ≤ 1 con una mejora al menos de 1 punto en comparación con el valor inicial.
Los índices de mejora del prurito con el paso del tiempo se presentan en la Figura 1.
Figura 1: Mejora en la SPS con el paso del tiempo en sujetos con dermatitis atópica de leve a moderada
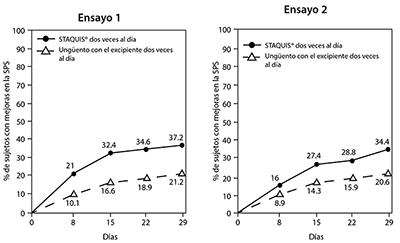
La mejora en la SPS se define como la obtención de un puntaje medio semanal en la escala de severidad de prurito (SPS) de ≤ 1 con una mejora al menos de 1 punto en comparación con el valor inicial.
El tiempo de mejora del prurito se definió como el tiempo transcurrido hasta obtener un puntaje de SPS medio diario ≤ 1 con una mejora al menos de 1 punto en comparación con el valor inicial. En el Ensayo 1 y en el Ensayo 2, los sujetos tratados con STAQUIS® dos veces al día tuvieron estadísticamente significativamente un menor tiempo mediano para obtener mejoras que los sujetos tratados con el excipiente. En el Ensayo 1, los sujetos tratados con STAQUIS® dos veces al día alcanzaron mejoras en los síntomas de prurito con una mediana de 4 días frente a 9 días en los sujetos tratados con el excipiente dos veces al día (p = 0.0008). En el Ensayo 2, los sujetos tratados con STAQUIS® dos veces al día alcanzaron mejoras en los síntomas de prurito con una mediana de 5 días frente a 9 días en los sujetos tratados con el excipiente dos veces al día p = 0.0042).
En los ensayos clínicos combinados (Ensayos 1 y 2), el grupo tratado con STAQUIS® dos veces al día mostró una mayor reducción en el ASC media tratable afectada con dermatitis atópica (-7.4% desde un valor inicial de 18.3%) en comparación con el grupo tratado con el excipiente dos veces al día (-4.4% desde un valor inicial de 18.1%) en el Día 29.
En un ensayo multicéntrico, abierto, no controlado, 137 sujetos pediátricos de 3 meses a menos de 2 años fueron tratados con STAQUIS® dos veces al día durante 4 semanas. La eficacia se consideró un objetivo exploratorio en este estudio. En el periodo inicial, el 38.0% de los sujetos tenían una ISGA de leve (2) y el 61.3% tenían una ISGA de moderada (3). En el Día 29, el 30.2% de los sujetos lograron una ISGA de limpia (0) o casi limpia (1) con al menos una mejora de 2 grados. Además, el 47.3% de los sujetos habían alcanzado una ISGA de limpia o casi limpia. El porcentaje medio del ASC tratable afectado con dermatitis atópica disminuyó del 28.1% en el valor inicial al 12.4% el Día 29. Los resultados de la ISGA y el porcentaje de ASC tratable fueron comparables a los observados entre los sujetos tratados con STAQUIS® en los Ensayos 1 y 2.
Terapia de mantenimiento:
En un ensayo de Fase 3, aleatorizado, doble ciego, controlado con excipiente (Ensayo 3) se evaluó la eficacia y seguridad de STAQUIS® una vez al día durante 52 semanas como tratamiento de mantenimiento para reducir la incidencia de brotes en pacientes adultos y pediátricos (3 meses a 17 años) con dermatitis atópica de leve a moderada, que respondieron a STAQUIS® dos veces al día durante un tratamiento abierto de hasta 8 semanas.
Un total de 497 sujetos de 5 meses a 79 años con una ASC tratable del 2% al 90% entraron en un periodo abierto para recibir tratamiento con STAQUIS® dos veces al día durante un máximo de 8 semanas. Al inicio, el 66.2% de los sujetos tenía una ISGA de moderada (3) y el 33.6% tenía una ISGA de leve (2), en la evaluación general de la dermatitis atópica (eritema, induración/papulación y supuración/formación de costras) en una escala de gravedad de 0 a 4.
De los 497, un total de 270 sujetos de 5 meses a 79 años, que lograron éxito tanto en la ISGA (puntuación de limpio [0] o casi limpio [1] con una mejora de ≥ 2 grados desde el periodo inicial) como en la respuesta de EASI50 (al menos el 50% de mejora desde el periodo inicial en las puntuaciones EASI) se aleatorizaron en una proporción de 1:1 en un periodo de mantenimiento doble ciego para recibir tratamiento con STAQUIS® una vez al día o el excipiente durante 52 semanas. Al inicio del periodo de mantenimiento, el 60.0% de los sujetos tenía una ISGA de casi limpio (1) y el 38.5% tenía un ISGA de limpio (0).
La eficacia se demostró mediante una duración prolongada estadísticamente significativa de días sin brotes al comparar sujetos tratados con STAQUIS® con sujetos tratados con el excipiente (Tabla 4, Figura 2).
Los criterios de valoración de eficacia secundarios incluyeron el número de días sin brotes y el número de brotes durante el periodo de mantenimiento de 52 semanas. Los sujetos tratados con la dosis de mantenimiento de STAQUIS® una vez al día de experimentaron más días sin brotes y menos brotes que los sujetos tratados con el excipiente durante el periodo de mantenimiento (Tabla 4).
Tabla 4: Resultados de eficacia en sujetos con dermatitis atópica leve a moderada durante el periodo de mantenimiento (hasta la semana 52)
|
Ensayo 3 |
||
|
STAQUIS® Una vez al día (N = 125) |
Excipiente una vez al día (N = 129) |
|
|
Días sin brotes hasta la aparición del primer brote (mediana de días [IC del 95%])* |
111 (56, 224) |
30 (28, 56) |
|
Cantidad de días sin brotes (media de LS [IC del 95%]) |
234 (209.7; 258.3) |
199 (176.1; 222.7) |
|
Cantidad de brotes (media) |
0.95 |
1.36 |
* Estimado mediante el método de Kaplan-Meier (límite del producto); IC: Intervalo de Confianza; LS: Mínimos Cuadrados.
Figura 2: Diagrama de Kaplan-Meier del ensayo 3 de días sin brotes durante el periodo de mantenimiento (52 semanas)
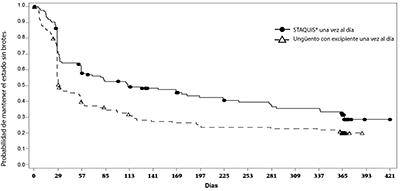
CONTRAINDICACIONES:
Hipersensibilidad al principio activo o a cualquiera de los excipientes. Embarazo y lactancia.
Menores de 3 meses de edad.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Existen datos limitados sobre la administración de STAQUIS® en mujeres embarazadas. Los estudios en animales no indican efectos perjudiciales directos o indirectos con respecto a la toxicidad reproductiva en dosis no tóxicas para la maternidad (consulte la sección Precauciones en relación con los efecto de carcinogénesis, mutagénesis, teratogénesis y sobre fertilidad). Debido a que los estudios de reproducción en animales no siempre predicen la respuesta humana, como medida de precaución, se debe considerar el beneficio clínico de STAQUIS® para la madre junto con cualquier riesgo potencial para el feto.
Es preferible evitar el uso de STAQUIS® durante el embarazo.
Lactancia:
No se realizaron estudios de excreción de leche en animales después de la aplicación cutánea, y no se estudió el uso de STAQUIS® en mujeres en periodo de lactancia. STAQUIS® se absorbe de manera sistemática. Se desconoce si crisaborol o los metabolitos se excretan en la leche humana después de la aplicación cutánea de STAQUIS®, así como los efectos del medicamento en el lactante o en la producción de leche materna. La falta de datos clínicos durante la lactancia impide una determinación clara del riesgo de STAQUIS® para un lactante. Por lo tanto, los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de STAQUIS® para la madre y cualquier posible efecto adverso en el lactante debido a STAQUIS® o debido a la afección subyacente de la madre. Debido a la posibilidad de reacciones adversas en el lactante, STAQUIS® no debe utilizarse en mujeres que estén dando pecho.
Para evitar que el recién nacido ingiera STAQUIS® accidentalmente, el producto no debe aplicarse en las mamas.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las reacciones adversas al medicamento más frecuentes observadas en los ensayos clínicos completados de STAQUIS® (Ensayos 1 y 2) fueron reacciones en el lugar de aplicación (5.6% y 3.6% para los grupos que recibieron STAQUIS® y el excipiente, respectivamente) y la mayoría de estos eventos se clasificaron como leves. De estas reacciones en el lugar de la aplicación relacionadas con el medicamento, el dolor en el lugar de aplicación (p. ej., ardor o picazón) fue la única reacción adversa que mostró una diferencia clínicamente relevante en los índices entre los grupos de tratamiento (4.4% y 1.2% para el grupo que recibió STAQUIS® y para el que recibió el excipiente, respectivamente). En general, al principio del periodo de tratamiento, se observó dolor en el sitio de aplicación de corta duración, el cual se resolvió de manera espontánea.
Tabla 5: Reacciones adversas al medicamento
|
Clasificación por órganos y sistemas |
Reacciones adversas al medicamento |
|
Trastornos generales y alteraciones en el lugar de la administración |
Reacciones en el lugar de aplicación (p. ej., dolor en el lugar de aplicación*, prurito en el lugar de aplicación) |
* Hace referencia a las sensaciones en la piel, como ardor o picazón
En el Ensayo 3, las reacciones adversas al medicamento observadas durante el periodo abierto de tratamiento dos veces al día durante un máximo de 8 semanas fueron consistentes con el perfil de seguridad conocido de STAQUIS® dos veces al día. Durante el periodo de mantenimiento doble ciego en el Ensayo 3 hasta la Semana 52, no se informaron casos de dolor en el lugar de aplicación en el grupo tratado con STAQUIS® una vez al día, en comparación con el 1.5% (2/135 sujetos) en el grupo tratado con excipiente una vez al día (ver sección Farmacocinética y farmacodinamia).
Ensayo clínico pediátrico:
En un ensayo multicéntrico, abierto, no controlado, 137 sujetos pediátricos de 3 meses a menos de 2 años fueron tratados con STAQUIS® dos veces al día durante 4 semanas. En general, el perfil de seguridad de STAQUIS® en este grupo etario fue consistente con el de los Ensayos 1 y 2 en sujetos de 2 años y mayores.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Los datos preclínicos no revelan riesgo especial alguno para los humanos, según estudios convencionales de farmacología de seguridad, toxicidad de dosis repetida, genotoxicidad, carcinogenicidad, toxicidad juvenil o toxicidad que afecta la reproducción y el desarrollo.
Los estudios toxicológicos de dosis repetida demostraron que la administración de crisaborol por vías tanto cutánea como oral en ratones, ratas y cerdos enanos en exposiciones plasmáticas hasta 11 veces mayores que en humanos no provocó una toxicidad significativa pertinente al uso en humanos.
Crisaborol no reveló evidencias de potencial mutagénico o clastogénico con base en los resultados de dos pruebas de genotoxicidad in vitro (ensayo Ames y ensayo de aberración cromosómica de linfocitos humanos) y una prueba de genotoxicidad in vivo (ensayo de micronúcleos de ratas).
En un estudio de carcinogenicidad oral en ratas Sprague-Dawley, se administraron dosis orales de 30, 100, o 300 mg/kg por día de crisaborol en ratas una vez al día. Se observó una mayor incidencia relacionada con crisaborol de tumores de células granulares benignos en útero, cuello uterino y vagina (combinados) en las ratas hembra tratadas con 300 mg/kg/día de crisaborol (2 veces la dosis humana máxima recomendada [MRHD, por sus siglas en inglés] sobre una base de comparación de área bajo la curva [ABC]). Se desconoce la relevancia clínica de este hallazgo; sin embargo, dado el tipo de tumor y el estado de benigno en una sola especie y en un solo sexo, la relevancia para los humanos se considera baja.
En un estudio de carcinogenicidad cutánea en ratones CD-1, se administraron dosis cutáneas de crisaborol ungüento al 2%, 5% o 7% una vez al día. No hubo hallazgos relacionados con crisaborol en las dosis cutáneas de crisaborol ungüento hasta al 7% (1 vez la MRHD sobre una base de comparación de ABC).
Según los hallazgos, crisaborol no es tóxico para la salud reproductiva ni es un teratógeno de acuerdo con los estudios de toxicología reproductiva en dosis no tóxicas para la maternidad que examinaron los efectos sobre la fertilidad, el desarrollo embriofetal y la generación de F1. Se observó toxicidad materna en ratas (asociada a una disminución del peso corporal fetal y a un retraso en la osificación esquelética), pero no se observaron malformaciones fetales relacionadas con el crisaborol después de la administración oral de crisaborol durante la organogénesis en dosis máximas de 600 mg/kg/día (13 veces la MRHD sobre una base de comparación de ABC). Crisaborol no causó efectos adversos en el feto con dosis orales hasta la dosis máxima analizada de 100 mg/kg/día en conejas preñadas, administrado durante el periodo de organogénesis (2 veces la MRHD sobre una base de comparación de ABC).
En un estudio de desarrollo prenatal/posnatal en ratas, crisaborol no tuvo efectos adversos sobre el desarrollo fetal en dosis máximas de hasta 300 mg/kg/día (3 veces la MRHD sobre una base de comparación de ABC). La toxicidad materna se produjo en la dosis alta de 600 mg/kg/día en ratas preñadas y se asoció a hallazgos de mortinatos, mortalidad de las crías y disminución de peso de las crías.
No se observaron efectos sobre la fertilidad en ratas macho o hembra que recibieron dosis de hasta 600 mg/kg/día de crisaborol (13 veces la MRHD sobre una base de comparación de ABC) antes del embarazo y durante la etapa inicial del embarazo.
Los estudios en ratas y cerdos enanos jóvenes no revelaron hallazgos relevantes que indicaran un riesgo específico de la administración en la población pediátrica.
Fertilidad:
Los estudios de reproducción en ratas hembra o macho a los cuales se administrada crisaborol por vía oral no revelaron efectos sobre la fertilidad.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Según se prevé, ni crisaborol ni sus 2 metabolitos principales causan interacciones medicamentosas por inducción o inhibición de las enzimas del citocromo P450 (CYP) según los datos in vitro e in vivo (consulte la sección Farmacocinética y farmacodinamia).
Uno de los 2 metabolitos mostró una inhibición moderada de la uridina difosfato (UDP)-glucuronosiltransferasa (UGT) 1A9 y puede provocar un aumento moderado en las concentraciones de sustratos de UGT1A9 sensibles (consulte la sección Farmacocinética y farmacodinamia).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
No se cuenta con datos sobre alteraciones en los resultados de pruebas de laboratorio.
PRECAUCIONES GENERALES:
STAQUIS® no es para uso oral, oftálmico o intravaginal. En casos de exposición accidental en estas zonas, el ungüento debe quitarse completamente con un paño y/o enjuagarse con agua.
Hipersensibilidad:
Se ha producido hipersensibilidad, incluyendo urticaria de contacto, en pacientes tratados con STAQUIS®. Se debe sospechar la presencia de hipersensibilidad en casos de prurito severo, hinchazón y eritema en el lugar de aplicación o en una zona alejada de éste. Si se presentan signos y síntomas de hipersensibilidad, suspenda el uso de STAQUIS® de inmediato e inicie un tratamiento adecuado.
No debe aplicarse en el cuero cabelludo.
Efectos sobre la capacidad de conducir o usar máquinas:
No se han realizado estudios con STAQUIS® sobre el efecto de la capacidad para conducir o utilizar máquinas, por lo tanto, STAQUIS® no tiene influencia conocida sobre la capacidad para conducir o utilizar máquinas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Cutánea.
Si persisten algunos signos y/o síntomas o aparecen nuevas zonas afectadas, se pueden aplicar más ciclos de tratamiento siempre que la aplicación no supere el 40% del ASC.
Se debe suspender la aplicación del ungüento si los signos y/o síntomas en las zonas tratadas persisten después de 3 ciclos de tratamiento consecutivos de 4 semanas cada uno o si los signos y/o síntomas empeoran durante el tratamiento.
Posología:
Adultos:
Se debe aplicar una capa de ungüento dos veces al día en las áreas afectadas para el tratamiento de la enfermedad activa; durante un máximo de 4 semanas por ciclo de tratamiento.
Para el mantenimiento, una vez que las áreas afectadas estén limpias o casi limpias, se debe aplicar una capa de ungüento una vez al día en las áreas más comúnmente afectadas. Si los signos y síntomas de la enfermedad empeoran, se debe aplicar una capa de ungüento dos veces al día en las áreas afectadas.
STAQUIS® puede aplicarse sobre la piel de todo el cuerpo, incluso en el rostro, el cuello y las zonas intertriginosas. No se ha estudiado el uso de STAQUIS® en el cuero cabelludo. Por lo tanto no debe aplicarse en el cuero cabelludo.
Únicamente se debe aplicar el ungüento en las zonas afectadas de la piel hasta un máximo del 40% del Área de Superficie Corporal (ASC).
Población pediátrica:
Para los niños y adolescentes (de 3 meses a 17 años), la posología es la misma que para los adultos.
Poblaciones especiales:
No se realizaron ensayos clínicos con sujetos que presentan insuficiencia renal o hepática. Sin embargo, no se espera que sea necesario el ajuste de la dosis en los sujetos con insuficiencia hepática leve a moderada o en los sujetos con insuficiencia renal.
Los estudios clínicos de STAQUIS® no incluyeron números suficientes de sujetos de 65 años y mayores como para determinar si responden de forma diferente en comparación con los sujetos más jóvenes. Sin embargo, no se espera que sea necesario el ajuste de la dosis en esta población de pacientes.
Método de administración:
STAQUIS® sólo es para uso cutáneo y no para uso oral, oftálmico o intravaginal.
STAQUIS® no se ha estudiado bajo oclusión. Sin embargo, la experiencia clínica disponible para el uso del ungüento bajo oclusión (es decir, pañales o ropa) no ha demostrado la necesidad de un ajuste de la dosis. No debe aplicarse en el cuero cabelludo.
Los pacientes deben recibir la instrucción de lavarse las manos después de aplicar STAQUIS®, a menos que el tratamiento deba aplicarse en las manos. Si otra persona aplica STAQUIS® al paciente, esa persona deberá lavarse las manos después de la aplicación.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No hubo experiencias de sobredosis con STAQUIS®. La sobredosis después de la administración cutánea es poco probable. Si se aplicó demasiado STAQUIS®, el exceso puede limpiarse con un paño, y lavar la zona con agua.
PRESENTACIONES:
Caja con tubo de 30 g o 60 g (2%) e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 30 °C. Consérvese el tubo bien cerrado.
LEYENDAS DE PROTECCIÓN:
Material exclusivo para profesionales de atención médica. No se deje al alcance de los niños. No se use en menores de 3 meses. No ingerible. No aplicar en el cuero cabelludo. No se use durante el embarazo y la lactancia.
Reporte las sospechas de reacción adversa a los correos electrónicos: farmacovigilancia@cofepris.gob.mx y
MEX.AEReporting@pfizer.com y
a la línea de Pfizer 800 401 2002.
PFIZER, S.A. de C.V.
Km. 63 Carretera México, Toluca,
Zona Industrial, C.P. 50140,
Toluca, México, México.
Reg. Núm. 175M2022 SSA IV