
TERIEM
TERIFLUNOMIDA
Comprimidos
1 Caja, 14 Comprimidos, 14 mg
1 Caja, 28 Comprimidos, 14 mg
1 Caja, 56 Comprimidos, 14 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada COMPRIMIDO contiene:
Teriflunomida 14 mg Excipiente cbp 1 comprimido
INDICACIONES TERAPÉUTICAS:
Está indicado para el tratamiento de pacientes con formas recurrentes de esclerosis múltiple (EM), incluyendo síndrome clínico aislado, enfermedad recurrente- remitente y la enfermedad activa secundaria progresiva para reducir la frecuencia de exacerbaciones clínicas y retrasa la incapacidad física.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades Farmacocinéticas:
Absorción:
El tiempo medio para alcanzar las concentraciones plasmáticas máximas se produce entre 1 y 4 horas después de la dosis tras la administración oral repetida de teriflunomida, con una alta biodisponibilidad (aproximadamente 100%) determinada mediante la comparación de estudios cruzados. Los alimentos produjeron una disminución estadísticamente significativa en la Cmáx (18%) y un aumento en el Tmáx (aproximadamente 3 horas), que no tiene un efecto clínicamente relevante sobre la farmacocinética de teriflunomida.
Con base en la predicción individual de los parámetros farmacocinéticos, utilizando el modelo de farmacocinética poblacional (PopPK) de teriflunomida en voluntarios sanos y pacientes con EM, la dosis no fue una covariable significativa de la farmacocinética de teriflunomida.
Hay un enfoque lento hacia la concentración en estado estacionario (es decir, aproximadamente 100 días (3.5 meses) para alcanzar el 95% de las concentraciones en estado estacionario, según la vida media terminal (t1/2z) de aproximadamente 19 días calculada a partir de la farmacocinética poblacional (PopPK) utilizando datos de voluntarios sanos y pacientes con EM) y el cociente estimado de acumulación del AUC es de aproximadamente 34 veces para 14 mg de teriflunomida.
Distribución:
La teriflunomida se une ampliamente a las proteínas plasmáticas (> 99%), probablemente a la albúmina, y se distribuye principalmente en el plasma, en lugar de los glóbulos rojos. El volumen de distribución es bajo (11 L) tras una sola administración intravenosa (IV).
Biotransformación:
La teriflunomida se metaboliza de forma moderada y es el único componente detectado en el plasma. La principal forma de biotransformación de la teriflunomida es la hidrólisis, siendo la oxidación una vía menor. Otras vías implican N-acetilación y la conjugación de sulfatos.
Eliminación:
La teriflunomida se excreta en el tracto gastrointestinal principalmente a través de la bilis como medicamento inalterado y probablemente por secreción directa. La teriflunomida es un sustrato del transportador de salida de proteína resistente al cáncer de mama (BCRP), que podría estar relacionado con la secreción directa. Durante 21 días, el 60.1% de la dosis administrada se excreta a través de las heces (37.5%) y la orina (22.6%). Después del procedimiento de eliminación acelerada con colestiramina, se recuperó un 23.1% adicional (principalmente en heces). Según la predicción individual de parámetros farmacocinéticos que utilizan el modelo PopPK de teriflunomida en voluntarios sanos y pacientes de EM, la vida media exponencial terminal t1/2z fue de aproximadamente 19 días tras dosis repetidas de 14 mg. Después de una única administración IV, la eliminación total de teriflunomida del cuerpo es de 30.5 ml/h. El reciclaje biliar es uno de los principales contribuyentes a la larga vida media de eliminación de la teriflunomida. Estudios con hemodiálisis y CAPD (crónica diálisis peritoneal ambulatoria) indican que la teriflunomida no es dializable.
Procedimiento de eliminación acelerada:
Colestiramina y carbón activado: La eliminación de la teriflunomida de la circulación se puede acelerar mediante la administración de colestiramina y carbón activado, presumiblemente mediante la interrupción de los procesos de reabsorción en el intestino. Las concentraciones de teriflunomida medidas durante un procedimiento de 11 días para acelerar la eliminación de la teriflunomida con 8 g de colestiramina tres veces al día, 4 g de colestiramina tres veces al día o 50 g de carbón activado dos veces al día tras el cese del tratamiento, han mostrado que estas dosificaciones fueron eficaces a la hora de acelerar la eliminación de la teriflunomida, provocando un descenso de más del 98% en las concentraciones de teriflunomida en plasma, siendo la colestiramina más rápida que el carbón. Tras la interrupción del tratamiento con teriflunomida y administrar colestiramina 8 g tres veces al día, la concentración en plasma de la teriflunomida se redujo al 52% al final del día 1, 91% al final del día 3, 99.2% al final del día 7 y 99.9% al final del día 11. La elección de uno de los 3 procedimientos de eliminación debe depender de la tolerabilidad del paciente. Si no se tolera bien la colestiramina 8 g tres veces al día, se puede utilizar colestiramina 4 g tres veces al día. De forma alternativa, también se puede utilizar carbón activado (no es necesario que los 11 días sean consecutivos a menos que haya necesidad de reducir la concentración de teriflunomida en plasma rápidamente).
Linealidad/no linealidad:
La exposición sistémica aumenta de forma proporcional a la dosis tras la administración oral de teriflunomida de 7 a 14 mg.
Características en grupos específicos de pacientes:
Sexo, personas de edad avanzada:
Se identificaron varias fuentes de variabilidad intrínseca en sujetos sanos y pacientes con EM según el análisis de PopPK: edad, peso corporal, sexo, raza y niveles de albúmina y bilirrubina. No obstante, el impacto sigue siendo limitado (< 31%).
Insuficiencia hepática:
La insuficiencia hepática leve y moderada no afectó a la farmacocinética de teriflunomida. Así, no es necesario anticipar un ajuste de dosis en pacientes con insuficiencia hepática leve y moderada. No obstante, teriflunomida está contraindicada en pacientes con insuficiencia hepática grave, ver dosis y vía de administración, y contraindicaciones.
Insuficiencia renal:
La insuficiencia renal grave no afecta a la farmacocinética de teriflunomida. Así, no es necesario anticipar un ajuste de dosis en pacientes con insuficiencia renal leve, moderada y grave, ver dosis y vía de administración, y contraindicaciones
Población pediátrica:
En pacientes pediátricos con peso corporal > 40 kg tratados con 14 mg una vez al día, las exposiciones en estado estacionario estuvieron en el rango observado en pacientes adultos tratados con el mismo régimen de dosificación.
En pacientes pediátricos con peso corporal ≤ 40 kg tratamiento con 7 mg una vez al día (basado en simulaciones y datos clínicos limitados) condujo a exposiciones en estado estacionario en el rango observado en pacientes adultos tratados con 14 mg una vez al día. Las concentraciones mínimas en estado estacionario observadas fueron muy variables entre individuos, como se observó en pacientes adultos con EM.
Propiedades farmacodinámicas:
La teriflunomida es un inhibidor oral de la síntesis de pirimidina de novo de la enzima dihidroorotato deshidrogenasa (DHO-DH). La teriflunomida es clase 2 en el sistema de clasificación biofarmacéutica.
Mecanismo de acción:
Teriflunomida es un agente inmunomodulador con propiedades antiinflamatorias que inhibe de forma selectiva y reversible la enzima mitocondrial dihidroorotato-deshidrogenasa (DHO-DH), necesaria para la síntesis de novo de la pirimidina. Como consecuencia, teriflunomida bloquea la proliferación de linfocitos estimulados que necesitan la síntesis de novo de la pirimidina para expandirse. Las células en reposo o que se dividen lentamente y que dependen de la vía de recuperación para la síntesis de pirimidina no se ven afectadas por la teriflunomida. El mecanismo exacto por el cual teriflunomida ejerce un efecto terapéutico en la EM no se comprende del todo, pero puede incluir una cantidad reducida de linfocitos activados en el SNC (sistema nervioso central). Es probable que la teriflunomida disminuya en la periferia el número de linfocitos activados disponibles para migrar al SNC.
Sistema inmunológico:
Efectos en el número de células inmunológicas en la sangre: En los estudios controlados con placebo, teriflunomida 14 mg una vez al día provocó una leve reducción media en el recuento de linfocitos, de 0.3 x 109/l, la mayoría de los cuales se produjeron durante los primeros 3 primeros meses de tratamiento, después de los cuales los niveles se mantuvieron hasta el final del tratamiento.
Potencial para prolongar el intervalo QT:
En un estudio QT exhaustivo controlado con placebo realizado en sujetos sanos, teriflunomida en concentraciones medias en estado estacionario no mostró ninguna capacidad para prolongar el intervalo QTcF en comparación con placebo: la mayor diferencia en el tiempo entre teriflunomida y placebo fue de 3.46 ms, con un límite máximo de 6.45 ms en el 90% CI. Además, ningún valor de QTcF fue ≥ 480 ms y ningún cambio desde el inicio fue > 60 ms.
Efecto en las funciones tubulares renales:
En los estudios controlados con placebo, se observaron disminuciones medias en el ácido úrico en suero en un rango de 20 a 30% en pacientes tratados con teriflunomida en comparación con placebo. La disminución media del fósforo sérico fue del 10% en el grupo de teriflunomida en comparación con el placebo. Se considera que estos efectos están relacionados con un aumento de la excreción tubular renal y no están relacionados con los cambios en las funciones glomerulares.
Ensayos clínicos:
La eficacia de teriflunomida se estableció mediante un estudio de fase 2 y dos de fase 3 controlados con placebo en pacientes con formas recurrentes de RMS, un estudio de comparación activa de fase 3 y un estudio de fase 3 controlado con placebo en pacientes con EM temprana (es decir, con un primer episodio clínico).
El estudio 1 (EFC6049/TEMSO) fue un estudio doble ciego controlado con placebo que evaluó dosis diarias de teriflunomida de 7 mg y 14 mg en pacientes con formas recurrentes de esclerosis múltiple (EMR) durante 108 semanas.
1,088 pacientes con EMR fueron aleatorizados para recibir 7 mg (n = 366) o 14 mg (n = 359) de teriflunomida o placebo (n = 363) durante 108 semanas. Todos los pacientes tenían un diagnóstico definitivo (basado en los criterios de McDonald) de EM que presentaba un curso clínico recidivante, con o sin progresión, y experimentaron al menos 1 recaída durante el año anterior al ensayo o al menos 2 recaídas durante los 2 años anteriores al ensayo. En el momento del ingreso, los pacientes tenían una puntuación en la Escala Expandida del Estado de Discapacidad (EDSS) ≤ 5.5. La edad media de la población de estudio fue de 37.9 años.
El criterio principal de valoración fue la tasa de recaída anualizada (ARR). La tasa de recaída anualizada fue significativamente menor en los pacientes tratados con teriflunomida que los pacientes que recibieron placebo. El criterio de valoración secundario fue el tiempo hasta la progresión de la discapacidad, sostenida durante 12 semanas. El riesgo de progresión de la discapacidad se redujo de forma estadísticamente significativa en el grupo de teriflunomida 14 mg en comparación con placebo. No se demostró una diferencia estadísticamente significativa en la progresión de la discapacidad sostenida durante 24 semanas. La proporción estimada de pacientes libres de recaídas en la semana 108 fue del 45.6% en el grupo placebo y 56.5% en el grupo teriflunomida 14 mg.
Se evaluaron los efectos de la teriflunomida en las variables de imágenes por resonancia magnética (RM) (carga de la enfermedad definida como el volumen total de todas las lesiones anormales del tejido cerebral y otras variables de resonancia RM). Los resultados indicaron que teriflunomida 14 mg es más eficaz en la progresión de la discapacidad y los parámetros de resonancia magnética que teriflunomida 7 mg. Los resultados de este estudio se muestran en la Tabla 1 y Figura 1.
Tabla 1. Resultados clínicos y de resonancia magnética del estudio EFC6049/TEMSO
|
Estudio TEMSO |
|||
|---|---|---|---|
|
Teriflunomida 14 mg (N = 358*) |
Placebo (N = 363) |
Teriflunomida 14 mg versus placebo |
|
|
Criterios de valoración clínicos |
|||
|
ARR: ajustado (variable principal) |
0.369 |
0.539 |
RRa (95 IC): 0.69 (0.55, 0.85) 0.0005b |
|
Probabilidad de progresión de la discapacidad en la semana 108 |
20.2% |
27.3% |
HRc (95 IC):0.70 (0.51, 0.97) 0.0279b |
|
Punto final de resonancia magnética |
|||
|
Carga de enfermedad (mL) |
|||
|
Cambio medio (DE) desde el valor inicial en la semana 108 |
0.723 (7.59) |
2.208 (7.00) |
|
|
Cambio medio (DE) [LSM (SE)] en el valor absoluto de la DBO transformada en raíz cúbica desde el inicio en la semana 108 |
0.045 (0.30) [0.043 (0.02)] |
0.111 (0.31) [0.132 (0.02)] |
Diferencia media LSM (SE) del placebo: - 0.089 (0.025) 0.0003b |
|
Número de lesiones T1 realzadas con Gd por Resonancia Magnética en la semana 108 |
0.261 |
1.331 |
RRa (95% CI): 0.196 |
|
Volumen de lesiones T1 hipointensas (ml) Cambio medio (DE) desde el inicio en la semana 108 |
0.331 (1.012) |
0.533 (1.063) |
|
|
Cambio medio de LS (SE) desde el inicio en la semana 108 |
0.066 (0.009) |
0.096 (0.009) |
Diferencia media LSM (SE) del placebo: -0.030 (0.013) |
* Un paciente aleatorizado a teriflunomida 14 mg no fue tratado.
a Riesgo relativo.
b Valor p.
c Relación de riesgo.
ARR: tasa de recaída anualizada.
DBO-Carga de enfermedad.
HR-Relación de riesgo.
Figura 1. Gráfica de Kaplan-Meier del tiempo hasta la progresión de la discapacidad sostenida durante 12 semanas-población ITT (EFC6049/TEMSO)
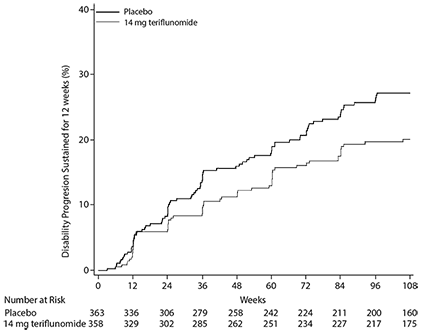 La probabilidad de progresión de la discapacidad a las 108 semanas (con IC del 90%) fue de 0.273 (0.223, 0.323) para el placebo y de 0.202 (0.156, 0.247) para la teriflunomida de 14 mg.
La probabilidad de progresión de la discapacidad a las 108 semanas (con IC del 90%) fue de 0.273 (0.223, 0.323) para el placebo y de 0.202 (0.156, 0.247) para la teriflunomida de 14 mg.
El estudio 2 (EFC10531/TOWER) fue un estudio doble ciego controlado con placebo que evaluó dosis diarias de teriflunomida de 7 mg y 14 mg en pacientes con formas recidivantes de esclerosis múltiple (EMR) con una duración variable del tratamiento (promedio de aproximadamente 18 meses). Todos los pacientes tenían un diagnóstico definitivo de EM que mostraba un curso clínico recidivante, con o sin progresión, y experimentaron al menos 1 recaída durante el año anterior al ensayo o al menos 2 recaídas durante los 2 años anteriores al ensayo. Los sujetos no habían recibido interferón-beta ni ningún otro medicamento preventivo para la EM durante al menos 3 meses antes de ingresar al estudio ni se permitieron estos medicamentos durante el estudio. Se realizaron evaluaciones neurológicas en la selección, cada 12 semanas hasta la finalización y en visitas no programadas por sospecha de recaída. El criterio principal de valoración fue la tasa de recaída anualizada (ARR).
Un total de 1169 pacientes fueron aleatorizados para recibir 7 mg (n = 408) o 14 mg (n = 372) de teriflunomida o placebo (n = 389). La edad media fue de 37.9 años, la duración media de la enfermedad fue de 5.16 años y la EDSS media al inicio del estudio fue de 2.69 para el placebo y de 2.71 para 14 mg. La mayoría de los pacientes tenían EM remitente recurrente (97.5%). El tiempo medio con placebo fue de 571 días y con 14 mg de teriflunomida de 567 días. La ARR se redujo significativamente en los pacientes tratados con 14 mg de teriflunomida en comparación con los pacientes que recibieron placebo (ver Tabla 2).
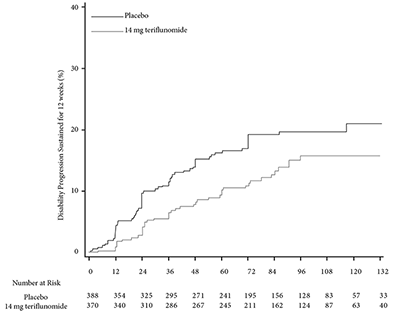
El riesgo de progresión de la discapacidad sostenida durante 12 semanas (medido por al menos un aumento de 1 punto desde el valor inicial de EDSS ≤ 5.5 o un aumento de 0.5 puntos para aquellos con un valor inicial de EDSS > 5.5) se redujo de forma estadísticamente significativa sólo en el grupo de teriflunomida 14 mg en comparación con el placebo (Tabla 2 y Figura 2).
Tabla 2. Resultados clínicos del estudio EFC10531/TOWER
|
Teriflunomida 14 mg (N = 370) |
Placebo |
Teriflunomida 14 mg versus placebo |
|
|
Criterios de valoración clínicos |
|||
|
ARR: ajustado (variable principal) |
0.319 |
0.501 |
RRa (95% CI): 0.64 |
|
Probabilidad de progresión de la discapacidad en la semana 108 |
15.80% |
19.70% |
HRc (95% CI): 0.68 |
a Riesgo relativo.
b Valor p.
c Cociente de riesgos instantáneos-Derivado usando el modelo de riesgos proporcionales de Cox con tratamiento, estratos EDSS al inicio y la región como covariables.
ARR: tasa de recaída anualizada.
HR Cociente de riesgos instantáneos.
Figure 2. Kaplan-Meier plot of time to disability progression sustained for 12 weeks-ITT population (EFC10531/TOWER)
El estudio 3 (EFC6260/TOPIC) fue un estudio doble ciego controlado con placebo que evaluó dosis diarias de teriflunomida de 7 mg y 14 mg durante un máximo de 108 semanas en pacientes con EM temprana (es decir, un primer episodio clínico). Los pacientes tuvieron un primer evento neurológico que ocurrió dentro de los 90 días posteriores a la aleatorización, con 2 o más lesiones T2 de al menos 3 mm de diámetro que eran características de la EM. El punto final primario fue el tiempo hasta un segundo episodio clínico (recaída).
Un total de 618 pacientes fueron aleatorizados para recibir 7 mg (n = 205) o 14 mg (n = 216) de teriflunomida o placebo (n = 197). La edad media de la población de estudio fue de 32.1 años y el tiempo medio desde el primer evento neurológico fue de 1.85 meses, el 59.1% de los pacientes ingresaron al estudio con episodio monofocal y el 40.9% con episodio multifocal. El tiempo medio con placebo fue de 464 días y con 14 mg de teriflunomida de 493 días.
El riesgo de un segundo episodio clínico se redujo de forma estadísticamente significativa en el grupo de 14 mg de teriflunomida en comparación con el grupo de placebo (Tabla 3).
Tabla 3. Resultados clínicos de EFC6260/TOPIC
|
Teriflunomida 14 mg |
Placebo |
Teriflunomida 14 mg versus placebo |
|
|
Criterios de valoración clínicos |
|
|
|
|
Porcentaje de pacientes que permanecen libres de un segundo episodio clínico en la semana 108 (variable principal) |
76.00% |
64.10% |
HRa (95% CI): 0.57 |
|
Porcentaje de pacientes que permanecen libres de un segundo episodio clínico y una nueva lesión en la RM en la semana 108 |
28.50% |
13.00% |
HRb (95% CI): 0.65 |
a Cociente de riesgo.
b Valor p.
La actividad de IRM de teriflunomida también se mostró en un estudio de fase 2 (estudio 4 [Estudio 2001]). Un total de 179 pacientes recibieron 7 mg (n = 61) o 14 mg (n = 57) de teriflunomida o placebo (n = 61) durante 36 semanas. Los datos demográficos iniciales fueron consistentes en todos los grupos de tratamiento. El número medio de lesiones activas únicas por resonancia magnética cerebral durante el periodo de tratamiento de 36 semanas fue menor en los pacientes tratados con teriflunomida 14 mg (0.98) en comparación con el placebo (2.69), siendo la diferencia estadísticamente significativa (p = 0.0052).
El estudio 5 (EFC10891/TENERE) fue un estudio metacéntrico, aleatorizado, de grupos paralelos, ciego para el evaluador, que comparó la eficacia y la seguridad de teriflunomida e interferón beta-1a en pacientes con esclerosis múltiple recidivante más un periodo de extensión a largo plazo. La eficacia de la teriflunomida se comparó con la de un interferón beta-1a subcutáneo (a la dosis recomendada de 44μg tres veces por semana) en 324 pacientes aleatorizados en un estudio con una duración mínima del tratamiento de 48 semanas (máximo 114 semanas). El riesgo de fracaso (recaída confirmada o interrupción permanente del tratamiento, lo que ocurriera primero) fue el criterio principal de valoración. La teriflunomida 14 mg/día no fue estadísticamente superior al interferón beta-1a en el criterio principal de valoración: el porcentaje estimado de pacientes con fracaso del tratamiento a las 96 semanas mediante el método de Kaplan-Meier fue del 41.1% frente al 44.4%.
Población pediátrica:
Niños y adolescentes (10 a 17 años):
El estudio EFC11759/TERIKIDS fue un estudio internacional doble ciego controlado con placebo en pacientes pediátricos de 10 a 17 años con EM remitente-recurrente que evaluó dosis de teriflunomida una vez al día (ajustadas para alcanzar una exposición equivalente a la dosis de 14 mg en adultos) por hasta 96 semanas seguidas de una extensión de etiqueta abierta. Todos los pacientes habían experimentado al menos 1 recaída durante 1 año o al menos 2 recaídas durante los 2 años anteriores al estudio. Se realizaron evaluaciones neurológicas en el cribado y cada 24 semanas hasta su finalización, y en visitas no programadas por sospecha de recaída. Los pacientes con recaída clínica promedio o alta actividad de resonancia magnética de al menos 5 lesiones T2 nuevas o en aumento en 2 exploraciones consecutivas se cambiaron antes de las 96 semanas a la extensión de etiqueta abierta para garantizar un tratamiento activo. El criterio principal de valoración fue el tiempo hasta la primera recaída clínica después de la aleatorización. El tiempo hasta la primera recaída clínica confirmada o alta actividad de resonancia magnética, lo que sucediera primero, se predefinió como análisis de sensibilidad porque incluye condiciones tanto clínicas como de resonancia magnética que califican para cambiar al periodo abierto.
Un total de 166 pacientes fueron aleatorizados en una proporción de 2:1 para recibir teriflunomida (n=109) o placebo (n = 57). Al ingreso, los pacientes del estudio tenían una puntuación EDSS ≤ 5.5; la edad media fue de 14.6 años; el peso medio fue de 58.1 kg; la duración media de la enfermedad desde el diagnóstico fue de 1.4 años; y la media de lesiones realzadas con Gd en T1 por resonancia magnética fue de 3.9 lesiones al inicio del estudio. Todos los pacientes tenían EM remitente recurrente con una puntuación EDSS media de 1.5 al inicio del estudio. El tiempo medio de tratamiento fue de 362 días con placebo y 488 días con teriflunomida. El cambio del periodo doble ciego al tratamiento abierto debido a la alta actividad de la resonancia magnética fue más frecuente de lo previsto, y más frecuente y más temprano en el grupo de placebo que en el grupo de teriflunomida (26% con placebo, 13% con teriflunomida).
La teriflunomida redujo el riesgo de recaída clínica en un 34% en relación con el placebo, sin alcanzar significación estadística (p = 0.29). En el análisis de sensibilidad predefinido, la teriflunomida logró una reducción estadísticamente significativa en el riesgo combinado de recaída clínica o alta actividad de resonancia magnética en un 43% en relación con el placebo (p = 0.04).
La teriflunomida redujo significativamente el número de lesiones T2 nuevas y en aumento por exploración en un 55% (p = 0.0006) (el análisis post-hoc también se ajustó para los recuentos T2 iniciales: 34%, p = 0.0446), y el número de lesiones T1 realzadas con gadolinio por exploración en un 75% (p < 0.0001).
Resultados clínicos y de resonancia magnética de EFC11759/TERIKIDS
|
EFC11759 ITT población |
Teriflunomida (N = 109) |
Placebo (N = 57) |
|
Criterios de valoración clínicos |
||
|
Tiempo hasta la primera recaída clínica confirmada, |
||
|
Probabilidad (IC del 95%) de recaída confirmada en la semana 96 |
0.39 (0.29, 0.48) |
0.53 (0.36, 0.68) |
|
Probabilidad (IC del 95%) de recaída confirmada en la semana 48 |
0.30 (0.21, 0.39) |
0.39 (0.30, 0.52) |
|
Cociente de riesgos instantáneos (IC del 95%) |
0.66 (0.39, 1.11)^ |
|
|
Tiempo hasta la primera recaída clínica confirmada o alta actividad de resonancia magnética, |
||
|
Probabilidad (IC del 95%) de recaída confirmada o alta actividad de resonancia magnética en la semana 96 |
0.51 (0.41, 0.60) |
0.72 (0.58, 0.82) |
|
Probabilidad (95% IC) de recaída confirmada o alta actividad de resonancia magnética en la semana 48 |
0.38 (0.29, 0.47) |
0.56 (0.42, 0.68) |
|
Cociente de riesgos instantáneos (IC del 95%) |
0.57 (0.37, 0.87)* |
|
|
Criterios de valoración clave de la RM |
||
|
Número ajustado de lesiones T2 nuevas o agrandadas |
||
|
Estimación(95% CI) |
4.74 (2.12, 10.57) |
10.52 (4.71, 23.50) |
|
Estimación (IC del 95%), análisis post-hoc también ajustado para los recuentos de T2 basales |
3.57 (1.97, 6.46) |
5.37 (2.84, 10.16) |
|
Riesgo Relativo (95% IC) |
0.45 (0.29, 0.71)** |
|
|
Riesgo relativo (IC del 95%), análisis post-hoc también ajustado para recuentos T2 basales |
0.67 (0.45, 0.99)* |
|
|
Número ajustado de lesiones realzadas con Gd T1 Estimación (95% IC) |
1.90 (0.66, 5.49) |
7.51 (2.48, 22.70) |
|
Riesgo Relativo (95% CI) |
0.25 (0.13, 0.51)*** |
|
^ p ≥ 0.05 comparado con placebo, * p < 0.05, ** p < 0.001, ***p < 0.0001.
La probabilidad se basó en el estimador de Kaplan-Meier y la semana 96 fue el final del tratamiento del estudio (EOT).
CONTRAINDICACIONES:
Hipersensibilidad al principio activo o a alguno de los excipientes.
Pacientes con insuficiencia hepática grave (Child-Pugh clase C).
Mujeres embarazadas o mujeres en edad fértil que no estén utilizando métodos anticonceptivos fiables durante el tratamiento con teriflunomida y posteriormente siempre que sus niveles plasmáticos estén por encima de 0.02 mg/l (ver Restricciones de uso durante el embarazo y la lactancia). El embarazo debe ser excluido antes del inicio del tratamiento (ver Restricciones de uso durante el embarazo y la lactancia).
Mujeres en periodo de lactancia (ver Restricciones de uso durante el embarazo y la lactancia).
Pacientes con estados de inmunodeficiencia grave, por ejemplo, síndrome de inmunodeficiencia adquirida (SIDA).
Pacientes con función significativamente alterada de la médula ósea o anemia significativa, leucopenia, neutropenia o trombocitopenia.
Pacientes con infección activa grave hasta su resolución (ver Precauciones generales).
Pacientes con insuficiencia renal grave sometidos a diálisis, porque no se dispone de experiencia clínica suficiente en este grupo de pacientes.
Pacientes con hipoproteinemia grave, por ejemplo, en el síndrome nefrótico.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Uso en hombres: El riesgo de toxicidad embriofetal mediada por el hombre a través del tratamiento con teriflunomida se considera bajo (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Embarazo: Hay una cantidad limitada de datos sobre el uso de teriflunomida en mujeres embarazadas. Los estudios en animales han mostrado toxicidad para la reproducción (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Teriflunomida puede causar defectos de nacimiento graves cuando se administra durante el embarazo. Teriflunomida está contraindicada durante el embarazo (ver Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante y después del tratamiento siempre que la concentración plasmática de teriflunomida sea superior a 0.02 mg/L. Durante este periodo, las mujeres deben discutir cualquier plan para suspender o cambiar la anticoncepción con el médico tratante.
Las niñas y/o padres/cuidadores de niñas deben ser informados sobre la necesidad de contactar al médico tratante una vez que la niña bajo tratamiento con teriflunomida inicie su menstruación.
Se debe proporcionar asesoramiento a las nuevas pacientes en edad fértil acerca de la anticoncepción y el riesgo potencial para el feto. Se debe considerar derivar con un ginecólogo.
Se debe advertir a la paciente de que, si hay algún retraso en el inicio de la menstruación o cualquier otra razón para sospechar un embarazo, debe suspender la teriflunomida y notificar al médico de inmediato para realizar una prueba de embarazo y, si fuera positiva, el médico y la paciente deben discutir el riesgo para el embarazo. Es posible que reducir rápido el nivel de teriflunomida en sangre, al instituir el procedimiento de eliminación acelerada que se describe a continuación, en el primer retraso de la menstruación, pueda reducir el riesgo para el feto
Para las mujeres que reciben tratamiento con teriflunomida y desean quedar embarazadas, se debe suspender el medicamento y se recomienda un procedimiento de eliminación acelerada para alcanzar más rápidamente una concentración inferior a 0.02 mg/L (ver a continuación).
Si no se utiliza un procedimiento de eliminación acelerada, se puede esperar que los niveles de teriflunomida en plasma pueden ser superiores a 0.02 mg/L durante una media de 8 meses, sin embargo, en algunos pacientes pueden pasar hasta 2 años para alcanzar una concentración por debajo de 0.02 mg/L. Por lo tanto, las concentraciones plasmáticas de teriflunomida deben medirse antes de que una mujer comience a intentar quedar embarazada. Una vez que se determine que la concentración plasmática de teriflunomida es inferior a 0.02 mg/L, la concentración plasmática debe determinarse nuevamente después de un intervalo al menos, 14 días. Si ambas concentraciones están por debajo de 0.02 mg/L, se espera que no haya riesgo para el feto.
Tras interrumpir el tratamiento con teriflunomida:
• Se administra colestiramina 8 g 3 veces al día durante un periodo de 11 días o, si esta dosificación no se tolera bien, se puede utilizar colestiramina 4 g tres veces al día.
• Alternativamente se puede utilizar 50 g de carbón activado en polvo cada 12 horas durante 11 días.
No obstante, después de realizar los procedimientos de eliminación acelerada, es necesario verificar mediante 2 pruebas separadas por un intervalo de, al menos, 14 días y esperar un mes y medio entre el primer resultado inferior a 0.02 mg/L y la fertilización.
Tanto la colestiramina como el carbón activado en polvo pueden influir en la absorción de estrógenos y progestágenos, de modo que es posible que no se garantice una anticoncepción fiable con anticonceptivos orales durante el procedimiento de eliminación acelerada con colestiramina o carbón activado en polvo. Se recomienda el uso de métodos anticonceptivos alternativos.
Lactancia: Los estudios realizados en animales han mostrado que teriflunomida se excreta en la leche materna. Por ello, no se debe administrar teriflunomida a las mujeres en periodo de lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad:
Un total de 2,267 pacientes fueron expuestos a teriflunomida (1,155 a teriflunomida 7 mg y 1,112 a teriflunomida 14 mg) una vez al día durante un periodo medio de unos 672 días en cuatro estudios controlados mediante placebo (1,045 y 1,002 pacientes para teriflunomida 7 mg y 14 mg, respectivamente) y un estudio comparativo activo (110 pacientes en cada uno de los grupos de tratamiento con teriflunomida) en pacientes con formas recurrentes de EM (esclerosis múltiple recurrente, EMR).
Teriflunomida es el metabolito principal de leflunomida. El perfil de seguridad de leflunomida en pacientes que sufren de artritis reumatoide y artritis psoriásica puede ser pertinente a la hora de prescribir teriflunomida en pacientes con EM.
El análisis combinado controlado mediante placebo se basó en 2,047 pacientes con esclerosis múltiple recurrente tratados con teriflunomida una vez al día. Con esta población de seguridad, las reacciones adversas notificadas con más frecuencia en los pacientes tratados con teriflunomida fueron: cefalea, diarrea, aumento de ALT, náuseas y alopecia. En general, la cefalea, la diarrea, las náuseas y la alopecia, fueron de leves a moderadas, transitorias e infrecuentemente condujeron a la interrupción del tratamiento.
A continuación, se muestran las reacciones adversas notificadas con teriflunomida en estudios controlados con placebo, notificadas para teriflunomida 7 mg o 14 mg en un rango superior al > 1% frente a placebo, se muestran a continuación. Las frecuencias se definieron según la siguiente convención: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1 000); muy raras (< 1/10.000); desconocido (no se puede estimar a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las reacciones adversas se clasifican en orden descendente de gravedad.
Infecciones e infestaciones:
Frecuentes: influenza, infección del tracto respiratorio superior, infección del tracto urinario, bronquitis, sinusitis, faringitis, cistitis, gastroenteritis viral, herpes bucal, infección dental, laringitis, tinea pedis.
Poco frecuentes: infecciones graves, incluida sepsisa.
Trastornos de la sangre y del sistema linfático:
Común: neutropeniab , anemia.
Poco frecuentes: trombocitopenia leve (plaquetas < 100G/l).
Trastornos del sistema inmunológico:
Común: reacciones alérgicas leves.
Poco común: reacciones de hipersensibilidad (inmediata o retardada) incluyendo anafilaxia y angioedema.
Trastornos psiquiátricos:
Frecuentes: ansiedad.
Trastornos del sistema nervioso:
Muy común: dolor de cabeza.
Común: parestesia, ciática, síndrome del túnel carpiano
Poco frecuentes: hiperestesia, neuralgia, neuropatía periférica.
Trastornos cardiacos:
Frecuentes: palpitaciones.
Trastornos vasculares:
Común: hipertensiónb.
Trastornos respiratorios, torácicos y mediastínicos:
Poco frecuentes: enfermedad pulmonar intersticial.
Frecuencia no conocida: hipertensión pulmonar.
Desórdenes gastrointestinales:
Muy comunes: diarrea, náuseas.
Frecuentes: pancreatitisb,c, dolor abdominal superior, vómitos, dolor de muelas.
Poco frecuentes: estomatitis, colitis.
Trastornos hepatobiliares:
Muy frecuentes: aumento de la alanina aminotransferasa (ALT)b.
Frecuentes: aumento de gammaglutamiltransferasa (GGT)b, aumento de aspartato aminotransferasab. Raros: hepatitis aguda.
No conocida: daño hepático inducido por medicamento. Trastornos de la piel y del tejido subcutáneo:
Muy común: alopecia.
Común: erupción, acné.
Poco frecuentes: trastornos de las uñas, psoriasis (incluyendo pustulosa)b, reacciones cutáneas gravesa.
Trastornos musculoesqueléticos y del tejido conjuntivo:
Frecuentes: dolor musculoesquelético, mialgia, artralgia.
Trastornos renales y urinarios:
Frecuentes: polaquiuria.
Trastornos del aparato reproductor y de la mama:
Frecuentes: menorragia.
Desórdenes generales y condiciones administrativas del sitio:
Frecuentes: dolor, asteniaa.
Investigaciones:
Frecuentes: disminución de peso, disminución del recuento de neutrófilosb, disminución del recuento de glóbulos blancosb, aumento de la creatina fosfoquinasa en sangre.
Lesiones, intoxicaciones y complicaciones de procedimientos:
Poco frecuentes: dolor postraumático.
Trastornos del metabolismo y de la nutrición:
Poco común: dislipidemia.
a Ver la sección de descripción detallada.
b Ver Precauciones Generales.
c La frecuencia es “común” en niños según un estudio clínico controlado en pediatría; la frecuencia es “poco común” en adultos.
Descripción de reacciones adversas seleccionadas:
Alopecia: La alopecia se notificó como adelgazamiento del pelo, dimisión de la densidad del pelo, pérdida del pelo, asociados o no al cambio en la textura del pelo, en el 13.9% de los pacientes tratados con 14 mg de teriflunomida frente al 5.1% de los pacientes tratados con placebo. La mayoría de los casos se describen como difusos o generalizados en el cuero cabelludo (no se informó pérdida completa del pelo) y ocurrieron con mayor frecuencia durante los primeros 6 meses y con una resolución en 121 de 139 (87.1%) pacientes tratados con teriflunomida 14 mg. La interrupción del tratamiento debido a la alopecia fue del 1.3% en los grupos de teriflunomida 14 mg, frente al 0.1% del grupo de placebo.
Efectos hepáticos:
Durante los estudios controlados con placebo se detectó lo siguiente:
Aumentos de ALT (basados en datos de laboratorio) según estatus basal-Seguridad en los pacientes de los estudios controlados con placebo
|
Placebo (N = 997) |
Teriflunomida 14 mg (N = 1,002) |
|
|---|---|---|
|
> 3 LSN |
66/994 (6,6%) |
80/999 (8,0%) |
|
> 5 LSN |
37/994 (3,7%) |
31/999 (3,1%) |
|
> 10 LSN |
16/994 (1,6%) |
9/999 (0,9%) |
|
> 20 LSN |
4/994 (0,4%) |
3/999 (0,3%) |
|
ALT > 3 LSN y TBILI > 2 LSN |
5/994 (0,5%) |
3/999 (0,3%) |
Se observaron con mayor frecuencia aumentos leves en la transaminasa, con ALT por debajo o igual a 3 veces el LSN, en los grupos tratados con teriflunomida en comparación con el placebo. La frecuencia de elevaciones encima de 3 veces el LSN y superiores se equilibró entre los grupos de tratamiento. Estas elevaciones de las transaminasas ocurrieron principalmente dentro de los 6 primeros meses de tratamiento y fueron reversibles después de la interrupción del tratamiento. El tiempo de recuperación varió entre meses y años.
Efectos de la presión sanguínea:
En los estudios controlados con placebo se estableció lo siguiente:
• La presión arterial sistólica fue de > 140 mm Hg en el 19.9% de los pacientes que recibieron 14 mg/día en comparación con el 15.5% de los pacientes que recibieron placebo;
• la presión arterial sistólica fue de > 160 mm Hg en el 3.8% de los pacientes que recibieron 14 mg/día en comparación con el 2.0% de los pacientes que recibieron placebo;
• la presión arterial diastólica fue de > 90 mm Hg en el 21.4% de los pacientes que recibieron 14 mg/día en comparación con el 13.6% de los pacientes que recibieron placebo.
Infecciones:
En estudios controlados con placebo, no se observó un aumento de infecciones graves con teriflunomida 14 mg (2.7%) en comparación con placebo (2.2%). Se presentaron infecciones oportunistas graves en el 0.2% de cada grupo. Se han notificado infecciones graves, incluyendo sepsis, en ocasiones mortales, posteriores a la comercialización.
Efectos hematológicos:
En ensayos controlados con teriflunomida, se observó una disminución media que afectaba al recuento de glóbulos blancos (< 15% respecto a los niveles basales, principalmente una disminución de neutrófilos y linfocitos), aunque se observó una disminución mayor en algunos pacientes. La disminución en el recuento medio desde el inicio ocurrió durante las primeras 6 semanas y luego se estabilizó con el tiempo durante el tratamiento, pero a niveles reducidos (menos de un 15% de disminución desde el inicio). El efecto sobre los recuentos de glóbulos rojos (< 2%) y plaquetas (< 10%) fue menos pronunciado.
Neuropatía periférica:
En estudios controlados con placebo, la neuropatía periférica, que incluye polineuropatía y mononeuropatía (por ejemplo, síndrome del túnel carpiano), se notificó con mayor frecuencia en pacientes que tomaban teriflunomida que en pacientes que tomaban placebo. En los estudios pivotales controlados con placebo, la incidencia de neuropatía periférica confirmada por estudios de conducción nerviosa fue del 1.9% (17 pacientes de 898) con 14 mg de teriflunomida, en comparación con el 0.4% (4 pacientes de 898) con placebo. El tratamiento se interrumpió en 5 pacientes con neuropatía periférica con teriflunomida 14 mg. La recuperación después de la suspensión del tratamiento se notificó en 4 de estos pacientes.
Neoplasias benignas, malignas y no especificadas (incl. quistes y pólipos):
No parece haber un mayor riesgo de malignidad con teriflunomida en la experiencia de los ensayos clínicos. El riesgo de malignidad, en particular de trastornos linfoproliferativos, aumenta con el uso de otros agentes que afectan el sistema inmunológico (efecto de clase).
Reacciones cutáneas graves:
Se han notificado casos de reacciones cutáneas graves con teriflunomida después de la comercialización.
Astenia:
En estudios controlados con placebo, las frecuencias de astenia fueron del 2.0% 1.6% y 2.2% en el grupo de placebo, teriflunomida 7 mg y teriflunomida grupo de 14 mg, respectivamente.
Psoriasis:
En estudios controlados con placebo, las frecuencias de psoriasis fueron del 0.3%, 0.3% y 0.4% en el grupo placebo, teriflunomida 7 mg y teriflunomida grupo de 14 mg, respectivamente.
Desórdenes gastrointestinales:
Se ha notificado pancreatitis con poca frecuencia en el periodo posterior a la comercialización con teriflunomida en adultos incluidos casos de pancreatitis necrotizante y seudoquiste pancreático. Los eventos pancreáticos pueden ocurrir en cualquier momento durante el tratamiento con teriflunomida, lo que puede conducir a la hospitalización y/o requerir tratamiento correctivo.
Población pediátrica:
El perfil de seguridad observado en pacientes pediátricos (de 10 a 17 años de edad) que recibieron teriflunomida diariamente fue en general similar al observado en pacientes adultos. Sin embargo, en el estudio pediátrico (166 pacientes: 109 en el grupo de teriflunomida y 57 en el de placebo grupo), se notificaron casos de pancreatitis en el 1.8% (2/109) de los pacientes tratados con teriflunomida en comparación con ninguno en el grupo placebo, en la fase de doble ciego. Uno de estos hechos condujo a la hospitalización y requirió tratamiento correctivo. En pacientes pediátricos tratados con teriflunomida en la fase abierta del estudio, 2 casos adicionales de pancreatitis (uno fue informado como evento grave, el otro fue un evento no grave de intensidad leve) y un caso de pancreatitis aguda grave (con pseudo-papiloma) fueron reportados. En dos de estos 3 pacientes, la pancreatitis motivó la hospitalización. Los síntomas clínicos incluyeron dolor abdominal, náuseas y/o vómitos, la amilasa y la lipasa séricas estaban elevadas en estos pacientes. Todos los pacientes se recuperaron después de la interrupción del tratamiento y el procedimiento de eliminación acelerada (ver Precauciones generales) y tratamiento correctivo.
Las siguientes reacciones adversas se notificaron con mayor frecuencia en la población pediátrica que en la población adulta:
• Se notificó alopecia en el 22.0% de los pacientes tratados con teriflunomida frente al 12.3% en pacientes tratados con placebo.
• Se notificaron infecciones en el 66.1% de los pacientes tratados con teriflunomida frente al 45.6% en pacientes tratados con placebo. Entre ellos, la nasofaringitis y las infecciones del tracto respiratorio superior se informaron con mayor frecuencia con teriflunomida.
• Se notificó un aumento de CPK en el 5.5% de los pacientes tratados con teriflunomida versus 0% en pacientes tratados con placebo. La mayoría de los casos se asociaron con ejercicio físico documentado.
• Se notificó parestesia en el 11.0% de los pacientes tratados con teriflunomida frente al 1.8% en pacientes tratados con placebo.
• Se notificó dolor abdominal en el 11.0% de los pacientes tratados con teriflunomida frente al 1.8% en pacientes tratados con placebo.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Los animales son más sensibles a la farmacología y, por tanto, a la toxicidad de la teriflunomida que los humanos. Así, se observó toxicidad en animales a exposiciones equivalentes o inferiores a los niveles terapéuticos humanos. Teriflunomida no fue mutagénica in vitro o clastogénica in vivo. La clastogenicidad observada in vitro se consideró un efecto indirecto relacionado con el desequilibrio de la combinación de nucleótidos provocado por la farmacología de la inhibición de DHO-DH.
El metabolito menor TFMA (4-trifluorometilanilina) provocó mutagenicidad y clastogenicidad in vitro, pero no in vivo.
No se observaron evidencias de carcinogenicidad en ratas y ratones.
La fertilidad no resultó afectada en las ratas a pesar de los efectos adversos de teriflunomida en los órganos reproductores masculinos, incluyendo el descenso del recuento de espermatozoides. No hubo malformaciones en la descendencia de las ratas macho a las que se administró teriflunomida antes de su apareamiento con ratas hembra sin tratar. Teriflunomida fue embriotóxica y teratogénica en ratas y conejos a dosis que están dentro del intervalo terapéutico humano. También se observaron efectos adversos en la descendencia tras la administración de teriflunomida a ratas preñadas durante la gestación y la lactancia. Se considera que el riesgo de toxicidad embriofetal mediada por el varón a través del tratamiento con teriflunomida es bajo. La exposición estimada del plasma femenino a través del semen de un paciente tratado se espera que sea 100 veces menor que la exposición del plasma tras 14 mg de teriflunomida oral.
Fertilidad:
Los resultados de los estudios en animales no han mostrado ningún efecto sobre la fertilidad (ver Farmacocinética y farmacodinamia). Aunque se carece de datos en humanos, no se anticipa ningún efecto sobre la fertilidad masculina y femenina.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones farmacocinéticas de otras sustancias en teriflunomida:
La principal vía de biotransformación de la teriflunomida es la hidrólisis, siendo la oxidación una vía secundaria.
Inductores potentes del citocromo P450 (CYP) y del transportador:
La coadministración de dosis repetidas (600 mg una vez al día durante 22 días) de rifampicina (un inductor de CYP2B6, 2C8, 2C9, 2C19, 3A), así como un inductor de los transportadores de salida de glicoproteína P [P-gp] y la proteína resistente al cáncer de mama [BCRP] con teriflunomida (dosis única de 70 mg) resultó en una disminución de aproximadamente 40% en la exposición a teriflunomida. La rifampicina y otros inductores potentes del CYP y del transportador conocido, como la carbamazepina, el fenobarbital, la fenitoína y la hierba de San Juan, deben utilizarse con precaución durante el tratamiento con teriflunomida.
Colestiramina o carbón activado:
Se recomienda que los pacientes que reciben teriflunomida no sean tratados con colestiramina o carbón activado porque esto conduce a una disminución rápida y significativa de la concentración plasmática a menos que se desee una eliminación acelerada. Se cree que el mecanismo es por interrupción del reciclaje enterohepático y/o diálisis gastrointestinal de teriflunomida.
Interacciones farmacocinéticas de teriflunomida sobre otras sustancias:
Efecto de la teriflunomida sobre el sustrato CYP2C8: repaglinida: Hubo un aumento en la media de Cmáx y ABC de repaglinida (1.7 y 2.4 veces, respectivamente), luego de dosis repetidas de teriflunomida, lo que sugiere que teriflunomida es un inhibidor de CYP2C8 in vivo. Por lo tanto, los medicamentos metabolizados por CYP2C8, como repaglinida, paclitaxel, pioglitazona o rosiglitazona, deben usarse con precaución durante el tratamiento con teriflunomida.
Efecto de teriflunomida en anticonceptivos orales:
0.03 mg de etinilestradiol y 0.15 mg de levonorgestrel:
Hubo un incremento de la Cmáx y AUC0-24 medios del etinilestradiol (1.58 y 1.54 veces, respectivamente) y Cmáx y AUC0-24 de levonorgestrel (1.33 y 1.41 veces, respectivamente) después de dosis repetidas de teriflunomida. Si bien no se espera que esta interacción de teriflunomida afecte negativamente la eficacia de los anticonceptivos orales, debe tenerse en cuenta al seleccionar o ajustar el tratamiento anticonceptivo oral usado en combinación con teriflunomida.
Efecto de la teriflunomida sobre el sustrato CYP1A2:
Cafeína: Las dosis repetidas de teriflunomida disminuyeron la Cmáx y el AUC medias de la cafeína (sustrato de CYP1A2) en un 18% y un 55%, respectivamente, lo que sugiere que la teriflunomida puede ser un inductor débil de CYP1A2 in vivo. Por lo tanto, los medicamentos metabolizados por CYP1A2 (como duloxetina , alosetrón, teofilina y tizanidina) deben usarse con precaución durante el tratamiento con teriflunomida, ya que podría reducir la eficacia de estos medicamentos.
Efecto de la teriflunomida sobre la warfarina:
Las dosis repetidas de teriflunomida no tuvieron efecto sobre la farmacocinética de la S-warfarina, lo que indica que la teriflunomida no es un inhibidor ni un inductor de CYP2C9. Sin embargo, se observó una disminución del 25% en el índice internacional normalizado (INR) máximo cuando se coadministró teriflunomida con warfarina en comparación con warfarina sola. Por lo tanto, cuando se coadministra warfarina con teriflunomida, se recomienda un estrecho seguimiento y control del INR.
Efecto de la teriflunomida en los sustratos del transportador de aniones orgánicos 3 (OAT3): Hubo un aumento en la media de Cmáx y AUC de cefaclor (1.43 y 1.54 veces, respectivamente), luego de dosis repetidas de teriflunomida, lo que sugiere que teriflunomida es un inhibidor de OAT3 in vivo. Por lo tanto, cuando se coadministra teriflunomida con sustratos de OAT3, como cefaclor, bencilpenicilina, ciprofloxacina, indometacina, ketoprofeno, furosemida, cimetidina, metotrexato, zidovudina, se recomienda precaución.
Efecto de la teriflunomida sobre sustratos de BCRP y/o polipéptido transportador de aniones orgánicos B1 y B3 (OATP1B1/B3):
Hubo un aumento en la media de Cmáx y AUC de rosuvastatina (2.65 y 2.51 veces, respectivamente), luego de dosis repetidas de teriflunomida. Sin embargo, no hubo un impacto aparente de este aumento en la exposición plasmática a la rosuvastatina en la actividad de la HMG-CoA reductasa. Para rosuvastatina, se recomienda una reducción de la dosis del 50% para la coadministración con teriflunomida. Para otros sustratos de BCRP (por ejemplo, metotrexato, topotecán, sulfasalazina, daunorrubicina, doxorrubicina) y la familia OATP, especialmente los inhibidores de la reductasa HMG-Co (por ejemplo, simvastatina, atorvastatina, pravastatina, metotrexato, nateglinida, repaglinida, rifampicina), se debe administrar concomitantemente teriflunomida también debe llevarse a cabo con cautela. Se debe controlar de cerca a los pacientes para detectar signos y síntomas de exposición excesiva a los medicamentos y se debe considerar la reducción de la dosis de estos medicamentos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Datos preclínicos de seguridad:
La administración oral repetida de teriflunomida en ratones, ratas y perros hasta 3, 6 y 12 meses, respectivamente, reveló que la toxicidad afecta principalmente a la médula ósea, los órganos linfoides, la cavidad oral/tracto gastrointestinal, los órganos reproductores y el páncreas. También se observaron evidencias de efecto oxidativo en los hematíes. Anemia, descenso en el recuento de plaquetas y efectos en el sistema inmunológico, incluyendo leucopenia, linfopenia e infecciones secundarias, estuvieron relacionados con los efectos en la médula ósea y/o los órganos linfoides. La mayoría de los efectos reflejan el modo básico de acción del compuesto (inhibición de la división celular).
Efectos hepáticos:
Durante los estudios controlados con placebo se detectó lo siguiente:
Aumentos de ALT (basados en datos de laboratorio) según estatus basal-Seguridad en los pacientes de los estudios controlados con placebo
|
Placebo (N = 997) |
Teriflunomida 14 mg (N = 1,002) |
|
|
> 3 LSN |
66/994 (6,6%) |
80/999 (8,0%) |
|
> 5 LSN |
37/994 (3,7%) |
31/999 (3,1%) |
|
> 10 LSN |
16/994 (1,6%) |
9/999 (0,9%) |
|
> 20 LSN |
4 /994 (0,4%) |
3/999 (0,3%) |
|
ALT > 3 LSN y TBILI > 2 LSN |
5/994 (0,5%) |
3/999 (0,3%) |
PRECAUCIONES GENERALES:
Monitorización:
Antes del tratamiento:
Antes de iniciar el tratamiento con teriflunomida, se debe evaluar lo siguiente:
• Presión arterial.
• Alanina aminotransferasa/transaminasa glutámico pirúvico sérica (ALT/SGPT).
• Recuento completo de glóbulos, incluido el recuento diferencial de glóbulos blancos y plaquetas.
Durante el tratamiento:
Durante el tratamiento con teriflunomida se debe dar seguimiento a:
• Presión arterial.
? Revisar periódicamente.
• Alanina aminotransferasa/transaminasa glutámico pirúvico sérica (ALT/SGPT).
? Las enzimas hepáticas se deben evaluar al menos cada cuatro semanas durante los primeros 6 meses de tratamiento y después regularmente.
? Considere un control adicional cuando se administre teriflunomida a pacientes con trastornos hepáticos preexistentes, junto con otros medicamentos potencialmente hepatotóxicos o según lo indiquen los signos y síntomas clínicos, como náuseas, vómitos, dolor abdominal, fatiga, anorexia o ictericia y/u orina oscura sin causa aparente, las enzimas hepáticas deben evaluarse cada dos semanas durante los primeros 6 meses de tratamiento y, a partir de entonces, al menos cada 8 semanas durante al menos 2 años desde el inicio del tratamiento.
? Para elevaciones de ALT (SGPT) entre 2 y 3 veces el límite superior de lo normal, la monitorización debe realizarse semanalmente.
• Se debe realizar biometría hemática completa en función de los signos y síntomas clínicos (por ejemplo, infecciones) durante el tratamiento.
• Se deben realizar recuentos sanguíneos completos
• Se debe realizar un recuento sanguíneo completo según signos y síntomas clínicos (por ejemplo, infecciones) durante el tratamiento.
Procedimiento de eliminación acelerada:
La teriflunomida se elimina lentamente del plasma. Sin un procedimiento de eliminación acelerada, toma un promedio de 8 meses alcanzar concentraciones plasmáticas inferiores a 0.02 mg/L, aunque debido a la variación individual en la eliminación de sustancias puede tardar hasta 2 años.
Se puede utilizar un procedimiento de eliminación acelerada en cualquier momento después de discontinuación de teriflunomida (ver Restricciones de uso durante el embarazo y la lactancia, Farmacocinética y farmacodinamia).
Efectos hepáticos: Se han observado elevaciones de las enzimas hepáticas en pacientes que reciben teriflunomida (ver Reacciones secundarias y adversas). Estas elevaciones ocurrieron principalmente dentro de los primeros 6 meses de tratamiento.
Se han observado casos de lesión hepática inducida por medicamentos (DILI por sus siglas en inglés) durante el tratamiento con teriflunomida, en ocasiones potencialmente mortales. La mayoría de los casos de DILI ocurrieron con un tiempo de inicio de varias semanas o varios meses después del inicio del tratamiento con teriflunomida, pero DILI también puede ocurrir con el uso prolongado.
El riesgo de aumento de las enzimas hepáticas y DILI con teriflunomida podría ser mayor en pacientes con trastornos hepáticos preexistente, concomitante con otros medicamentos hepatotóxicos, y/o consumo de cantidades sustanciales de alcohol. Por lo tanto, los pacientes deben ser monitoreados de cerca para detectar signos y síntomas de daño hepático.
El tratamiento con teriflunomida debe suspenderse si se sospecha daño hepático; considere interrumpir el tratamiento con teriflunomida si se confirman enzimas hepáticas elevadas (más de 3 veces del LSN [Límite Superior de lo Normal]).
Los pacientes con enfermedad hepática preexistente y/o que consumen cantidades sustanciales de alcohol pueden tener un mayor riesgo de desarrollar enzimas hepáticas elevadas cuando toman teriflunomida y deben ser monitoreados de cerca para detectar signos de enfermedad hepática.
Hipoproteinemia: Ya que la teriflunomida está altamente ligada a las proteínas y su unión depende de las concentraciones de albúmina, se espera que las concentraciones de teriflunomida libre en plasma aumenten en pacientes con hipoproteinemia, por ejemplo, con síndrome nefrótico. Teriflunomida no debe utilizarse en pacientes con situaciones de hipoproteinemia grave. Presión arterial: Puede producirse una elevación de la presión arterial durante el tratamiento con teriflunomida (ver la sección Efectos no deseados). Debe comprobarse la presión arterial antes de comenzar el tratamiento con teriflunomida y, de forma periódica, a partir de entonces. Se debe tratar adecuadamente el aumento de la presión arterial antes y durante el tratamiento con teriflunomida.
Infecciones: En pacientes con una infección activa grave, el inicio del tratamiento con teriflunomida se debe retrasar hasta su resolución.
En estudios controlados mediante placebo, no se observó un aumento de las infecciones graves con teriflunomida (ver Reacciones adversas). No obstante, debido al efecto inmunomodulador de teriflunomida, si un paciente desarrolla una infección grave, se debe considerar la interrupción del tratamiento con teriflunomida y se deben volver a valorar los beneficios y los riesgos antes de volver a iniciarlo. Debido a su prolongada semivida, se puede considerar la eliminación acelerada con colestiramina o carbón activado.
Se debe indicar a los pacientes en tratamiento con teriflunomida que notifiquen a un médico si sufren síntomas de infección. Los pacientes con infecciones activas agudas o crónicas no deben iniciar el tratamiento con teriflunomida hasta su resolución.
Se desconoce la seguridad de teriflunomida en pacientes con tuberculosis latente, ya que no se realizó de forma sistemática un cribado de tuberculosis en los estudios clínicos. En pacientes positivos en las pruebas de diagnóstico de cribado de la tuberculosis, se debe realizar un tratamiento médico estándar antes de empezar el tratamiento con teriflunomida.
Reacciones respiratorias: Se han notificado casos de Enfermedad pulmonar intersticial (EPI) y casos de hipertensión pulmonar con teriflunomida posterior a la comercialización.
El riesgo puede aumentar en pacientes con antecedentes de EPI.
La EPI puede ocurrir de forma aguda en cualquier momento durante la terapia con una presentación clínica variable.
La EPI puede ser fatal. Los síntomas pulmonares de nueva aparición o que empeoran, como tos persistente y disnea, pueden ser motivo para interrumpir el tratamiento y para realizar una mayor investigación, según corresponda. Si es necesario suspender el medicamento, debe considerar el inicio de un procedimiento de eliminación acelerada.
Efectos hematológicos: Se observó un descenso medio del recuento de leucocitos (< 15% de los niveles basales [ver Reacciones adversas]), como precaución, debe haber un hemograma completo reciente disponible, que incluya fórmula leucocitaria y plaquetas, antes de iniciar el tratamiento con teriflunomida y se debe valorar el hemograma completo durante el tratamiento con teriflunomida según lo indiquen los signos y síntomas clínicos (por ejemplo, infecciones).
En pacientes con anemia, leucopenia y/o trombocitopenia preexistentes, así como en pacientes con deterioro de la función de la médula ósea o con riesgo de supresión de médula ósea, aumenta el riesgo de alteraciones hematológicas. Si tales efectos se producen, se debe considerar el procedimiento de eliminación acelerada (ver anteriormente) para reducir los niveles de teriflunomida en plasma.
En caso de reacciones hematológicas graves, incluyendo pancitopenia, se debe interrumpir el tratamiento con teriflunomida y cualquier tratamiento mielosupresor simultáneo y se debe considerar un procedimiento de eliminación acelerada.
Reacciones cutáneas: Se han notificado casos de reacciones cutáneas graves con teriflunomida post-comercialización (incluyendo síndrome de Stevens Johnson y necrólisis epidérmica tóxica).
En pacientes tratados con leflunomida, el compuesto original, también se han notificado casos muy raros de reacciones al medicamento con eosinofilia y síntomas sistémicos (RMESS).
En caso de estomatitis ulcerosa, se debe interrumpir la administración de teriflunomida. Si se observan reacciones cutáneas y/o de las mucosas que aumentan la sospecha de reacciones graves generalizadas de la piel (síndrome de Stevens Johnson o necrólisis epidérmica tóxica-síndrome de Lyell), se debe interrumpir el tratamiento con teriflunomida y otros posibles tratamientos asociados, e iniciar un procedimiento de eliminación acelerada de inmediato. En tales casos, los pacientes no se deben reexponer a teriflunomida (ver Contraindicaciones).
Se han notificado nuevos casos de psoriasis (incluida la psoriasis pustulosa) y empeoramiento de la psoriasis preexistente durante el uso de teriflunomida. La retirada del tratamiento y el inicio de un procedimiento de eliminación acelerada pueden considerarse teniendo en cuenta la enfermedad y el historial médico del paciente.
Neuropatía periférica: Se han notificado casos de neuropatía periférica en pacientes en tratamiento con teriflunomida (ver Reacciones adversas). La mayoría de los pacientes mejoraron tras interrumpir el tratamiento con teriflunomida. Sin embargo, hubo una amplia variabilidad en el desenlace final, es decir, en algunos pacientes la neuropatía se resolvió y algunos pacientes tuvieron síntomas persistentes. Si un paciente en tratamiento con teriflunomida desarrolla una neuropatía periférica confirmada, se debe considerar la interrupción del tratamiento con teriflunomida y realizar un procedimiento de eliminación acelerada.
Vacunación: Dos estudios clínicos han mostrado que la vacunación con neoantígenos inactivados (primera vacunación), o antígeno de recuerdo (reexposición) fue segura y eficaz durante el tratamiento con teriflunomida. El uso de vacunas atenuadas vivas puede conllevar un riesgo de infecciones y, por tanto, se debe evitar.
Tratamientos inmunosupresores o inmunomoduladores: Ya que la leflunomida es el componente original de la teriflunomida, no se recomienda su administración simultánea.
No se ha evaluado la administración conjunta de tratamientos antineoplásicos o inmunosupresores utilizados para el tratamiento de la EM. Los estudios de seguridad, en los que la teriflunomida se administró de forma simultánea con interferón beta o acetato de glatirámero durante periodos de hasta un año, no revelaron ningún problema de seguridad específico, pero se observó un mayor índice de reacciones adversas en comparación con la monoterapia con teriflunomida. No se ha establecido la seguridad a largo plazo de estas combinaciones en el tratamiento de la esclerosis múltiple.
Cambio desde o a teriflunomida: Según los datos clínicos relacionados con la administración simultánea de teriflunomida con interferón beta o acetato de glatirámero, no se requiere un periodo de espera al iniciar teriflunomida tras interferón beta o acetato de glatirámero, o al iniciar interferón beta o acetato de glatirámero tras teriflunomida. Debido a la larga semivida de natalizumab, la exposición simultánea y, por tanto, los efectos inmunes simultáneos, pueden darse hasta 2-3 meses después de la interrupción de natalizumab si teriflunomida se inició de forma inmediata. Por tanto, se requiere precaución a la hora de cambiar pacientes de natalizumab a teriflunomida.
Según la semivida de fingolimod, es necesario un intervalo de 6 semanas sin tratamiento para su eliminación de la circulación y un periodo de 1 a 2 meses para que los linfocitos vuelvan a sus niveles normales tras la interrupción de fingolimod. Si se inicia teriflunomida durante este intervalo se provocará una exposición simultánea al fingolimod. Esto puede provocar un efecto aditivo en el sistema inmunológico y, por tanto, se requiere precaución. En pacientes con EM, la mediana de t½z fue de aproximadamente 19 días tras dosis repetidas de 14 mg. Si se decide interrumpir el tratamiento con teriflunomida, durante el intervalo de 5 semividas (aproximadamente 3.5 meses, aunque puede ser más en algunos pacientes), comenzar otros tratamientos provocará una exposición simultánea a teriflunomida. Esto puede provocar un efecto aditivo en el sistema inmunológico y, por tanto, se requiere precaución.
Interferencia con la determinación de los niveles de calcio ionizado: La medición de los niveles de calcio ionizado puede mostrar valores falsamente disminuidos durante el tratamiento con leflunomida y/o teriflunomida (el metabolito activo de la leflunomida) según el tipo de analizador de calcio ionizado utilizado (por ejemplo, analizador de gases en sangre).
Por lo tanto, debe cuestionarse la plausibilidad de la disminución observada de los niveles de calcio ionizado en pacientes en tratamiento con leflunomida o teriflunomida. En caso de mediciones dudosas, se recomienda determinar la concentración de calcio sérico ajustado por albúmina total.
Población pediátrica:
Pancreatitis:
En el ensayo clínico pediátrico se han observado casos de pancreatitis, algunos agudos, en pacientes que recibían teriflunomida (ver Reacciones secundarias y adversas).
Los síntomas clínicos incluyeron dolor abdominal, náuseas y/o vómitos. La amilasa y lipasa séricas estaban elevadas en estos pacientes. El tiempo de aparición osciló entre unos pocos meses y tres años. Los pacientes deben ser informados de los síntomas característicos de pancreatitis Si se sospecha pancreatitis, se deben obtener las enzimas pancreáticas y los parámetros de laboratorio relacionados. Si se confirma la pancreatitis, se debe suspender la teriflunomida y se debe iniciar un procedimiento de eliminación acelerada (ver Farmacocinética y farmacodinamia).
Lactosa: Ya que los comprimidos teriflunomida contienen lactosa, los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o malabsorción de glucosa o galactosa, no deben tomar este medicamento.
Efectos en la capacidad para conducir y utilizar maquinaria: La influencia de teriflunomida sobre la capacidad para conducir es nula o insignificante. En el caso de reacciones adversas como mareo, que se ha informado con leflunomida, el compuesto original, la capacidad del paciente para concentrarse y reaccionar correctamente puede verse afectada. En tales casos, los pacientes deben abstenerse de conducir y el uso de máquinas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Un médico con experiencia en el tratamiento de la esclerosis múltiple debe iniciar y supervisar el tratamiento.
La dosis recomendada de teriflunomida es de 14 mg por vía oral una vez al día.
Poblaciones especiales:
Población pediátrica (10 años y mayores):
En pacientes pediátricos (10 años de edad y mayores), la dosis recomendada depende del peso corporal:
• Pacientes pediátricos con peso corporal > 40 kg: 14 mg una vez al día.
• Pacientes pediátricos con peso corporal ≤ 40 kg: 7 mg una vez al día.
Pacientes pediátricos que alcanzan un peso corporal estable superior a 40 kg debe cambiarse a 14 mg una vez al día.
Población de edad avanzada:
TERIEM® se debe utilizar con precaución en pacientes de 65 o más años debido a la falta de datos suficientes sobre seguridad y eficacia.
Insuficiencia renal:
No será necesario ajustar la dosis en pacientes con insuficiencia renal leve, moderada o grave que no estén en diálisis.
No se evaluó a los pacientes con insuficiencia renal grave sometidos a diálisis. Teriflunomida está contraindicada en esta población (ver Contraindicaciones).
Insuficiencia hepática:
No será necesario ajustar la dosis en pacientes con insuficiencia hepática leve y moderada. Teriflunomida está contraindicada en pacientes con insuficiencia hepática grave (ver Contraindicaciones).
Población pediátrica:
No se ha establecido todavía la seguridad y la eficacia de TERIEM® en pacientes menores de 18 años. No existe una recomendación de uso específica para teriflunomida en niños de 0 a 10 años para el tratamiento de esclerosis múltiple. No se dispone de datos.
Vía de administración:
Los comprimidos recubiertos se administran por vía oral. Los comprimidos deben tragarse enteros con agua. TERIEM® puede tomarse con o sin comida.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Síntomas:
No hay experiencia relacionada con la sobredosis o intoxicación en humanos con teriflunomida. Se administraron 70 mg al día de teriflunomida hasta 14 días en sujetos sanos. Las reacciones adversas concordaron con el perfil de seguridad para teriflunomida en pacientes con EM.
Manejo:
En caso de toxicidad o sobredosis importante, se recomienda el tratamiento con colestiramina o carbón activado para acelerar la eliminación. El procedimiento de eliminación recomendado es colestiramina 8 g tres veces al día durante 11 días. Si esta dosificación no se tolera bien, se puede utilizar colestiramina 4 g tres veces al día durante 11 días. De forma alternativa, en caso de que no hubiera colestiramina disponible, también se pueden utilizar 50 g de carbón activado dos veces al día durante 11 días. Además, si fuera necesario por motivos de tolerabilidad, la administración de colestiramina o carbón activado no necesita hacerse en días consecutivos (ver Propiedades farmacocinéticas).
PRESENTACIONES:
Caja con 14, 28 o 56 comprimidos de 14 mg e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 25 °C.
Consérvese la caja bien cerrada.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Léase instructivo anexo. No se deje al alcance de los niños. Su venta requiere receta médica. Prohibida la venta fraccionada del producto. No se use en el embarazo y la lactancia. Contiene lactosa los pacientes con intolerancia hereditaria a galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx
Propiedad de:
Sandoz GmbH
Biochemiestraße 10, 6250 Kundl
Austria
Representante Legal:
SANDOZ, S.A. de C.V.
La Candelaria No. 186, Col. Atlántida, C.P. 04370, Coyoacán, Ciudad de México, México.
Reg. Núm. 106M2022 SSA IV
22330022130787/07Jun2023/IPPA_DRA-Sandoz
®Marca Registrada


