

TRAYENTA DUO
LINAGLIPTINA, METFORMINA
Tabletas
1 Caja, 30 Tabletas, 2.5/500 mg/mg
1 Caja, 30 Tabletas, 2.5/850 mg/mg
1 Caja, 30 Tabletas, 2.5/1000 mg/mg
1 Caja, 60 Tabletas, 2.5/500 mg/mg
1 Caja, 60 Tabletas, 2.5/850 mg/mg
1 Caja, 60 Tabletas, 2.5/1000 mg/mg
1 Caja, 1 Frasco(s), 60 Tabletas, 2.5/500 mg/mg
1 Caja, 1 Frasco(s), 60 Tabletas, 2.5/850 mg/mg
1 Caja, 1 Frasco(s), 60 Tabletas, 2.5/1000 mg/mg
1 Caja, 1 Frasco(s), 180 Tabletas, 2.5/500 mg/mg
1 Caja, 1 Frasco(s), 180 Tabletas, 2.5/850 mg/mg
1 Caja, 1 Frasco(s), 180 Tabletas, 2.5/1000 mg/mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Linagliptina 2.5 mg 2.5 mg 2.5 mg
Clorhidrato de Metformina 500 mg 850 mg 1000 mg
Excipiente cbp 1 tableta 1 tableta 1 tableta
INDICACIONES TERAPÉUTICAS:
TRAYENTA DUO® está indicado para:
• Como tratamiento adjunto a la alimentación y la actividad física para mejorar el control de la glucemia en los adultos con diabetes mellitus tipo 2 (DMT2) para quienes el tratamiento con linagliptina y metformina es apropiado, en los pacientes para los que la metformina sola resulta insuficiente, y en los que están en tratamiento y logran buen control con la combinación libre de linagliptina y metformina.
• Para administrarlo combinado con una sulfonilurea (SU) (por ejemplo, tratamiento combinado triple) como tratamiento adjunto de la alimentación y la actividad física a los pacientes para quienes la dosis tolerada máxima de metformina y una sulfonilurea resultan insuficientes.
• En combinación con un inhibidor del SGLT2 (es decir, tratamiento combinado triple) como tratamiento complementario de la dieta y el ejercicio en pacientes en los que no se logra un control adecuado con su dosis máxima tolerada de metformina y un inhibidor del SGLT2.
• Como terapia coadyuvante de la insulina (es decir, tratamiento de triple combinación) como complemento de la alimentación y la actividad física para mejorar el control glucémico en los pacientes quienes con la insulina y metformina solas no logran el control glucémico adecuado.
FARMACOCINÉTICA Y FARMACODINAMIA:
Modo de acción: Linagliptina es un inhibidor de la enzima DPP-4 (Dipeptidil peptidasa 4, EC 3.4.14.5) una enzima que participa en la inactivación de las hormonas incretinas GLP-1 y GIP (péptido 1 semejante al glucagón, polipéptido insulinotrópico dependiente de la glucosa). Estas hormonas son degradadas rápidamente por la enzima DPP-4. Ambas hormonas incretinas participan en la regulación fisiológica del homeostasis de la glucosa. Las incretinas son secretadas en una baja concentración basal durante todo el día y los niveles aumentan inmediatamente después de la ingesta de alimentos. Las hormonas GLP-1 y GIP aumentan la biosíntesis y secreción de insulina de las células beta pancreáticas en presencia de concentraciones sanguíneas de glucosa normales y elevadas. Por otra parte, el GLP-1 también reduce la secreción de glucagón de las células alfa pancreáticas, lo cual resulta en una reducción de la producción hepática de glucosa. Linagliptina se fija muy eficazmente a la DPP-4 de una manera reversible y, de este modo, produce un aumento sostenido y una prolongación de las concentraciones activas de incretinas. Linagliptina aumenta, de una manera dependiente de la glucosa, la secreción de insulina y reduce la secreción de glucagón, lo cual resulta en una mejoría global de la homeostasis de la glucosa. Linagliptina se fija selectivamente a la DPP-4 y exhibe una selectividad > 10,000 veces mayor en comparación con la actividad de DPP-8 o DPP-9 in vitro.
El clorhidrato de metformina es una biguanida con efectos antihiperglucémicos que disminuye el nivel de glucosa en ayunas y posprandial. No estimula la secreción de insulina y, por consiguiente, no causa hipoglucemia.
Puede actuar por medio de tres mecanismos:
1) Disminución de la producción de glucosa hepática mediante supresión de gluconeogénesis y glucogenólisis.
2) En el músculo, aumento de sensibilidad a la insulina y de captación periférica y utilización de la glucosa.
3) Retardo de la absorción de glucosa en el intestino.
Estimula la síntesis intracelular de glucógeno actuando en el glucógeno-sintasa. Aumenta la capacidad de transporte de todos los tipos de transportadores de glucosa a través de las membranas (GLUTs) conocidos a la fecha. En humanos, independientemente de la acción en la glucemia, tiene efectos favorables en el metabolismo de los lípidos, lo que se ha demostrado a dosis terapéuticas en estudios comparativos a mediano y a largo plazo: disminuye el colesterol total, el colesterol LDL y los triglicéridos.
Estudios clínicos:
Linagliptina como complemento del tratamiento con metformina: Se evaluó la eficacia y la seguridad de la linagliptina combinada con metformina en pacientes tratados con metformina sola y control insuficiente de la glucemia en un estudio doble ciego, comparativo con placebo, de 24 semanas.
Linagliptina agregada a la metformina disminuyó significativamente el valor de HbA1c (-0.64% de cambio frente al placebo) desde un valor inicial medio de 8%, el nivel de glucosa plasmática en ayunas (GPA) en -21.1 mg/dL (-1.2 mmol/L) y el nivel de glucosa posprandial a las 2 horas (GPP) en -67.1 mg/dL (-3.7 mmol/L) frente al placebo, y logró que una mayor cantidad de pacientes tuviera un valor objetivo de HbA1c < 7.0% (28.3% en el grupo linagliptina frente al 11.4% en el grupo placebo). La incidencia de hipoglucemia en los pacientes tratados con linagliptina fue similar a la de los que recibieron placebo. El peso no mostró diferencias significativas entre los grupos.
En un estudio de diseño factorial de tratamiento inicial de 24 semanas con control de placebo, la administración de 2.5 mg de linagliptina 2 veces por día junto con metformina (500 o 1000 mg 2 veces por día) disminuyó significativamente los parámetros glucémicos comparada con cualquier monoterapia, según se resume en la Tabla 1 (HbA1c inicial media: 8.65%).
Tabla 1. Parámetros glucémicos a la última consulta (estudio de 24 semanas) respecto de linagliptina y metformina solas y combinadas, en pacientes con diabetes mellitus tipo 2 con control insuficiente, dieta y ejercicio
|
Placebo |
Linagliptina 5 mg 1 vez/día* |
Metformina 500 mg 2 veces/día |
Linagliptina 2.5 mg 2 veces/día* + metformina 500 mg 2 veces/día |
Metformina 1000 mg 2 veces/día |
Linagliptina 2.5 mg 2 veces/día* + metformina 1000 mg 2 veces/día |
|
|
HbA1c (%) |
||||||
|
Cantidad de pacientes |
n = 65 |
n = 135 |
n = 141 |
n = 137 |
n = 138 |
n = 140 |
|
Valor inicial (media) |
8.7 |
8.7 |
8.7 |
8.7 |
8.5 |
8.7 |
|
Cambios desde el valor inicial (media ajustada) |
0.1 |
-0.5 |
-0.6 |
-1.2 |
-1.1 |
-1.6 |
|
Diferencia con placebo (media ajustada) (95% IC) |
- |
-0.6 (-0.9, -0.3) |
-0.8 (-1.0; -0.5) |
-1.3 (-1.6; -1.1) |
-1.2 (-1.5, 0.9) |
-1.7 (-2.0, -1.4) |
|
Pacientes (n,%) que alcanzaron HbA1c <7% |
7 (10.8) |
14 (10.4) |
27 (19.1) |
42 (30.7) |
43 (31.2) |
76 (54.3) |
|
Pacientes (%) que recibieron medicación de rescate |
29.2 |
11.1 |
13.5 |
7.3 |
8.0 |
4.3 |
|
GPA (mg/dL) |
||||||
|
Cantidad de pacientes |
n = 61 |
n = 134 |
n = 136 |
n = 135 |
n = 132 |
n = 136 |
|
Valor inicial (media) |
203 |
195 |
191 |
199 |
191 |
196 |
|
Cambios desde el valor inicial (media ajustada) |
10 |
-9 |
-16 |
-33 |
-32 |
-49 |
|
Diferencia con placebo (media ajustada) (95% IC) |
- |
-19 (-31; -6) |
-26 (-38; -14) |
-43 (-56; -31) |
-42 (-55; -30) |
-60 (-72; -47) |
* Dosis diaria total de linagliptina es igual a 5 mg.
La disminución media a partir del valor inicial de la HbA1c en general fue mayor en los pacientes con los valores iniciales más altos. En términos generales, los efectos en los lípidos plasmáticos fueron neutros. La disminución del peso con la combinación de linagliptina y metformina fue similar al valor observado en los pacientes que recibieron metformina sola o placebo; no se observaron modificaciones del valor inicial en los que recibieron linagliptina sola. La incidencia de hipoglucemia fue similar en todos los grupos de tratamiento (placebo 1.4%, linagliptina 5 mg 0%, metformina 2.1%, y linagliptina 2.5 mg + metformina 2 veces por día 1.4%).
Además, este estudio incluyó pacientes (n = 66) con hiperglucemia más severa (HbA1c inicial > 11%) tratados con linagliptina 2.5 mg + metformina 1000 mg 2 veces por día en un estudio abierto. En este grupo, el valor inicial medio de HbA1c era 11.8% y la GPA, 261.8 mg/dL (14.5 mmol/L). Se observó una disminución media de -3.74% en el valor de HbA1c (n = 48), y de -81.2 mg/dL (-4.5 mmol/L) en el valor de GPA (n = 41) en los pacientes que terminaron el ensayo de 24 semanas sin tratamiento de rescate (n = 48). En el análisis de la última observación, que incluyó a todos los pacientes a los que se habían medido los criterios de valoración primarios (n = 65) a la última observación sin medicación de rescate, las modificaciones de los valores iniciales fueron -3.19% HbA1c y -73.6 mg/dL (-4.1 mmol/L) GPA.
Se evaluó la eficacia y la seguridad de 2.5 mg de linagliptina administrada 2 veces por día frente a 5 mg administrados 1 vez por día junto con metformina en pacientes tratados con metformina sola y control insuficiente de la glucemia en un estudio comparativo de 12 semanas doble ciego con control de placebo. Linagliptina (2.5 mg 2 veces por día y 5 mg 1 vez por día) como complemento de la metformina disminuyó significativamente los parámetros glucémicos comparada con el placebo. Disminuyó significativamente (IC: -0.07; 0.19) los valores de HbA1c: -0.80% (inicial 7.98%), y -0.74 (inicial 7.96%) comparada con el placebo.
La incidencia de hipoglucemia en los pacientes tratados con linagliptina fue similar a la de los que recibieron el placebo (2.2% linagliptina 2.5 mg 2 veces por día, 0.9% linagliptina 5 mg 1 vez por día, y 2.3% placebo). El peso no mostró diferencias significativas entre los grupos.
Linagliptina como complemento del tratamiento combinado con metformina y sulfonilurea: Se hizo un estudio comparativo de 24 semanas para evaluar la eficacia y la seguridad de linagliptina 5 mg frente al placebo en pacientes tratados con una combinación de metformina y una sulfonilurea y control insuficiente de la glucemia. Linagliptina disminuyó significativamente los valores de HbA1c (-0.62% de modificación frente al placebo) inicial media de 8.14%.
También produjo disminuciones significativas en los pacientes que tenían un valor objetivo de HbA1c < 7.0% (31.2% linagliptina frente a 9.2% placebo), y en la disminución de la GPA con -12.7 mg/dL (-0.7 mmol/L) frente al placebo. El peso no mostró diferencias significativas entre los grupos.
Linagliptina como complemento de un tratamiento combinado de metformina y empagliflozina: En pacientes en los que no se logró un control adecuado con metformina y empagliflozina (10 mg [n = 247] o 25 mg [n = 217], el tratamiento durante 24 semanas con terapia de adición de linagliptina 5 mg brindó una media ajustada de reducciones en los valores de HbA1 respecto del nivel basal de -0.53% (diferencia significativa respecto a la adición de placebo -0.32% (IC 95%, -0.25, -0.13) y -0.58% (diferencia significativa respecto a la adición de placebo -0.47% (IC 95%, -0.66, -0.28), respectivamente. Una proporción mayor de pacientes con un valor basal de HbA1c ≥ 7.0% y tratados con linagliptina 5 mg lograron un valor objetivo de HbA1c de < 7% en comparación con placebo, lo cual fue estadísticamente significativo.
En subgrupos preespecificados de pacientes con un nivel basal de HbA1c de 8.5% o más (n = 66 y n = 42 pacientes tratados con metformina más empagliflozina 10 mg o 25 mg, respectivamente), la media ajustada de reducciones en los valores de HbA1c desde el nivel basal hasta las 24 semanas de tratamiento complementario con linagliptina 5 mg fue de -0.97% (p = 0.0875, para la diferencia respecto a la adición de placebo) y -1.16% (p = 0.0046 para la diferencia respecto a la adición de placebo), respectivamente.
Linagliptina en combinación con metformina e insulina: Un estudio de 24 semanas controlado con placebo fue realizado para evaluar la eficacia y seguridad de linagliptina (5 mg una vez al día) agregado a insulina con o sin metformina. 83% de los pacientes fueron tratados con metformina en combinación con insulina en este estudio. Linagliptina en combinación con metformina más insulina brindó mejoras significativas en HbA1c en este subgrupo con -0.68 (IC-0.78; -0.57) promedio ajustado desde el inicio (inicio promedio HbA1c 8.28%) comparado con placebo en combinación con metformina más insulina. No hubo un cambio significativo desde el inicio en el peso corporal de cada grupo.
Datos de 24 meses de tratamiento con linagliptina como complemento de metformina comparada con glimepirida: En un estudio comparativo de la eficacia y la seguridad de 5 mg de linagliptina o glimepirida (una sulfonilurea) en pacientes tratados con metformina como monoterapia y control insuficiente de la glucemia, la disminución de HbA1c por la linagliptina fue similar a la causada por la glimepirida, con una diferencia media a partir del valor inicial de 104 semanas con linagliptina frente a + 0.20% con glimepirida.
En este estudio, la relación proinsulina-insulina, marcador de la eficacia de la síntesis y liberación de insulina, mostró incremento estadísticamente significativo del tratamiento con linagliptina frente al tratamiento con glimepirida. La incidencia de hipoglucemia en el grupo linagliptina (7.5%) fue significativamente inferior a la del grupo glimepirida (36.1%).
Los pacientes tratados con linagliptina mostraron disminución media significativa del peso inicial comparados con un aumento significativo del peso en los pacientes que recibieron glimepirida (-1.39 frente a + 1.29 kg).
Linagliptina como tratamiento adicional en pacientes ancianos (edad ≥ 70 años) con diabetes tipo 2: La eficacia y seguridad de linagliptina en pacientes ancianos (≥ 70 años) con diabetes tipo 2 fue evaluada en un estudio doble ciego de 24 semanas de duración. Los pacientes recibían metformina y/o sulfonilurea y/o insulina como tratamiento de base. La dosis del tratamiento antidiabético de base se mantuvo estable durante las primeras 12 semanas, luego de las cuales, se permitieron ajustes. Linagliptina brindó una mejora significativa en HbA1c de -0.64% (95% IC -0.81, -0.48; p < 0.0001) comparado con placebo luego de 24 semanas, partiendo de una medición basal de HbA1c de 7.8%. Linagliptina también demostró mejoras significativas en la glucosa plasmática de ayuno (GPA) de -20.7 mg/dL (95% IC -30.2, -11.2; p < 0.0001) comparado con placebo (-1.1 mmol/L). El peso corporal no difirió de manera significativa entre los grupos. En general, la incidencia de hipoglicemia fue comparable entre linagliptina (2 de 45 pacientes, 4.4%) y placebo (ninguno de los 22 pacientes, 0%) sobre la base de metformina sola. Las tasas de hipoglicemia también fueron comparables con tratamiento basal insulina con o sin metformina (13 de 35 pacientes, 37.1% tratados con linagliptina y 6 de 15 pacientes, 40.0% tratados con placebo). Sin embargo, con un tratamiento basal de sulfonilurea con o sin metformina, se reportó hipoglicemia en una mayor proporción de pacientes tratados con linagliptina (24 de 82 pacientes, 29.3%) comparando con placebo (7 de 42 pacientes, 16.7%). No hubo diferencia entre linagliptina y placebo en eventos hipoglicémicos severos.
En un análisis agrupado de pacientes ancianos (edad ≥ 70 años) con diabetes tipo 2 (n = 183) quienes estaban siendo tratados con metformina e insulina basal como tratamiento de base, el tratamiento con linagliptina combinada con metformina e insulina brindó mejoras significativas en parámetros de HbA1c con cambios de -0.81 (IC -1.01; -0.61) promedio ajustado desde el inicio (promedio inicial de HbA1c 8.13%) comparado con placebo en combinación con metformina más insulina. No se observó una diferencia clínica significativa en la incidencia de eventos hipoglucémicos, en pacientes de ≥ 70 años (37.2% tratados con linagliptina en combinación con metformina más insulina vs 39.8% tratados con placebo combinado con metformina más insulina).
Linagliptina y combinación inicial con linagliptina y metformina en pacientes diagnosticados recientemente y sin tratamiento previo con hiperglucemia marcada: La eficacia y la seguridad de la combinación inicial de linagliptina 5 mg una vez al día y metformina dos veces al día (ajustada mediante incrementos graduales durante las primeras 6 semanas a 1500 mg o 2000 mg/d) en comparación con linagliptina 5 mg en una toma han sido evaluadas en un estudio de 24 semanas de duración en pacientes con DMT2 diagnosticada recientemente sin tratamiento previo y con hiperglucemia marcada (HbA1c basal, 8.5-12.0%). Al cabo de 24 semanas, tanto la monoterapia de linagliptina como la combinación inicial de linagliptina y metformina redujeron significativamente los niveles de HbA1c a razón de -2.0% y 2.8%, respectivamente, a partir de un valor basal de HbA1c de 9.9% y 9.8%, respectivamente. Esta diferencia entre los tratamientos de -0.8% (IC 95%, -1.1 a -0.5) demostró la superioridad de la combinación inicial por sobre la monoterapia (p < 0.0001). Es de destacar que el 40% y el 61% de los pacientes de la rama de monoterapia y de la rama de la combinación alcanzaron valores de HbA1c < 7.0%.
Estudio de seguridad cardiovascular y renal con linagliptina (CARMELINA): CARMELINA fue un estudio aleatorizado realizado sobre 6979 pacientes con diabetes tipo 2, con riesgo cardiovascular elevado, evidenciado por la presencia de antecedentes de enfermedad renal o macrovascular establecida, que habían recibido 5 mg de linagliptina (3494) o placebo (3485) agregados al tratamiento protocolar regionalmente establecido para lograr los objetivos de HbA1c, para controlar los factores de riesgo cardiovascular y la enfermedad renal. La población del estudio incluyó 1211 (17.4%) pacientes ≥ 75 años de edad y 4348 (62.3%) pacientes con insuficiencia renal. Aproximadamente el 19% de la población tenía una TFGe ≥ 45 y < 60 ml/min/1.73 m2, 28% de la población tenía una TFGe ≥ 30 y < 45 ml/min/1.73 m2 y 15% tenía una TFGe < 30 ml/min/1.73 m2.
La HbA1c media al inicio del estudio fue del 8.0%.
El estudio estuvo diseñado para demostrar la no inferioridad en función del criterio de valoración cardiovascular primario, que era un compuesto de la primera aparición de: o bien muerte cardiovascular, o un infarto de miocardio no mortal (IM), o un accidente cerebrovascular no fatal (3P-MACE). El criterio de valoración compuesto renal se definió como muerte renal o enfermedad renal en etapa terminal sostenida, o disminución sostenida del 40% o en la TFGe.
Después de un seguimiento de 2.2 años (mediana), la linagliptina, agregada al tratamiento estándar, no aumentó el riesgo de eventos cardiovasculares graves o eventos de desenlace renal (Tabla 2 y Figura 1). No hubo un aumento en el riesgo de hospitalización por insuficiencia cardiaca, que fue un criterio de valoración adjudicado adicional observado, en comparación con el tratamiento estándar sin linagliptina en pacientes con diabetes tipo 2 (Tabla 3).
Tabla 2. Eventos adversos graves cardiovasculares (MACE) y eventos de desenlace renal estratificados por grupo de tratamiento en el estudio CARMELINA
|
Linagliptina 5 mg |
Placebo |
Razón de riesgos |
|||
|
Cantidad de sujetos |
Tasa de incidencia por cada 1000 años-paciente* |
Cantidad de sujetos (%) |
Tasa de incidencia por cada 1000 años-paciente* |
(IC del 95%) |
|
|
3494 |
3485 |
||||
|
Compuesto cardiovascular primario (muerte cardiovascular, infarto de miocardio no mortal, accidente cerebrovascular no mortal) |
434 (12.4) |
57.7 |
420 (12.1) |
56.3 |
1.02 (0.89, 1.17)** |
|
Compuesto renal secundario (muerte renal, enfermedad renal terminal, disminución sostenida del 40% de la TFGe) |
327 (9.4) |
48.9 |
306 (8.8) |
46.6 |
1.04 (0.89, 1.22) |
* PY = años paciente.
** Prueba de no inferioridad para demostrar que el límite superior del IC del 95% para la razón de riesgos instantáneos es menor a 1.3.
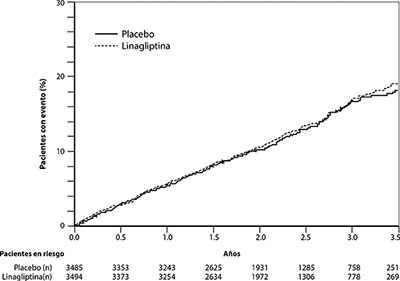
Tabla 3. Hospitalización por insuficiencia cardiaca y mortalidad estratificados por grupo de tratamiento, en el estudio CARMELINA
|
Linagliptina 5 mg |
Placebo |
Razón de riesgos |
|||
|
Cantidad de sujetos (%) |
Tasa de incidencia por cada 1000 años-paciente* |
Cantidad de sujetos (%) |
Tasa de incidencia por cada 1000 años-paciente* |
(IC del 95%) |
|
|
Cantidad de pacientes |
3494 |
3485 |
|||
|
Mortalidad por todas las causas |
367 (10.5) |
46.9 |
373 (10.7) |
48.0 |
0.98 (0.84, 1.13) |
|
Muerte cardiovascular |
255 (7.3) |
32.6 |
264 (7.6) |
34 |
0.96 (0.81, 1.14) |
|
Hospitalización por insuficiencia cardiaca |
209 (6.0) |
27.7 |
226 (6.5) |
30.4 |
0.90 (0.74, 1.08) |
* PY = años-paciente.
En los análisis de progresión de la albuminuria (cambio de normoalbuminuria a micro o macroalbuminuria, o de microalbuminuria a macroalbuminuria), la razón de riesgos instantáneos estimada para la linagliptina, en comparación con placebo, fue 0.86 (IC del 95%: 0.78 a 0.95). El criterio de valoración microvascular se definió como el compuesto de muerte renal, enfermedad renal crónica sostenida, reducción sostenida ≥50% de la TFGe, progresión de la albuminuria, tratamiento de la retinopatía diabética con fotocoagulación retineana o inyecciones intravítreas de inhibidores del factor de crecimiento endotelial vascular, hemorragia vítrea o ceguera provocada por la diabetes. La razón de riesgos instantáneos estimada para el tiempo transcurrido hasta la primera aparición del criterio de valoración microvascular compuesto fue 0.86 (IC del 95%: 0.78; 0.95) para la linagliptina, comparada con el placebo, y se debió principalmente a la progresión de la albuminuria.
Estudio de seguridad cardiovascular con linagliptina (CAROLINA): CAROLINA es un estudio aleatorizado que se llevó a cabo con 6033 pacientes con diabetes tipo 2 temprana y con mayor riesgo cardiovascular o complicaciones conocidas tratados con linagliptina 5 mg (3023) o glimepirida 1-4 mg (3010) en combinación al tratamiento estándar (incluido tratamiento de b se con metformina en 83% de los pacientes) conforme a las normas regionales para abordar la HbA1c y los factores de riesgo cardiovasculares. La media de edad de la población del estudio fue de 64 años e incluyó a 2030 (34%) pacientes ≥ 70 años. La población del estudio incluyó 2089 (35%) pacientes con enfermedad cardiovascular y 1130 (19%) pacientes con insuficiencia renal y una TFGe < 60 ml/min/1.73 m2 al inicio. La HbA1c media basal fue de 7.15%.
El estudio fue diseñado para demostrar la no inferioridad del criterio de valoración cardiovascular primario, el cual consistía en la combinación de la primera aparición de muerte cardiovascular o infarto de miocardio (IM) no mortal o accidente cerebrovascular no mortal (3P-MACE).
Luego de una mediana de seguimiento de 6.25 años, la linagliptina, administrada junto con el tratamiento estándar, no incrementó el riesgo de eventos cardiovasculares graves (Tabla 4) en comparación con la glimepirida. Los resultados fueron consistentes para los pacientes tratados con metformina o sin ella.
Tabla 4. Episodios cardiovasculares graves (MACE) y mortalidad por grupo de tratamiento en el estudio CAROLINA
|
Linagliptina 5 mg |
Glimepirida (1-4 mg) |
Razón de riesgos |
|||
|
Cant. de participantes (%) |
Tasa de incidencia por 1000 AP* |
Cant. de participantes (%) |
Tasa de incidencia por 1000 AP* |
(IC 95%) |
|
|
Cant. de pacientes |
3023 |
3010 |
|||
|
CV primario combinado (muerte cardiovascular, IM no mortal, accidente cerebrovascular no mortal) |
356 (11.8) |
20.7 |
362 (12.0) |
21.2 |
0.98 (0.84; 1.14)** |
* AP = años-paciente.
** Prueba de no inferioridad para demostrar que el límite superior del IC 95% de la razón de riesgos es menor que 1.3.
La combinación de sostenibilidad del tratamiento, un criterio de valoración secundario clave, definido como el porcentaje de pacientes en el tratamiento del estudio luego del periodo de ajuste de dosis inicial (16 semanas) que mantenían el control glucémico (HbA1c ≤ 7.0%) en la última visita sin necesidad de tratamiento antidiabético adicional (medicación de rescate), sin episodios de hipoglucemia moderada (sintomática con valores de glucosa ≤ 70 mg/mL) o grave (que requirieran asistencia) y sin aumento de peso > 2%. La cantidad de pacientes que alcanzaron este criterio de valoración secundario clave fue mayor para los tratados con linagliptina (481; 16.0%) en comparación con los que recibieron glimepirida (305; 10.2%).
Para todo el periodo de tratamiento (mediana de tratamiento 5.9 años) el porcentaje de pacientes con hipoglucemia moderada o grave fue de 6.5% para linagliptina en comparación con 30.9% para glimepirida. La hipoglucemia grave se observó en 0.3% de los pacientes tratados con linagliptina y 2.2% de los que recibieron glimepirida.
Farmacocinética: Los estudios de bioequivalencia en sujetos sanos demostraron que el comprimido combinado de TRAYENTA DUO® (linagliptina/clorhidrato de metformina) guarda bioequivalencia con la administración conjunta de comprimidos individuales de linagliptina y clorhidrato de metformina.
La administración de TRAYENTA DUO® 2.5/1000 mg con la comida no modificó la exposición global a la linagliptina. Con metformina no se observaron modificaciones en el ABC, si bien la concentración sérica media máxima de metformina disminuyó el 18% cuando se administró con la comida. Se observó una demora de 2 horas en la concentración sérica máxima respecto de la metformina con la comida. No es probable que estos cambios sean clínicamente significativos.
Los siguientes párrafos reflejan las propiedades farmacocinéticas de cada principio activo de TRAYENTA DUO®.
Linagliptina: La farmacocinética de linagliptina se caracterizó ampliamente en sujetos sanos y pacientes con diabetes tipo 2. Después de la administración oral de 5 mg a voluntarios sanos, se absorbió rápidamente, con una concentración plasmática máxima (mediana de Tmáx.) presente a 1.5 horas posterior a la dosis.
La concentración plasmática disminuye en 2 etapas con una vida media terminal prolongada (vida media terminal de linagliptina mayor a 100 horas) que se relaciona principalmente con la unión saturable y estrecha de linagliptina a la DPP-4 que no promueve la acumulación del fármaco. La vida media efectiva de acumulación es de alrededor de 12 horas, según se determinó con la administración oral de varias dosis de 5 mg de linagliptina. Con la administración 1 vez por día, el equilibrio dinámico de la concentración plasmática de 5 mg de linagliptina se alcanza con la tercera dosis.
El ABC plasmática aumentó alrededor del 33% después de administrar dosis de 5 mg en equilibrio comparada con la primera dosis. Los coeficientes intrasujetos e intersujetos de variación del ABC de linagliptina fueron bajos (12.6% y 28.5%, respectivamente).
El ABC plasmática aumentó menos que proporcionalmente a la dosis. La farmacocinética de linagliptina fue, en general, similar en sujetos sanos y en pacientes con diabetes tipo 2.
Absorción: La biodisponibilidad absoluta de linagliptina es de alrededor del 30%. La comida con alto contenido de grasa no tuvo efectos clínicamente relevantes en la farmacocinética, por lo que linagliptina se puede administrar con o sin alimentos. Los estudios in vitro indicaron que linagliptina es un sustrato de gp P y de CYP3A4. El ritonavir, poderoso inhibidor de gp P y CYP3A4, produjo un aumento del doble de la exposición (ABC), y la coadministración múltiple de linagliptina y rifampicina, poderoso inductor de gp P y CYP3A, disminuyó el ABC en equilibrio alrededor del 40%, probablemente aumentando/disminuyendo la biodisponibilidad de la linagliptina por inhibición/inducción de la gp P.
Distribución: Como resultado de la unión a los tejidos, el volumen aparente de distribución medio en equilibrio después de una dosis intravenosa única de 5 mg de linagliptina a sujetos sanos es de alrededor de 1,110 litros, lo que indica que linagliptina se distribuye extensamente a los tejidos. La unión de linagliptina a las proteínas plasmáticas depende de la concentración y disminuye desde alrededor del 99% a 1 nmol/l hasta 75-89% a ≥30 nmol/l, lo que refleja la saturación de la unión a la DPP-4 con mayor concentración de linagliptina. A altas concentraciones, donde la DPP-4 está totalmente saturada, el 70 al 80% de linagliptina se unió a otras proteínas plasmáticas, y el 20 al 30% estuvo libre en plasma.
Biotransformación/metabolismo: Después de la administración oral de 10 mg de [14C]linagliptina, alrededor del 5% de la radioactividad se eliminó en la orina. El metabolismo es una vía de eliminación menor. Se detectó un metabolito principal con exposición relativa de 13.3% de linagliptina en equilibrio, que se determinó como farmacológicamente inactivo y, por lo tanto, no promueve la actividad inhibidora de la DPP-4 plasmática por la linagliptina.
Excreción: Después de la administración oral de [14C] linagliptina a sujetos sanos, alrededor del 85% de la radioactividad se eliminó en heces (80%) u orina (5%) a los 4 días. La depuración renal en equilibrio fue aproximadamente de 70 mL/min.
Poblaciones especiales:
Insuficiencia renal: Se hizo un estudio abierto de varias dosis para evaluar la farmacocinética de linagliptina (5 mg) en pacientes con insuficiencia renal crónica de varios grados comparada con la farmacocinética en sujetos sanos. Se incluyeron pacientes con insuficiencia renal clasificada según la depuración de creatinina como leve (50 a < 80 mL/min), moderada (30 a < 50 mL/min) o severa (< 30 mL/min), y pacientes con ERT con hemodiálisis. Además, los pacientes con DMT2 e insuficiencia renal severa (< 30 mL/min) se compararon con pacientes con DMT2 con función renal normal.
La depuración de creatinina se midió por depuración en orina de 24 horas o por estimación a partir de la creatinina en suero basada en la fórmula de Cockcroft-Gault: CrCl=[140-edad (años)] x peso (kg) {x 0.85 para mujeres}/[72 x creatinina sérica (mg/dL)].
En equilibrio, la exposición a linagliptina en pacientes con insuficiencia renal leve fue comparable a la de los sujetos sanos. En pacientes con insuficiencia renal moderada, la exposición a la linagliptina aumentó en grado moderado alrededor de 1.7 veces frente al control.
Los pacientes con DMT2 e insuficiencia renal grave mostraron una exposición aproximadamente 1.4 veces superior a la de los pacientes con DMT2 y función renal normal. Las predicciones en estado de equilibro sobre el ABC de linagliptina en los pacientes con ERT indicaron una exposición comparable a la de los pacientes con insuficiencia renal moderada o grave.
No se prevé la eliminación de linagliptina en grado terapéuticamente significativo por hemodiálisis o diálisis peritoneal; por lo tanto, no es necesario modificar la dosis en los pacientes con insuficiencia renal de cualquier grado.
La insuficiencia renal leve no tuvo efectos en la farmacocinética de los pacientes con diabetes tipo 2, según se evaluó en los análisis farmacocinéticos poblacionales.
Insuficiencia hepática: En los pacientes con insuficiencia hepática leve, moderada y grave (clasificación de Child-Pugh), el ABC media y la Cmáx. de linagliptina fueron similares a las de los controles sanos pareados, después de administrar varias dosis de 5 mg. En los pacientes con insuficiencia hepática leve, moderada y grave no es necesario modificar la dosis.
Índice de masa corporal (IMC): No es necesario modificar la dosis según el IMC. El IMC no tuvo efectos clínicamente significativos en la farmacocinética de linagliptina tomando como base un análisis farmacocinético poblacional de los datos de las fases I y II.
Género: No es necesario modificar la dosis según el género. El género no tuvo efectos clínicamente significativos en la farmacocinética de linagliptina tomando como base un análisis farmacocinético poblacional de los datos de las fases I y II.
Población geriátrica: No es necesario modificar la dosis según la edad debido a que la edad no tuvo efectos clínicamente significativos en la farmacocinética de linagliptina tomando como base un análisis farmacocinético poblacional de los datos de las fases I y II. Los sujetos geriátricos (de 65 a 80 años) mostraron concentración de linagliptina plasmática comparable a la de los sujetos más jóvenes.
Población pediátrica: Aún no se han realizado los estudios para caracterizar la farmacocinética de la linagliptina en pacientes pediátricos.
Raza: No es necesario modificar la dosis según la raza. La raza no tuvo efectos evidentes en la concentración plasmática de linagliptina tomando como base el análisis compuesto de los datos farmacocinéticos disponibles, es decir, sujetos blancos, hispanoamericanos de ascendencia india o mestiza, afroamericanos y asiáticos. Las características farmacocinéticas de linagliptina fueron similares, en los estudios de fase I dedicados, a las de voluntarios sanos japoneses, chinos y blancos, y de pacientes con diabetes tipo 2 afroamericanos.
Metformina:
Absorción: Después de administrar una dosis oral de metformina, la Tmáx. se alcanza a las 2.5 horas. La biodisponibilidad absoluta de un comprimido de 500 u 850 mg de clorhidrato de metformina es aproximadamente del 50-60% en sujetos sanos. Después de la dosis oral, la fracción no absorbida recuperada en heces fue del 20-30%.
Después de la administración oral, la absorción del clorhidrato de metformina es saturable e incompleta. Se asume que la farmacocinética de la absorción no es lineal.
A la dosis recomendada y siguiendo el esquema posológico, la concentración plasmática en equilibrio se alcanza a las 24 a 48 horas, y por lo general es menor que 1 microgramo/mL. En ensayos clínicos comparativos, la concentración plasmática máxima (Cmáx.) no superó los 5 microgramos/mL aun a las dosis máximas.
La comida disminuye el alcance y demora levemente la absorción. Después de administrar una dosis de 850 mg, se observó un 40% menos de concentración plasmática máxima, un 25% de reducción del ABC y una prolongación de 35 minutos del tiempo transcurrido hasta alcanzar la concentración plasmática máxima. Se desconoce la relevancia clínica de estas disminuciones y reducciones.
Distribución: La unión a las proteínas plasmáticas es insignificante. El clorhidrato de metformina se compartimentaliza hacia los eritrocitos. La distribución máxima en sangre es inferior a la plasmática y se produce casi en el mismo momento. Los glóbulos rojos probablemente representen un segundo compartimiento de distribución. El volumen medio de distribución (Vd) osciló entre 63 y 276 L.
Biotransformación/Metabolismo: El clorhidrato de metformina se elimina sin modificaciones en la orina. No se identificaron metabolitos en humanos.
Eliminación: La depuración renal del clorhidrato de metformina es > 400 mL/min, lo que indica eliminación por filtración glomerular y secreción tubular. Después de la administración oral, la vida media terminal aparente es de alrededor de 6.5 horas.
En los casos de deterioro de la función renal, su depuración renal disminuye proporcionalmente a la de la creatinina y, por consiguiente, la vida media de eliminación se prolonga y aumenta el nivel de clorhidrato de metformina plasmático.
Poblaciones específicas:
Población pediátrica:
Estudio de dosis única: Después de administrar dosis únicas de 500 mg de metformina, los pacientes pediátricos mostraron un perfil farmacocinético similar al observado en adultos sanos.
Estudio de varias dosis: Los datos se limitan a un estudio. Después de administrar dosis de 500 mg de metformina 2 veces por día durante 7 días a pacientes pediátricos, la concentración plasmática máxima y la exposición sistémica (AUC0-t) disminuyeron aproximadamente el 33 y el 40%, respectivamente comparadas con los valores observados en los pacientes diabéticos adultos que recibieron dosis de 500 mg 2 veces por día durante 14 días. Como cada dosis se titula en forma individual según el control glucémico por lo cual estos resultados tienen relevancia clínica limitada.
Insuficiencia renal: Son escasos los datos disponibles sobre sujetos con insuficiencia renal moderada y no es posible estimar con certeza la exposición sistémica a la metformina en este subgrupo en comparación con los sujetos con función renal normal. Por consiguiente, la adaptación de la dosis debe hacerse teniendo en consideración la eficacia clínica/tolerabilidad (Ver Dosis y vía de administración).
CONTRAINDICACIONES:
• Hipersensibilidad a los principios activos linagliptina o clorhidrato de metformina, a ambos, o a alguno de los excipientes.
• Cualquier tipo de acidosis metabólica aguda (como acidosis láctica, cetoacidosis diabética).
• Precoma diabético.
• Insuficiencia renal grave o (CrCl< 30 mL/min o eGFR<30 mL/min/1.73 m2).
• Afecciones agudas que pueden alterar la función renal: deshidratación, infección grave, shock, administración intravascular de agentes de contraste yodados (Ver Precauciones generales).
• Condiciones que puedan causar hipoxia (en especial, afección aguda o empeoramiento de enfermedad crónica), como: insuficiencia cardiaca descompensada, insuficiencia respiratoria, infarto de miocardio reciente, shock.
• Insuficiencia hepática.
• Alcoholismo agudo.
• Alcoholismo.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No se han hecho estudios comparativos suficientes con TRAYENTA DUO® ni con cada uno de sus componentes en embarazadas. Los estudios de reproducción en ratas preñadas con los productos combinados en TRAYENTA DUO® no indicaron efectos teratogénicos atribuidos a la administración conjunta de linagliptina y metformina.
Son escasos los datos existentes sobre la administración de linagliptina en embarazadas. Los estudios no clínicos no indican la presencia de efectos perjudiciales directos ni indirectos respecto de la toxicidad en la reproducción.
Son escasos los datos existentes sobre la administración de metformina en embarazadas. El principio activo no mostró efectos teratogénicos en ratas a razón de 200 mg/kg/día con relación a la exposición humana cuadruplicada. A dosis mayores (500 y 1000 mg/kg/día, con relación a la exposición humana 11 y 23 veces mayor), se observó teratogenicidad en ratas.
Como medida preventiva, se aconseja no administrar TRAYENTA DUO® durante el embarazo.
En las pacientes que planean un embarazo, y en las embarazadas, la diabetes no se debe tratar con TRAYENTA DUO®, sino con insulina para mantener el nivel de glucosa en sangre lo más próximo posible al nivel normal y así disminuir el riesgo de malformación fetal relacionada con el nivel anormal de glucosa en sangre.
Lactancia: No se han hecho estudios en animales lactantes con la combinación de metformina y linagliptina. Los estudios no clínicos con cada principio activo mostraron excreción de ambos en la leche de ratas que amamantaban. La metformina se excreta en la leche humana, pero en el caso de la linagliptina, no se tiene certeza sobre este particular. Se recomienda no administrar TRAYENTA DUO® durante la lactancia.
REACCIONES SECUNDARIAS Y ADVERSAS: Se evaluó la seguridad de 2.5 mg de linagliptina administrada 2 veces por día (o la dosis bioequivalente de 5 mg 1 vez por día) más metformina en más de 6,800 pacientes con DMT2.
Se trató a más de 1,800 pacientes en estudios comparativos con placebo administrándoles la dosis terapéutica de 2.5 mg de linagliptina 2 veces por día (o la dosis bioequivalente de 5 mg 1 vez por día) combinada con metformina durante ≥ 12/24 semanas.
En el análisis de ensayos comparativos con placebo agrupados, la incidencia global de eventos adversos (EA) en los pacientes tratados con placebo y metformina fue similar a la de los tratados con linagliptina 2.5 mg y metformina (54.3% y 49.0%). La interrupción del tratamiento debido a los EA fue similar en los pacientes que recibieron placebo y metformina y en los que recibieron linagliptina y metformina (3.8% y 2.9%).
Debido al efecto del tratamiento de base en los EA (por ejemplo, en la hipoglucemia), los EA se analizaron y mostraron según el tratamiento respectivo, complementario de metformina, y complementario de metformina más sulfonilurea.
Los estudios comparativos con placebo incluyeron siete en los que se administró linagliptina como complemento de metformina, y uno en el que se administró linagliptina como complemento de metformina + sulfonilurea.
Resumen tabulado de reacciones adversas: En la tabla 5 que se presenta a continuación se incluyen las reacciones adversas surgidas con el uso de la combinación linagliptina/metformina o con el uso de los monocomponentes (linagliptina o metformina) en los ensayos clínicos o durante la farmacovigilancia. Los efectos no deseados informados previamente con uno de los componentes individuales pueden ser potenciales efectos no deseados de TRAYENTA DUO® incluso si no se observaron en los estudios clínicos con este medicamento.
|
Terminología de la Clasificación por sistema y órgano (SOC) del MedDRA |
Reacciones adversas a la linagliptina y metformina |
|
Infecciones e infestaciones |
Nasofaringitis1; 3 |
|
Trastornos del sistema inmunológico |
Hipersensibilidad1; 3 Angioedema4 Urticaria2; 4 |
|
Trastornos del metabolismo y de la nutrición |
Acidosis láctica2 Resultado anormal en la prueba de absorción de vitamina B12*; 2 Hipoglucemia (cuando la linagliptina y la metformina se combinaron con una sulfonilurea) |
|
Trastornos del sistema nervioso |
Alteración del gusto2 |
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos1; 3 |
|
Trastornos gastrointestinales** |
Disminución del apetito3; 5 Diarrea3; 5 Estreñimiento (cuando la linagliptina y la metformina se combinaron con insulina) Náuseas3; 5 Pancreatitis3 Vómitos3; 5 Dolor abdominal2 Ulceración en cavidad bucal4 |
|
Trastornos hepatobiliares |
Resultado anormal en la prueba de función hepática2 Hepatitis2 |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito3; 5 Eritema2 Exantema4 Penfigoide ampolloso4; § |
|
Exploraciones complementarias |
Aumento de la lipasa3; † Aumento de la amilasa? |
1 Reacciones adversas informadas también en pacientes tratados con linagliptina como monoterapia.
2 Reacciones adversas de la metformina como monoterapia; sírvase consultar el Resumen de Características del Producto correspondiente a la metformina para obtener información adicional.
3 Reacciones adversas de la combinación a dosis fija de linagliptina + metformina (análisis combinado de los estudios comparados con placebo).
4 Reacciones adversas identificadas durante la farmacovigilancia con linagliptina.
5 Reacciones adversas informadas en pacientes que recibieron la combinación a dosis fija de linagliptina + metformina y también en pacientes que recibieron metformina como monoterapia.
* El tratamiento prolongado con metformina se ha asociado con una disminución de la absorción de la vitamina B12, lo cual en casos muy raros puede provocar una deficiencia de la vitamina B12 clínicamente significativa (p. ej., anemia megaloblástica).
** Los trastornos gastrointestinales, como el dolor abdominal y las náuseas, vómitos, diarrea y disminución del apetito presentan su mayor frecuencia durante el inicio del tratamiento con clorhidrato de metformina y se resuelven espontáneamente en la mayoría de los casos. Para prevenirlos, se recomienda tomar el clorhidrato de metformina en 2 dosis diarias durante las comidas o después de ellas si se lo administra como monoterapia.
§ Véase también el estudio de seguridad cardiovascular y renal con linagliptina (CARMELINA) a continuación.
† Sobre la base de las elevaciones de lipasa > 3 veces el límite normal superior.
? En el estudio CAROLINA de linagliptina vs. el comparador activo glimepirida (véase la sección Ensayos clínicos) los análisis de laboratorio de la amilasa revelaron aumento de > 3 veces el límite normal superior en 0.99% de los pacientes tratados con linagliptina y 0.54% de la pacientes tratados con glimepirida.
En los estudios comparativos con placebo, la reacción adversa relacionada con linagliptina + metformina informada con mayor frecuencia fue diarrea (1.6%) frente a un índice comparable para metformina + placebo (2.4%).
Reacciones adversas de la combinación linagliptina y metformina con SU: Al administrar linagliptina y metformina junto con una SU, se informó hipoglucemia como el EA más común (linagliptina + metformina + SU 23.9% frente a 16.0% en el grupo placebo) y se lo identificó como reacción adversa agregada en esas condiciones. Ninguno de los episodios de hipoglucemia se clasificó como severa (que requirieran asistencia).
Reacciones adversas reportadas cuando se combinaron linagliptina y metformina con insulina: Al administrar linagliptina y metformina junto con insulina, se informó hipoglucemia como el EA más común, pero ocurrió en proporción comparable cuando se administró placebo y metformina combinados con insulina (linagliptina más metformina más insulina 29.5% vs 30.9% en el grupo placebo más metformina más insulina) con una baja proporción de episodios severos (que requirieran asistencia) (1.5% vs 0.9%).
Estudio de seguridad renal y desenlaces cardiovasculares con linagliptina (CARMELINA): En el estudio CARMELINA, se evaluó la seguridad cardiovascular y renal de linagliptina comparada con un placebo en pacientes con diabetes tipo 2 y con un aumento del riesgo cardiovascular evidenciado por la presencia de antecedentes de enfermedad renal o macrovascular establecida (ver la sección Estudios clínicos). Se incluyeron en el estudio 3494 pacientes tratados con linagliptina (5 mg) y 3485 pacientes tratados con placebo. Ambos tratamientos se agregaron al tratamiento estándar empleado para alcanzar la meta de HbA1c, según los criterios regionales, y controlar los factores de riesgo cardiovascular. Al inicio del estudio, 57% de los pacientes recibían insulina, 54% metformina y 32%, una sulfonilurea. La incidencia global de eventos adversos y eventos adversos graves en pacientes que recibieron linagliptina fue similar a la de los pacientes que recibieron placebo. Los datos de seguridad de este estudio estaban en línea con el perfil de seguridad conocido anterior de linagliptina.
En la población tratada, se informaron eventos hipoglucémicos graves (que requirieron asistencia) en 3.0% de los pacientes tratados con linagliptina y en 3.1% de los pacientes tratados con placebo. Entre los pacientes que estaban recibiendo una sulfonilurea al inicio del estudio, la incidencia de la hipoglucemia grave fue 2.0% con linagliptina y 1.7% con placebo. Entre los pacientes que estaban recibiendo insulina al inicio del estudio, la incidencia de la hipoglucemia grave fue 4.4% con linagliptina y 4.9% con placebo.
En el periodo de observación total del estudio, se notificaron eventos adjudicados como pancreatitis aguda en 0.3% de los pacientes tratados con linagliptina y en 0.1% de los pacientes tratados con placebo.
En el estudio CARMELINA, se informó penfigoide ampolloso en 0.2% de los pacientes tratados con linagliptina y en ningún paciente tratado con placebo.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Se hicieron estudios generales de toxicidad en ratas de hasta 13 semanas de vida con los productos combinados en TRAYENTA DUO®. La única interacción entre linagliptina y metformina observada fue la disminución del aumento de peso. No se observó otro tipo de toxicidad causado por la combinación de linagliptina y metformina.
Un estudio experimental de reproducción en ratas preñadas indicó ausencia de efectos teratogénicos atribuidos a la administración conjunta de linagliptina y metformina.
Los siguientes datos surgieron de estudios realizados con linagliptina o metformina por separado.
Linagliptina: Se observaron efectos en estudios no clínicos solamente en casos de exposición muy por encima comparada con la máxima exposición en humanos, lo que indica baja relevancia clínica.
Genotoxicidad: Linagliptina no tuvo efectos mutagénicos ni clastogénicos con o sin activación metabólica como resultado de la prueba de Ames de mutagenicidad bacteriana, un ensayo de aberraciones cromosómicas en linfocitos humanos, y en un ensayo de micronúcleos in vivo.
Carcinogenicidad: Se hizo un estudio de carcinogenicidad durante 2 años en ratas machos y hembras a los que se administraron dosis orales de 6, 18 y 60 mg/kg/día de linagliptina. No se observó aumento de la incidencia tumoral en ningún órgano a dosis máximas de 60 mg/kg/día. Esta dosis provoca exposición de aproximadamente 418 veces la exposición humana a la dosis máxima recomendada en humanos (DMRH) de 5 mg/día basada en la comparación del ABC. Se hizo un estudio de carcinogenicidad durante 2 años en ratones machos y hembras a los que se administraron dosis orales de 8, 25 y 80 mg/kg/día. No se observaron indicios de potencial carcinogénico a dosis máximas de 80 mg/kg/día, aproximadamente 242 veces la exposición humana a la DMRH.
Toxicidad en la reproducción: En los estudios de fertilidad en ratas con dosis orales de 10, 30 y 240 mg/kg/día administradas por sonda los machos se trataron durante 4 semanas antes y durante el apareamiento, y las hembras, durante 2 semanas antes del apareamiento y hasta el sexto día de gestación. No se observaron efectos adversos en el desarrollo embrionario precoz, el apareamiento, la fertilidad ni las crías recién nacidas hasta la máxima dosis, 240 mg/kg/día (aproximadamente 943 veces la exposición humana a la DMRH de 5 mg/día basada en la comparación del ABC). En los estudios del desarrollo embriofetal en ratas y conejos se demostró que linagliptina no es teratogénica a dosis máximas de 240 mg/kg/día (943x DMRH) en ratas, y 150 mg/kg/día (1943x DMRH) en conejos. Estos resultados derivaron en un nivel sin efectos adversos observados (NOAEL) de 30 mg/kg/día (49x DMRH) y 25 mg/kg (78x DMRH) para analizar la toxicidad embriofetal en ratas y conejos, respectivamente.
Metformina: Los datos no clínicos indican ausencia de riesgo para humanos sobre la base de estudios convencionales de seguridad farmacológica, genotoxicidad y potencial carcinogénico. En un estudio de toxicidad de 13 semanas en ratas la toxicidad relacionada con la metformina se observó en corazón, hígado, riñones, glándulas salivales, ovarios, timo, tracto gastrointestinal y glándulas suprarrenales a dosis relacionadas con exposición sistémica 7 veces mayor que la DMRH o más.
La metformina no tuvo efectos teratogénicos en ratas a dosis de 200 mg/kg/día relacionadas con exposición sistémica 4 veces mayor que la DMRH (2000 mg de metformina). A dosis mayores (500 y 1000 mg/kg/día, relacionadas con exposición sistémica 11 y 23 veces mayor que la DMRH), se observó teratogenicidad en ratas.
Fertilidad: No se han hecho estudios sobre el efecto de TRAYENTA DUO® en la fertilidad humana. No se observaron efectos adversos de linagliptina en la fertilidad en estudios no clínicos con la mayor dosis probada de 240 mg/kg/día (> 900 veces la exposición humana).
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones de farmacocinética:
Generales: La coadministración de varias dosis de linagliptina (10 mg 1 vez por día) y metformina (850 mg 2 veces por día) no alteró significativamente la farmacocinética de ninguno de los principios activos en voluntarios sanos.
Si bien no se han hecho estudios de interacción farmacocinética con TRAYENTA DUO®, sí se han hecho con cada uno de los principios activos de TRAYENTA DUO®: linagliptina y metformina.
Linagliptina:
Evaluación in vitro de las interacciones farmacológicas: La linagliptina es un competidor débil y un inhibidor entre débil y moderado, basado en el mecanismo, de la isoenzima CYP3A4 del citocromo CYP, pero no inhibe otras isoenzimas del CYP. No es un inductor de isoenzimas del CYP.
La linagliptina es un sustrato de la glucoproteína P (gp P), e inhibe el transporte de digoxina provocado por la gp P a baja potencia. Es sobre la base de estos resultados y de estudios farmacológicos in vivo que se considera improbable que linagliptina tenga interacciones con otros sustratos de la gp P.
Evaluación in vivo de las interacciones farmacológicas: Los datos clínicos incluidos más adelante indican que el riesgo de interacciones farmacológicas clínicamente significativas provocadas por la administración conjunta de fármacos es bajo. No se observaron interacciones clínicamente significativas que exigieran modificar la dosis.
Linagliptina no tuvo efectos clínicamente relevantes en la farmacocinética de metformina, glibenclamida, simvastatina, pioglitazona, warfarina, digoxina y anticonceptivos orales, lo que indica in vivo bajo riesgo de interacción farmacológica con sustratos de CYP3A4, CYP2C9, CYP2C8, gp P y transportador de cationes orgánicos (TCO).
Metformina: La coadministración de varias dosis de 850 mg de metformina 3 veces por día con una dosis supraterapéutica de 10 mg de linagliptina 1 vez por día no alteró en forma clínicamente significativa la farmacocinética de ninguno de los principios activos en voluntarios sanos. Por lo tanto, la linagliptina no es un inhibidor del transporte producido por TCO.
Sulfonilureas: La farmacocinética en equilibrio de 5 mg de linagliptina no se modificó con la administración de una dosis única de 1.75 mg de glibenclamida junto con varias dosis orales de linagliptina 5 mg, si bien se observó una disminución del 14% del ABC y de la Cmáx de glibenclamida que no fue clínicamente relevante. Al ser la glibenclamida metabolizada principalmente por la CYP2C9, estos datos también justifican la conclusión de que linagliptina no es un inhibidor de la CYP2C9. No se esperan interacciones clínicamente significativas con otras sulfonilureas (por ejemplo, glipizida, tolbutamida y glimepirida) que son eliminadas principalmente por la CYP2C9, como la glibenclamida.
Tiazolidinadionas: La coadministración de varias dosis diarias de 10 mg de linagliptina (supraterapéutica) y varias dosis diarias de 45 mg de pioglitazona, sustrato de CYP2C8 y CYP3A4, no alteró en forma clínicamente significativa la farmacocinética de linagliptina o pioglitazona ni los metabolitos activos de la pioglitazona, lo que indica que linagliptina no es un inhibidor del metabolismo provocado por la CYP2C8 in vivo, y justifica la conclusión de que la inhibición in vivo de la CYP3A4 por la linagliptina es insignificante.
Ritonavir: Se llevó a cabo un estudio para evaluar el efecto del ritonavir, un potente inhibidor de la glucoproteína P y la isoenzima CYP3A4, sobre la farmacocinética de la linagliptina. La coadministración de una sola dosis oral de 5 mg de linagliptina y dosis orales repetidas de 200 mg de ritonavir aumentaron el ABC y la Cmáx., de la linagliptina aproximadamente dos y tres veces, respectivamente. Las simulaciones de las concentraciones plasmáticas en estado estable de linagliptina con y sin ritonavir indicaron que el aumento de la exposición no estará asociado con un aumento de la acumulación. Estos cambios en la farmacocinética de la linagliptina no se consideraron clínicamente relevantes. Por lo tanto, no se esperan interacciones clínicamente relevantes con otros inhibidores de la glucoproteína P / CYP3A4 y no se requiere ajuste de la dosis.
Rifampicina: Se llevó a cabo un estudio para evaluar el efecto de la rifampicina, un potente inductor de la glucoproteína P y de CYP3A4, sobre la farmacocinética de 5 mg de linagliptina. La coadministración repetida de linagliptina con rifampicina resultó en decrementos de 39.6% y 43.8% del ABC y la Cmáx en estado estable de linagliptina y un decremento de aproximadamente 30% de la inhibición de la dipeptidil peptidasa 4 (DPP-4) en concentraciones basales. Por lo tanto, se espera que linagliptina en combinación con inductores potentes de la P-gp sea clínicamente eficaz, aunque podría no lograrse la eficacia completa.
Digoxina: La coadministración de dosis diarias repetidas de 5 mg de linagliptina con dosis repetidas de 0.25 mg de digoxina no tuvo efectos sobre la farmacocinética de la digoxina en voluntarios sanos. Por lo tanto, linagliptina no es un inhibidor del transporte mediado por la glucoproteína P in vivo.
Warfarina: Las dosis diarias repetidas de 5 mg de linagliptina no alteraron la farmacocinética de la S-warfarina y la R-warfarina, un sustrato de CYP2C9, lo cual demuestra que linagliptina no es un inhibidor de la isoenzima CYP2C9.
Simvastatina: Las dosis diarias repetidas de 10 mg de linagliptina (supraterapéuticas) tuvieron un efecto mínimo sobre la farmacocinética en estado estable de la simvastatina, un sustrato sensible de la isoenzima CYP3A4, en voluntarios sanos. Después de la administración de 10 mg de linagliptina concomitantemente con 40 mg de simvastatina diariamente durante 6 días, el ABC plasmática de la simvastatina aumentó en 34%, y la Cmáx plasmática aumentó en 10%. Por lo tanto, se considera que linagliptina es un inhibidor débil del metabolismo mediado por la isoenzima CYP3A4, y se considera innecesario el ajuste de la dosis de las sustancias administradas concomitantemente metabolizados por la isoenzima CYP3A4.
Anticonceptivos orales: La coadministración con 5 mg de linagliptina no alteró la farmacocinética en estado estable del levonorgestrel o el etinilestradiol.
La biodisponibilidad absoluta de linagliptina es de aproximadamente 30%. Debido a que la coadministración de una comida con alto contenido de grasa con linagliptina no tuvo efectos clínicamente relevantes sobre la farmacocinética, linagliptina puede administrarse con o sin alimentos.
Metformina: Uso concomitante no recomendado.
Alcohol: La intoxicación alcohólica aguda está asociada con un mayor riesgo de acidosis láctica, especialmente en presencia de ayuno, desnutrición o insuficiencia hepática).
Medios de contraste yodados: La administración de TRAYENTA DUO® debe suspenderse con anterioridad al procedimiento de diagnóstico por imágenes, o al momento de su realización, y podrá reanudarse después de que hayan transcurrido por lo menos 48 horas siempre que se haya evaluado la función renal y comprobado que es estable (Ver Dosis y vía de administración y Precauciones generales”).
Combinaciones que requieren precauciones de uso: Algunos medicamentos pueden afectar de forma adversa la función renal, lo que puede incrementar el riesgo de acidosis láctica, por ejemplo, los AINE, incluidos los inhibidores selectivos de la ciclooxigenasa (COX) II, los inhibidores de la ECA, los antagonistas del receptor de la angiotensina II y los diuréticos, en especial, los diuréticos del asa. Cuando se inicie un tratamiento con estos productos o se usen en combinación con metformina, es necesario un monitoreo estrecho de la función renal.
La metformina es un sustrato de ambos transportadores OCT1 y OCT2. La coadministración de metformina con:
• Los inhibidores de OCT1 (como el verapamilo) pueden reducir la eficacia de la metformina.
• Los inductores de OCT1 (como la rifampicina) pueden aumentar la absorción gastrointestinal y la eficacia de la metformina.
• Los inhibidores de OCT2 (como cimetidina, dolutegravir, ranolazina, trimetoprima, vandetanib, isavuconazol) pueden disminuir la eliminación renal de metformina y, por lo tanto, conducir a un aumento de la concentración plasmática de metformina.
• Los inhibidores de OCT1 y OCT2 (como crizotinib, olaparib) pueden alterar la eficacia y la eliminación renal de la metformina.
Por lo tanto, se recomienda precaución, especialmente en pacientes con insuficiencia renal, cuando estos fármacos son coadministrados con metformina, ya que la concentración plasmática de metformina puede aumentar. Si es necesario, se puede considerar el ajuste de la dosis de metformina, ya que los inhibidores/inductores de los OCT pueden alterar la eficacia de la metformina.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No se han reportado cambios clínicamente significativos en los parámetros de laboratorio y de ECG.
PRECAUCIONES GENERALES: TRAYENTA DUO® no se debe administrar a los pacientes con diabetes tipo 1 ni se debe utilizar para el tratamiento de la cetoacidosis diabética.
Pancreatitis: Se ha observado pancreatitis aguda en pacientes que estaban tomando linagliptina. Ante la sospecha de pancreatitis debe suspenderse la administración de TRAYENTA DUO®.
Hipoglucemia: Linagliptina sola mostró incidencia de hipoglucemia similar al placebo. En los ensayos clínicos en los que linagliptina era parte del tratamiento combinado con agentes no considerados causantes de hipoglucemia (metformina, tiazolidinedionas), los índices de hipoglucemia informados con linagliptina fueron similares a los informados por los pacientes que recibieron el placebo.
Se sabe que las sulfonilureas son causantes de hipoglucemia; por lo tanto, se advierte que se deben tomar precauciones cuando se administra TRAYENTA DUO® junto con una sulfonilurea. Se puede considerar disminuir la dosis de la sulfonilurea.
Se sabe que la insulina es causante de hipoglucemia. Por lo tanto, se advierte que se deben tomar precauciones cuando se administra TRAYENTA DUO® junto con insulina. Se puede considerar disminuir la dosis de insulina.
La metformina sola no es causante de hipoglucemia en condiciones normales de administración, pero en los casos de deficiencia en la ingesta de calorías, de actividad física muy exigente sin compensación con suplementos calóricos, o durante la administración concomitante de otros agentes reductores de la glucosa (como las sulfonilureas y la insulina) o de etanol, puede aparecer hipoglucemia.
Acidosis láctica: La acidosis láctica es una complicación metabólica muy esporádica, pero grave. Se produce con mayor frecuencia durante el empeoramiento agudo de la función renal, en caso de enfermedad cardiorrespiratoria o septicemia. La acumulación de metformina se produce durante el empeoramiento agudo de la función renal e incrementa el riesgo de acidosis láctica.
En caso de deshidratación (diarrea o vómitos intensos, fiebre o reducción de la ingesta de líquidos), la metformina se debe interrumpir de forma temporal y se recomienda contactar a un profesional sanitario.
El uso de medicamentos que puedan alterar de manera aguda la función renal (como antihipertensivos, diuréticos y AINE) se debe iniciar con precaución en los pacientes tratados con metformina.
Otros factores de riesgo para la acidosis láctica son la ingesta excesiva del alcohol, la insuficiencia hepática, diabetes mal controlada, cetosis, ayuno prolongado y cualquier afección relacionada con hipoxia, así como el uso concomitante de medicamentos que puedan causar acidosis láctica (Ver Contraindicaciones e Interacciones medicamentosas y de otro género).
Se debe informar a los pacientes y/o a las personas que los cuidan acerca del riesgo de acidosis láctica.
La acidosis láctica se caracteriza por la presencia de disnea acidótica, dolor abdominal, calambres musculares, astenia e hipotermia seguida de coma. En caso de que se sospeche de la presencia de síntomas, el paciente debe dejar de tomar metformina y buscar atención médica inmediata.
Los valores de laboratorio para establecer un diagnóstico son disminución del pH sanguíneo (< 7.35), aumento de los niveles de lactato plasmático (> 5 mmol/L), con aumento de la brecha aniónica y de la relación lactato/piruvato.
Administración de un agente de contraste yodado: La administración intravascular de agentes de contraste yodados puede provocar nefropatía inducida por el contraste, lo que ocasiona la acumulación de metformina y aumenta el riesgo de acidosis láctica. La administración de metformina se debe interrumpir con anterioridad al procedimiento de diagnóstico por imágenes, o al momento de su realización, y deben dejarse transcurrir como mínimo 48 horas antes de reanudarlo, siempre que se haya reevaluado la función renal y comprobado que es estable, véanse las secciones “Dosis y vía de administración” e “Interacciones medicamentosas y de otro género”.
Función renal: Se debe evaluar la TFG antes de iniciar el tratamiento y periódicamente a partir de entonces, véase la sección Dosis y vía de administración. TRAYENTA DUO® está contraindicado en pacientes con TFG < 30 ml/min y se debe interrumpir de forma temporal en presencia de trastornos que alteren la función renal, véase la sección Contraindicaciones.
Función cardiaca: Los pacientes con insuficiencia cardiaca presentan mayor riesgo de hipoxia e insuficiencia renal. En los pacientes con insuficiencia cardiaca crónica estable, se podrá administrar TRAYENTA DUO® si se realiza un control regular de la función cardiaca y renal.
En los pacientes con insuficiencia cardiaca aguda e inestable, TRAYENTA DUO® está contraindicado debido a la presencia del componente metformina (ver Contraindicaciones).
Intervención quirúrgica: La metformina se debe interrumpir en el momento de la realización de una intervención quirúrgica con anestesia general, raquídea o epidural. Se puede retomar a las 48 horas de la cirugía o tras la recuperación de la ingesta oral y siempre que se haya revaluado la función renal y comprobado que es estable.
Penfigoide ampolloso: Se han observado casos de penfigoide ampolloso en pacientes que toman linagliptina. Si se sospecha de penfigoide ampolloso, el tratamiento con TRAYENTA DUO® debe interrumpirse.
Efectos sobre la capacidad de conducir y utilizar maquinarias: No se han estudiado los efectos sobre la capacidad para conducir vehículos y operar maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Oral.
Adultos con función renal normal (tasa de filtración glomerular [TFG] ≥ 90 mL/min).
La dosis recomendada es de 2.5/500 mg, 2.5/850 mg o 2.5/1000 mg 2 veces por día.
La dosis debe individualizarse con base en el régimen actual, la eficacia y la tolerabilidad de cada sujeto. La dosis máxima recomendada de TRAYENTA DUO® es de 5 mg de linagliptina y 2000 mg de metformina. (Ver Tabla 6 para acceder a información posológica adicional).
Se recomienda administrar TRAYENTA DUO® junto con las comidas para disminuir los efectos gastrointestinales no deseados de la metformina.
Pacientes actualmente no tratados con metformina: En el caso de los pacientes que no son tratados actualmente con metformina, la dosis inicial recomendada es de 2.5 mg de linagliptina/500 mg de clorhidrato de metformina, dos veces al día.
Pacientes para los que la dosis tolerada máxima de metformina como monoterapia resulta insuficiente: Para los pacientes en tratamiento con metformina sola cuyo control glucémico no es el adecuado, la dosis inicial de rutina de TRAYENTA DUO® debe aportar 2.5 mg de linagliptina 2 veces por día (dosis total diaria: 5 mg) más la dosis de metformina que ya se administra.
Pacientes que cambian el tratamiento combinado con linagliptina y metformina: Para los pacientes que cambian la administración conjunta de linagliptina y metformina por la combinación a dosis fija, el tratamiento con TRAYENTA DUO® se debe comenzar con la dosis de linagliptina y metformina que ya se administra.
Pacientes en tratamiento combinado doble con la dosis tolerada máxima de metformina y una sulfonilurea y control insuficiente: La dosis de TRAYENTA DUO ® debe aportar 2.5 mg de linagliptina 2 veces por día (dosis total diaria: 5 mg) y una dosis de metformina similar a la que ya se administra. Si se administra TRAYENTA DUO® junto con una sulfonilurea, puede ser necesario administrar una dosis inferior de la sulfonilurea para disminuir el riesgo de hipoglucemia (Ver Precauciones generales).
Para pacientes inadecuadamente controlados con el tratamiento de combinación doble con insulina y la dosis máxima tolerada de metformina.
La dosis de TRAYENTA DUO® debe aportar 2.5 mg de linagliptina 2 veces por día (dosis total diaria: 5 mg) y una dosis de metformina similar a la dosis que ya se administra. Si se administra TRAYENTA DUO® junto con insulina, puede ser necesario administrar una dosis inferior de insulina para reducir el riesgo de hipoglucemia (Ver sección de Precauciones generales).
TRAYENTA DUO® se comercializa con diferentes concentraciones de metformina: 2.5 mg de linagliptina más 500 mg de clorhidrato de metformina, 850 mg de clorhidrato de metformina o 1,000 mg de clorhidrato de metformina.
Insuficiencia renal: Se debe evaluar la TFG antes de iniciar el tratamiento con productos que contengan metformina y, al menos, una vez al año a partir de entonces.
En los pacientes expuestos a un mayor riesgo de progresión de la insuficiencia renal y en pacientes de edad avanzada, se debe evaluar la función renal con mayor frecuencia, por ej. cada 3-6 meses.
Se deben revisar los factores que puedan aumentar el riesgo de acidosis láctica (ver Precauciones generales) antes de considerar el inicio del tratamiento con metformina en los pacientes con TFG < 60 ml/min.
Tabla 6. Posología para pacientes con insuficiencia renal*
|
TFGe ml/min |
Metformina |
Linagliptina |
|
60-89 |
La dosis diaria máxima es de 3000 mg*. Se puede considerar la reducción de la dosis en relación con el deterioro de la función renal. |
No se requiere un ajuste de la dosis |
|
45-59 |
La dosis diaria máxima es de 2000 mg. La dosis inicial es, a lo sumo, la mitad de la dosis máxima. |
No se requiere un ajuste de la dosis |
|
30-44 |
La dosis diaria máxima es de 1000 mg*. La dosis inicial es, a lo sumo, la mitad de la dosis máxima. |
No se requiere un ajuste de la dosis |
|
<30 |
El uso de metformina está contraindicado. |
No se requiere un ajuste de la dosis |
* Si no se dispone de la dosis adecuada de TRAYENTA DUO®, se deben utilizar los monocomponentes individuales en lugar de la combinación a dosis fija.
Insuficiencia hepática: TRAYENTA DUO® está contraindicado para los pacientes con insuficiencia hepática debido a la presencia de metformina (ver Contraindicaciones).
Pacientes geriátricos: Como la metformina se elimina por vía renal, y el paciente geriátrico muestra tendencia a la disminución de la función renal, si se administra TRAYENTA DUO® a esta población, se debe monitorear la función renal periódicamente (Ver Precauciones generales).
Pacientes pediátricos y adolescentes: TRAYENTA DUO® no está recomendado para menores de 18 años porque no existen datos de seguridad y eficacia al respecto.
Olvido de una dosis: Si el paciente se olvida de tomar una dosis, debe tomarla en cuanto se dé cuenta, pero no debe tomar dos dosis juntas. En ese caso, debe saltar la dosis que se olvidó.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Síntomas: Durante los ensayos clínicos comparativos en sujetos sanos la dosis única máxima de 600 mg de linagliptina (equivalente a 120 veces la dosis recomendada) fue bien tolerada. No existen antecedentes con dosis superiores a 600 mg en humanos.
No se observó hipoglucemia al administrar clorhidrato de metformina en dosis máximas de 85 g, aunque se observó acidosis láctica. La acidosis láctica puede ser causada por alta sobredosis de clorhidrato de metformina o por riesgos concomitantes. Es una urgencia que se debe tratar en un hospital.
Tratamiento: En caso de sobredosificación, se recomienda aplicar los métodos usuales: eliminar del tracto gastrointestinal el material no absorbido, realizar monitoreo clínico y administrar el tratamiento de apoyo que indique el estado clínico del paciente. La hemodiálisis es el método más eficaz para eliminar el lactato y el clorhidrato de metformina.
PRESENTACIONES:
Caja con 30 y 60 tabletas de 2.5 mg/500 mg, 2.5 mg/850 mg o 2.5 mg/1000 mg en envase de burbuja.
Caja de cartón con frasco con 60 o 180 tabletas de 2.5 mg/500 mg, 2.5 mg/850 mg o 2.5 mg/1000 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 25 °C.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo, en la lactancia ni en niños menores de 18 años.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx y
farmacovigilancia.mex@boehringer-ingelheim.com
BOEHRINGER INGELHEIM PROMECO, S.A. de C.V.
Calle del Maíz No. 49, Col. Barrio Xaltocan,
C.P. 16090, Xochimilco, Ciudad de México, México
Reg. Núm. 118M2012 SSA
®Marca Registrada


