TRECTYL - Tableta
Sustancia(s):
- Sildenafil
Presentaciones:
- 1 Caja, 1 Tabletas, 50 Miligramos
- 1 Caja, 4 Tabletas, 50 Miligramos
- 1 Caja, 8 Tabletas, 50 Miligramos
- 1 Caja, 1 Tabletas, 100 Miligramos
- 1 Caja, 4 Tabletas, 100 Miligramos
- 1 Caja, 8 Tabletas, 100 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
Forma farmacéutica: Polvo
Consideración de uso: Para inhalación
Cada dosis contiene:
Furoato de Fluticasona 100 mcg
Bromuro de Umeclidinio equivalente a 62.5 mcg de Umeclidinio
Trifenatato de Vilanterol equivalente a 25 mcg de Vilanterol
Excipiente cbp 25 mg
Cada dosis contiene:
Furoato de Fluticasona 200 mcg
Bromuro de Umeclidinio equivalente a 62.5 mcg de Umeclidinio
Trifenatato de Vilanterol equivalente a 25 mcg de Vilanterol
Excipiente cbp 25 mg
INDICACIONES TERAPÉUTICAS:
ASMA: TRELEGY® está indicado para el tratamiento de mantenimiento de asma en pacientes mayores de 18 años.
EPOC: TRELEGY® está indicado para el tratamiento de mantenimiento para prevenir y aliviar síntomas asociados con la Enfermedad Pulmonar Obstructiva Crónica (EPOC).
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacodinamia:
Código ATC:
Grupo farmacoterapéutico: Fármacos para enfermedades obstructivas de las vías respiratorias, Adrenérgicos en combinación con anticolinérgicos que incluyen combinaciones triples con corticosteroides.
Código ATC: R03AL08.
Mecanismo de acción: El Furoato de Fluticasona, Umeclidinio y Vilanterol representan tres clases de fármacos: un corticosteroide sintético, un antagonista de los receptores muscarínicos de larga acción (también denominado LAMA o anticolinérgico) y un agonista selectivo de los receptores beta2 de larga acción (LABA), respectivamente.
Furoato de Fluticasona: El Furoato de Fluticasona es un corticosteroide con una potente actividad antiinflamatoria. Se desconoce el mecanismo preciso a través del cual el Furoato de Fluticasona impacta al asma y a los síntomas de EPOC. Los corticosteroides han demostrado tener un amplio rango de acciones en múltiples tipos de células (ej. eosinófilos, macrófagos, linfocitos) y mediadores (ej. citosinas y quimiocinas) que participan en la inflamación.
Umeclidinio: El Umeclidinio es un antagonista de los receptores pan-muscarínicos de larga acción (también denominados anticolinérgicos). El Umeclidinio ejerce su actividad broncodilatadora al inhibir competitivamente la unión de la acetilcolina con los receptores colinérgicos muscarínicos en el músculo liso de las vías aéreas. Demuestra una lenta reversibilidad en el subtipo de receptor muscarínico M3 humano in vitro y una larga duración de acción in vivo cuando se administra directamente a los pulmones en los modelos preclínicos.
Vilanterol: Vilanterol es un LABA selectivo. Los efectos farmacológicos de los fármacos agonistas de los adrenorreceptores beta2 , incluyendo Vilanterol, son al menos en parte atribuibles a la estimulación de la adenilato ciclasa intracelular, la enzima que cataliza la conversión de adenosina trifosfato (ATP) a adenosina monofosfato 3’,5’ cíclico (AMP cíclico). Los niveles elevados de AMP cíclico provocan la relajación del músculo liso bronquial y la inhibición de la liberación de los mediadores de hipersensibilidad inmediata de las células, especialmente de las células mastoides.
Efectos farmacodinámicos:
Efectos cardiovasculares: El efecto de TRELEGY® sobre el intervalo QT no se ha evaluado en un estudio detallado QT (TQT).
Los estudios TQT para Furoato de Fluticasona/Vilanterol y Umeclidinio/Vilanterol no mostraron efectos clínicamente relevantes sobre el intervalo QT con dosis clínicas de Furoato de Fluticasona, Umeclidinio y Vilanterol (véase a continuación).
El efecto de Umeclidinio/Vilanterol sobre el intervalo QT se evaluó en un estudio de QT controlado con placebo y Moxifloxacino que incluyó una administración una vez al día de 125/25 microgramos o 500/100 microgramos de Umeclidinio/Vilanterol por 10 días en 103 voluntarios sanos. La media de la diferencia máxima en las prolongaciones del intervalo QT (corregido usando el método de Fridericia, QTcF) del placebo después de la corrección inicial fue 4.3 (CI de 90%: 2.2, 6.4) milisegundos observada 10 minutos después de la administración con 125/25 microgramos de Umeclidinio/Vilanterol y 8.2 (CI de 90%: 6.2, 10.2) milisegundos 30 minutos después de la administración con 500/100 microgramos de Umeclidinio/Vilanterol.
No se observó ningún efecto clínicamente relevante sobre la prolongación del intervalo QT (corregido usando el método de Fridericia) con la dosis de 125/25 microgramos de Umeclidinio/Vilanterol. Además, no se observaron efectos clínicamente significativos de Umeclidinio/Vilanterol sobre el ritmo cardiaco en un monitoreo Holter de 24 horas en 281 pacientes que recibieron 125/25 microgramos de Umeclidinio/Vilanterol una vez al día por hasta 12 meses.
El efecto de Furoato de Fluticasona/Vilanterol sobre el intervalo QT se evaluó en un estudio cruzado, doble ciego, de múltiples dosis, controlado con placebo y fármaco positivo en 85 voluntarios sanos. La media de la diferencia máxima (límite de confianza superior de 95%) en el QTcF a partir del placebo después de la corrección inicial fue 4.9 (7.5) milisegundos y 9.6 (12.2) milisegundos observada 30 minutos después de la dosificación con 200/25 microgramos de Furoato de Fluticasona/Vilanterol y 800/100 microgramos de Furoato de Fluticasona/Vilanterol, respectivamente. También se observó un incremento dependiente de la dosis en la frecuencia cardiaca. La media de la diferencia máxima (límite superior de confianza de 95%) en la frecuencia cardiaca a partir del placebo después de la corrección inicial fue 7.8 (9.4) latidos/min y 17.1 (18.7) latidos/min observada 10 minutos después de la dosificación con 200/25 microgramos de Furoato de Fluticasona/Vilanterol y 800/100 microgramos de Furoato de Fluticasona/Vilanterol, respectivamente.
No se observaron efectos clínicamente relevantes en el intervalo QTc en la revisión de las lecturas centrales de ECG de 1,519 sujetos con asma expuestos a TRELEGY® hasta por 24 semanas, o en un subgrupo de 364 sujetos expuestos hasta por 52 semanas.
No se observaron efectos clínicamente relevantes sobre el intervalo QTc en la revisión de los ECG que tuvieron una lectura central de 911 sujetos con EPOC expuestos a TRELEGY® por hasta 24 semanas o en el subconjunto de 210 sujetos expuestos por hasta 52 semanas.
Farmacocinética: Cuando se administraron Furoato de Fluticasona, Umeclidinio y Vilanterol en combinación por vía inhalada a partir de un inhalador único en sujetos sanos, la farmacocinética de cada componente fue similar a la observada cuando se administró cada sustancia activa ya sea como combinación de Furoato de Fluticasona/Vilanterol (FF/VI), como combinación de Umeclidinio/Vilanterol (UMEC/VI), o cada componente como monoterapia.
Se realizaron análisis farmacocinéticos (PK) de la población para evaluar la exposición sistémica de Furoato de Fluticasona, Umeclidinio y Vilanterol en sujetos con asma. En estos análisis, los niveles sistémicos de los fármacos Furoato de Fluticasona y Vilanterol (Cmáx y AUC0-24 en estado estable) después de Furoato de Fluticasona /Umeclidinio/Vilanterol (100/62.5/25 microgramos y 200/62.5/25 microgramos) en un inhalador (combinación triple) se encontraron dentro del rango de aquellos observados después de la combinación dual de FF/VI con los respectivos 100 microgramos y 200 microgramos de dosis de FF; la exposición sistémica de 62.5 microgramos de Umeclidinio después de Furoato de Fluticasona/Umeclidinio/Vilanterol en un inhalador estuvo dentro del rango de aquellos observados después de la administración de 62.5 microgramos de Umeclidinio como monoterapia.
Se realizaron análisis PK de la población para Furoato de Fluticasona/Umeclidinio/Vilanterol 100/62.5/25 microgramos utilizando un conjunto de datos de PK combinados de tres estudios fase III en 821 sujetos con EPOC. En estos análisis, los niveles sistémicos de fármaco (Cmáx y AUC0-24 en estado estable) de Furoato de Fluticasona, Umeclidinio, y Vilanterol después de la administración de Furoato de Fluticasona/Umeclidinio/Vilanterol en un inhalador (combinación triple) se encontraron dentro del rango de los observados después de la administración de Furoato de Fluticasona/Vilanterol más Umeclidinio a través de dos inhaladores, combinaciones dobles Furoato de Fluticasona/Vilanterol y Umeclidinio/Vilanterol), o con inhaladores individuales (Furoato de Fluticasona, Umeclidinio y Vilanterol).
Absorción:
Furoato de Fluticasona: Después de la administración inhalada de TRELEGY® en sujetos sanos, la Cmax de Furoato de Fluticasona ocurrió a los 15 minutos. La biodisponibilidad absoluta de Furoato de Fluticasona al administrarse como Furoato de Fluticasona/Vilanterol por inhalación fue en promedio 15.2%, principalmente debido a la absorción de la porción inhalada de la dosis administrada al pulmón, con una contribución insignificante a partir de la absorción oral. Después de la dosificación repetida de Furoato de Fluticasona/Vilanterol inhalados, el estado estable se alcanzó en 6 días con una acumulación de hasta 1.6 veces.
Umeclidinio: Después de la administración inhalada de TRELEGY® en sujetos sanos, la Cmax de Umeclidinio ocurrió a los 5 minutos. La biodisponibilidad absoluta de Umeclidinio inhalado fue en promedio 13%, con una contribución insignificante a partir de la absorción oral. Después de la dosificación repetida de Umeclidinio inhalado, el estado estable se alcanzó en 7 a 10 días con una acumulación de 1.5 a 2 veces.
Vilanterol: Después de la administración inhalada de TRELEGY® en sujetos sanos, la Cmax de Vilanterol ocurrió a los 7 minutos. La biodisponibilidad absoluta de Vilanterol inhalado fue en promedio 27%, con una contribución insignificante a partir de la absorción oral. Después de la dosificación repetida de Furoato de Fluticasona/Vilanterol inhalados, el estado estable se alcanzó en 6 días con una acumulación de hasta 1.5 veces.
Distribución:
Furoato de Fluticasona: Después de la administración intravenosa de Furoato de Fluticasona a sujetos sanos, el volumen de distribución medio fue de 661 litros. La unión a proteínas plasmáticas in vitro en el plasma humano fue > 99.6%.
Umeclidinio: Después de la administración intravenosa de Umeclidinio a sujetos sanos, el volumen de distribución medio fue 86 litros. La unión a proteínas plasmáticas in vitro en el plasma humano fue en promedio 89%.
Vilanterol: Después de la administración intravenosa de Vilanterol a voluntarios sanos, el volumen de distribución medio en estado estable fue 165 litros. La unión a proteínas plasmáticas in vitro en el plasma humano fue en promedio 94%.
Metabolismo:
Furoato de Fluticasona: Los estudios in vitro mostraron que el Furoato de Fluticasona se metaboliza principalmente por la enzima CYP3A4 y es un sustrato para el transportador de P-glicoproteína (P-gp). El Furoato de Fluticasona se metaboliza principalmente a través de hidrólisis del grupo S-fluorometil carbotioato a los metabolitos con una actividad de corticosteroides significativamente reducida. La exposición sistémica a los metabolitos es baja.
Umeclidinio: Los estudios in vitro mostraron que Umeclidinio se metaboliza principalmente por CYP2D6 y es un sustrato para el transportador de P-gp. Las vías metabólicas primarias para Umeclidinio son oxidación (hidroxilación, O-desalquilación) seguida de conjugación (glucuronidación, etc.), lo que resulta en un rango de metabolitos ya sea con una actividad farmacológica reducida o para la cual la actividad farmacológica no se ha establecido. La exposición sistémica a los metabolitos es baja.
Vilanterol: Los estudios in vitro mostraron que el Vilanterol se metaboliza principalmente por medio de CYP3A4 y es un sustrato para el transportador de P-gp. Las vías metabólicas primarias son O-desalquilación a un rango de metabolitos con una actividad agonista beta1 y beta2 significativamente reducida. Los perfiles metabólicos plasmáticos después de la administración oral de Vilanterol en un estudio radiomarcado humano fueron consistentes con un alto metabolismo de primer paso. La exposición sistémica a los metabolitos es baja.
Interacciones medicamentosas: Se realizó un estudio de dosis repetidas en sujetos sanos con la combinación de Furoato de Fluticasona/Vilanterol (200/25 microgramos) y ketoconazol (400 miligramos, un fuerte inhibidor de CYP3A4 e inhibidor de Pgp). La coadministración incrementó el valor medio de la AUC (0-24) y Cmax del Furoato de Fluticasona en 36% y 33%, respectivamente. El incremento en la exposición a Furoato de Fluticasona se asoció con una reducción del 27% en la media ponderada de cortisol sérico de 0-24 horas. La coadministración incrementó las medias de AUC(0-t) y Cmax de Vilanterol en 65% y 22%, respectivamente. El incremento en la exposición al Vilanterol no se asoció con un incremento en los efectos sistémicos relacionados a los agonistas beta en la frecuencia cardiaca o el potasio sanguíneo.
El Furoato de Fluticasona, Umeclidinio y Vilanterol son sustratos de la P-gp. Un estudio de interacción medicamentosa de dosis repetidas realizado en sujetos sanos que recibieron Umeclidinio/Vilanterol o Umeclidinio, y el inhibidor de P-gp y el inhibidor moderado de CYP3A4 verapamilo (240 miligramos), no mostró ningún efecto clínicamente significativo sobre la farmacocinética de Vilanterol o Umeclidinio.
El efecto de un genotipo metabolizador deficiente de CYP2D6 sobre la farmacocinética en estado estable de Umeclidinio se evaluó en voluntarios sanos (metabolizadores normales de CYP2D6 y metabolizadores deficientes de CYP2D6). No se observó ninguna diferencia clínicamente importante en la exposición sistémica a Umeclidinio (500 microgramos lo cual es ocho veces mayor que la dosis terapéutica) después de una dosificación inhalada diaria repetida en sujetos metabolizadores normales y deficientes de la CYP2D6.
Eliminación:
Furoato de Fluticasona: La vida media de la eliminación plasmática aparente de Furoato de Fluticasona después de la administración inhalada de Furoato de Fluticasona/Vilanterol fue, en promedio, 24 horas. Después de la administración intravenosa, la vida media de la fase de eliminación promedió 15.1 horas. La depuración plasmática después de la administración intravenosa fue 65.4 litros/hora. La excreción urinaria representó aproximadamente 2 % de la dosis administrada por vía intravenosa. Después de la administración oral, el Furoato de Fluticasona se eliminó en humanos principalmente por metabolismo con los metabolitos excretándose casi exclusivamente en las heces, con < 1% de la dosis radioactiva recuperada eliminada en la orina.
Umeclidinio: La vida media de eliminación plasmática de Umeclidinio después de una dosificación inhalada por 10 días promedió 19 horas, con 3% a 4% del fármaco excretado sin cambios en la orina en estado estable. La depuración plasmática después de la administración intravenosa fue 151 litros/hora. Después de la administración intravenosa, alrededor de 58% de la dosis radiomarcada administrada se excretó en las heces y aproximadamente 22% de la dosis radiomarcada administrada se excretó en la orina. La excreción del material relacionado al fármaco en las heces después de la dosificación intravenosa indicó una secreción en la bilis. Posterior a la administración oral, 92% de la dosis radiomarcada administrada se excretó principalmente en las heces. Menos de 1% de la dosis administrada por vía oral (1% de la radioactividad recuperada) se excretó en la orina, lo que indica una absorción insignificante después de la administración oral.
Vilanterol: La vida media de eliminación plasmática de Vilanterol después de la administración inhalada por 10 días promedió 11 horas. La depuración plasmática de Vilanterol después de la administración intravenosa fue 108 litros/hora. Después de la administración oral de Vilanterol radiomarcado, 70% de la radiomarca se excretó en la orina y 30% en las heces. La eliminación primaria de Vilanterol fue por metabolismo seguido de la excreción de los metabolitos en la orina y las heces.
Poblaciones especiales de pacientes: En los análisis farmacocinéticos (PK) de población con asma (1,265 sujetos para Furoato de Fluticasona; 1,263 sujetos para Vilanterol; 634 sujetos para Umeclidinio), se evaluó el efecto de las covariables demográficas (raza/etnia, edad, género, peso) sobre la farmacocinética de Furoato de Fluticasona, Umeclidinio y Vilanterol. En un análisis farmacocinético (PK) de población con EPOC (n = 821), se evaluó el efecto de las covariables demográficas (raza/etnia, edad, género, peso) sobre la farmacocinética de Furoato de Fluticasona, Umeclidinio y Vilanterol. La insuficiencia renal y la insuficiencia hepática se evaluaron en estudios separados.
Raza: No se observaron diferencias clínicamente relevantes que hayan requerido ajustar la dosis para asma o EPOC con base en la raza en la exposición sistémica a Furoato de Fluticasona, Umeclidinio o Vilanterol.
En datos farmacocinéticos de población de sujetos con asma del este de Asia (descendientes de japoneses y de personas nacidas en el este y sureste de Asia) (n = 92) que recibieron Furoato de Fluticasona/Umeclidinio/Vilanterol (100/62.5/25 microgramos y 200/62.5/25 microgramos) los estimados de la Cmáx de Vilanterol en estado estable fue aproximadamente 3-veces más alta que sujetos fuera del este de Asia. No hubo efecto de la raza sobre la farmacocinética de Furoato de Fluticasona o Umeclidinio en sujetos con asma.
En sujetos del este de Asia con EPOC, (descendientes de japoneses y de personas nacidas en el este de Asia) (n = 113) que recibieron Furoato de Fluticasona/Umeclidinio/Vilanterol 100/62.5/25 microgramos, los estimados de AUC ss de Furoato de Fluticasona fueron en promedio 30% mayores en comparación con los sujetos caucásicos. No obstante, no se espera que estas exposiciones sistémicas más altas tengan un efecto clínicamente relevante sobre la excreción de cortisol sérico o urinario de 24 horas. No hubo ningún efecto de la raza sobre la farmacocinética de Umeclidinio o Vilanterol en los sujetos con EPOC.
Pacientes de edad avanzada: En sujetos con asma o EPOC no se observaron efectos clínicamente relevantes que hayan requerido ajustes de la dosis con base en la edad.
Insuficiencia renal: TRELEGY® no se ha evaluado en sujetos con insuficiencia renal. No obstante, se han realizado estudios con Furoato de Fluticasona/Vilanterol y Umeclidinio/Vilanterol.
Un estudio de farmacología clínica de Furoato de Fluticasona/Vilanterol mostró que la insuficiencia renal severa (depuración de creatinina < 30 ml/min) no resultó en una exposición significativamente mayor a Furoato de Fluticasona o Vilanterol o en unos efectos sistémicos más notorios de los corticosteroides o los agonistas beta2 en comparación con los sujetos sanos.
Un estudio en sujetos con insuficiencia renal severa que fueron administrados con Umeclidinio/Vilanterol no mostró evidencia de un incremento en la exposición sistémica ya sea a Umeclidinio o Vilanterol (Cmax y AUC). Se realizaron estudios de unión a proteínas in vitro entre los sujetos con insuficiencia renal severa y voluntarios sanos y no se observó ninguna evidencia clínicamente significativa de una unión proteica alterada.
No se han estudiado los efectos de la hemodiálisis.
Insuficiencia hepática: TRELEGY® no se ha evaluado en sujetos con insuficiencia hepática. No obstante, se han realizado estudios con Furoato de Fluticasona/Vilanterol y Umeclidinio/Vilanterol.
Después de la dosificación repetida de Furoato de Fluticasona/Vilanterol por 7 días, hubo un incremento en la exposición sistémica de Furoato de Fluticasona (hasta de tres veces conforme a la medición del AUC (0-24)) en sujetos con insuficiencia hepática (Child-Pugh A, B o C) en comparación con sujetos sanos. No se observaron efectos clínicamente relevantes en la media ponderada de cortisol sérico en sujetos con insuficiencia hepática ligera (Child-Pugh A). El incremento en la exposición sistémica de Furoato de Fluticasona (200/25 microgramos de Furoato de Fluticasona/Vilanterol) en sujetos con insuficiencia hepática moderada (Child-Pugh B) se asoció con una reducción promedio de 34% en el cortisol sérico en comparación con los sujetos sanos. En sujetos con insuficiencia hepática severa (Child-Pugh C) que recibieron 100/12.5 microgramos de Furoato de Fluticasona/Vilanterol no hubo ninguna reducción en el cortisol sérico (incremento de 10% en el cortisol sérico).
En pacientes con insuficiencia hepática moderada o severa la dosis máxima es 100/62.5/25 microgramos (véase Dosificación y Administración).
Después de la dosificación repetida de Furoato de Fluticasona/Vilanterol por 7 días, no hubo un incremento significativo en la exposición sistémica al Vilanterol (Cmax y AUC) en los sujetos con insuficiencia hepática leve, moderada o severa (Child-Pugh A, B o C).
No hubo efectos clínicamente relevantes de la combinación de Furoato de Fluticasona/Vilanterol sobre los efectos sistémicos betaadrenérgicos (frecuencia cardiaca o potasio sérico) en los sujetos con insuficiencia hepática leve o moderada (Vilanterol, 25 microgramos) o con insuficiencia hepática severa (Vilanterol, 12.5 microgramos) en comparación con los sujetos sanos.
Los sujetos con insuficiencia hepática moderada no mostraron evidencia de un incremento en la exposición sistémica ya sea a Umeclidinio o Vilanterol (Cmax y AUC). Se realizaron estudios de unión proteica in vitro entre sujetos con insuficiencia hepática moderada y voluntarios sanos, y no se observó ninguna evidencia clínicamente significativa de unión proteica alterada. Umeclidinio no se ha evaluado en sujetos con insuficiencia hepática severa.
Otras características de los pacientes: No se observaron diferencias clínicamente relevantes que hayan requerido ajuste de la dosis con base en el efecto del género, el peso o el índice de masa corporal en sujetos con asma o EPOC.
Sujetos con metabolismo deficiente de CYP2D6 no mostraron ninguna evidencia de un efecto clínicamente significativo de polimorfismo genético de CYP2D6 sobre la exposición sistémica a Umeclidinio.
Estudios clínicos:
Asma: La seguridad y eficacia de TRELEGY® (FF/UMEC/VI) fueron evaluadas en 2,436 sujetos en un ensayo clínico aleatorizado, multicéntrico, con control del activo, doble-ciego de 24 a 52 semanas de duración en sujetos adultos con asma controlados inadecuadamente en sus tratamientos actuales de terapia combinada (ICS más un LABA), (estudio 205715, CAPTAIN).
El estudio evaluó la eficacia de TRELEGY® en la función pulmonar, la tasa anualizada de exacerbaciones de asma moderadas y severas, el control de los síntomas de asma y la calidad de vida relacionada con la salud en comparación con el Furoato de Fluticasona/Vilanterol . El criterio de valoración primario fue el cambio desde el valor basal en el punto mínimo del volumen espiratorio forzado en 1 segundo (FEV1) en la semana 24. El criterio de valoración secundario clave fue la tasa anualizada de exacerbación de asma moderada/severa.
Este estudio tuvo un periodo de inducción/estabilización de 5 semanas que se describe a continuación: sujetos controlados de manera inadecuada [Cuestionario de Control de Asma (ACQ-6) ≥1.5] en su tratamiento actual de asma de corticosteroides inhalados (mayor que o equivalente a 250 microgramos por día de propionato de Fluticasona) más LABA entraron en un período de inducción de 3 semanas de tratamiento con 250/50 microgramos dos veces al día de propionato de Fluticasona/salmeterol. Los sujetos que permanecían controlados de manera inadecuada (ACQ-6 ≥1.5) después del período de inducción fueron transferidos al período de estabilización con 100/25 microgramos una vez al día de Furoato de Fluticasona/Vilanterol (FF/VI) por 2 semanas. En todos los grupos de tratamiento, la demografía basal fue similar.
En la evaluación, el porcentaje medio de prebroncodilatador predicho FEV1 fue del 58.5% (SD: 12.8%); el porcentaje medio de reversibilidad fue del 29.9% (SD: 18.1%), con una reversibilidad absoluta media de 0.484 L (SD: 0.274 L), y el puntaje medio de ACQ-6 fue 2.5 (SD: 0.6). Durante el período de 5 semanas de inducción/estabilización, los sujetos tuvieron mejoras sustanciales tanto en la función pulmonar (mejoría en el punto mínimo del FEV1 de 0.287 L) como en el control del asma (la puntuación media de ACQ-6 disminuyó en 0.6). A pesar de estas mejorías, la mayoría de los sujetos (93%) no estaban bien controlados (puntuación media ACQ-6 de 1.9), lo que demuestra la necesidad de una terapia adicional. En la aleatorización, el porcentaje medio de prebroncodilatador predicho FEV1 fue del 68.2% (SD: 14.8%).
Después del período de inducción/estabilización de 5-semanas, los sujetos elegibles recibieron aleatoriamente inhalaciones de una vez al día TRELEGY® de 100/62.5/25 microgramos (n = 406), TRELEGY® de 200/62.5/25 microgramos (n = 408), FF/UMEC/VI de 100/31.25/25 microgramos (n = 405), FF/UMEC/VI de 200/31.25/25 microgramos (n = 404), FF/VI de 100/25 microgramos (n = 407), o FF/VI 200/25 microgramos (n = 406).
Mientras que 4 dosis de TRELEGY® se estudiaron en el ensayo, los resultados de los datos de eficacia mostrados son para TRELEGY® de 100/62.5/25 microgramos y TRELEGY® de 200/62.5/25 microgramos, las dosis recomendadas para el tratamiento de asma. En la evaluación de la eficacia, los análisis de criterio de valoración de la función no pulmonar incluyeron comparaciones agrupadas preespecificadas de TRELEGY® (100/62.5/25 y 200/62.5/25 microgramos) con FF/VI (100/25 y 200/25 microgramos).
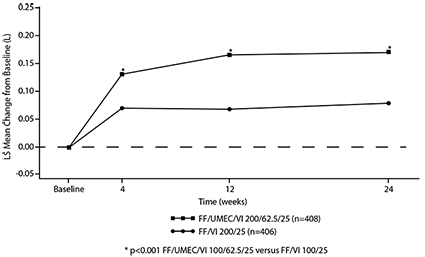
El cambio de la línea basal en el punto mínimo del FEV1 en la semana 24 (criterio de valoración primario de eficacia) mostró mejorías estadísticamente significativas en la función pulmonar para ambos TRELEGY® de 100/62.5/25 microgramos y TRELEGY® de 200/62.5/25 microgramos comparado con FF/VI 100/25 microgramos y FF/VI 200/25 microgramos, respectivamente. (véase Tabla 2, Figuras 1 y 2).
Tabla 2. Criterios de valoración de la función pulmonar (Estudio 205715)
|
FF/VI 100/25 (n = 407) |
TRELEGY® FF/UMEC/VI 100/62.5/25 (n = 406) |
FF/VI 200/25 (n = 406) |
TRELEGY® FF/UMEC/VI 200/62.5/25 (n = 408) |
|
|
Punto mínimo del FEV1 (L) en la semana 24 |
||||
|
Media de LS del cambio a partir del valor inicial (SE) |
0.024 (0.0157) |
0.134 (0.0155) |
0.076 (0.0156) |
0.168 (0.0155) |
|
Diferencia entre el tratamiento FF/UMEC/VI 100/62.5/25 vs. FF/VI 100/25 CI 95% Valor de p |
Referencia |
0.110 0.066, 0.153 p < 0.001 |
--- |
--- |
|
Diferencia entre el tratamiento FF/UMEC/VI 200/62.5/25 vs. FF/VI 200/25 CI 95% Valor de p |
--- |
--- |
Referencia |
0.092 0.049, 0.135 p<0.001 |
|
FF/UMEC/VI 200/62.5/25 100/62.5/25ª Diferencia de tratamiento CI 95% |
--- |
Referencia |
--- |
0.034 -0.009 0.077 |
|
FF/UMEC/VI 100/62.5/25 vs FF/VI 200/25ª Diferencia de tratamiento CI 95% |
--- |
0.059 0.015, 0.102 |
Referencia |
--- |
|
FEV1 a las 3 horas después de la dosisb (L) en la semana 24 |
||||
|
Media de LS del cambio a partir del valor inicial (SE) |
0.132 (0.0160) |
0.243 (0.0158) |
0.168 (0.0159) |
0.286 (0.0158) |
|
Diferencia entre el tratamiento FF/UMEC/VI 100/62.5/25 vs. FF/VI 100/25 CI 95% |
Referencia |
0.111 0.067, 0.155 |
--- |
--- |
|
Diferencia entre el tratamiento FF/UMEC/VI 200/62.5/25 vs. FF/VI 200/25 CI 95% |
--- |
--- |
Referencia |
0.118 0.074, 0.162 |
|
FF/UMEC/VI 200/62.5/25 vs 100/62.5/25 Diferencia de tratamiento CI 95% |
--- |
Referencia |
--- |
0.044 -0.087 |
|
FF/UMEC/VI 100/62.5/25 vs FF/VI 200/25 Diferencia de tratamiento CI 95% |
--- |
0.075 0.031, 0.119 |
Referencia |
--- |
|
FF/UMEC/VI 200/62.5/25 vs FF/VI 100/25 Diferencia de tratamiento CI 95% |
Referencia |
--- |
--- |
0.155 0.110, 0.199 |
CI= intervalo de confianza; FEV1= Volumen Espiratorio Forzado en 1 segundo; L=litros; LS=mínimos cuadrados; n=número en la población con intención de tratar; SE=error estándarl.
a Estas comparaciones no estaban en la jerarquía de pruebas predefinidas y no fueron ajustadas por multiplicidad.
b El criterio de valoración no estaba en la jerarquía de prueba predefinida, por lo tanto, no se ajustó por multiplicidad.
Figura 1. Media de los Mínimos Cuadrados (LS) del cambio a partir del valor inicial en el punto mínimo del FEV1 (L) para TRELEGY® de 100/62.5/25 microgramos
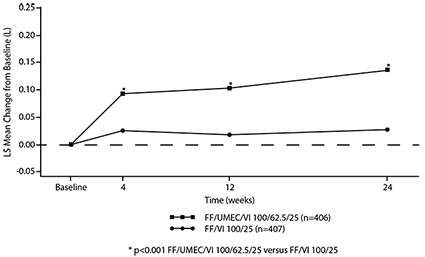
Figura 2. Media de los Mínimos Cuadrados (LS) del cambio a partir del valor inicial en el punto mínimo del FEV1 (L) para TRELEGY® de 200/62.5/25 microgramos
Las exacerbaciones de asma moderada/severa se evaluaron durante el periodo de tratamiento de 52 semanas (ver Tabla 3). En el análisis agrupado, la tasa anualizada de exacerbaciones moderadas/severas fue numéricamente menor con TRELEGY® (100/62.5/25 y 200/62.5/25 microgramos) en comparación con FF/VI (100/25 y 200/25 microgramos) (13 % de reducción en la tasa; CI 95%: -5.2, 28.1). También se proporcionan análisis descriptivos de las comparaciones de tratamiento no agrupadas para la tasa anualizada de exacerbaciones moderadas/severas.
Tabla 3. Tasa anualizada de exacerbaciones moderadas/severasa (hasta 52 semanas) (Estudio 205715)
|
FF/VI 100/25 (n = 407) |
TRELEGY® FF/UMEC/VI 100/62.5/25 (n = 406) |
FF/VI 200/25 (n = 406) |
TRELEGY® FF/UMEC/VI 200/62.5/25 (n = 408) |
|
|
Tasa Media Anualizada |
0.87 |
0.68 |
0.57 |
0.55 |
|
FF/UMEC/VI 100/62.5/25 vs. FF/VI 100/25 Reducción de la Tasa (%) CI 95% |
Referencia |
21.8% 1.1, 39.5 |
--- |
--- |
|
FF/UMEC/VI 200/62.5/25 vs. FF/VI 200/25 Reducción de la Tasa (%) CI 95% |
--- |
--- |
Referencia |
3.2% 28.2, 27.0 |
|
FF/UMEC/VI 200/62.5/25 vs. 100/62.5/25 Reducción en la Tasa (%) CI 95% |
--- |
Referencia |
--- |
19.1% -6.4, 38.5 |
|
FF/UMEC/VI 100/62.5/25 vs. FF/VI 200/25 Cambio en la Tasa (%) CI 95% |
--- |
-19.6%b -57.2, 9.0 |
Referencia |
--- |
|
FF/UMEC/VI 200/62.5/25 vs. FF/VI 100/25 Reducción en la Tasa (%) CI 95% |
Referencia |
--- |
--- |
36.7% 17.6, 51.5 |
CI = Intervalo de confianza; n = Número en la población por intención de tratar.
a Estas comparaciones no estaban en la jerarquía de prueba predefinida y no se ajustaron por multiplicidad.
b El porcentaje negativo refleja un aumento en la tasa de exacerbación para FF/UMEC/VI 100/62.5/25 vs. FF/VI 200/25.
Además, se evaluaron las exacerbaciones severas de asma. En un análisis agrupado descriptivo, no se observó una diferencia en la tasa media anualizada de exacerbaciones severas para TRELEGY® (100/62.5/25 y 200/62.5/25 microgramos) en comparación con FF/VI (100/25 y 200/25 microgramos) (2.6% de reducción en la tasa; CI 95%: -26.2, 24.9). Las tasas anuales medias de exacerbaciones severas fueron 0.41 y 0.23 para TRELEGY® 100/62.5/25 microgramos y TRELEGY® 200/62.5/25 microgramos, respectivamente. Las tasas anuales medias de exacerbaciones severas fueron 0.38 y 0.26 para FF/VI 100/25 microgramos y FF/VI 200/25 microgramos, respectivamente.
Los síntomas del paciente y la calidad de vida relacionada con la salud se evaluaron mediante el ACQ, la Evaluación de los síntomas respiratorios en el asma (E-RS: Asma) y el Cuestionario respiratorio de St. George (SGRQ) (ver Tabla 4). En un análisis descriptivo agrupado la tasa de respuesta ACQ-7 fue del 63% para TRELEGY® (100/62.5/25 y 200/62.5/25 microgramos) en comparación con el 55% para FF/VI (100/25 y 200/25 microgramos) en la semana 24 (OR: 1.43; CI 95%: 1.16, 1.76). También se proporcionan análisis descriptivos de las comparaciones de tratamientos no agrupados.
Tabla 4. Resultados del Cuestionario de control del asma (ACQ) -7a en la semana 24 (Estudio 205715)
|
FF/VI 100/25 (n = 407) |
TRELEGY® FF/UMEC/VI 100/62.5/25 (n = 406) |
FF/VI 200/25 (n = 406) |
TRELEGY® FF/UMEC/VI 200/62.5/25 (n = 408) |
|
|
Sujeto con respuestab (%) |
52% |
62% |
58% |
64% |
|
FF/UMEC/VI 100/62.5/25 vs. FF/VI 100/25 Índice de probabilidad CI 95% |
Referencia |
1.59 1.18, 2.13 |
--- |
--- |
|
FF/UMEC/VI 200/62.5/25 vs. FF/VI 200/25 Índice de probabilidad 95% CI |
--- |
--- |
Referencia |
1.28 0.95, 1.72 |
|
FF/UMEC/VI 200/62.5/25 vs. 100/62.5/25 Índice de probabilidad CI 95% |
--- |
Referencia |
--- |
1.08 0.80, 1.45 |
|
FF/UMEC/VI 100/62.5/25 vs. FF/VI 200/25 Índice de probabilidad CI 95% |
--- |
1.19 0.88, 1.60 |
Referencia |
--- |
|
FF/UMEC/VI 200/62.5/25 vs. FF/VI 100/25 Índice de probabilidad CI 95% |
Referencia |
--- |
--- |
1.71 1.27, 2.30 |
CI= Intervalo de confianza; n= Número en la población con intención de tratar.
a Estas comparaciones no estaban en la jerarquía de pruebas predefinidas y no fueron ajustadas por multiplicidad.
b Definida como una puntuación de ACQ-7 ≥0.5 debajo del valor basal.
Las tasas de respuesta de ACQ-5 (que comprende las 5 preguntas sobre síntomas de ACQ-7) en la semana 24 fueron similares a los resultados de ACQ-7. En un análisis descriptivo agrupado, la tasa de respuesta ACQ-5 fue del 64% para TRELEGY® (100/62.5/25 y 200/62.5/25 microgramos) en comparación con el 60% para FF/VI (100/25 y 200/25 microgramos) (OR: 1.23; CI 95%: 1.00, 1.52) en la semana 24.
En un análisis descriptivo no agrupado, la tasa de respuesta ACQ-5 fue del 63% para TRELEGY® 100/62.5/25 microgramos en comparación con el 58% para FF/VI 100/25 microgramos (OR: 1.28; CI 95%: 0.96, 1.72) en la Semana 24. La tasa de respuesta ACQ-5 fue de 66% para TRELEGY® 200/62.5/25 microgramos en comparación con 62% para FF/VI 200/25 microgramos (OR: 1.19; CI 95%: 0.88, 1.60) en la semana 24.
En un análisis descriptivo agrupado, la tasa de respuesta de E-RS: Asma (definida como una disminución en la puntuación de ≥ 2 desde la línea basal) fue del 45% con TRELEGY® (100/62.5/25 y 200/62.5/25 microgramos) en comparación con 41% para FF/VI (100/25 y 200/25 microgramos) (OR: 1.18; CI 95%: 0.96, 1.45) (Semanas 21-24).
En un análisis descriptivo no agrupado, la tasa de respuesta de E-RS: asma fue del 42% para TRELEGY® 100/62.5/25 microgramos en comparación con el 38% para FF/VI 100/25 microgramos (OR: 1.22; CI 95%: 0.91, 1.63 ) (Semanas 21-24). La tasa de respuesta E-RS: Asma fue del 47% para TRELEGY® 200/62.5/25 microgramos en comparación con el 44% para FF/VI 200/25 microgramos (OR: 1.15; CI 95%: 0.86, 1.53) (Semanas 21-24).
En un análisis descriptivo agrupado, la tasa de respuesta SGRQ (definida como una disminución en el puntaje ≥ 4 desde la línea basal) fue del 69% para TRELEGY® (100/62.5/25 y 200/62.5/25 microgramos) en comparación con el 66% para FF/VI (100/25 y 200/25 microgramos) (OR: 1.14; CI 95%: 0.92, 1.42) en la semana 24.
En un análisis descriptivo no agrupado, la tasa de respuesta SGRQ fue del 68% para TRELEGY® 100/62.5/25 microgramos en comparación con el 64% para FF/VI 100/25 microgramos (OR: 1.26; CI 95%: 0.93, 1.70) en la semana 24. La tasa de respuesta SGRQ fue del 69% para TRELEGY® 200/62.5/25 microgramos en comparación con el 68% para FF/VI 200/25 microgramos (OR: 1.04; CI 95%: 0.76, 1.41) en la semana 24.
EPOC:
Estudio 1: La eficacia de TRELEGY® (100/62.5/25 microgramos de FF/UMEC/VI) administrado como un tratamiento una vez al día en pacientes con un diagnóstico clínico de EPOC se ha evaluado en un estudio de 24 semanas controlado con fármaco activo con una extensión de hasta 52 semanas en un subconjunto de pacientes (estudio CTT116853, FULFIL).
TRELEGY® de 100/62.5/25 microgramos administrado una vez al día demostró una mejoría estadísticamente significativa en la función pulmonar (como lo define el cambio a partir del valor mínimo inicial de VEF1 en la Semana 24; criterio de valoración co-primario) en comparación con 400/12 microgramos de budesonida/formoterol (BUD/FOR) administrados dos veces al día (véase la Tabla 5). Los efectos broncodilatadores con TRELEGY® fueron evidentes en el primer día del tratamiento y se mantuvieron durante el periodo de tratamiento de 24 semanas.
TRELEGY® demostró una mejoría estadísticamente significativa en comparación con BUD/FOR en la Semana 24 para la Calidad de Vida relacionada a la salud (HRQoL) medida por la puntuación total (criterio de valoración co-primario) del Cuestionario Respiratorio St. George (SGRQ), el análisis de sujetos con respuesta de SGRQ, la puntuación de la Prueba de evaluación de EPOC (CAT) y el análisis de los sujetos con respuesta de CAT y también para síntomas respiratorios medidos utilizando la puntuación de Evaluación de Síntomas Respiratorios en EPOC (E-RSTM: EPOC) y las puntuaciones de las subescalas durante las Semanas 21-24, la dificultad respiratoria medida usando la puntuación focal del Índice de Disnea Transicional (TDI) en la Semana 24, y el uso de medicamento de rescate medido por el número medio de ocasiones por día durante las Semanas 1-24 (véase la Tabla 5).
TRELEGY® demostró una reducción estadísticamente significativa en la tasa anual de exacerbaciones moderadas/severas (es decir, que requieren tratamiento con antibióticos o corticosteroides u hospitalización; extrapolado a partir de los datos hasta la Semana 24) en comparación con BUD/FOR. Se observó una reducción en el riesgo de una exacerbación moderada/severa con TRELEGY® en comparación con BUD/FOR (con base en el análisis del tiempo para la primera exacerbación) (véase la Tabla 5).
Tabla 5. Criterios de valoración de eficacia claves hasta la Semana 24 (Estudio CTT116853)
|
TRELEGY® FF/UMEC/VI 100/62.5/25 mcg OD (n = 911) |
BUD/FOR 400/12 mcg BID (n = 899) |
Comparación con BUD/FOR |
||
|
Diferencia de los tratamientos (CI de 95%) valor p |
Relación del tratamiento (CI de 95%) valor p |
|||
|
VEF1 valle (L) en la Semana 24, cambio promedio por LS desde el valor basal (SE)a, e |
0.142 (0.0083) |
-0.029 (0.0085) |
0.171 (0.148, 0.194) p < 0.001 |
- |
|
Puntuación total del SGRQ en la Semana 24, cambio promedio por LS desde el valor basal (SE)a, f |
-6.6 (0.45) |
-4.3 (0.46) |
-2.2 (-3.5, -1.0) p < 0.001 |
- |
|
Sujetos con respuesta de acuerdo a la Puntuación total del SGRQ en la Semana 24, %f, h |
50% |
41% |
- |
1.41b (1.16, 1.70) p < 0.001 |
|
Tasa anual de exacerbación moderada/severa de EPOC en tratamiento (con base en los datos hasta la Semana 24) |
0.22 |
0.34 |
- |
0.65c (0.49, 0.86) p = 0.002 |
|
Incidencia de exacerbación moderada/severa de EPOC hasta la Semana 24, % |
10% |
14% |
- |
0.67d (0.52, 0.88) p = 0.004 |
|
Puntuación total de E-RS: EPOC durante las Semanas 21-24, cambio promedio por LS desde el valor basal (SE)g |
-2.31 (0.157) |
-0.96 (0.160) |
-1.35 (-1.79, -0.91) p < 0.001 |
- |
|
Sujetos con respuesta de acuerdo a la Puntuación total de E-RS: EPOC durante las Semanas 21-24, % g, h |
47% |
37% |
- |
1.59b (1.30, 1.94) p < 0.001 |
|
Puntuación focal de TDI en la Semana 24, media de LS (SE)f |
2.29 (0.096) |
1.72 (0.099) |
0.57 (0.30, 0.84) p < 0.001 |
- |
|
Sujetos con respuesta de acuerdo a la puntuación focal del TDI en la Semana 24, % f, h |
61% |
51% |
- |
1.61b (1.33, 1.95) p < 0.001 |
|
Porcentaje de actividad diaria de días con una puntuación de 2 (capaz de realizar más actividades de lo usual) durante las Semanas 1-24, cambio promedio por LS desde el valor basal (SE) |
0.0 (0.38) |
-0.1 (0.39) |
0.1 (-0.9, 1.1) p = 0.817 |
- |
|
Número promedio de ocasiones de uso de medicamento de rescate por día durante las Semanas 1-24, cambio promedio por LS desde el valor basal (SE) |
-0.1 (0.04) |
0.1 (0.04) |
-0.2 (-0.3, -0.1) p < 0.001 |
- |
|
Puntuación CAT en la Semana 24, cambio promedio por LS desde el valor basal (SE) |
-2.5 (0.18) |
-1.6 (0.19) |
-0.9 (-1.4, -0.4) p < 0.001 |
- |
|
Sujetos con respuesta de acuerdo a la Puntuación CAT en la Semana 24, %h |
53% |
45% |
- |
1.44b (1.19, 1.75) p < 0.001 |
BID = dos veces al día; BUD = budesonida; FOR = formoterol; CI = intervalo de confianza; FEV1= volumen espiratorio forzado en 1 segundo; L = litros; LS = mínimos cuadrados; mcg = microgramos; n = número en la población con intención de tratar; OD = una vez al día; SE = error estándar; SGRQ =Cuestionario Respiratorio de St. George; CAT = Prueba de evaluación de EPOC; E-RS = Evaluación de Síntomas Respiratorios; TDI = Índice de Disnea Transicional.
a Criterios de valoración co-primarios.
b Cociente de probabilidades.
c Relación de incidencia.
d Índice de riesgo con base en el análisis del tiempo para el primer evento.
e Diferencia entre los tratamientos estadísticamente significativa para FF/UMEC/VI vs. BUD/FOR también observada en las Semanas 2, 4 y 12.
f Diferencia entre los tratamientos estadísticamente significativa para FF/UMEC/VI vs. BUD/FOR también observada en la Semana 4.
g Diferencia entre los tratamientos estadísticamente significativa para FF/UMEC/VI vs. BUD/FOR también observada durante cada periodo de 4 semanas a lo largo de la duración del estudio.
h La respuesta se definió como una reducción de ≥ 4 unidades desde el valor inicial para SGRQ, una reducción de ≥ 2 unidades desde la situación inicial para la puntuación total de E-RS y para CAT y una puntuación ≥ 1 unidad para TDI.
La función pulmonar, el HRQoL, los síntomas y los resultados de las exacerbaciones hasta 52 semanas de tratamiento en un subconjunto de pacientes (n = 430) fueron consistentes con los resultados hasta 24 semanas.
Estudio 2: La eficacia a largo plazo de TRELEGY® (100/62.5/25 microgramos de FF/UMEC/VI) administrado una vez al día en pacientes con EPOC y antecedentes de exacerbaciones moderadas o severas dentro de los 12 meses anteriores se ha evaluado en un estudio controlado con activo de 52 semanas en comparación con la combinación de dosis fija de Furoato de Fluticasona/Vilanterol (100/25 microgramos de FF/VI) y Umeclidinio/Vilanterol (62.5/25 microgramos de UMEC/VI) (aleatorización 2:2:1) (estudio CTT116855, IMPACT).
Los pacientes tratados con TRELEGY® demostraron una reducción estadísticamente significativa en la tasa anual de exacerbaciones moderadas/severas durante el tratamiento (criterio de valoración primario) en comparación con FF/VI y en comparación con UMEC/VI. Véase los resultados del criterio de valoración de eficacia en la Tabla 6.
Tabla 6. Criterios de valoración clave de eficacia (Estudio CTT116855)
|
TRELEGY® FF/UMEC/VI (n = 4,151) |
FF/VI (n = 4,134) |
UMEC/VI (n = 2,070) |
TRELEGY® FF/UMEC/VI vs. FF/VI |
TRELEGY® FF/UMEC/VI vs. UMEC/VI |
|
|
Tasa de exacerbaciones moderadas/severasa |
|||||
|
Exacerbaciones al año |
0.91 |
1.07 |
1.21 |
||
|
Reducción en la tasa (%) CI del 95% valor p |
15% 10, 20 p < 0.001 |
25% 19,30 p < 0.001 |
|||
|
Tiempo hasta la primera exacerbación moderada/severa |
|||||
|
Pacientes con un evento (%) |
47% |
49% |
50% |
||
|
Reducción en el riesgo (%) CI del 95% valor p |
14.8% 9.3, 19.9 p < 0.001 |
16.0% 9.4, 22.1 p < 0.001 |
|||
|
Tasa de exacerbaciones severas |
|||||
|
Exacerbaciones al año |
0.13 |
0.15 |
0.19 |
||
|
Reducción en la tasa (%) CI del 95% valor p |
13% -1,24 p = 0.064 |
34% 22,44 p < 0.001 |
|||
|
FEV1 valle (L) en la semana 52 |
|||||
|
Media de LS del cambio a partir del valor inicial (SE) |
0.094 (0.004) |
-0.003 (0.004) |
0.040 (0.006) |
||
|
Diferencia entre tratamientos CI del 95% valor p |
0.097 0.085, 0.109 p < 0.001 |
0.054 0.039, 0.069 p < 0.001 |
|||
|
Puntuación total del SGRQ en la Semana 52 |
|||||
|
Media de LS del cambio a partir del valor inicial (SE) |
-5.5 (0.23) |
-3.7 (0.24) |
-3.7 (0.35) |
||
|
Diferencia entre tratamientos CI del 95% valor p |
-1.8 -2.4, -1.1 p < 0.001 |
-1.8 -2.6, -1.0 p < 0.001 |
|||
|
Sujetos con respuesta de acuerdo con la Puntuación total del SGRQ en la Semana 52 |
|||||
|
Sujetos con respuestab (%) |
42% |
34% |
34% |
||
|
Índice de probabilidad CI del 95% valor p |
1.41 1.29, 1.55 p < 0.001 |
1.41 1.26, 1.57 p < 0.001 |
|||
CI = intervalo de confianza; FEV1 = volumen espiratorio forzado en 1 segundo; L = litros; LS = mínimos cuadrados; n = número en la población con intención de tratar; SE = error estándar; SGRQ = Cuestionario Respiratorio de St. George.
a Criterio de valoración primario.
b Definido como una puntuación total del SGRQ de 4 unidades debajo del valor inicial o inferior.
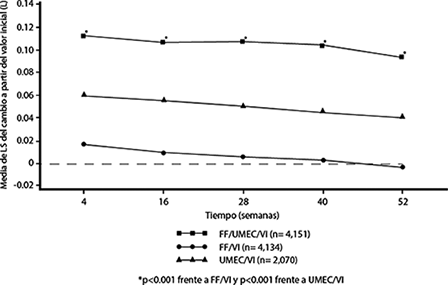
Los efectos de la función pulmonar (cambio desde la línea base de FEV1) de TRELEGY® en comparación con FF/VI y UMEC/VI para el punto mínimo del FEV1 se observaron en todos los momentos de valoración a lo largo del estudio de 52 semanas (véase la Figura 3).
Figura 3. Media de mínimos cuadrados (LS) del cambio a partir de la línea base en el punto mínimo FEV1 (L)
El tratamiento con TRELEGY® redujo significativamente el riesgo de mortalidad por todas las causas, incluyendo los datos con y sin tratamiento, por 27.7% comparado con UMEC/VI (estado vital confirmado en 99.6% de los pacientes en la semana 52) (ver Tabla 7). La reducción del riesgo de mortalidad por todas las causas fue 11.3% con TRELEGY® comparado con FF/VI; sin embargo, este resultado no fue significativo estadísticamente.
Tabla 7. Reducción de mortalidad por todas las causas (Estudio CTT116855)
|
Tratamiento |
N |
Índice de Riesgo vs. Comparador (95% CI) |
Reducción en el Riesgo (95% CI) |
valor p |
|
TRELEGY® FF/UMEC/VI |
4,151 |
|||
|
UMEC/VI |
2,070 |
0.72 (0.53, 0.99) |
27.7% (1.2, 47.1) |
0.042 |
|
FF/VI |
4,134 |
0.89 (0.67, 1.16) |
11.3% (-16.5, 32.5) |
0.387 |
CI= intervalo de confianza.
También fue llevado a cabo el análisis de la mortalidad por todas las causas con el tratamiento, y los resultados fueron consistentes con los resultados anteriores. El tratamiento con TRELEGY® redujo significativamente el riesgo de mortalidad por todas las causas con el tratamiento por 42.1% (95% CI: 11.9, 61.9; p=0.011) comparado con UMEC/VI. La reducción en el riesgo de mortalidad por todas las causas fue 5.5% (95% CI: -40.2, 36.3) con TRELEGY® comparado con FF/VI; sin embargo, este resultado no fue significativo estadísticamente.
La reducción del número medio de ocasiones/día del uso del medicamento de rescate de agonista beta2 y el porcentaje de periodos de 24 horas sin necesidad de medicamento de rescate fue estadísticamente significativo en pacientes que recibieron TRELEGY® en comparación con FF/VI o UMEC/VI en las semanas 49 a 52 (véase la Tabla 8) y estas diferencias se observaron en el trascurso del estudio de 52 semanas.
Los pacientes que recibieron TRELEGY® tuvieron estadísticamente una reducción significativamente mayor en el número de veces que despertaron durante la noche debido a síntomas de EPOC en comparación con FF/VI o UMEC/VI en las semanas 49 a 52 (véase la Tabla 8) y estas diferencias se observaron durante el transcurso del estudio de 52 semanas para UMEC/VI y para la mayoría de los momentos de valoración para FF/VI.
Tabla 8. Otros criterios de valoración (Estudio CTT116855)
|
TRELEGY® FF/UMEC/VI (n = 4,151) |
FF/VI (n = 4,134) |
UMEC/VI (n = 2,070) |
TRELEGY® FF/UMEC/VI vs. FF/VI |
TRELEGY® FF/UMEC/VI vs. UMEC/VI |
|
|
Media del número de ocasiones/día de uso de medicamento de rescate en las semanas 49 a 52 |
|||||
|
Media de LS del cambio a partir el valor inicial (SE) |
0.16 (0.031) |
0.44 (0.032) |
0.46 (0.045) |
||
|
Diferencia entre tratamientos CI del 95% valor p |
-0.28 -0.37, -0.19 p < 0.001 |
-0.30 -0.41, -0.19 p < 0.001 |
|||
|
Porcentaje de periodos de 24 horas sin necesidad de medicamento de rescate en las semanas 49 a 52 |
|||||
|
Media de LS del cambio a partir el valor inicial (SE) |
-1.9 (0.61) |
-7.1 (0.62) |
-6.3 (0.89) |
||
|
Diferencia entre tratamientos CI del 95% valor p |
5.2 3.5, 6.9 p < 0.001 |
4.4 2.3, 6.5 p < 0.001 |
|||
|
Despertares nocturnos debidos a síntomas de EPOC en las semanas 49 a 52 |
|||||
|
Media de LS del cambio a partir el valor inicial (SE) |
-0.21 (0.012) |
-0.16 (0.013) |
-0.12 (0.018) |
||
|
Diferencia entre tratamientos CI del 95% valor p |
-0.05 -0.08, -0.01 p = 0.005 |
-0.10 -0.14, -0.05 p < 0.001 |
|||
CI = intervalo de confianza; LS =mínimos cuadrados; n = número en la población con intención de tratar; SE = error estándar.
El tratamiento con TRELEGY® demostró una mejoría clínicamente significativa de -2.0 puntos en la Prueba de evaluación de EPOC (CAT) a partir del valor inicial en la semana 52. Las diferencias fueron estadísticamente significativas en comparación con FF/VI (-0.5; CI del 95%: -0.8, -0.2; p < 0.001) y con UMEC/VI (-0.4; CI del 95%: -0.8, -0.1; p = 0.021). La tasa de sujetos con respuesta en CAT (definida como 2 unidades por debajo del valor inicial o inferior) en la semana 52 fue estadística y significativamente mayor en los pacientes tratados con TRELEGY® (42%) en comparación con FF/VI (37%; índice de probabilidad 1.24; CI del 95%: 1.14, 1.36; p < 0.001) y con UMEC/VI (36%; índice de probabilidad 1.28; CI del 95%: 1.15, 1.43; p < 0.001).
La dificultad respiratoria, medida usando la puntuación focal del Índice de Disnea Transicional (TDI) en la semana 52, se midió en un subconjunto de pacientes (N = 5,058 de 10 países: Bélgica, Canadá, República Checa, Dinamarca, Alemania, Países Bajos, Polonia, España, Reino Unido, Estados Unidos). El tratamiento con TRELEGY® (n = 2,029) demostró una mejoría estadísticamente significativa en comparación con FF/VI (n = 2,014), media de LS de la puntuación focal del TDI de 0.98 y 0.71, respectivamente, una diferencia de 0.27 (CI del 95%: 0.04, 0.49; p = 0.020). No se observó un efecto estadísticamente significativo entre TRELEGY® y UMEC/VI (n = 1,015), media de LS de la puntuación focal del TDI de 0.98 y 0.89, respectivamente, una diferencia de 0.09 (CI del 95%: -0.19, 0.37; p = 0.522). La proporción de sujetos con respuesta mediante TDI (definida como al menos 1 unidad) fue estadística y significativamente mayor para TRELEGY® (36%) en comparación con FF/VI (29%; índice de probabilidad 1.36; CI del 95%: 1.19, 1.55; p < 0.001) y UMEC/VI (30%; índice de probabilidad 1.33; CI del 95%: 1.13, 1.57; p<0.001) en la semana 52.
Otros estudios que respaldan la eficacia:
El 200812 fue un estudio de 24 semanas de no inferioridad (N = 1,055) que comparó TRELEGY® (FF/UMEC/VI 100/62.5/25 microgramos), administrado como inhalador único, con Furoato de Fluticasona/Vilanterol (100/25 microgramos) + Umeclidinio (62.5 microgramos), co- administrados como terapia con inhaladores múltiples, una vez al día a pacientes con antecedentes de exacerbaciones moderadas o severas en los 12 meses previos. En este estudio, TRELEGY® no fue inferior en comparación con FF/VI + UMEC en cuanto a la mejoría a partir de la línea basal en el punto mínimo del FEV1 en la semana 24. El margen de no inferioridad previamente especificado fue de 50 mL.
Umeclidinio con Furoato de Fluticasona/Vilanterol: En dos estudios de 12 semanas, controlados con placebo (200109 y 200110), la suma de Umeclidinio (62.5 microgramos) a Furoato de Fluticasona/Vilanterol (FF/VI) (100/25 microgramos) una vez al día en pacientes adultos con un diagnóstico clínico de EPOC, resultó en mejorías estadísticamente significativas y clínicamente importantes en el criterio de valoración primario del punto mínimo del VEF1 mínimo en el Día 85 en comparación con el placebo más FF/VI (124 ml [CI de 95%: 93, 154, p < 0.001] en el estudio 200109 y 122 mL [CI de 95%: 91, 152, p < 0.001] en el estudio 200110).
Estudios de 12 meses con Furoato de Fluticasona/Vilanterol: Dos estudios de 52 semanas, aleatorizados, doble ciego, de grupos paralelos (HZC102970 y HZC102871) compararon la tasa anual de exacerbaciones moderadas/severas en pacientes adultos con un diagnóstico clínico de EPOC, tratados con FF/VI o con Vilanterol una vez al día.
Los resultados de un análisis integrado de ambos estudios mostraron que el tratamiento con 100/25 microgramos de FF/VI una vez al día resultó en una reducción de 27% en la tasa anual de exacerbaciones de EPOC moderada/severa en comparación con Vilanterol (CI de 95%: 16, 37, p < 0.001). Las reducciones en el riesgo de exacerbación moderada/severa (con base en el análisis del tiempo para la primera exacerbación) y la tasa de exacerbaciones que requirieron uso de corticosteroides también se observaron con 100/25 microgramos de FF/VI una vez al día en comparación con Vilanterol.
CONTRAINDICACIONES: TRELEGY® está contraindicado en pacientes con alergia severa a las proteínas de la leche o que han demostrado hipersensibilidad al Furoato de Fluticasona, Umeclidinio, Vilanterol o cualquiera de los componentes de la fórmula.
Contraindicado durante síntomas agudos de asma o exacerbaciones agudas de EPOC.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Fertilidad: No existen datos sobre los efectos de TRELEGY® sobre la fertilidad humana. Los estudios en animales no indican ningún efecto sobre la fertilidad masculina o femenina (véase Precauciones generales y Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Embarazo: Existen datos insuficientes derivados del uso de TRELEGY® en mujeres embarazadas. Los estudios en animales han demostrado toxicidad reproductiva después de la administración de agonistas beta2 o corticosteroides (véase Precauciones generales y Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
TRELEGY® debe utilizarse durante el embarazo únicamente si el beneficio esperado para la madre justifica el riesgo potencial para el feto.
Lactancia: Se desconoce si el Furoato de Fluticasona, Umeclidinio, Vilanterol o sus metabolitos se excretan en la leche humana. Sin embargo, otros corticosteroides, antagonistas muscarínicos y agonistas beta2 se detectan en la leche humana. No puede excluirse un riesgo para los recién nacidos en lactancia/lactantes.
Se debe tomar una decisión respecto a suspender la lactancia o suspender la terapia con TRELEGY® considerando el beneficio de la lactancia para el niño y el beneficio de la terapia para la mujer.
REACCIONES SECUNDARIAS Y ADVERSAS:
Datos de estudios clínicos: Los datos de un estudio clínico de asma fase III y tres estudios clínicos de EPOC fase III se utilizaron para determinar la frecuencia de las reacciones adversas asociadas con TRELEGY® (ver tabla 1). En el programa de desarrollo clínico de asma, un total de 1,623 sujetos adultos fueron evaluados en reacciones adversas. En el programa de desarrollo clínico de EPOC, un total de 5,589 sujetos adultos fueron incluidos en una evaluación integrada de reacciones adversas.
Se informa la frecuencia más alta, cuando las frecuencias de reacción adversa difirieron entre estudios y poblaciones.
Las reacciones adversas se enlistan por clase de sistema orgánico MedDRA y frecuencia (véase la Tabla 5). Se ha utilizado la siguiente convención para la clasificación de reacciones adversas:
Muy común: ≥ 1/10.
Común: ≥ 1/100 a < 1/10.
Poco común: ≥ 1/1000 a < 1/100.
Rara: ≥ 1/10000 a < 1/1000.
Muy rara: < 1/10000.
Tabla 1. Reacciones adversas.
|
Clase de sistema y órganos |
Reacción(es) adversa(s) |
Frecuencia |
|
Infecciones e infestaciones |
Nasofaringitis |
Muy común |
|
Neumonía Infección de vías respiratorias superiores Bronquitis Faringitis Rinitis Sinusitis Influenza Candidiasis de boca y garganta Infección de las vías urinarias Infección viral de las vías respiratorias |
Común |
|
|
Trastorno del sistema nervioso |
Cefalea |
Común |
|
Disgeusia |
Poco común |
|
|
Trastornos cardiacos |
Taquiarritmia supraventricular Taquicardia Fibrilación auricular |
Poco común |
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos Dolor bucofaríngeo |
Común |
|
Disfonía |
Común |
|
|
Trastornos gastrointestinales |
Estreñimiento |
Común |
|
Boca seca |
Poco común |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Artralgia Dolor de espalda |
Común |
|
Fracturas |
Poco común |
Descripción de las reacciones adversas seleccionadas:
Neumonía (véase Precauciones generales):
EPOC: En un total de 1,810 pacientes con EPOC avanzada (media de la selección de VEF1 posterior a broncodilatador de 45% de lo pronosticado en la visita de evaluación, desviación estándar [SD] 13%), 65% de los cuales tuvieron una experiencia de exacerbación moderada/severa de EPOC en el año previo al ingreso al estudio (estudio CTT116853), se notificó una mayor incidencia de eventos de neumonía en pacientes que recibieron TRELEGY® (20 pacientes, 2%) que en los pacientes que recibieron Budesonida/Formoterol (7 pacientes, < 1%). Neumonías que requirieron hospitalización se presentaron en 1% de los pacientes que recibieron TRELEGY® y < 1% de los pacientes que recibieron budesonida/formoterol hasta 24 semanas. Se notificó un caso letal de neumonía en un paciente que recibió TRELEGY®. En el subconjunto de 430 pacientes tratados por hasta 52 semanas, la incidencia de eventos de neumonía reportados tanto en el grupo de TRELEGY® y budesonida/formoterol fue igual a 2%.
En un estudio de 52 semanas, un total de 10,355 pacientes con EPOC y antecedentes de 1 o más exacerbaciones moderadas o severas dentro de los 12 meses anteriores (media del FEV1 post-broncodilatador del 46% predictivo, SD 15%) (estudio CTT116855), la incidencia de neumonía fue de 8% para TRELEGY® (n = 4,151), 7% para Furoato de Fluticasona/Vilanterol (n = 4,134), y 5% para Umeclidinio/Vilanterol (n = 2,070). Se presentó neumonía fatal en 12 de 4,151 pacientes (3.5 por 1,000 años-paciente) que recibieron TRELEGY®, 5 de 4,134 pacientes (1.7 por 1,000 años-paciente) que recibieron Furoato de Fluticasona/Vilanterol y 5 de 2,070 pacientes (2.9 por 1,000 años-paciente) que recibieron Umeclidinio/Vilanterol.
La incidencia de eventos de neumonía con TRELEGY® es comparable con aquella observada con Fluticasona Furoato/Vilanterol 100/25 microgramos en estudios clínicos en EPOC.
Asma: En pacientes con asma (estudio 205715) tratados hasta por 52 semanas, la incidencia de neumonía fue del 1% (5 de 406 pacientes) para TRELEGY® 100/62.5/25 microgramos y del < 1% (4 de 408 pacientes) para TRELEGY® 200/62.5/25 microgramos. La incidencia de neumonía fue del 2% en los grupos de Furoato de Fluticasona/Vilanterol 100/25 microgramos (7 de 407 pacientes) y Furoato de Fluticasona/Vilanterol 200/25 microgramos (7 de 406 pacientes). La incidencia de eventos de neumonía que requieren hospitalización fue similar en los grupos TRELEGY® y Furoato de Fluticasona/Vilanterol (< 1% para todos los grupos). No hubo eventos de neumonía fatal.
Datos posteriores a la comercialización:
|
Clase de sistema orgánico |
Reacción(es) adversa(s) |
Frecuencia |
|
Trastornos del sistema inmunológico |
Reacciones de hipersensibilidad, que incluyen anafilaxia, angioedema, urticaria y erupción cutánea |
Raro |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Los efectos farmacológicos y toxicológicos observados con Furoato de Fluticasona, Umeclidinio o Vilanterol en los estudios no clínicos fueron aquellos típicamente asociados con los glucocorticoides, los antagonistas de los receptores muscarínicos o los agonistas de los receptores beta2 adrenérgicos. La administración de la combinación de Furoato de Fluticasona, Umeclidinio y Vilanterol a perros no resultó en ninguna toxicidad significativa nueva o ninguna exacerbación mayor de los hallazgos esperados asociados con el Furoato de Fluticasona, Umeclidinio o Vilanterol solos.
Carcinogénesis/mutagénesis: El Furoato de Fluticasona no fue genotóxico en un conjunto de estudios estándar y no fue carcinogénico en los estudios de inhalación de por vida en ratas o ratones con exposiciones AUC 0.6 o 1.3 veces, respectivamente, mayores a aquellas en los humanos que recibieron 200 microgramos de Furoato de Fluticasona.
El Umeclidinio no fue genotóxico en un conjunto de estudios estándar y no fue carcinogénico en estudios de inhalación de por vida en ratones o ratas a exposiciones ≥ 20 o ≥ 17 veces la exposición clínica humana a 62.5 microgramos de Umeclidinio, con base en el AUC, respectivamente.
Los estudios de toxicidad genética indican que el Vilanterol no representa un riesgo genotóxico para los humanos. Consistente con los hallazgos para otros agonistas beta2, en los estudios de inhalación de por vida, el Vilanterol causó efectos proliferativos en las ratas hembra y el tracto reproductivo de los ratones y de la glándula pituitaria en ratas. No hubo ningún incremento en la incidencia tumoral en ratas o ratones con exposiciones 0.9 o 22 veces, respectivamente, la exposición clínica humana de Vilanterol a 25 microgramos con base en el AUC.
Toxicología reproductiva: Ni el Furoato de Fluticasona ni el Umeclidinio ni el Vilanterol tuvieron un efecto adverso sobre la fertilidad masculina o femenina en ratas.
El Furoato de Fluticasona no fue teratogénico en ratas o conejos, pero retrasó el desarrollo en las ratas y causó aborto en conejos con dosis inhaladas tóxicas maternas. No hubo ningún efecto sobre el desarrollo en ratas a exposiciones 3.0 veces la exposición clínica humana a 200 microgramos, con base en el AUC. El Furoato de Fluticasona no tuvo ningún efecto adverso sobre el desarrollo pre o postnatal en ratas.
El Umeclidinio no fue teratogénico en ratas o conejos. En un estudio pre y postnatal, la administración subcutánea de Umeclidinio a ratas resultó en una menor ganancia de peso corporal materno y consumo de alimentos y redujo ligeramente los pesos corporales de los cachorros antes del destete en las madres que recibieron la dosis de 180 microgramos/kg/día (alrededor de 61 veces la exposición clínica humana a 62.5 microgramos de Umeclidinio, con base en el AUC).
El Vilanterol no fue teratogénico en ratas. En estudios de inhalación en conejos, el Vilanterol causó efectos similares a los observados con otros agonistas beta2 (paladar hendido, párpados abiertos, fusión esternebral y mal rotación/flexión de extremidades). Cuando se administró subcutáneamente no hubo efectos con exposiciones de 62 veces la exposición clínica humana a 25 microgramos, con base en el AUC. El Vilanterol no tuvo ningún efecto adverso sobre el desarrollo pre o postnatal en ratas.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: Las interacciones medicamentosas clínicamente significativas mediadas por Furoato de Fluticasona, Umeclidinio o Vilanterol en dosis clínicas se consideran poco probables debido a las bajas concentraciones plasmáticas alcanzadas después de una administración inhalada.
Interacción con beta bloqueadores: Los bloqueadores beta adrenérgicos pueden debilitar o antagonizar el efecto de los agonistas beta2-adrenérgicos como el Vilanterol. En caso de que se requieran beta bloqueadores, se deben considerar los beta bloqueadores cardioselectivos; no obstante, se debe tener precaución durante el uso simultáneo de beta bloqueadores no selectivos y selectivos.
Interacción con inhibidores de CYP3A4: El Furoato de Fluticasona y Vilanterol, ambos componentes de TRELEGY®, se eliminan rápidamente por metabolismo de primer paso extensivo mediado por la enzima CYP3A4.
Se recomienda tener precaución al coadministrarse con fuertes inhibidores de CYP3A4 (ej. Ketoconazol, Ritonavir) ya que existe el potencial de una exposición sistémica elevada tanto a Furoato de Fluticasona como a Vilanterol, lo que podría conducir a un incremento en el potencial de reacciones adversas (véase Farmacocinética).
Otros antimuscarínicos de larga acción y agonistas beta2-adrenérgicos de larga acción: La coadministración de TRELEGY® con otros antagonistas muscarínicos de larga acción o agonistas beta2 adrenérgicos de larga acción no se ha estudiado y no se recomienda ya que puede potenciar las reacciones adversas (véase Reacciones secundarias y adversas, así como Manifestaciones y manejo de la sobredosificación o ingesta accidental).
Inhibidores de la monoaminooxidasa y antidepresivos tricíclicos: El Vilanterol, al igual que otros agonistas beta2, debe administrarse con extrema precaución a los pacientes tratados con inhibidores de la monoaminooxidasa, antidepresivos tricíclicos o fármacos que se sabe que prolongan el intervalo QTc o dentro de las 2 semanas de la discontinuación de dichos agentes, debido a que el efecto de los agonistas adrenérgicos en el sistema cardiovascular puede ser potenciado por estos agentes. Los fármacos que prolongan el intervalo QTc tienen un mayor riesgo de arritmias ventriculares.
Agentes bloqueadores de los receptores beta-adrenérgicos: Los beta bloqueantes no solo bloquean el efecto pulmonar de los beta-agonistas, como el Vilanterol, sino que también pueden producir broncoespasmo severo en pacientes con EPOC o asma. Por tanto, los pacientes con EPOC o asma normalmente no deben ser tratados con Beta-bloqueadores. Sin embargo, en determinadas circunstancias, puede que no existan alternativas aceptables al uso de agentes bloqueadores beta-adrenérgicos para estos pacientes; se podrían considerar los Beta-bloqueadores cardioselectivos, aunque deben administrarse con precaución.
Diuréticos no ahorradores de potasio: Los cambios electrocardiográficos y/o la hipocalemia que pueden resultar de la administración de diuréticos no ahorradores de potasio (como diuréticos de asa o tiazida) pueden empeorar de forma aguda por los agonistas beta, especialmente cuando se excede la dosis recomendada del agonista beta. Aunque se desconoce la importancia clínica de estos efectos, se recomienda precaución en la administración concomitante de agonistas beta con diuréticos no ahorradores de potasio.
Anticolinérgicos: Existe la posibilidad de una interacción aditiva con los medicamentos anticolinérgicos utilizados de forma concomitante. Por lo tanto, evite la coadministración de TRELEGY® con otros fármacos que contengan anticolinérgicos, ya que esto puede provocar un aumento de los efectos adversos anticolinérgicos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No se cuenta con información.
PRECAUCIONES GENERALES:
Exacerbaciones: TRELEGY® no debe utilizarse para tratar síntomas agudos de asma o una exacerbación aguda en EPOC para lo cual se requiere un broncodilatador de acción corta.
Incrementar el uso de broncodilatadores de acción corta para aliviar los síntomas indica un deterioro en el control y un médico debe evaluar a los pacientes.
Los pacientes con asma o EPOC no deben suspender la terapia con TRELEGY®, sin la supervisión de un médico ya que los síntomas pueden recurrir después de la interrupción. Los eventos adversos relacionados con asma y las exacerbaciones pueden ocurrir durante el tratamiento con TRELEGY®. A los pacientes debe pedírseles continuar con el tratamiento, pero consultar a un médico si los síntomas de asma permanecen sin control o empeoran después del inicio con TRELEGY®.
Eventos relacionados - Asma severa - Hospitalizaciones, Intubaciones, Muerte: El uso de agonistas de los receptores beta2 de larga acción (LABA) como monoterapia [sin corticoesteroides inhalados (ICS)] para asma está asociado con un incremento en el riesgo de muerte relacionada al asma. Los datos disponibles de estudios clínicos controlados también sugieren que el uso de LABA como monoterapia incrementa el riesgo de hospitalización relacionada con asma en pacientes pediátricos y adolescentes. Estos efectos son considerados un efecto de clase de la monoterapia de LABA. Cuando LABA es usado en una combinación de dosis fija con ICS, datos de estudios clínicos prolongados no muestran un incremento significativo en el riesgo de eventos serios relacionados con asma (hospitalizaciones, intubaciones, muerte) en comparación con ICS solo.
Eventos graves relacionados con el asma con corticosteroides inhalados/agonistas adrenérgicos beta2 de acción prolongada: Se llevaron a cabo cuatro (4) ensayos de seguridad clínica grandes, aleatorios, doble ciego, controlados con activos, de 26 semanas de duración, para evaluar el riesgo de eventos graves relacionados con el asma cuando se utilizaron LABA en combinación de dosis fija con ICS en sujetos con asma. Tres (3) ensayos incluyeron sujetos adultos y adolescentes de 12 años o más: un ensayo comparó budesonida/formoterol con budesonida, un ensayo comparó propionato de Fluticasona/salmeterol en polvo para inhalación con propionato de Fluticasona en polvo para inhalación, y un ensayo comparó Furoato de mometasona/formoterol con Furoato de mometasona. El cuarto ensayo incluyó sujetos pediátricos de 4 a 11 años y comparó el polvo para inhalación de propionato de Fluticasona/salmeterol con el polvo para inhalación de propionato de Fluticasona. El criterio principal de valoración de seguridad para los 4 ensayos fue eventos graves relacionados con el asma (hospitalizaciones, intubaciones, muerte). Un comité de adjudicación ciego determinó si los eventos estaban relacionados con el asma.
Los 3 ensayos de adultos y adolescentes se diseñaron para descartar un margen de riesgo de 2,0, y el ensayo pediátrico se diseñó para descartar un margen de riesgo de 2,7. Cada ensayo individual cumplió con su objetivo preestablecido y no hay inferioridad demostrada de ICS/LABA a ICS solo. Un metanálisis de los 3 ensayos de adultos y adolescentes no mostró un aumento significativo en el riesgo de un evento grave relacionado con el asma con la combinación de dosis fija de ICS/LABA en comparación con ICS solo (Tabla 1). Estos ensayos no fueron diseñados para descartar todos los riesgos de eventos graves relacionados con el asma con CSI/LABA en comparación con CSI.
Tabla 1. Meta-análisis de eventos graves relacionados con el asma en sujetos con asma de 12 años o más
|
ICS/LABA (n = 17,537)ª |
ICS (n = 17,552)ª |
ICS/LABA vs. ICS Cociente de riesgo (95% CI)b |
|
|
Evento grave relacionado con el asmac |
116 |
105 |
1.10 (0.85, 1.44) |
|
Muerte relacionada con el asma |
2 |
0 |
|
|
Intubación relacionada con el asma (endotraqueal) |
1 |
2 |
|
|
Hospitalización relacionada con el asma (estancia ≥ 24 horas) |
115 |
105 |
CI = Corticosteroide inhalado, LABA = Agonista beta2-adrenérgico de acción prolongada.
a Sujetos aleatorizados que habían tomado al menos 1 dosis del fármaco del estudio. Tratamiento planificado utilizado para el análisis.
b Estimado utilizando un modelo de riesgos proporcionales de Cox para el tiempo hasta el primer evento con los riesgos de referencia estratificados por cada uno de los 3 ensayos.
b Número de sujetos con un evento que ocurrió dentro de los 6 meses posteriores al primer uso del fármaco del estudio o 7 días después de la última fecha del fármaco del estudio, lo que sea posterior. Los sujetos pueden tener 1 o más eventos, pero solo se contó el primer evento para el análisis. Un único comité de adjudicación independiente, ciego, determinó si los eventos estaban relacionados con el asma.
El ensayo de seguridad pediátrica incluyó a 6.208 sujetos pediátricos de 4 a 11 años que recibieron ICS/LABA (polvo para inhalación de propionato de Fluticasona/salmeterol) o ICS (polvo para inhalación de propionato de Fluticasona).
En este ensayo, 27/3,107 (0,9%) sujetos asignados al azar a ICS / LABA y 21/3,101 (0,7%) sujetos asignados al azar a ICS experimentaron un evento grave relacionado con el asma. No hubo muertes ni intubaciones relacionadas con el asma. Los ICS/LABA no mostraron un riesgo significativamente mayor de un evento grave relacionado con el asma en comparación con los ICS según el margen de riesgo preespecificado (2,7), con una razón de riesgo estimada de tiempo hasta el primer evento de 1,29 (del 95%: 0,73, 2,27 ).
TRELEGY® no está indicado para su uso en pacientes pediátricos de 17 años o menos.
Ensayo multicéntrico de investigación sobre el asma con salmeterol (SMART): Un ensayo estadounidense de 28 semanas controlado con placebo que comparó la seguridad del salmeterol con el placebo, cada uno agregado a la terapia habitual para el asma, mostró un aumento en las muertes relacionadas con el asma en sujetos que recibieron salmeterol (13/13,176 en sujetos tratados con salmeterol versus 3/13,179 en sujetos tratados con placebo; riesgo relativo: 4,37 [CI del 95%: 1,25, 15,34]). El uso de ICS de fondo no era necesario en SMART. El mayor riesgo de muerte relacionada con el asma se considera un efecto de clase de la monoterapia con LABA.
Deterioro de la enfermedad y episodios agudos: TRELEGY® no debe iniciarse en pacientes durante episodios de EPOC o asma con rápido deterioro o potencialmente mortales. TRELEGY® no se ha estudiado en sujetos con EPOC o asma con deterioro agudo. El inicio de TRELEGY® en este caso no es apropiado.
Existen marcadores de deterioro de la enfermedad como son: a) Si TRELEGY® 100/62.5/25 mcg ya no controla los síntomas de broncoconstricción. B) Si el agonista Beta2 de acción corta inhalado del paciente se vuelve menos eficaz; c) Si el paciente necesita más agonistas beta2 de acción corta de lo habitual. En este contexto, reevalúe al paciente y al régimen de tratamiento de EPOC de una vez. Para EPOC, no se debe aumentar la dosis diaria de TRELEGY® 100/62.5/25 mcg.
El uso cada vez mayor de agonistas beta2 de acción corta inhalados es un marcador de deterioro del asma. En esta situación, el paciente requiere una reevaluación inmediata con una reevaluación del régimen de tratamiento, prestando especial atención a la necesidad de opciones terapéuticas adicionales. Los pacientes no deben usar más de 1 inhalación una vez al día de TRELEGY®.
TRELEGY® no debe utilizarse para el alivio de síntomas agudos, es decir, como terapia de rescate para el tratamiento de episodios agudos de broncoespasmo. TRELEGY® no se ha estudiado para el alivio de los síntomas agudos y no se deben usar dosis adicionales para ese propósito. Los síntomas agudos se deben tratar con un agonista beta2 inhalado de acción corta.
Al comenzar el tratamiento con TRELEGY®, los pacientes que hayan estado recibiendo agonistas beta2 de acción corta orales o inhalados de forma regular (p. ej., 4 veces al día) deben recibir instrucciones de interrumpir el uso regular de estos fármacos y usarlos únicamente para el alivio sintomático de los síntomas respiratorios agudos.
Al prescribir TRELEGY®, el médico también debe prescribir un agonista beta2 de acción corta inhalado e instruir al paciente sobre cómo debe usarse.
Uso excesivo de TRELEGY® y su uso con otros agonistas beta2 de acción prolongada: TRELEGY® no debe usarse con más frecuencia de la recomendada, ni en dosis superiores a las recomendadas, o en combinación con otros medicamentos que contengan LABA, ya que puede producirse una sobredosis. Se han informado efectos cardiovasculares clínicamente significativos y muertes en asociación con el uso excesivo de fármacos simpaticomiméticos inhalados. Los pacientes que usan TRELEGY® no deben usar otro medicamento que contenga un LABA (p. e., salmeterol, fumarato de formoterol, Tartrato de arformoterol, indacaterol) por ningún motivo.
Candidiasis orofaríngea: TRELEGY® contiene Furoato de Fluticasona, un corticosteroide inhalado (ICS). Se han producido infecciones localizadas de la boca y la faringe con Candida albicans en sujetos tratados con medicamentos inhalados oralmente que contienen Furoato de Fluticasona. Cuando se desarrolla una infección de este tipo, debe tratarse con terapia antifúngica local o sistémica (es decir, oral) apropiada mientras continúa el tratamiento con TRELEGY® pero a veces puede ser necesario interrumpir el tratamiento con TRELEGY®. Aconseje al paciente que se enjuague la boca con agua sin tragar después de la inhalación para ayudar a reducir el riesgo de candidiasis orofaríngea.
Neumonía: Los médicos deben permanecer atentos al posible desarrollo de neumonía en pacientes con EPOC ya que las características clínicas de la neumonía y las exacerbaciones se superponen con frecuencia. Se han reportado infecciones del tracto respiratorio inferior, incluida neumonía, después de la administración inhalada de corticosteroides.
En dos ensayos de 12 semanas de pacientes con EPOC (N = 824), la incidencia de neumonía fue inferior al 1% para ambos grupos de tratamiento: Umeclidinio 62.5 mcg + Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg o placebo + Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg. Se produjo neumonía fatal en 1 sujeto que recibió placebo + Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg.
En un estudio de 52 semanas en sujetos con EPOC (N= 10,355), la incidencia de neumonía fue 8% para TRELEGY® 100/62.5/25 mcg (n=4,151), 7% para Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg (n=4,134), y 5% para Umeclidinio/Vilanterol 62.5 mcg/25 mcg (n=2,070). Neumonía fatal ocurrió en 12 de 4,151 pacientes (0.35 por cada 100 pacientes/año) recibiendo TRELEGY® 100/62.5/25 mcg; 5 de 4,134 pacientes (0.17 por cada 100 pacientes/año) recibiendo Fluticasona Furoato/Vilanterol 100/62.5/25 mcg; y 5 de 2,070 pacientes (0.29 por cada 100 pacientes/año) recibiendo Umeclidinio/Vilanterol.
En un ensayo de mortalidad con Furoato de Fluticasona/Vilanterol 100/25 mcg con una duración mediana del tratamiento de 1.5 años en 16,568 sujetos con EPOC moderada y enfermedad cardiovascular, la tasa de incidencia anualizada de neumonía fue de 3.4 por cada 100 años-pacientes para el Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg, 3.2 para placebo, 3.3 para Furoato de Fluticasona 100 mcg y 2.3 para Vilanterol 25 mcg. Las muertes adjudicadas durante el tratamiento debido a neumonía se produjeron en 13 sujetos que recibieron Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg, 9 sujetos que recibieron placebo, 10 sujetos que recibieron Furoato de Fluticasona 100 mcg y 6 sujetos que recibieron Vilanterol 25 mcg (menos de 0.2 por cada 100 años-pacientes para cada grupo de tratamiento).
Inmunosupresión y Riesgo de Infecciones: La varicela y el sarampión pueden tener un curso más grave o incluso fatal en niños o adultos susceptibles que usan corticosteroides. En estos niños o adultos que no han tenido estas enfermedades o no han sido debidamente inmunizados, se debe tener especial cuidado para evitar la exposición. Se desconoce cómo la dosis, la vía y la duración de la administración de corticosteroides afectan el riesgo de desarrollar una infección diseminada. Tampoco se conoce la contribución de la enfermedad subyacente y/o el tratamiento previo con corticosteroides al riesgo. Si un paciente está expuesto a la varicela, puede estar indicada la profilaxis con inmunoglobulina de varicela zóster (VZIG). Si un paciente está expuesto al sarampión, puede estar indicada la profilaxis con inmunoglobulina intramuscular (IG) combinada. (Consulte los prospectos respectivos para obtener información completa de prescripción de VZIG e IG). Si se desarrolla varicela, se puede considerar el tratamiento con agentes antivirales.
Los ICS deben usarse con precaución, en su caso, en pacientes con infecciones del tracto respiratorio por tuberculosis, activas o quiescentes; infecciones sistémicas por hongos, bacterias, virus o parásitos; o herpes simple ocular.
Transferencia de pacientes de la terapia con corticosteroides sistémicos: Se necesita un cuidado especial para los pacientes que han sido transferidos de corticosteroides sistémicamente activos a ICS debido a que se han producido muertes por insuficiencia suprarrenal en pacientes con asma durante y después de la transferencia de corticosteroides sistémicos a ICS menos disponibles sistémicamente. Después de la retirada de los corticosteroides sistémicos, se requieren varios meses para la recuperación de la función hipotalámica-hipofisaria-suprarrenal (HPA).
Los pacientes que se han mantenido previamente con la dosis de 20 mg o más de prednisona (o su equivalente) pueden ser los más susceptibles, especialmente cuando sus corticosteroides sistémicos se han retirado casi por completo. Durante este período de supresión de HPA, los pacientes pueden exhibir signos y síntomas de insuficiencia suprarrenal cuando se exponen a traumatismos, cirugía o infección (en particular, gastroenteritis) u otras afecciones asociadas con la pérdida severa de electrólitos. Aunque TRELEGY® puede controlar los síntomas de la EPOC o asma durante estos episodios, en las dosis recomendadas suministra menos de las cantidades fisiológicas normales de glucocorticoides sistémicamente y NO proporciona la actividad mineralocorticoide que es necesaria para hacer frente a estas emergencias.
Durante los períodos de estrés o una exacerbación grave de la EPOC o ataque severo de asma, los pacientes a quienes se les han retirado corticosteroides sistémicos deben recibir instrucciones de reanudar los corticosteroides orales (en dosis grandes) inmediatamente y ponerse en contacto con sus médicos para recibir más instrucciones. A estos pacientes también se les debe indicar que lleven una tarjeta de advertencia que indique que pueden necesitar corticosteroides sistémicos suplementarios durante períodos de estrés o una exacerbación grave de la EPOC o ataque severo de asma.
Los pacientes que requieren corticosteroides orales deben interrumpir paulatinamente el uso de corticosteroides sistémicos después de cambiarlos al tratamiento con TRELEGY®. La reducción de la prednisona se puede lograr reduciendo la dosis diaria de prednisona en 2.5 mg semanalmente durante el tratamiento con TRELEGY®. La función pulmonar (volumen espiratorio forzado en 1 segundo [FEV1]), el uso de agonistas beta y los síntomas de la EPOC o asma deben controlarse cuidadosamente durante la retirada de los corticosteroides orales. Además, los pacientes deben ser observados para detectar signos y síntomas de insuficiencia suprarrenal, como fatiga, cansancio, debilidad, náuseas y vómitos e hipotensión.
Desenmascaramiento de afecciones alérgicas previamente suprimidas por corticosteroides sistémicos: La transferencia de pacientes de la terapia sistémica con corticosteroides a TRELEGY® puede desenmascarar afecciones alérgicas previamente suprimidas por la terapia con corticosteroides sistémicos (p. ej., rinitis, conjuntivitis, eccema, artritis, afecciones eosinofílicas).
Síntomas de abstinencia de corticosteroides: Durante la retirada de los corticosteroides orales, algunos pacientes pueden experimentar síntomas de abstinencia de corticosteroides sistémicamente activos (p. ej., dolor articular y/o muscular, lasitud, depresión) a pesar del mantenimiento o incluso de la mejora de la función respiratoria.
Hipercortisolismo y supresión suprarrenal: El Furoato de Fluticasona inhalado se absorbe en la circulación y puede ser sistémicamente activo. Los efectos del Furoato de Fluticasona en el eje HPA no se observan con las dosis terapéuticas de Furoato de Fluticasona en TRELEGY®. Sin embargo, exceder la dosis recomendada o la administración concomitante con un inhibidor fuerte del citocromo P450 3A4 (CYP3A4) puede resultar en disfunción HPA (consulte Precauciones generales e Interacciones medicamentosas y de otro género).
Debido a la posibilidad de absorción sistémica significativa de ICS en pacientes sensibles, los pacientes tratados con TRELEGY® deben observarse cuidadosamente para detectar cualquier evidencia de efectos sistémicos de corticosteroides. Se debe tener especial cuidado al observar a los pacientes en el contexto postoperatorio o durante los períodos de estrés para detectar una respuesta suprarrenal inadecuada.
Es posible que los efectos sistémicos de los corticosteroides, como el hipercortisolismo y la supresión suprarrenal (incluida la crisis suprarrenal), puedan aparecer en un pequeño número de pacientes que son sensibles a estos efectos. Si se producen tales efectos, reduzca la dosis de TRELEGY® lentamente, de acuerdo con los procedimientos aceptados para reducir los corticosteroides sistémicos, y considere otros tratamientos para el manejo de la EPOC o los síntomas del asma.
Interacciones farmacológicas con inhibidores potentes del citocromo P450 3A4: Se debe tener precaución al considerar la administración concomitante de TRELEGY® con Ketoconazol y otros inhibidores potentes conocidos del CYP3A4 (incluyendo, pero no limitado a ritonavir, claritromicina, conivaptán, indinavir, itraconazol, lopinavir, nefazodona, nelfinavir, saquinavir, telitromicina, troleandomicina, voriconazol) porque puede haber un aumento de efectos adversos de corticosteroides sistémicos y efectos adversos cardiovasculares (véase Interacciones medicamentosas y de otro género).
Broncoespasmo paradójico: Al igual que con otros medicamentos inhalados, TRELEGY® puede producir broncoespasmo paradójico, que puede ser potencialmente mortal. Si se produce broncoespasmo paradójico después de la administración de TRELEGY® , debe tratarse inmediatamente con un broncodilatador inhalado de acción corta; TRELEGY® debe discontinuarse inmediatamente; y se debe instituir una terapia alternativa.
Reacciones de hipersensibilidad incluyendo anafilaxia: Las reacciones de hipersensibilidad como anafilaxia, angioedema, exantema y urticaria pueden ocurrir después de la administración de TRELEGY® . Suspenda TRELEGY® si ocurren tales reacciones. Ha habido informes de reacciones anafilácticas en pacientes con alergia severa a la proteína láctea después de la inhalación de otros medicamentos en polvo que contienen lactosa; por lo tanto, los pacientes con alergia severa a la proteína de la leche no deben usar TRELEGY® (véase Contraindicaciones).
Efectos cardiovasculares: El Vilanterol, como otros agonistas beta2, puede producir un efecto cardiovascular clínicamente significativo en algunos pacientes, medido por aumentos en la frecuencia cardíaca, presión arterial sistólica o diastólica, y también arritmias cardíacas, como taquicardia supraventricular y extrasístoles. Si se producen tales efectos, puede ser necesario interrumpir TRELEGY®. Además, se ha informado que los agonistas beta producen cambios electrocardiográficos, como aplanamiento de la onda T, prolongación del intervalo QTc y depresión del segmento ST, aunque se desconoce el significado clínico de estos hallazgos (véase Farmacocinética y farmacodinamia).
Se han reportado muertes en asociación con el uso excesivo de fármacos simpaticomiméticos inhalados.
TRELEGY®, como otras aminas simpaticomiméticas, debe usarse con precaución en pacientes con trastornos cardiovasculares, especialmente insuficiencia coronaria, arritmias cardíacas e hipertensión.
En un estudio de 52 semanas en sujetos con EPOC, las tasas ajustadas a la exposición para cualquier evento cardíaco adverso mayor durante el tratamiento, incluidas hemorragias del sistema nervioso central y afecciones cerebrovasculares no mortales, infarto de miocardio (IM) no mortal, IM agudo no mortal, y la muerte en tratamiento debida a eventos cardiovasculares fue de 2.2 por cada 100 pacientes/año para TRELEGY® (n = 4,151), 1.9 por cada 100 pacientes/año para Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg (n = 4,134 ), y 2.2 por cada 100 pacientes/año para umeclidinio/Vilanterol 62.5 mcg/ 25 mcg (n = 2,070). La muerte en tratamiento debida a eventos cardiovasculares ocurrió en 20 de 4,151 pacientes (0.54 por cada 100 pacientes/año) que recibieron TRELEGY®, 27 de 4,134 pacientes (0.78 por cada 100 pacientes/año) que recibieron Furoato de Fluticasona/Vilanterol, y 16 de 2,070 pacientes (0.94 por cada 100 pacientes/año) que recibieron umeclidinium/Vilanterol.
En un ensayo de mortalidad con Furoato de Fluticasona/ Vilanterol con una duración mediana del tratamiento de 1.5 años en 16,568 sujetos con EPOC moderada y enfermedad cardiovascular, la tasa de incidencia anualizada de eventos cardiovasculares adjudicados (compuesto de infarto de miocardio, accidente cerebrovascular, angina inestable, ataque isquémico transitorio, o muerte durante el tratamiento debido a eventos cardiovasculares) fue de 2.5 por cada 100 años-pacientes para Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg, 2.7 para placebo, 2.4 para Furoato de Fluticasona 100 mcg y 2.6 para Vilanterol 25 mcg. Las muertes adjudicadas durante el tratamiento debido a eventos cardiovasculares ocurrieron en 82 sujetos que recibieron Furoato de Fluticasona/Vilanterol 100 mcg/25 mcg, 86 sujetos que recibieron placebo, 80 sujetos que recibieron Furoato de Fluticasona 100 mcg y 90 sujetos que recibieron Vilanterol 25 mcg (la tasa de incidencia anualizada varió de 1.2 a 1.3 por cada 100 años-pacientes para los grupos de tratamiento).
Reducción de la densidad mineral ósea: Se han observado disminuciones en la Densidad Mineral Ósea (DMO) con la administración a largo plazo de productos que contienen ICS. Se desconoce la importancia clínica de los pequeños cambios en la (DMO) con respecto a las consecuencias a largo plazo, como la fractura. Los pacientes con factores de riesgo importantes de disminución del contenido mineral óseo, como inmovilización prolongada, antecedentes familiares de osteoporosis, estado posmenopáusico, consumo de tabaco, edad avanzada, mala nutrición o uso crónico de fármacos que pueden reducir la masa ósea (p. ej., anticonvulsivos, corticosteroides orales) deben ser monitoreados y tratados con estándares de cuidado establecidos. Dado que los pacientes con EPOC a menudo tienen múltiples factores de riesgo para una (DMO) reducida, se recomienda evaluar la (DMO) antes de iniciar TRELEGY® y periódicamente a partir de entonces. Si se observan reducciones significativas en la (DMO) y TRELEGY® aún se considera médicamente importante para el tratamiento de la EPOC de ese paciente, se debe considerar seriamente el uso de medicamentos para tratar o prevenir la osteoporosis.
Glaucoma y cataratas, empeoramiento del glaucoma de ángulo estrecho y coriorretinopatía serosa central: Se ha reportado glaucoma, aumento de la presión intraocular y cataratas en pacientes con EPOC o asma después de la administración a largo plazo de ICS o con el uso de anticolinérgicos inhalados. Otro efecto sistémico posible es la coriorretinopatía serosa central. TRELEGY® debe usarse con precaución en pacientes con glaucoma de ángulo estrecho. Los médicos y los pacientes también deben estar alerta para detectar los signos y síntomas del glaucoma agudo de ángulo estrecho (p. ej., dolor o malestar ocular, visión borrosa, halos visuales o imágenes coloreadas en asociación con ojos rojos por congestión conjuntival y edema corneal). Indique a los pacientes que consulten a su médico de inmediato si se desarrolla alguno de estos signos o síntomas. Considere referir con un oftalmólogo a los pacientes que desarrollan síntomas oculares o que usan TRELEGY® a largo plazo.
Empeoramiento de la retención urinaria: TRELEGY®, como todos los medicamentos que contienen un anticolinérgico, debe usarse con precaución en pacientes con retención urinaria. Los médicos y los pacientes deben estar alertas para detectar signos y síntomas de retención urinaria (p. ej., dificultad para orinar, dolor al orinar), especialmente en pacientes con hiperplasia prostática u obstrucción del cuello de la vejiga. Indique a los pacientes que consulten a un proveedor de atención médica de inmediato si se desarrolla alguno de estos signos o síntomas.
Condiciones coexistentes: TRELEGY®, como todos los medicamentos que contienen aminas simpaticomiméticas, debe usarse con precaución en pacientes con trastornos convulsivos o tirotoxicosis y en aquellos que son excepcionalmente sensibles a las aminas simpaticomiméticas. Se ha informado que las dosis del agonista albuterol del adrenoreceptor beta2 relacionado, cuando se administran por vía intravenosa, agravan la diabetes mellitus y la cetoacidosis, preexistentes.
Hipocalemia e hiperglucemia: Los agonistas adrenérgicos beta pueden producir hipocalemia significativa en algunos pacientes, posiblemente a través de la derivación intracelular, que tiene el potencial de producir efectos cardiovasculares adversos. La disminución del potasio sérico suele ser transitoria y no requiere suplementos. Los medicamentos beta-agonistas pueden producir hiperglucemia transitoria en algunos pacientes.
Efecto sobre el crecimiento: Los corticosteroides inhalados por vía oral pueden reducir la velocidad de crecimiento cuando se administran a niños y adolescentes.
Efectos sobre la capacidad de conducir y utilizar maquinaria: No existen estudios para investigar el efecto de TRELEGY® sobre la capacidad de realizar tareas que requieran juicio, o capacidades motrices o cognitivas.
No se anticiparía un efecto perjudicial sobre dichas actividades a partir de la farmacología de Furoato de Fluticasona, Umeclidinio o Vilanterol en dosis clínicas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Vía de administración: Bucal.
Consideración de uso: Para inhalación.
TRELEGY® debe administrarse una vez al diariamente, ya sea en la mañana o por la tarde, pero siempre a la misma hora cada día.
Después de la inhalación, el paciente debe enjuagar su boca con agua sin tragarla.
Poblaciones:
ASMA: Los pacientes deberán estar conscientes que TRELEGY® debe utilizarse con regularidad, aún cuando no presentan síntomas.
Si aparecen los síntomas en el período entre la dosis, debe inhalarse un agonista-beta2 de acción corta para el alivio inmediato.
Los pacientes deben ser reevaluados periódicamente por un médico para que la concentración que reciben de TRELEGY®, permanezca siendo la óptima y solo sea cambiada por recomendación médica.
Adultos: La dosis recomendada de TRELEGY® es:
La dosis recomendada y máxima es una inhalación de TRELEGY® de 100/62.5/25 microgramos una vez al día o una inhalación de TRELEGY® de 200/62.5/25 microgramos una vez al día.
Una dosis inicial de TRELEGY® de 100/62.5/25 microgramos debe considerarse para los pacientes que requieren una dosis baja a mediana del corticosteroide inhalado en combinación con un antagonista de los receptores muscarínicos de acción larga y un agonista de los beta2 de acción larga.
TRELEGY® de 200/62.5/25 microgramos debe considerarse para pacientes que requieren una dosis más alta de corticosteroides inhalados en combinación con un antagonista de los receptores muscarínicos de acción larga y agonista de los beta2 de acción larga.
Si los pacientes son controlados de manera inadecuada con TRELEGY® de 100/62.5/25 microgramos, considerar incrementar la dosis a 200/62.5/25 microgramos, lo que puede proporcionar una mejoría adicional en el control del asma.
Niños y adolescentes: La seguridad y eficacia de TRELEGY® no se ha establecido en niños y adolescentes menores de 18 años de edad.
EPOC:
Adultos: La dosis recomendada y máxima es una inhalación de TRELEGY® de 100/62.5/25 microgramos una vez al día, a la misma hora todos los días.
Niños y adolescentes: El uso en pacientes menores de 18 años de edad no es relevante, para la indicación de EPOC en este producto.
ASMA y EPOC:
Pacientes de edad avanzada: No se requiere ningún ajuste de la dosis en los pacientes mayores de 65 años (véase Farmacocinética-Poblaciones especiales de pacientes).
Insuficiencia renal: No se requiere ningún ajuste de la dosis para los pacientes con insuficiencia renal (véase Farmacocinética-Poblaciones especiales de pacientes
Insuficiencia hepática: Debe tenerse precaución al administrar la dosis a pacientes con insuficiencia hepática quienes pueden estar en mayor riesgo de reacciones adversas sistémicas asociadas con corticosteroides. Para los pacientes con insuficiencia hepática moderada o severa la dosis máxima es 100/62.5/25 microgramos (véase Farmacocinética – Poblaciones especiales en pacientes).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No se cuenta con datos disponibles derivados de los estudios clínicos relativos a sobredosis de TRELEGY®.
Síntomas y signos: Una sobredosis de TRELEGY® puede producir signos, síntomas o efectos adversos asociados con las acciones farmacológicas de los componentes individuales (véase Precauciones generales, así como Farmacodinamia).
Tratamiento: No existe un tratamiento específico para una sobredosis con TRELEGY®. En caso de que se presente sobredosis, el paciente debe recibir tratamiento de soporte con el monitoreo apropiado según sea necesario.
El bloqueo beta cardio selectivo debe considerarse únicamente para los efectos de sobredosis profunda con Vilanterol que sean clínicamente preocupantes y no respondan a medidas de soporte. Los fármacos beta bloqueadores cardio selectivos deben utilizarse con precaución en pacientes con una historia de broncoespasmo.
Se debe dar tratamiento adicional cuando esté clínicamente indicado, o conforme a lo recomendado por el centro nacional de intoxicaciones cuando esté disponible.
PRESENTACIONES:
Caja con dispositivo inhalador con 30 dosis de 100/62.5/25 mcg e instructivo anexo.
Caja con dispositivo inhalador con 30 dosis de 200/62.5/25 mcg e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: 100/62.5/25 MCG: Consérvese a no más de 30°C.
Después de retirar el dispositivo inhalador de la bandeja, el producto se conserva por un periodo máximo de 6 semanas cuando se almacena a no más de 30°C. 200/62.5/25 mcg: Consérvese a no más de 25°C.
Después de retirar el dispositivo inhalador de la bandeja, el producto se conserva por un periodo máximo de 6 semanas cuando se almacena a no más de 25°C.
Consérvese el dispositivo inhalador bien cerrado.
Contiene un desecante NO INGERIBLE, consérvese dentro del envase.
Si se mantiene en refrigeración, permitir que el inhalador regrese a la temperatura ambiente por lo menos una hora antes de usarlo.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en menores de 18 años. El uso de este medicamento durante el embarazo y la lactancia queda bajo la responsabilidad del médico. Este medicamento contiene lactosa monohidratada, que puede producir reacciones de hipersensibilidad.
Reporte las sospechas de reacción adversa a los correos:
farmacovigilancia@cofepris.gob.mx y
farmacovigilancia.mx@gsk.com
Titular:
Glaxo Wellcome, S.A.
Avda. Extremadura, 3, Pol. Ind. Allendeduero,
Aranda de Duero, 09400 Burgos, España.
Representante Legal:
GLAXOSMITHKLINE MÉXICO, S.A. de C.V.
Autopista México-Querétaro Km. 41.5 Edif. TR9
Interior 5-C, Ex. Hacienda San Miguel,
C.P. 54715, Cuautitlán Izcalli, México, México.
Reg. Núm. 374M2017, SSA IV
Clave IPP: GDS09/IPI10 04-marzo-2020
ACTUALIZACIÓN: 04-marzo-2022
®Marca Registrada