TREMFYA - Solución inyectable
Sustancia(s):
- Guselkumab
Presentaciones:
- 1 Caja, 1 Jeringa(s) prellenada(s), 100 mg/ml
- 1 Caja, 1 Pluma precargada, 1 mL, 100 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
La jeringa prellenada contiene:
Guselkumab 100 mg
Vehículo cbp 1 mL
La pluma precargada contiene:
Guselkumab 100 mg
Vehículo cbp 1 mL
Guselkumab, un bloqueador de la interleucina 23, es un anticuerpo monoclonal de inmunoglobulina humana G1 lambda (IgG1?). Guselkumab se produce en una línea celular de mamíferos, mediante la tecnología de ADN recombinante.
TREMFYA® (guselkumab) Inyectable es una solución estéril, libre de conservadores, transparente, incolora a amarillo claro, que puede contener pequeñas partículas translúcidas. Cada jeringa prellenada de dosis única para uso subcutáneo contiene 100 mg de guselkumab en 1 mL. TREMFYA® se presenta como una solución de dosis única en una jeringa de vidrio de 1 mL con una aguja fija de calibre 27G, de media pulgada, montada en un sistema de dosificación con protector pasivo de la aguja o una pluma precargada controlado por el paciente para uso subcutáneo.
Cada jeringa prellenada o pluma precargada de TREMFYA® suministra 1 mL de solución que contiene guselkumab (100 mg), L-histidina (0.6 mg), monoclorhidrato de L-histidina monohidratada (1.5 mg), polisorbato 80 (0.5 mg), sacarosa (79 mg) y agua para inyección a pH 5.8.
INDICACIONES TERAPÉUTICAS:
Psoriasis en placa: TREMFYA® está indicado para el tratamiento de adultos con psoriasis en placa moderada a severa que son candidatos para terapia sistémica o fototerapia.
Artritis psoriásica: TREMFYA® está indicado para el tratamiento de pacientes adultos con artritis psoriásica activa.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacocinética: Guselkumab presentó una farmacocinética lineal en sujetos sanos y sujetos con psoriasis en placa después de su aplicación en inyecciones subcutáneas. En los sujetos con psoriasis en placa, después de la administración subcutánea de 100 mg de TREMFYA® en las semanas 0 y 4, y luego cada 8 semanas, la concentración sérica media de guselkumab en estado estacionario fue de aproximadamente 1.2 mcg/mL.
La farmacocinética de guselkumab en sujetos con artritis psoriásica fue similar a la de sujetos con psoriasis en placa. Después de la administración subcutánea de 100 mg de TREMFYA® en las semanas 0 y 4, y luego cada 8 semanas, la concentración sérica media de guselkumab en estado estacionario fue de aproximadamente 1.2 mcg/mL
Absorción: Después de una inyección subcutánea única de 100 mg en sujetos sanos, guselkumab alcanzó una concentración sérica máxima media (± DE) de 8.09 ± 3.68 μg/mL, por aproximadamente 5.5 días después de la dosis. La biodisponibilidad absoluta de guselkumab después de una inyección subcutánea única de 100 mg, se estimó en aproximadamente 49% en sujetos sanos.
Distribución: En sujetos con psoriasis en placa, el volumen aparente de distribución fue de 13.5 L.
Eliminación: La depuración aparente en sujetos con psoriasis en placa fue de 0.516 L/día. La vida media de guselkumab fue de aproximadamente 15 a 18 días en sujetos con psoriasis en placa en todos los estudios.
Metabolismo: No se ha caracterizado la ruta exacta a través de la cual se metaboliza guselkumab. Como anticuerpo monoclonal IgG humano, se espera que guselkumab se degrade en péptidos pequeños y aminoácidos, mediante rutas catabólicas, de la misma manera que las IgG endógenas.
Poblaciones específicas: No se observaron diferencias aparentes en la depuración en sujetos ≥ 65 años, en comparación con sujetos < 65 años, lo que sugiere que no es necesario ajustar la dosis en sujetos de edad avanzada. La depuración y el volumen de distribución de guselkumab, aumentan a medida que aumenta el peso corporal; sin embargo, los datos de estudios clínicos observados indican que no se justifica ajustar la dosis por el peso corporal. No se han realizado estudios específicos para determinar el efecto de la insuficiencia renal o hepática sobre la farmacocinética de guselkumab.
Interacciones farmacológicas: Los análisis de farmacocinética poblacional indicaron que el uso concomitante de AINE, corticosteroides orales y FARME convencionales como metotrexato, no afectó la depuración de guselkumab.
Sustratos del citocromo P450: Se evaluaron los efectos de guselkumab sobre la farmacocinética de midazolam (metabolizado por CYP3A4), warfarina (metabolizada por CYP2C9), omeprazol (metabolizado por CYP2C19), dextrometorfano (metabolizado por CYP2D6) y cafeína (metabolizada por CYP1A2), en un estudio exploratorio con 6 a 12 sujetos evaluables con psoriasis en placa moderada a severa. Los cambios en el ABCinf de midazolam, S-warfarina, omeprazol y cafeína después de una dosis única de guselkumab, no fueron clínicamente relevantes. Para dextrometorfano, los cambios en el ABCinf después de la administración de guselkumab no fueron clínicamente relevantes en 9 de 10 sujetos; sin embargo, se observó un cambio de 2.9 veces en el ABCinf en un sujeto (ver Interacciones medicamentosas y de otro género).
Farmacodinamia: En los sujetos con psoriasis en placa evaluados, guselkumab redujo los niveles séricos de IL-17A, IL-17F e IL-22 en relación con los niveles previos al tratamiento con base en los análisis exploratorios de marcadores farmacodinámicos.
En los sujetos con artritis psoriásica evaluados, los niveles séricos de proteínas de fase aguda proteína C reactiva, amiloide sérico A e IL-6, y citocinas Th17 efectoras, IL-17A, IL-17F e IL-22, se encontraron elevadas en la basal. Los niveles séricos de estas proteínas medidos en la semana 4 y la semana 24 disminuyeron en comparación con los valores iniciales después del tratamiento con guselkumab en la semana 0, la semana 4 y cada 8 semanas a partir de entonces.
No se conoce la relación entre estos marcadores farmacodinámicos y los mecanismos por los cuales guselkumab ejerce sus efectos clínicos.
Mecanismo de acción:
Guselkumab es un anticuerpo IgG1? monoclonal humano que se une selectivamente a la subunidad p19 de la interleucina 23 (IL-23) e inhibe su interacción con el receptor IL-23. IL-23 es una citocina presente en el organismo de manera natural, que está implicada en respuestas inflamatorias e inmunes normales. Guselkumab inhibe la liberación de citocinas y de quimiocinas proinflamatorias.
Estudios clínicos:
Psoriasis en placa: Cuatro estudios multicéntricos, aleatorizados, doble ciego (PsO1 [NCT02207231], PsO2 [NCT02207244], PsO3 [NCT02203032] y PsO4 [NCT02905331) reclutaron sujetos de 18 años y mayores con psoriasis en placa moderada a severa, que eran elegibles para terapia sistémica o fototerapia. Los sujetos tenían una puntuación en la Evaluación Global del Médico (IGA, por sus siglas en inglés) ≥ 3 (“moderada”) en una escala de cinco puntos de severidad; una puntuación ≥ 12 en el Índice de Severidad y Actividad de Psoriasis (PASI) y una Superficie Corporal Afectada (BSA, por sus siglas en inglés) mínima de 10%. Se excluyeron los sujetos con psoriasis guttata, eritrodérmica o pustular.
Estudios PsO1 y PsO2: En PsO1 y PsO2, se aleatorizaron 1443 sujetos a TREMFYA® (100 mg a las semanas 0 y 4, y luego cada 8 semanas), placebo o adalimumab aprobado en EUA (80 mg a la semana 0 y 40 mg a la semana 1, seguidos de 40 mg cada dos semanas).
Ambos estudios evaluaron las respuestas la semana 16, en comparación con el placebo, para los dos criterios de valoración coprimarios:
• La proporción de sujetos que alcanzaron una puntuación IGA de 0 (“aclarada”) o 1 (“mínima”);
• La proporción de sujetos que alcanzaron por lo menos una reducción de 90% en la puntuación PASI compuesta basal (PASI 90).
Las comparaciones entre TREMFYA® y adalimumab aprobado en EUA, fueron criterios de valoración secundarios a los siguientes tiempos:
• A la semana 16 (PsO1 y PsO2), la proporción de sujetos que alcanzaron una puntuación IGA de 0 o 1, una respuesta PASI 90 y una respuesta PASI 75;
• A la semana 24 (PsO1 y PsO2) y a la semana 48 (PsO1), la proporción de sujetos que alcanzaron una puntuación IGA de 0, una puntuación IGA de 0 o 1 y una respuesta PASI 90.
Otros resultados incluyeron la mejoría en los síntomas de la psoriasis evaluados en el Diario de Síntomas y Signos de Psoriasis (PSSD) y mejorías en la psoriasis de piel cabelluda a la semana 16.
En ambos estudios, los sujetos eran predominantemente hombres y blancos, con una edad media de 44 años y un peso promedio de 90 kg. En la basal, los sujetos tenían una mediana de BSA de aproximadamente 21%, una puntuación mediana PASI de 19, y 18% tenían una historia de artritis psoriásica. Aproximadamente 24% de los sujetos tenían una puntuación severa en el IGA. En ambos estudios, 23% de los sujetos habían recibido tratamiento sistémico biológico previo.
Respuesta clínica:
La Tabla 1 presenta los resultados de eficacia a la semana 16 en PsO1 y PsO2.
Tabla 1. Resultados de eficacia a la semana 16 en adultos con psoriasis en placa (NRIª)
|
Criterio de valoración |
PsO1 |
PsO2 |
||
|
TREMFYA® (N = 329) n (%) |
Placebo (N = 174) n (%) |
TREMFYA® (N = 496) n (%) |
Placebo (N = 248) n (%) |
|
|
Respuesta IGA de 0/1b,c |
280 (85) |
12 (7) |
417 (84) |
21 (8) |
|
Respuesta PASI 90b |
241 (73) |
5 (3) |
347 (70) |
6 (2) |
a NRI = Imputación para no Respondedores.
b Criterios de valoración co-primarios.
c Respuesta IGA de 0 (aclarada) o 1 (mínima).
La Tabla 2 presenta los resultados de un análisis de todos los sitios de Norteamérica (EUA y Canadá), demostrando la superioridad de TREMFYA® sobre adalimumab aprobado en EUA.
Tabla 2. Resultados de eficacia en adultos con psoriasis en placa (NRIª)
|
Criterio de valoración |
PsO1 |
PsO 2 |
||
|
TREMFYA® (N = 115)b n (%) |
Adalimumabc (N = 115)b n (%) |
TREMFYA® (N = 160)b n (%) |
Adalimumabc (N = 81)b n (%) |
|
|
Respuesta IGA de 0/1 (aclarada o mínima) |
||||
|
Semana 16 |
97 (84) |
70 (61) |
119 (74) |
50 (62) |
|
Semana 24 |
97 (84) |
62 (54) |
119 (74) |
46 (57) |
|
Semana 48 |
91 (79) |
62 (54) |
NA |
NA |
|
Respuesta IGA de 0/1 (aclarada) |
||||
|
Semana 24 |
61 (53) |
27 (23) |
76 (48) |
23 (28) |
|
Semana 48 |
54 (47) |
28 (24) |
NA |
NA |
|
Respuesta PASI 75 |
||||
|
Semana 16 |
105 (91) |
80 (70) |
132 (83) |
51 (63) |
|
Respuesta PASI 90 |
||||
|
Semana 16 |
84 (73) |
47 (41) |
102 (64) |
34 (42) |
|
Semana 24 |
92 (80) |
51 (44) |
113 (71) |
41 (51) |
|
Semana 48 |
84 (73) |
53 (46) |
NA |
NA |
a NRI = Imputación para no Respondedores.
b Sujetos de sitios de Estados Unidos y de Canadá.
c Adalimumab aprobado en EUA
Se observó una mejoría en la psoriasis, que involucraba la piel cabelluda en sujetos aleatorizados a TREMFYA®, en comparación con placebo a la semana 16.
El análisis del efecto con la edad, el sexo, la raza, el peso corporal y el tratamiento previo con agentes sistémicos o biológicos, no identificó diferencias en la respuesta a TREMFYA® entre estos subgrupos.
Mantenimiento y durabilidad de la respuesta: Para evaluar el mantenimiento y la durabilidad de la respuesta (PsO2), los sujetos aleatorizados a TREMFYA® en la semana 0 y que obtuvieron respuesta PASI 90 en la semana 28, fueron re aleatorizados para continuar el tratamiento con TREMFYA® cada 8 semanas o fueron retirados de la terapia (es decir, recibieron placebo).
En la semana 48, el 89% de los sujetos que continuaron con TREMFYA® mantuvieron la respuesta PASI 90 en comparación con el 37% de los sujetos que fueron retirados de TREMFYA® y re aleatorizados a placebo. Para los sujetos respondedores en la semana 28 que fueron re aleatorizados a placebo y retirados de TREMFYA®, la mediana de tiempo para perder la respuesta PASI 90 fue de aproximadamente 15 semanas.
Se observó mejoría en los síntomas de psoriasis (comezón, dolor, escozor, ardor y rigidez de la piel) en la semana 16 con TREMFYA® comparados con placebo, en ambos estudios clínicos con base en el PSSD. Una mayor proporción de sujetos con TREMFYA® comparados con adalimumab aprobado en EUA alcanzaron un valor en PSSD de cero (sin síntomas) en la semana 24 en ambos estudios.
Estudio PsO3:
PsO3 [NCT02203032] evaluó la eficacia de 24 semanas de tratamiento con TREMFYA® en sujetos (N = 268) que no habían alcanzado una respuesta adecuada, definida como IGA ≥ 2 a la semana 16 después del tratamiento inicial con ustekinumab aprobado en EUA (dosis de 45 mg o 90 mg según el peso basal del sujeto, a la semana 0 y la semana 4). Estos sujetos fueron aleatorizados a continuar con el tratamiento con ustekinumab aprobado en EUA cada 12 semanas o cambiar a TREMFYA® 100 mg a las semanas 16, 20 y luego cada 8 semanas. Las características basales de los sujetos aleatorizados fueron similares a las observadas en PsO1 y PsO2.
En los sujetos con una respuesta inadecuada (IGA ≥ 2 a la semana 16 con ustekinumab aprobado en EUA), una mayor proporción de sujetos en TREMFYA® comparado con ustekinumab aprobado en EUA alcanzó una puntuación IGA de 0 o 1 con una mejoría ≥ 2 grados a la semana 28 (31% contra 14%, respectivamente, 12 semanas después de la aleatorización).
Estudio PsO4:
PsO4 [NCT02905331] evaluó la eficacia, seguridad y farmacocinética de TREMFYA®. En este estudio, se aleatorizaron 78 sujetos para recibir TREMFYA® (100 mg en las semanas 0 y 4 y cada 8 semanas a partir de entonces) [N = 62] o placebo [N = 16]. Las características basales de los sujetos fueron comparables a las observadas en PsO1 y PsO2. Los criterios de valoración coprimarios fueron los mismos que para PsO1 y PsO2. Los puntos finales secundarios incluyeron la proporción de sujetos que alcanzaron una puntuación IGA de 0 en la semana 16 y la proporción de sujetos que lograron una respuesta PASI 100 en la semana 16.
Una mayor proporción de sujetos en el grupo de guselkumab logró una puntuación IGA de 0 o 1 o una respuesta PASI 90 en la semana 16 (81% y 76%, respectivamente) que en el grupo placebo (0% para ambos puntos finales). La proporción de sujetos que alcanzaron una puntuación IGA de 0 en la semana 16 fue mayor en el grupo de guselkumab en comparación con el grupo de placebo (56% frente a 0%). La proporción de sujetos que alcanzaron una respuesta PASI 100 en la semana 16 fue mayor en el grupo de guselkumab en comparación con el grupo de placebo (50% frente a 0%).
Artritis psoriásica (PsA):
La seguridad y eficacia de TREMFYA® se evaluaron en 1120 pacientes en 2 estudios aleatorizados, doble ciego, controlados con placebo (PsA1 [NCT03162796] y PsA2 [NCT03158285]) en pacientes adultos con artritis psoriásica activa (≥ 3 articulaciones inflamadas, ≥ 3 articulaciones dolorosas y un nivel de proteína C-reactiva (PCR) de ≥ 0.3 mg/dL en PsA1 y ≥ 5 articulaciones inflamadas, ≥ 5 articulaciones dolorosas y un nivel de PCR de ≥ 0.6 mg/dL en PsA2) que tuvieron una respuesta inadecuada a las terapias estándar (por ejemplo, FARME [fármaco antirreumático modificador de la enfermedad] convencionales [cFAMRE]), apremilast o antiinflamatorios no esteroideos (AINE). Los pacientes en estos estudios tenían un diagnóstico de PsA durante al menos 6 meses según los Criterios de Clasificación para la Artritis Psoriásica (CASPAR, por sus siglas en inglés) y una mediana de duración de PsA de 4 años en la basal.
En PsA1, aproximadamente el 31% de los sujetos habían sido tratados previamente con hasta 2 agentes anti-factor de necrosis tumoral alfa (anti-TNFα), mientras que en PsA2 todos los sujetos eran naïve al tratamiento biológico. Aproximadamente el 58% de los sujetos de ambos estudios tenían uso concomitante de metotrexato (MTX). En ambos estudios se incluyeron pacientes con diferentes tipos de PsA, incluyendo artritis poliarticular con ausencia de nódulos reumatoides (40%), espondilitis con artritis periférica (30%), artritis periférica asimétrica (23%), compromiso interfalángico distal (7%) y artritis mutilante (1%). En la basal, más del 65% y del 42% de los pacientes tenían entesitis y dactilitis, respectivamente, y el 79% tenían ≥ 3% de compromiso de la piel con psoriasis en el área de la superficie corporal (BSA).
PsA1 evaluó a 381 sujetos que fueron tratados con placebo SC, TREMFYA® 100 mg SC en las semanas 0, 4 y cada 8 semanas (q8w) a partir de entonces, o TREMFYA 100 mg SC cada 4 semanas (q4w). PsA2 evaluó a 739 sujetos que fueron tratados con placebo SC, TREMFYA® 100 mg SC en las semanas 0, 4 y q8w a partir de entonces, o TREMFYA 100 mg SC q4w. El criterio de valoración primario en ambos estudios fue el porcentaje de pacientes que lograron una respuesta ACR20 en la semana 24.
Respuesta clínica:
En ambos estudios, los pacientes tratados con TREMFYA® 100 mg q8w demostraron una mayor respuesta clínica que incluía ACR20, en comparación con el placebo en la semana 24 (Tablas 3 y 4). Se observaron respuestas similares independientemente de la exposición previa a anti-TNFα en PsA1, y en ambos estudios se observaron respuestas similares independientemente del uso concomitante de cFARME, el tratamiento previo con cFARME el género, y el peso corporal.
Tabla 3. Porcentaje de sujetos con respuestas ACR en PsA1)
|
Placebo (N = 126) |
TREMFYA® 100 mg q8w (N = 127) |
||
|
Tasa de respuesta |
Tasa de respuesta |
Diferencia con el placebo (IC 95%) |
|
|
Respuesta ACR 20ª |
|||
|
Semana 16 |
25% |
52% |
27 (15, 38) |
|
Semana 24 |
22% |
52% |
30 (19, 41) |
|
Respuesta ACR 50ª |
|||
|
Semana 16 |
13% |
23% |
10 (1, 19) |
|
Semana 24 |
9% |
30% |
21 (12, 31) |
|
Respuesta ACR 70ª |
|||
|
Semana 16 |
6% |
8% |
2 (-4, 8) |
|
Semana 24 |
6% |
12% |
6 (-0.3, 13) |
a Los sujetos con datos faltantes en una visita fueron imputados como no respondedores en esa visita. A los sujetos que cumplieron los criterios de escape (menos del 5% de mejoría en los recuentos de articulaciones sensibles e inflamadas) en la semana 16 se les permitió iniciar o aumentar la dosis de la medicación concomitante permitida y permanecieron en el grupo aleatorizado. Los sujetos que iniciaron o aumentaron la dosis de FARME no biológicos o corticosteroides orales sobre el valor inicial, interrumpieron el estudio/medicación del estudio o iniciaron medicamentos/terapias prohibidas por el protocolo para PsA antes de una visita se consideraron no respondedores en esa visita.
Tabla 4. Porcentaje de pacientes con respuestas ACR en PsA2
|
Placebo (N = 246) |
TREMFYA® 100 mg q8w (N = 248) |
||
|
Tasa de respuesta |
Tasa de respuesta |
Diferencia con el placebo (IC 95%) |
|
|
Respuesta ACR 20ª |
|||
|
Semana 16 |
34% |
55% |
22 (13, 30) |
|
Semana 24 |
33% |
64% |
31 (23,40) |
|
Respuesta ACR 50ª |
|||
|
Semana 16 |
9% |
29% |
19 (13, 26) |
|
Semana 24 |
14% |
32% |
17 (10, 24) |
|
Respuesta ACR 70ª |
|||
|
Semana 16 |
1% |
14% |
13 (9, 17) |
|
Semana 24 |
4% |
19% |
15 (9, 20) |
a Los sujetos con datos faltantes en una visita fueron imputados como no respondedores en esa visita. A los sujetos que cumplieron los criterios de escape (menos del 5% de mejoría en los recuentos de articulaciones sensibles e inflamadas) en la semana 16 se les permitió iniciar o aumentar la dosis de la medicación concomitante permitida y permanecieron en el grupo aleatorizado. Los sujetos que iniciaron o aumentaron la dosis de FARME no biológicos o corticosteroides orales sobre el valor inicial, interrumpieron el estudio/medicación del estudio o iniciaron medicamentos/terapias prohibidas por el protocolo para PsA antes de una visita se consideraron no respondedores en esa visita.
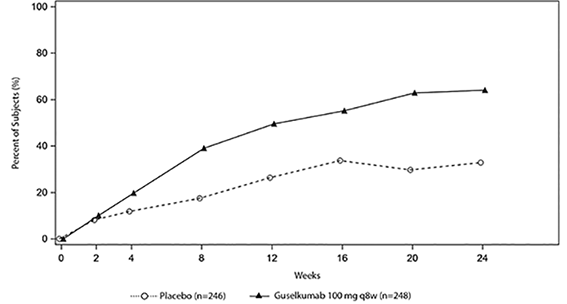
El porcentaje de sujetos que alcanzó la respuesta ACR20 en PsA2 por visita se muestra en la figura 1.
Figura 1. Sujetos que alcanzaron la respuesta ACR 20 por visita hasta la semana 24 en PsA2.
Los resultados de los componentes de los criterios de respuesta ACR se muestran en la tabla 5.
Tabla 5. Cambio medio (DEa) desde la basal en las puntuaciones de los componentes ACR en las semanas 16 y 24 en los datos observados
|
PsA1 |
PsA2 |
|||
|
Placebo (N = 126) |
TREMFYA® 100 mg q8w (N = 127) |
Placebo (N = 246) |
TREMFYA® 100 mg q8w (N = 248) |
|
|
No. de articulaciones inflamadas |
||||
|
Basal |
10.1 (7.1) |
10.9 (9.3) |
12.3 (6.9) |
11.7 (6.8) |
|
Cambio medio en la semana 16 |
-4.2 (7.0) (3%) |
-7.3 (7.0) |
-5.8 (7.1) |
-7.2 (6.0) |
|
Cambio medio en la semana 24 |
-5.1 (6.9) |
-7.3 (8.0) |
-6.4 (7.2) |
-8.1 (6.1) |
|
No. de articulaciones sensibles |
||||
|
Basal |
19.8 (14.4) |
20.2 (14.5) |
21.6 (13.1) |
19.8 (11.9) |
|
Cambio medio en la semana 16 |
-4.5 (10.8) |
-10.2 (10.4) |
-6.8 (10.5) |
-9.0 (9.4) |
|
Cambio medio en la semana 24 |
-6.8 (13.0) |
-10.5 (12.0) |
-7.3 (11.2) |
-10.4 (9.5) |
|
Evaluación del dolor del pacienteb |
||||
|
Basal |
5.8 (2.2) |
6.0 (2.1) |
6.3 (1.8) |
6.3 (2.0) |
|
Cambio medio en la semana 16 |
-0.8 (2.3) |
-1.7 (2.4) |
-0.9 (2.3) |
-2.2 (2.5) |
|
Cambio medio en la semana 24 |
-0.7 (2.4) |
-2.2 (2.6) |
-1.1 (2.4) |
-2.5 (2.5) |
|
Evaluación general del pacienteb |
||||
|
Basal |
6.1 (2.2) |
6.5 (2.0) |
6.5 (1.8) |
6.5 (1.9) |
|
Cambio medio en la semana 16 |
-1.0 (2.3) |
-2.0 (2.6) |
-1.0 (2.3) |
-2.3 (2.6) |
|
Cambio medio en la semana 24 |
-0.9 (2.5) |
-2.5 (2.7) |
-1.2 (2.6) |
-2.5 (2.5) |
|
Evaluación general del médicob |
||||
|
Basal |
6.3 (1.7) |
6.2 (1.7) |
6.7 (1.5) |
6.6 (1.6) |
|
Cambio medio en la semana 16 |
-1.9 (2.2) |
-2.9 (2.4) |
-2.1 (2.2) |
-3.5 (2.3) |
|
Cambio medio en la semana 24 |
-2.2 (2.3) |
-3.5 (2.4) |
-2.5 (2.3) |
-3.8 (2.3) |
|
Índice de discapacidad (HAQ-DI)c |
||||
|
Basal |
1.2 (0.7) |
1.2 (0.6) |
1.3 (0.6) |
1.3 (0.6) |
|
Cambio medio en la semana 16 |
-0.1 (0.5) |
-0.3 (0.5) |
-0.1 (0.5) |
-0.3 (0.5) |
|
Cambio medio en la semana 24 |
-0.1 (0.5) |
-0.3 (0.6) |
-0.2 (0.5) |
-0.4 (0.5) |
|
PCR (mg/dL) |
||||
|
Basal |
1.4 (1.9) |
1.6 (2.4) |
2.1 (2.7) |
2.0 (2.4) |
|
Cambio medio en la semana 16 |
-0.2 (1.5) |
-0.6 (2.2) |
-0.6 (2.5) |
-1.0 (2.2) |
|
Cambio medio en la semana 24 |
-0.0 (2.8) |
-0.7 (2.1) |
-0.5 (2.5) |
-1.1 (2.2) |
a DE = desviación estándar.
b Evaluación basada en la escala visual analógica (cm) con el extremo izquierdo que indica "sin dolor" (para la evaluación del dolor del paciente), "muy bien" (para la evaluación global del paciente) o "sin actividad de artritis" (para la evaluación global del médico) y el extremo derecho indica "el peor dolor posible" (para la evaluación del dolor del paciente), "pobre" (para la evaluación global del paciente) o "artritis extremadamente activa” (para la evaluación global del médico).
c Índice de discapacidad del Cuestionario de evaluación de la salud; 0 = sin dificultad a 3 = incapacidad para realizar, mide la capacidad del paciente para realizar lo siguiente: vestirse, levantarse, comer, caminar, higiene, alcanzar, agarrar y actividades de la vida diaria.
El tratamiento con TREMFYA® resultó en una mejora en las manifestaciones cutáneas de la psoriasis en sujetos con PsA.
El tratamiento con TREMFYA® dio como resultado una mejoría de la dactilitis y la entesitis en pacientes con dactilitis o entesitis preexistentes.
Función física: Los sujetos tratados con TREMFYA® en los grupos de TREMFYA® 100 mg q8w en PsA1 y PsA2 mostraron una mejoría media mayor desde la basal en la función física en comparación con los pacientes tratados con placebo según lo evaluado por el Índice de discapacidad del cuestionario de evaluación de salud (HAQ-DI) en las semanas 16 y 24. En ambos estudios, la proporción de respondedores HAQ-DI (mejora ≥0.35 en la puntuación HAQ-DI) fue mayor en el grupo de dosis de TREMFYA® q8w en comparación con el placebo en las semanas 16 y 24.
Otros resultados relacionados con la salud: El estado de salud general se evaluó mediante la encuesta de salud Short Form (SF-36). En la semana 24, los sujetos en los grupos de dosis TREMFYA® 100 mg q8w en PsA1 y PsA2 mostraron una mayor mejoría desde la basal en el resumen del componente físico (PCS, por sus siglas en inglés) del SF-36 en comparación con el placebo. No se observó una mejoría estadísticamente significativa en el SF-36 MCS. En la semana 24, hubo una mejora numérica en los dominios de funcionamiento físico, rol físico, dolor corporal, salud general, funcionamiento social y vitalidad, pero no en los dominios de rol emocional y salud mental. La fatiga se evaluó mediante la puntuación de la Evaluación Funcional de la Terapia de Enfermedades Crónicas-Fatiga (FACIT-F, por sus siglas en inglés) en los estudios PsA1 y PsA2. El tratamiento con TREMFYA® resultó en una mejora en la fatiga medida por FACIT-F.
CONTRAINDICACIONES:
• Hipersensibilidad a guselkumab o a cualquiera de los componentes de la fórmula.
• Embarazo y lactancia.
• Menores de 18 años de edad.
• Infecciones activas clínicamente importantes (p.ej. tuberculosis activa).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
TREMFYA® está contraindicando durante el embarazo y lactancia.
Embarazo:
Registro de exposición en el embarazo: Hay un registro de embarazo que monitoriza los resultados del embarazo en mujeres expuestas a TREMFYA® durante el embarazo.
Resumen del riesgo: No hay datos disponibles sobre el uso de TREMFYA® en mujeres embarazadas, para determinar un riesgo de eventos adversos en el desarrollo, asociados con el fármaco. Se sabe que los anticuerpos IgG humanos cruzan la barrera placentaria; por lo tanto, TREMFYA® puede transmitirse de la madre al feto en desarrollo. En un estudio de desarrollo embriofetal y pre- y postnatal combinado, no se observaron eventos adversos en el desarrollo en infantes nacidos de primates gestantes después de la administración subcutánea de guselkumab durante la organogénesis hasta el parto, a dosis de hasta 30 veces la Dosis Humana Máxima Recomendada (DHMR). Se observaron muertes neonatales de 6 a 30 veces la Dosis Humana Máxima Recomendada (ver Datos). Se desconoce la importancia clínica de estos hallazgos no clínicos.
Todos los embarazos tienen un riesgo basal de defectos de nacimiento, abortos u otros desenlaces adversos. Se desconoce el riesgo basal estimado de defectos congénitos y de abortos espontáneos en la población indicada. En la población general de Estados Unidos, el riesgo basal de defectos congénitos y de abortos espontáneos en embarazos clínicamente reconocidos, es de 2% a 4%, y de 15% a 20%, respectivamente.
Lactancia:
Resumen del riesgo: No hay datos acerca de la presencia de guselkumab en la leche materna, sus efectos sobre el infante lactando o los efectos sobre la producción de leche. No se detectó guselkumab en la leche de los primates cynomolgus lactantes. Se sabe que la IgG materna está presente en la leche humana. Se deben considerar los beneficios de la lactancia materna sobre la salud y el desarrollo, junto con la necesidad clínica de TREMFYA® para la madre y cualquier efecto adverso potencial sobre el infante amamantado, causado por TREMFYA® o por la condición materna subyacente.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las siguientes reacciones adversas se discuten con mayor detalle en otras secciones de la Información para prescribir.
• Infecciones (ver Precauciones generales).
• Reacciones de hipersensibilidad (ver Contraindicaciones y Precauciones generales).
Experiencia en estudios clínicos: Debido a que los estudios clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los estudios clínicos de un fármaco, no pueden compararse directamente con las tasas en los estudios clínicos de otro fármaco, y podrían no reflejar las tasas observadas en la práctica.
Psoriasis en placa: En estudios clínicos, un total de 1823 sujetos con psoriasis en placa moderada a severa, recibieron TREMFYA®. De estos, 1393 sujetos fueron expuestos a TREMFYA® por al menos 6 meses y 728 sujetos fueron expuestos, por al menos 1 año.
Los datos de dos estudios controlados con placebo y con activo (PsO1 y PsO2) se acumularon en 1441 sujetos (edad media 44 años, 70% hombres, 82% blancos) para evaluar la seguridad de TREMFYA® (100 mg administrados por vía subcutánea en las semanas 0 y 4, seguidos por cada 8 semanas).
Semanas 0 a 16:
En el periodo controlado con placebo de 16 semanas de los estudios clínicos acumulados (PsO1 y PsO2), ocurrieron eventos adversos en 49% de los sujetos del grupo de TREMFYA®, en comparación con 47% de los sujetos del grupo de placebo y 49% en el grupo de adalimumab, aprobado en EUA. Ocurrieron eventos adversos serios en 1.9% del grupo de TREMFYA® (6.3 eventos por 100 sujetos-años de seguimiento), en comparación con 1.4% del grupo de placebo (4.7 eventos por 100 sujetos-años de seguimiento) y en 2.6% del grupo de adalimumab aprobado en EUA (9.9 eventos por 100 sujetos-años de seguimiento).
La Tabla 7 resume las reacciones adversas que ocurrieron a una tasa de al menos 1% y a una tasa mayor en el grupo de TREMFYA® que en el grupo de placebo, durante el periodo controlado con placebo de 16 semanas.
Tabla 7. Reacciones adversas que ocurrieron en ≥ 1% de los sujetos hasta la semana 16 en PsO1 y PsO2
|
TREMFYA® 100 mg N = 823 N (%) |
Adalimumabb N = 196 N (%) |
Placebo N = 422 N (%) |
|
|
Infecciones del tracto respiratorio superiorc |
118 (14.3) |
21 (10.7) |
54 (12.8) |
|
Cefalead |
38 (4.6) |
2 (1.0) |
14 (3.3) |
|
Reacciones en el sitio de la inyeccióne |
37 (4.5) |
15 (7.7) |
12 (2.8) |
|
Artralgia |
22 (2.7) |
4 (2.0) |
9 (2.1) |
|
Diarrea |
13 (1.6) |
3 (1.5) |
4 (0.9) |
|
Gastroenteritisf |
11 (1.3) |
4 (2.0) |
4 (0.9) |
|
Infecciones por tiñag |
9 (1.1) |
0 |
0 |
|
Infecciones por herpes simpleh |
9 (1.1) |
0 |
2 (0.5) |
a Sujetos que recibieron 100 mg de TREMFYA® en la semana 0, semana 4 y cada 8 semanas después.
b Adalimumab aprobado en EUA.
c Infecciones respiratorias superiores incluyen nasofaringitis, infección del tracto respiratorio superior (ITRS), faringitis e ITRS viral.
d Cefalea incluye cefalea y cefalea tensional.
e Reacciones en el sitio de inyección incluyen eritema, equimosis, hematoma, hemorragia, hinchazón, edema, prurito, dolor, coloración, induración, inflamación y urticaria.
f Gastroenteritis incluye gastroenteritis y gastroenteritis viral.
g Infecciones por tiña incluyen tinea pedis, tinea cruris, infección por tiña e infecciones de tinea manuum.
h Infecciones por herpes simple incluyen herpes oral, herpes simple, herpes genital, herpes simple genital y herpes simple nasal.
Las reacciones adversas que ocurrieron en < 1%, pero > 0.1% de los sujetos en el grupo de TREMFYA®, y con una mayor tasa que en el grupo de placebo hasta, la semana 16 en PsO1 y PsO2, fueron migraña, infecciones por Candida y urticaria.
Reacciones adversas específicas:
Infecciones: Ocurrieron infecciones en 23% del grupo de TREMFYA®, en comparación con 21% del grupo de placebo.
Las infecciones más frecuentes (≥ 1%) fueron infecciones del tracto respiratorio superior, gastroenteritis, infecciones por tiña e infecciones por herpes simple; todos los casos fueron de intensidad leve a moderada y no condujeron a la descontinuación de TREMFYA®.
Enzimas hepáticas elevadas: Se reportaron elevaciones de las enzimas hepáticas con más frecuencia en el grupo de TREMFYA® (2.6%) que en el grupo de placebo (1.9%). De los 21 sujetos que reportaron enzimas hepáticas elevadas en el grupo de TREMFYA®, todos los eventos excepto uno, fueron de intensidad leve a moderada, y ninguno de los eventos condujo a la descontinuación de TREMFYA®.
Seguridad hasta la Semana 48: Hasta la semana 48, no se identificaron nuevas reacciones adversas con el uso de TREMFYA® y la frecuencia de las reacciones adversas fue similar al perfil de seguridad observado durante las primeras 16 semanas de tratamiento.
Artritis psoriásica: TREMFYA® se estudió en dos estudios controlados con placebo en pacientes con artritis psoriásica (748 pacientes con TREMFYA® y 372 pacientes con placebo). De los 748 pacientes que recibieron TREMFYA®, 375 pacientes recibieron TREMFYA® 100 mg en la semana 0, semana 4 y cada 8 semanas a partir de entonces, y 373 pacientes recibieron TREMFYA® 100 mg cada 4 semanas. El perfil de seguridad general observado en pacientes con artritis psoriásica tratados con TREMFYA® generalmente es consistente con el perfil de seguridad en pacientes con psoriasis en placas con la adición de bronquitis y disminución del recuento de neutrófilos. En el periodo controlado con placebo de 24 semanas, combinado en los dos estudios, se produjo bronquitis en el 1.6% de los sujetos del grupo de TREMFYA® cada 8 semanas y el 2.9% de los sujetos del grupo de TREMFYA® cada 4 semanas en comparación con el 1.1% de los sujetos del grupo de placebo. Se produjo una disminución del recuento de neutrófilos en el 0.3% de los sujetos del grupo de TREMFYA® cada 8 semanas y el 1.6% de los sujetos del grupo de TREMFYA® cada 4 semanas en comparación con el 0% de los sujetos del grupo de placebo. La mayoría de los eventos de neutropenia fueron leves, transitorios, no asociados con infección y no condujeron a la interrupción.
Inmunogenicidad: Como con todas las proteínas terapéuticas, existe el potencial de inmunogenicidad con TREMFYA®. La detección de la formación de anticuerpos depende sustancialmente de la sensibilidad y especificidad con la que se hace el estudio. Además, la incidencia observada de positividad de anticuerpos (incluyendo anticuerpos neutralizantes), puede estar influenciada por varios factores, incluyendo la metodología del estudio, el manejo de la muestra, el tiempo de recolección de la muestra, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones, la comparación de la incidencia de anticuerpos contra guselkumab entre las indicaciones con la incidencia de anticuerpos contra otros productos, puede ser confusa.
Psoriasis en placa: Hasta la semana 52, aproximadamente 6% de los sujetos tratados con TREMFYA® desarrollaron anticuerpos contra el fármaco. Entre los sujetos que desarrollaron anticuerpos contra el fármaco, aproximadamente 7% tenían anticuerpos que fueron clasificados como neutralizantes. Entre los 46 sujetos que desarrollaron anticuerpos contra guselkumab y con datos evaluables, 21 sujetos tuvieron concentraciones menores de guselkumab, incluyendo un sujeto que experimentó pérdida de eficacia después de desarrollar altos títulos de anticuerpos. Hasta la semana 156, aproximadamente el 9% de los sujetos tratados con TREMFYA® desarrollaron anticuerpos contra el fármaco y de estos sujetos, aproximadamente el 6% fueron clasificados como anticuerpos neutralizantes. Sin embargo, los anticuerpos contra guselkumab, generalmente no estaban asociados con cambios en la respuesta clínica, ni desarrollo de reacciones en el sitio de la inyección.
Artritis psoriásica: Hasta la semana 24, el 2% (n = 15) de los sujetos tratados con TREMFYA® desarrollaron anticuerpos contra el fármaco. De estos sujetos, 1 tenía anticuerpos que se clasificaron como anticuerpos neutralizantes. En general, el pequeño número de sujetos que fueron positivos para anticuerpos contra guselkumab limita la conclusión definitiva del efecto de la inmunogenicidad en la farmacocinética, eficacia y seguridad de guselkumab.
Experiencia postcomercialización: Se han reportado las siguientes reacciones adversas posterior a la aprobación de TREMFYA®. Debido a que estas reacciones se reportan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición a TREMFYA®.
Trastornos del sistema inmune: Hipersensibilidad, incluyendo anafilaxis (ver Precauciones generales).
Trastornos de la piel y del tejido subcutáneo: Erupción (ver Precauciones generales).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
No se han realizado estudios en animales para evaluar el potencial carcinogénico o mutagénico de TREMFYA®.
No se observaron efectos sobre los parámetros de fertilidad, después de administrar guselkumab por vía subcutánea a conejillos de indias machos a una dosis de 25 mg/kg dos veces por semana (15 veces la DHMR basada en una comparación en mg/kg).
No se observaron efectos sobre los parámetros de fertilidad después de administrar guselkumab por vía subcutánea a conejillos de indias hembras a dosis de hasta 100 mg/kg dos veces por semana (60 veces la DHMR basada en una comparación en mg/kg).
Datos:
Datos en animales: En un estudio del desarrollo embriofetal y pre y posnatal combinado, se les administró a primates cynomolgus gestantes dosis semanales de guselkumab de hasta 50 mg/kg (30 veces la DHMR basada en una comparación en mg/kg) desde el principio de la organogénesis hasta la resolución del embarazo. Se presentaron muertes en primates recién nacidos, uno del grupo control, en 3 que recibieron guselkumab en dosis de 10 mg/kg/semana (6 veces la DHMR basada en una comparación mg/kg) y en 3 primates que recibieron guselkumab en dosis de 50 mg/kg/semana (30 veces la DHMR basada en una comparación mg/kg). Se desconoce la importancia clínica de estos hallazgos. No se observaron efectos relacionados a guselkumab en el desarrollo funcional o inmunológico en los infantes desde el nacimiento hasta los 6 meses de edad.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Vacunas vivas: Evitar el uso de vacunas vivas en pacientes tratados con TREMFYA® (ver Precauciones generales).
Sustratos de CYP450: La formación de enzimas CYP450 puede ser alterada por niveles elevados de ciertas citocinas (por ejemplo, IL-1, IL-6, IL-10, FNTα, interferón) durante la inflamación crónica.
Los resultados de un estudio exploratorio de interacción farmacológica en sujetos con psoriasis en placa moderada a severa sugirieron un bajo potencial de interacciones farmacológicas, clínicamente relevantes para fármacos metabolizados por CYP3A4, CYP2C9, CYP2C19 y CYP1A2, pero no es posible descartar el potencial de interacción con fármacos metabolizados por CYP2D6. Sin embargo, los resultados fueron muy variables, debido al bajo número de sujetos en el estudio.
Después del inicio de TREMFYA® en pacientes que reciben sustratos de CYP450 de forma concomitante, particularmente aquéllos con un índice terapéutico estrecho, considerar el monitoreo del efecto terapéutico o la concentración del fármaco y considerar un ajuste de la dosis, según sea necesario (ver Farmacocinética y farmacodinamia).
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Se reportaron elevaciones de las enzimas hepáticas (ver Reacciones secundarias y adversas).
PRECAUCIONES GENERALES:
Reacciones de hipersensibilidad: Se han reportado reacciones de hipersensibilidad, incluyendo anafilaxis, graves con el uso de TREMFYA® después de la comercialización. Algunos casos requirieron hospitalización. Si se produce una reacción de hipersensibilidad grave, suspenda TREMFYA® e inicie la terapia adecuada.
Infecciones: TREMFYA® puede aumentar el riesgo de infección. En estudios clínicos en sujetos con psoriasis en placa, ocurrieron infecciones en 23% de los sujetos del grupo de TREMFYA® contra 21% de los sujetos en el grupo de placebo a través de 16 semanas de tratamiento. Las infecciones del tracto respiratorio superior, gastroenteritis, infecciones por tiña e infecciones por herpes simple ocurrieron con mayor frecuencia en el grupo de TREMFYA® que en el grupo placebo (ver Reacciones secundarias y adversas). La tasa de infecciones graves para el grupo TREMFYA® y el grupo de placebo fue ≤ 0.2%. Se observó un riesgo similar de infección en estudios controlados con placebo en sujetos con artritis psoriásica. El tratamiento con TREMFYA® no se debe iniciar en pacientes con cualquier infección activa clínicamente importante hasta que la infección se resuelva o sea tratada adecuadamente.
En pacientes con una infección crónica o antecedentes de infección recurrente, considere los riesgos y beneficios antes de prescribir TREMFYA®. Se deberá instruir a los pacientes para que busquen ayuda médica si presentan signos o síntomas de infección crónica o aguda clínicamente importante. Si un paciente desarrolla Infección grave o no responde a la terapia estándar, se deberá monitorizar de cerca al paciente y descontinuar TREMFYA® hasta que la infección se resuelva.
Evaluación de tuberculosis previa al tratamiento: Se deberá evaluar a los pacientes para detectar la infección de tuberculosis (TB) antes de iniciar el tratamiento con TREMFYA®. Inicie el tratamiento para la tuberculosis latente antes de administrar TREMFYA®. En estudios clínicos, 105 sujetos con psoriasis en placa y 71 sujetos con artritis psoriásica con TB latente, que fueron tratados simultáneamente con TREMFYA® y profilaxis apropiada para la TB, no desarrollaron TB activa. Se deberá monitorizar a los pacientes en busca de signos y síntomas de TB activa durante y después del tratamiento con TREMFYA®. Considere la terapia antiTB antes de iniciar TREMFYA®, en pacientes con antecedentes de TB latente o activa, en quienes no se puede confirmar un curso de tratamiento adecuado. No administre TREMFYA® a pacientes con infección de TB activa.
Inmunizaciones:
Antes de iniciar la terapia con TREMFYA®, considere completar todas las inmunizaciones apropiadas para la edad, de acuerdo con los lineamientos actuales de inmunización. Evite el uso de vacunas vivas en pacientes tratados con TREMFYA®. No hay datos disponibles sobre la respuesta a vacunas vivas o inactivas.
Uso pediátrico:
No se ha establecido la seguridad y la eficacia de TREMFYA® en pacientes pediátricos (menores de 18 años de edad) por lo que su uso está contraindicado en esta población.
Uso geriátrico:
De los 3406 sujetos con psoriasis en placa o artritis psoriásica expuestos a TREMFYA®, un total de 185 sujetos tenían 65 años o más, y 13 sujetos tenían 75 años o más. No se observaron diferencias globales en la seguridad o eficacia entre sujetos mayores y jóvenes que recibieron TREMFYA®. Sin embargo, el número de sujetos mayores de 65 años de edad no fue suficiente para determinar si responden de manera diferente a los sujetos más jóvenes (ver Farmacocinética y farmacodinamia).
INFORMACIÓN DE ASESORAMIENTO PARA PACIENTES:
Aconseje al paciente y/o cuidador que lea el instructivo de uso antes de comenzar la terapia con TREMFYA®, y cada vez que se renueve la receta, ya que puede haber nueva información que necesitan saber.
Reacciones de hipersensibilidad:
Aconseje a los pacientes que suspendan el tratamiento con TREMFYA® y busque atención médica inmediata si experimentan algún síntoma de reacciones de hipersensibilidad graves (ver arriba).
Infecciones:
Indique a los pacientes la importancia de comunicar cualquier historial de infecciones al proveedor de atención médica y contactar a su proveedor de atención médica si desarrollan algún síntoma de una infección (ver arriba).
Instrucción sobre técnica de inyección:
Indique a los pacientes o cuidadores que realicen la primera inyección bajo la supervisión y orientación de un profesional de la salud calificado para la capacitación adecuada en la técnica de inyección subcutánea. Indique a los pacientes que se administran la inyección por sí mismos, que se inyecten la dosis completa de TREMFYA® (consulte el instructivo de uso).
Instruya a los pacientes o cuidadores en la técnica adecuada de desecho de agujas y jeringas. Las agujas y jeringas deben desecharse en un recipiente resistente a los pinchazos. Aconseje a los pacientes y cuidadores que no reutilicen agujas o jeringas.
Recuerde a los pacientes que si olvidan administrar su dosis de TREMFYA®, deben inyectarse la dosis tan pronto como lo recuerden. La siguiente dosis debe administrarse con base en el calendario programado originalmente.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis:
Psoriasis en placa: TREMFYA® se administra por inyección subcutánea. La dosis recomendada es de 100 mg en la semana 0, semana 4 y en adelante, cada 8 semanas.
Artritis psoriásica: TREMFYA® se administra por inyección subcutánea. La dosis recomendada es de 100 mg en la semana 0, semana 4 y en adelante, cada 8 semanas.
TREMFYA® puede administrarse solo o en combinación con un fármaco antirreumático modificador de la enfermedad convencional (cFARME) (por ejemplo, metotrexato).
Evaluación de tuberculosis antes de iniciar TREMFYA®.
Evaluar a los pacientes para infección por tuberculosis (TB) antes de iniciar el tratamiento con TREMFYA® (ver Precauciones generales).
Instrucciones de administración importantes: Administrar TREMFYA® por vía subcutánea. Cada jeringa prellenada o pluma precargada es para dosis única solamente. Instruir a los pacientes de inyectarse la cantidad completa (1 mL), que aporta 100 mg de TREMFYA®.
No se inyecte TREMFYA® en áreas donde la piel esté sensible, maltratada, enrojecida, endurecida, engrosada, escamosa o afectada por la psoriasis [ver Instrucciones de Uso].
TREMFYA® está diseñado para su uso bajo la guía y supervisión de un médico. TREMFYA® puede ser administrado por un profesional de la salud, o el paciente puede auto-inyectarse después de un entrenamiento adecuado en la técnica de inyección subcutánea.
Las Instrucciones de Uso de TREMFYA® contienen instrucciones más detalladas para los pacientes sobre la preparación y administración de TREMFYA® (ver el Instructivo para el paciente que se encuentra dentro de la caja del producto).
Preparación para el uso de la jeringa prellenada o pluma precargada de TREMFYA®: Antes de la inyección, saque la jeringa prellenada de TREMFYA® del refrigerador y permita que TREMFYA® alcance la temperatura ambiente (30 minutos) sin retirar la tapa de la aguja.
Inspeccione la jeringa prellenada de TREMFYA® visualmente para ver si hay partículas y decoloración antes de la administración. TREMFYA® es una solución transparente e incolora a amarillo claro, que puede contener pequeñas partículas translúcidas. No use, si el líquido contiene partículas grandes, o si está decolorado o turbio. TREMFYA® no contiene conservadores; por lo tanto, se debe desechar cualquier producto no usado que permanezca en la jeringa prellenada o pluma precargada.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
En caso de sobredosis, monitorear al paciente para cualquier signo o síntoma de reacciones adversas y administre el tratamiento sintomático apropiado inmediatamente.
PRESENTACIONES:
Caja de cartón con una jeringa prellenada con 100 mg/mL de guselkumab e instructivo anexo.
Caja de cartón con una pluma precargada con 1 mL (100 mg/mL) e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese en refrigeración de 2 °C a 8 °C.
NO SE CONGELE.
Protéjase de la luz hasta su uso.
No se agite.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. Léase instructivo anexo. No se deje al alcance de los niños. No se use en el embarazo y la lactancia. No se use en menores de 18 años. Si no se administra todo el producto, deséchese el sobrante. No se administre si el cierre ha sido violado.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y atencionaclientes@its.jnj.com
JANSSEN-CILAG, S.A. de C.V.
Carretera Federal México-Puebla Km. 81.5,
San Mateo Capultitlán, C.P. 74160,
Huejotzingo, Puebla, México.
Reg. Núm. 290M2018, SSA IV
®Marca registrada