
UPTRAVI - Tabletas
Sustancia(s):
- Selexipag
Presentaciones:
- 1 Caja, 10 Tabletas, 200 µg
- 1 Caja, 140 Tabletas, 200 µg
- 1 Caja, 60 Tabletas, 200 µg
- 1 Caja, 60 Tabletas, 400 µg
- 1 Caja, 60 Tabletas, 600 µg
- 1 Caja, 60 Tabletas, 800 µg
- 1 Caja, 60 Tabletas, 1,000 µg
- 1 Caja, 60 Tabletas, 1,200 µg
- 1 Caja, 60 Tabletas, 1,400 µg
- 1 Caja, 60 Tabletas, 1,600 µg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Selexipag 200 μg
Excipiente c.b.p 1 tableta
Selexipag 400 μg
Excipiente c.b.p 1 tableta
Selexipag 600 μg
Excipiente c.b.p 1 tableta
Selexipag 800 μg
Excipiente c.b.p 1 tableta
Selexipag 1000 μg
Excipiente c.b.p 1 tableta
Selexipag 1200 μg
Excipiente c.b.p 1 tableta
Selexipag 1400 μg
Excipiente c.b.p 1 tableta
Selexipag 1600 μg
Excipiente c.b.p 1 tableta
INDICACIONES TERAPÉUTICAS: UPTRAVI® está indicado para el tratamiento a largo plazo de la hipertensión arterial pulmonar (HAP, Grupo I de la OMS), en pacientes adultos en clase funcional de la OMS 11-111, ya sea como terapia combinada en pacientes que se encuentran mal controlados con el tratamiento a base de antagonistas de los receptores de endotelina (ARE) y/ó inhibidores de la 5 fosfodiesterasa (PDE-5), o bien, como monoterapia en pacientes que no son candidatos a recibir otras terapias.
Su eficacia ha sido demostrada en población con HAP incluyendo HAP idiopática y HAP hereditaria, HAP asociada a enfermedad del tejido conectivo y, HAP asociada a enfermedad cardiaca congénita con corto circuitos ya reparados.
FARMACOCINÉTICA Y FARMACODINAMIA:
Mecanismo de Acción:
UPTRAVI® es un agonista selectivo oral de los receptores IP, el cual es estructuralmente y farmacológicamente diferente a la prostaciclina y sus análogos. Selexipag es hidrolizado por carboxilesterasas para dar paso a su metabolito activo, el cual es aproximadamente 37 veces más potente que selexipag. Selexipag y su metabolito activo son altamente afines a los agonistas del receptor IP, con una alta selectividad por el receptor IP versus otros receptores de prostanoides (EP1- EP4, DP, FP y TP). Su selectividad en contra de los receptores EP1, EP3, FP y TP es importante ya que éstos son receptores bien conocidos por su efecto contráctil a nivel del tracto gastrointestinal y los vasos sanguíneos. La selectividad en contra de los receptores EP2, EP4 y DP1 es importante debido a que estos receptores son mediadores de los efectos depresores en el sistema inmune.
La estimulación del receptor IP producida por selexipag y su metabolito activo dan origen a un efecto vasodilatador así como anti-proliferativo y anti-fibrótico. Selexipag mejoró las variables hemodinámicas y previno la remodelación cardiaca y pulmonar en un modelo de ratas con HAP. En estas ratas con HAP, existe una correlación entre la vasodilatación pulmonar y la periferia en respuesta a selexipag, indicando así que la vasodilatación periférica, refleja la eficacia en la farmacodinamia pulmonar. Selexipag no causa desensibilización del receptor IP in vitro así como tampoco taquifilaxia en el modelo de ratas.
Propiedades farmacodinámicas:
Electrofisiología Cardiaca: En un riguroso estudio QT en sujetos sanos, dosis repetidas de 800 y 1600 microgramos de selexipag dos veces al día, no mostraron efecto sobre la re-polarización cardiaca (intervalo QTc) o sobre la conducción (intervalos PR y QRS) y tuvieron un efecto acelerado leve en la frecuencia cardiaca (la frecuencia cardiaca placebo-corregida, ajustada al inicio alcanzó 6-7 Ipm a las 1.5 a 3 horas posteriores a una dosis de 800 microgramos de selexipag y 9-10 lpm en el mismo periodo de tiempo posterior a una dosis de 1,600 microgramos de selexipag).
Factores de Coagulación: En estudios fase 1 y fase 2 se observo una ligera disminución de los niveles plasmáticos del factor de von Willebrand factor (vWF) producida por selexipag. Los valores del vWF permanecieron por arriba del límite inferior del rango normal.
Hemodinámica Pulmonar: Se realizó un estudio clínico fase 2, doble ciego, placebo-controlado para evaluar las variables hemodinámicas después de 17 semanas de tratamiento en pacientes con HAP CF 11-111 de la OMS que recibían de manera concomitante ERAs y/ó inhibidores de la PDE-5. Los pacientes que fueron titulados de manera individual con selexipag hasta la dosis tolerada (200 microgramos dos veces al día con incremento hasta 800 microgramos dos veces al día; N=33), alcanzaron una reducción promedio estadísticamente significativa de 30.3% en la resistencia vascular pulmonar (95% de intervalo de confianza [IC]: -44.7%, -12.2%; p=0.0045) y un incremento en el índice cardiaco (efecto del tratamiento promedio) de 0.48 L/min/m2 (95% IC: 0.13, 0.83) comparado con placebo (N=10).
Propiedades farmacocinéticas: La farmacocinética de UPTRAVI® y su metabolito activo han sido estudiadas primordialmente en sujetos sanos. La farmacocinética de selexipag y la de su metabolito activo, posterior a dosis única y dosis repetidas, fue proporcional a la dosis hasta una dosis única de 800 microgramos y, de dosis repetidas hasta 1800 microgramos dos veces al día. Posterior a una administración de dosis múltiples, las condiciones de estado estable para selexipag y su metabolito activo fueron alcanzadas en 3 días. No se observó acumulación en plasma, tanto del compuesto principal como del metabolito activo, posterior a la administración de dosis múltiples.
En sujetos sanos, la inter-variabilidad de los sujetos a la exposición (área bajo la curva sobre un intervalo de dosis) en estado estable fue de 43% y 39% para selexipag y su metabolito activo, respectivamente. La variabilidad inter-sujetos en la exposición fue de 24% y 19% para selexipag y su metabolito activo, respectivamente.
La exposición a selexipag y su metabolito activo en estado estable en pacientes con HAP y en sujetos sanos fue similar. La farmacocinética de selexipag y su metabolito activo en pacientes con HAP no fue influenciada por la severidad de la enfermedad y no cambio con el tiempo.
Absorción: UPTRAVI® se absorbe rápidamente y es hidrolizado por carboxilesterasas hacia su metabolito activo.
Las concentraciones plasmáticas máximas observadas con selexipag y su metabolito activo posterior a la administración oral se alcanzaron 1-3 horas y 3-4 horas respectivamente.
La biodisponibilidad absoluta de selexipag en los humanos es de aproximadamente 49%. Esto se debe probablemente al efecto del primer paso de selexipag, dado que las concentraciones plasmáticas del metabolito activo son similares posteriores a la misma dosis administrada por vía oral o intravenosa.
En la presencia de alimento, la exposición a selexipag posterior a una dosis única de 400 microgramos fue incrementada en un 10% en sujetos caucásicos y disminuyó en un 15% en sujetos japoneses; mientras que la exposición al metabolito activo disminuyó en un 27% (sujetos caucásicos) y 12% (sujetos japoneses). Un mayor número de sujetos reportaron eventos adversos en estado de ayuno versus en pacientes que habían recibido alimento.
Distribución: UPTRAVI® y su metabolito activo tienen una alta unión a las proteínas plasmáticas (aproximadamente 99% en total y, en la misma proporción a la albumina y a la glicoproteína acida alfa-1). El volumen de distribución de selexipag en estado estable es de 11.7 L.
Biotransformación: UPTRAVI® es hidrolizada a su metabolito activo en el hígado y en el intestino mediante carboxilesterasas. El metabolismo oxidativo catalizado principalmente por el CYP2C8 y en una menor proporción por el CYP3A4, da como resultado la formación de productos hidrolizados y desalquilados. El UGT1A3 y el UGT2B7 están involucrados en la glucoronidación del metabolito activo. Con excepción del metabolito activo, ninguno de los metabolitos circulantes en el plasma humano excede el 3% del material total relacionado con medicamentos. Tanto en sujetos sanos como en pacientes con HAP, tras la administración oral, la exposición al estado estable del metabolito activo es aproximadamente 3 a 4 veces mayor a la del compuesto base.
Eliminación: La eliminación de UPTRAVI® se lleva a cabo predominantemente via metabólica con una vida media terminal de 0.8 - 2.5 horas. El metabolito activo tiene una vida media de 6.2 - 13.5 horas. El aparente aclaramiento oral de selexipag es en promedio de 35 L/h. La excreción en sujetos sanos se completo 5 días después de la administración y se dio principalmente vía heces (siendo hasta por el 93% de la dosis administrada) comparada con un 12% en orina.
Farmacocinética en poblaciones especiales: No se ha observado un efecto clínicamente relevante del género, edad, peso corporal u origen étnico sobre la farmacocinética de UPTRAVI® y su metabolito activo en sujetos sanos o en pacientes con HAP
Insuficiencia Renal: Se observó un incremento de 1.4 a 1.7 veces ante la exposición (concentración plasmática máxima y concentración plasmática del área bajo la curva) a selexipag y su metabolito activo en sujetos con insuficiencia renal severa (con una filtración glomerular estimada < 30 ml/ min/1.73 m2).
Insuficiencia hepática: En sujetos con insuficiencia hepática leve (Child-Pugh clase A) o moderada (Child-Pugh clase B), la exposición a selexipag fue 2 a 4 veces mayor, respectivamente, al compararse con sujetos sanos. La exposición al metabolito activo permaneció casi inalterada en sujetos con insuficiencia hepática leve y se duplico en sujetos con daño hepático moderado. Sólo dos sujetos con insuficiencia hepática severa (Child-Pugh clase C) fueron medicados con selexipag. La exposición a selexipag y su metabolito activo en estos dos sujetos fue similar a la de los sujetos con insuficiencia hepática moderada (Child-Pugh clase B).
Basados en datos de un estudio en modelado y estimulación en sujetos con insuficiencia hepática, la exposición a selexipag en estado estable en sujetos con insuficiencia hepática moderada (Child-Pugh clase B) posterior a un régimen de una toma al día, se calculó que es aproximadamente 2 veces mayor que la de los sujetos sanos durante un régimen de dos tomas al día. La exposición al metabolito activo en estado estable en estos pacientes durante un régimen de una toma al día se prevé que sea similar al de los sujetos sanos en un régimen de dos veces al día. Los sujetos con insuficiencia hepática severa (Child-Pugh cales C) mostraron una exposición prevista familiar en estado estable a la de los sujetos con insuficiencia hepática moderada en un regimen de una toma al día.
Eficacia y seguridad Clínica:
Eficacia en pacientes con HAP: El efecto de UPTRAVI® en la progresión de la HAP se demostró en un estudio fase 3, multicéntrico a largo plazo (duración máxima de la exposición aproximadamente de 4.2 años), doble ciego, placebo controlado, de grupos paralelos, conducido por eventos en 1156 pacientes con HAP sintomática (CF de la OMS I-IV). Los pacientes fueron aleatorizados a recibir placebo (N=582) o selexipag (N=574) dos veces al día. La dosis se incrementó en intervalos semanales con incrementos de 200 mg dos veces al día para determinar así la dosis de mantenimiento individualizada (200 - 1600 microgramos dos veces al día).
El objetivo primario de estudio fue el tiempo transcurrido al primer evento de morbilidad o mortalidad hasta el final del tratamiento, definida como: muerte (todas las causas); hospitalización por HAP; progresión de la HAP resultando en la necesidad de trasplante pulmonar o septostomía con balón auricular, el inicio de tratamiento con prostanoides parenterales, el uso de oxígeno de manera crónica o bien, otros eventos de progresión de la enfermedad (modificación de la clase funcional de la OMS/NYHA - New York Heart Association- 11 o 111 al inicio del estudio) confirmados por una disminución 15% en la prueba de caminata de 6 minutos (PC6M) a partir de la basal, y deterioro de la clase funcional de la OMS/NYHA que requirieron terapia específica adicional.
Todos los eventos fueron confirmados por un comité de evaluación independiente, cegado al tratamiento recibido.
La edad promedio fue de 48.1 años (rango de 18-80 años de edad), siendo la mayoría sujetos caucásicos (65%) y sexo femenino (79.8%). Aproximadamente el 1%, 46%, 53% y 1% de los pacientes se encontraban en las clases funcionales de la OMSI, II, III y IV respectivamente al inicio del estudio (basal).
La HAP idiopática o familiar fue la etiología más común en la población de estudio (58%), seguida por HAP secundaria a enfermedad del tejido conectivo (29%), HAP asociada a cardiopatía congénita con reparación del corto circuito (10%) y, HAP asociada con otras etiologías (medicamentos y toxinas [2%] y HIV [1%]).
En la basal, la mayoría de los pacientes reclutados (80%) estaban recibiendo tratamiento con dosis estables de medicamentos para la HAP, ya sea ERAs (15%) o con inhibidores de la PDE-5 (32%) o bien, con ambos medicamentos de forma combinada, es decir, un ERA y un inhibidor de la PDE-5 (33%).
La duración total promedio del tratamiento doble ciego fue de 63.7 semanas para el grupo placebo y de 70.7 semanas para el grupo de selexipag. El 23% de los pacientes en el grupo de selexipag alcanzaron su dosis de mantenimiento en el rango de 200 - 400 microgramos, 31% en el rango de 600 - 1,000 microgramos y, el 43% alcanzaron la dosis de mantenimiento en el rango de 1,200 -1,600 microgramos.
El tratamiento con selexipag 200-1600 microgramos dos veces al día resultó en una disminución del 40% (hazard ratio [HR] 0.60; 99% CI: 0.46, O.78; one-sided log-rank p-value < 0.0001) de la presentación de un evento de morbilidad o mortalidad hasta 7 días después de la última dosis en comparación con placebo. El efecto benéfico de selexipag fue atribuido primordialmente a la disminución en hospitalizaciones por HAP y a la disminución de otros eventos de progresión de la enfermedad (Tabla 1).
Figura 1 Estimado de Kaplan-Meier para el primer evento de morbilidad-mortalidad
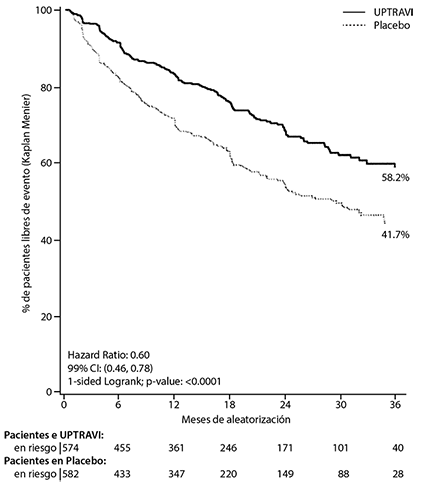
Tabla 1. Resumen de eventos de desenlace.
|
Desenlace y Estadísticas |
Pacientes con un evento |
Comparación de tratamientos: Selexipag vs placebo |
||||
|
Placebo (N=582) |
Selexipag (N=574) |
Reducción del Riesgo Absoluto |
Reducción del Riesgo Relativo (99% IC) |
HR (99% IC) |
Valor-p |
|
|
Evento de Morbilidad - Mortalidadª |
58.2% |
41.7% |
16.5% |
40% (22%; 54%) |
0.60 (0.46; 0.78) |
< 0.0001 |
|
Hospitalización por HAPb n (%) |
109 (18.7%) |
78 (13.6%) |
5.1% |
33% (2%; 54%) |
0.67 (0.46; 0.98) |
0.04 |
|
Progresión de la enfermedadb n (%) |
100 (17.2%) |
38 (6.6%) |
10.6% |
64% (41%; 78%) |
0.36 (0.22; 0.59) |
< 0.0001 |
|
Inicio de prostanoides i.v./s.c o empleo de terapia con oxígenobc n (%) |
15 (2.6%) |
11 (1.9%) |
0.7% |
32% (-90%; 76%) |
0.68 (0.24; 1.90) |
0.53 |
|
Muerte hasta el fin del tratamiento (EOT) + 7 díasd n (%) |
37 (6.4%) |
46 (8.0%) |
- 1.7% |
-17% (- 107%;34%) |
1.17 (0.66, 2.07) |
0.77 |
|
Muerte hasta el cierre del estudiod n (%) |
105 (18.0%) |
100 (17.4%) |
0.6% |
3% (- 39%; 32%) |
0.97 (0.68; 1.39) |
0.42 |
IC: Intervalo de confianza; EOT: fin del tratamiento; HR: Hazard Ratio; i.v.: intravenoso; HAP: Hipertensión arterial pulmonar; s.c.: subcutáneo.
(a) % de pacientes con un evento al mes 36 = 100 x (1 - estimado Kaplan Meier); el hazard ratio se calculo empleando el modelo de riesgo proporcional de Cox; valor de p sin estratificar lag Rank unilateral.
(b) % de pacientes con un evento como parte del objetivo primario hasta el fin del tratamiento+ 7 días; el hazard ratio se estimó empleando el método de Aalen Johansen; el valor de p de dos lados empleando la prueba de Gray.
(c) Incluye: necesidad de trasplante pulmonar o septostomía atrial (1 paciente en selexipag y 2 en placebo).
(d) % de pacientes con un evento hasta el fin del tratamiento+ 7 días o hasta el cierre del estudio; el hazard ratio se calculo empleando el modelo de riesgo proporcional de Cox; valor de p sin estratificar lag Rank unilateral.
El incremento numérico en las muertes hasta el final del tratamiento + 7 días pero no hasta el cierre (del estudio fue investigado a través de modelos matemáticos, demostrando que el desequilibrio en las muertes es consistente con la premisa de un efecto neutral de la mortalidad por HAP y la reducción de eventos no-fatales.
El efecto observado de selexipag versus placebo en el objetivo primario fue consistente entre los grupos de dosis de mantenimiento, tal como se muestra con el hazard ratio para las tres categorías predeterminadas (0.60 para 200-400 microgramos dos veces al día; 0.53 para 600-1,000 microgramos dos veces al día y, 0.64 para 1,200 - 1,600 microgramos dos veces al día), lo cual fue consistente con el efecto total del tratamiento (0.60).
La eficacia de selexipag en el objetivo primario fue consistente entre los subgrupos de edad, sexo, raza, etiología, región geográfica, clase funcional de la OMS y, ya sea como monoterapia o en combinación con un ERA o un inhibidor de la PDE-5 o una triple combinación de un ERA más un inihibidor de la PDE-5.
El tiempo para la muerte relacionada con HAP u hospitalización por HAP, fueron evaluadas como objetivo secundario. El riesgo de presentar un evento para este desenlace se redujo en un 30% en los pacientes recibiendo selexipag comparado con placebo (HR 0.70, 99% IC; 0.50, 0.98; log Rank unilateral p= 0.0031). Los porcentajes de pacientes con un evento al mes 36 fueron 28.9% y 41.3% en el grupo de selexipag y placebo, respectivamente, con una reducción del riesgo absoluta del 12.4%.
El número de pacientes que experimentaron como primer evento, muerte por HAP u hospitalización por HAP hasta el fin del tratamiento fueron 102 (17.8%) en el grupo de selexipag y 137 (23.5%) en el grupo placebo. Muerte por HAP como componente del objetivo fue observado en 16 (2.8%) pacientes en el grupo de selexipag y 14 (2.4%) en el grupo placebo. La hospitalización por HAP se observó en 86 (15%) de los pacientes con selexipag y 123 (21.1%) en los pacientes con placebo. Selexipag disminuyó el riesgo de hospitalización por HAP como evento del primer desenlace comparado con placebo (HR 0.67; 99% IC; 0.46; 0.98; log-rank unilateral, p=0.04).
El número total de muertes por cualquier causa hasta el cierre del estudio fue de 100 (17.4%) para el grupo de selexipag y 105 (18%) para el grupo de placebo (HR 0.97; 99% IC: 0.68; 1.39). El número de muertes por HAP hasta el cierre del estudio fue de 70 (12.2%) para el grupo de selexipag y 83 (14.3%) para el grupo de placebo.
Objetivos relacionados con la sintomatología:
La capacidad de ejercicio fue evaluada como objetivo secundario. La mediana basal en la prueba de caminata de 6 minutos (PC6M) fue de 376 m (rango: 90-482 mts.) y 369 mts. (rango: 50-515 mts.) en los pacientes con selexipag y placebo, respectivamente. El tratamiento con selexipag resultó en un efecto mediano placebo-corregido en la PC6M (ej., aproximadamente 12 hrs post-dosis) de 12 mts. a la semana 26 (99% IC: 1.24 mts; valor p unilateral =0.0027). En los pacientes sin tratamiento específico para HAP, el efecto del tratamiento placebo-corregido medido fue de 34 mts (99% IC: 10; 63 mts.).
La calidad de vida fue valorada en un sub-grupo de pacientes en el estudio GRIPHON empleando el cuestionario de CAMPHOR (Cambridge Pulmonary Hypertension Outcome- Review), sin que se encontrara un efecto del tratamiento significativo desde la basal a la semana 26.
CONTRAINDICACIONES:
• Hipersensibilidad al principio activo o a cualquiera de los excipientes.
• Angina inestable o enfermedad coronaria severa.
• Infarto al Miocardio en los 6 meses previos.
• Insuficiencia cardiaca descompensada cuando ésta no se encuentre bajo supervisión médica estrecha.
• Arritmias severas.
• Eventos cerebrovasculares (isquemia cerebral transitoria o infarto) durante los últimos 3 meses.
• Defectos valvulares congénitos o adquiridos con alteración relevante en la función miocárdica no relacionados con hipertensión pulmonar.
• Uso concomitante de inhibidores potentes de la CYP2C8 (ej., gemfibrozilo).
• Insuficiencia hepática severa (Child-Pugh clase C).
• Pacientes en diálisis.
• Embarazo.
• Lactancia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: Mujeres en edad reproductiva deberán emplear algún método efectivo de anticoncep estén bajo tratamiento con UPTRAVI®.
Embarazo: No existe información sobre el uso de selexipag en mujeres embarazadas. Los estudios en animales no indican un efecto dañino directo o indirecto con relación a la toxicidad reproductiva. Selexipag y su metabolito principal mostraron 20-80 veces menor potencia del receptor de prostaciclina (IP) in vitro para las especies animales empleadas en las pruebas de toxicidad reproductiva comparada con humanos. Por tanto, los márgenes de seguridad para efectos potenciales mediados por el receptor IP en la reproducción son menores que los efectos que no tienen relación con el receptor IP.
UPTRAVI® no debe ser recomendado durante el embarazo y en mujeres con edad reproductiva que no estén empleando métodos de anticoncepción eficaces.
Lactancia: Se desconoce si selexipag o sus metabolitos son excretados hacia la leche maternal. En ratas, selexipag y sus metabolitos fueron excretados a la leche. La lactancia no esta recomendada durante el tratamiento con UPTRAVI®.
REACCIONES SECUNDARIAS Y ADVERSAS: Los eventos adversos más comúnmente reportados con los efectos farmacológicos de UPTRAVI® son cefalea, diarrea, náusea y vómito, dolor mandibular, mialgias, dolor de las extremidades, rubincudez y artralgias. Estas reacciones son más frecuentes durante la fase de titulación. La mayoría de estas reacciones son de intensidad leve a moderada.
La seguridad de selexipag ha sido evaluada en un estudio clínico fase 3 a largo plazo, placebo controlado que enrolo a 1156 pacientes con HAP sintomática. La duración media del tratamiento fue de 76.4 semanas (mediana 70.7 semanas) para pacientes recibiendo selexipag versus 71.2 semanas (mediana 63.7 semanas) para pacientes en placebo. La exposición a selexipag fue de hasta 4.2 años.
Las reacciones adversas asociadas a selexipag en el estudio clínico pivote se muestran en la siguiente tabla. Con cada grupo de frecuencia, los efectos indeseables son presentados en orden decreciente de severidad. Tabla 2. Eventos Adversos
|
Sistema / Organo Clase |
Muy común (≥ 1/10) |
Común (≥ 1/100 a < 1/10) |
Poco común (≥ 1/1,000 a < 1/100) |
|
Trastornos Sanguíneos y del Sistema Linfático |
Anemia Disminución en la hemoglobina |
||
|
Trastornos Endocrinos |
Hipertiroidismo Disminución de la hormona estimulante de la tiroides |
||
|
Trastornos del metabolismo y nutrición |
Disminución del apetito Perdida de peso |
||
|
Trastornos del sistema nervioso |
Cefalea* |
||
|
Trastornos cardiacos |
Taquicardia sinusal |
||
|
Trastornos Vasculares |
Rubicundez* |
Hipotensión |
|
|
Trastornos respiratorios, torácicos de mediastino |
Nasofaringitis (de origen no infeccioso) |
Congestión nasal |
|
|
Trastornos Gastrointestinales |
Diarrea* Vómito* Nausea* |
Dolor Abdominal |
|
|
Trastornos de la piel y tejido subcutáneo |
Rash Urticaria Eritema |
||
|
Trastornos músculo esqueléticos y del Tejido Conectivo |
Dolor Mandibular* Dolor en las extremidades* Mialgias* Artralgias* |
||
|
Trastornos generales y condiciones en el sitio de administración |
Dolor |
*Revisar sección de eventos adversos seleccionados.
Descripción de los Eventos Adversos Seleccionados:
Efectos farmacológicos asociados con la titulación y mantenimiento dertratamiento: Las reacciones adversas asociadas con el mecanismo de acción de selexipag han sido frecuentemente observadas, particularmente durante la fase de titulación hacia la dosis individualizada y son descritas a continuación:
Tabla 3. Reacciones adversas asociadas con el mecanismo de acción de selexipag durante las fases de titulación y mantenimiento.
|
Reacción Adversa asociada al uso de prostaciclinas |
Fase de titulación |
Fase de mantenimiento |
||
|
Selexipag |
Placebo |
Selexipag |
Placebo |
|
|
Cefalea |
64% |
28% |
40% |
20% |
|
Diarrea |
36% |
12% |
30% |
13% |
|
Náusea |
29% |
13% |
20% |
10% |
|
Dolor Mandibular |
26% |
4% |
21% |
4% |
|
Mialgia |
15% |
5% |
9% |
3% |
|
Dolor en las extremidades |
14% |
5% |
13% |
6% |
|
Vómito |
14% |
4% |
8% |
6% |
|
Rubicundez |
11% |
4% |
10% |
3% |
|
Artralgias |
7% |
5% |
9% |
5% |
Estos efectos usualmente son transitorios y manejables con tratamiento sintomático. El 7.5% de los pacientes bajo tratamiento con selexipag descontinuaron el tratamiento debido a un evento adverso. La tasa aproximada de reacciones adversas consideradas como serias fue de 2.3% en el grupo de selexipag y 0.5% en el grupo de placebo. En la practica clinica, se observó que los eventos gastrointestinales responden al empleo de antidiarreicos, antieméticos y medicamentos para disminuir las náuseas y/o medicamentos para los trastornos de la función gastrointestinal. Los eventos asociados con dolor frecuentemente fueron tratados con analgésicos (ej., paracetamol).
Reporte de cualquier sospecha de reacción adversa: Es importante reportar cualquier sospecha de reacciones adversa una vez que se inicia la comercialización de cualquier medicamento. Esto permite un monitoreo continua del balance riesgo/beneficio del medicamento. Se invita a los profesionales de la salud a reportar cualquier sospecha de reacción adversa a través del sistema nacional de reporte al correo electrónico farmacovigilancia@cofepris.gob.mx y drugsafetymx@actelion.com
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: En el estudio de toxicidad de dosis repetidas en roedores, se identificó una importante disminución de la presión arterial como resultado de un efecto farmacológico exagerado, originando síntomas clínicos transitorios y disminuyendo el consumo de alimentos y la ganancia de peso corporal. En perros adultos y jóvenes, intestino, hueso y médula ósea fueron identificados como los órganos objetivo posterior al tratamiento con selexipag. En perros de menos de 1 año de edad, la intususcepción como efecto relacionado a la prostaciclina o efectos sobre la motilidad intestinal fueron observados de manera esporádica. Este efecto ocurrió en exposiciones 5 veces por arriba de la exposición en humanos (ej. Potencia corregida; 415 veces basado en exposición total -metabolito activo-). Los márgenes de seguridad basados en niveles de eventos adversos no observados para el metabolito activo, corregidos por diferencias en la potencia del receptor entre humanos y perros, fue 2 veces mayor (ej., potencia corregida; 180 veces basada en la exposición total) en relación con la exposición humana a una dosis de 1600 microgramos de selexipag dos veces al día. Este hallazgo no se observó en estudios de toxicidad en ratones o ratas. Debido a la sensibilidad especie por especies de los perros a desarrollar intususcepción y al margen de seguridad, este hallazgo es considerado no relevante para humanos adultos.
En estudios realizados en perros, se observó un incremento en la osificación y cambios relacionados con la médula ósea, que son considerados como resultado de la activación de los receptores EP4 en los perros. Dado que en los humanos los receptores EP4 no son activados por el efecto de selexipag o su metabolito activo, este efecto es específico a la especie y, por tanto, no es relevante par los humanos.
Selexipag y su metabolito activo no son genotóxicos con base en toda la evidencia generada en los estudios de genotoxicidad realizados.
En el estudio de carcinogenicidad de 2 años de duración, selexipag causo un incremento en la incidencia de adenomas tiroideos en ratones y, adenomas de células de Leydig en ratas. El mecanismo fue roedor específico. Se observó también tortuosidad de las arteriolas retinianas posterior a 2 años de tratamiento sólo en ratas. Mecánicamente, se considera que el efecto es provocado por la duradera vasodilatación y cambios subsecuentes en la hemodinámica ocular.
Hallazgos histopatológicos adicionales de selexipag se observaron sólo ante exposiciones excesivas a la exposición máxima en humanos, indicando su poca relevancia en humanos.
En un estudio de fertilidad realizado en ratas, una prolongación del ciclo de celo dio como resultado en un incremento de días hasta la copulación con exposiciones 173 veces por arriba de la exposición terapéutica (basado en exposiciones totales) y, el efecto no observado siendo 30 veces por debajo de la exposición terapéutica. Adicional a esto, ningún otro parámetro de fertilidad se vio afectado.
Selexipag no fue teratogénico en ratas y conejos (márgenes de exposición por arriba de exposición terapéutica en 13 veces para selexipag y en 43 veces para su metabolito activo, basado en exposición total). Los márgenes de seguridad para efectos potencialmente relacionados con el receptor IP en la reproducción fueron 20 para fertilidad y 5 y 1 (basados en libre exposición) para el desarrollo embriofetal en ratas y conejos, respectivamente, cuando se adapto por diferencias en la potencia del receptor. En el estudio de desarrollo en ratas pre-/post natal, selexipag no indujo efecto alguno en la función reproductiva materna.
No existe información disponible sobre los efectos de selexipag en fertilidad. Estudios en ratas mostraron que, selexipag en dosis elevadas causa alteraciones transitorias en los ciclos de celo que no afectaban la fertilidad. La relevancia de este hallazgo en humanos es desconocida.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Efectos de otros medicamentos en selexipag: Selexipag es hidrolizado a su metabolito activo por la carboxilesterasa hepática. Tanto selexipag como su metabolito activo son sometidos al metabolismo oxidativo principalmente por el CYP2C8 y, en menor proporción, por el CYP3A4. La glucuronidación del metabolito activo es catalizada por la UGT1A3 y la UGT2B7. Selexipag y su metabolito activo son sustratos de la OATP1B1 y la OATP1B, 3 Selexipag es un sustrato de la P-gp, y su metabolito activo es un sustrato de la proteína transportadora de resistencia al cáncer de mama (BCRP).
La farmacocinética de selexipag y su metabolito activo no se ven afectadas por la warfarina.
lnhibidores de CYP2CB: Ante la presencia de un potente inhibidor de la CYP2C8 como gemfibrozil 600 mg dos veces al día, la exposición a selexipag se incrementó en aproximadamente dos veces, mientras que la exposición a su metabolito activo, principal responsable de la eficacia del medicamento, se incremento en aproximadamente 11 veces. Por tanto, la administración concomitante de UPTRAVI® potentes de la CYP2C8 está contraindicada.
El efecto de inhibidores moderados de la CYP2C8 (ej., clopidrogel, deferasirox, teriflunomida) en la exposición a selexipag y su metabolito activo no ha sido estudiado. Se debe considerar un ajuste en la dosis de UPTRAVI® cuando un inhibidor moderado de la CYP2C8 se administre de manera conjunta o bien, considerar su descontinuación, dado que una interacción farmacocinética potente no puede ser descartada.
Inductores de la CYP2CB: Ante la presencia de un inductor de la CYP2C8 (y enzimas UGT) como 600 mg de rifampicina una vez al día, la exposición a selexipag no se altero, mientras que la exposición a su metabolito activo disminuyo a la mitad. Un ajuste de dosis de UPTRAVI® puede ser necesario ·cuando se administra de manera concomitante con inductores de la CYP2C8 (ej., rifampicina, carbamacepina, fenitoína).
lnhibidores de la UGT1A3 y UGT287: El efecto de inhibidores potentes de la UGT1A3 y UGT2B7 (ácido valproico, probenecida y fluconazol), en la exposición a selexipag y su metabolito activo no ha sido estudiada. Se recomienda tener precaución cuando se administran estos productos de manera concomitante con UPTRAVI® dado que una interacción farmacocinética potencia con los inhibidores potentes de la UGT1A3 y UGT2B7 no puede excluirse.
lnhibidores e inductores de CYP3A4: En la presencia de un inhibidor potente como 400/100 mg de lopinavir/ritonavir dos veces al día, la exposición a selexipag se incrementó aproximadamente 2 veces, mientras que la exposición al metabolito activo de selexipag permaneció sin cambios. Dada la potencia 37 veces mayor del metabolito activo, este efecto no es clínicamente relevante. Dado que un inhibidor de la CYP3A4 potente no afectó la farmacocinética del metabolito activo, indicando que la vía del CYP3A4 no es relevante para la eliminación del metabolito activo, no se espera efecto farmacocinético alguno de los inductores del CYP3A4.
Tratamientos específicos para HAP: En el estudio fase 3, placebo controlado en pacientes con HAP, el uso de selexipag en combinación tanto de ERAs como de inhibidores de la PDE-5, resultaron en un 30% menos exposición al metabolito activo.
lnhibidores de transportadores (lopinavir/ritonavir): En la presencia de 400/100 mg de lopinavir/ritonavir dos veces al día, un inhibidor potente de OATP (OATP1B1 y OATP1B3) y de P-gp, la exposición a selexipag se incremento aproximadamente 2 veces, mientras que la exposición al metabolito activo no se modifico. Dado que la mayoría del efecto farmacológico esta dado por el metabolito activo, este efecto no se considera clínicamente relevante.
Efecto de Selexipag en otros medicamentos: Selexipag y su metabolito activo no inhiben o inducen las enzimas del citocromo P450 y el transporte de proteínas en concentraciones clínicamente relevantes. Selexipag y su metabolito activo no inhiben a las proteínas transportadoras. No se espera que Selexipag o su metabolito activo induzcan a las enzimas del citocromo P450 en hígado y riñón en concentraciones clínicamente relevantes. Datos in vitro indican que selexipag puede ser un inductor tanto del CYP3A4 como del CYP2C9 en intestino.
Anticoagulantes o inhibidores de la agregación plaquetaria: Selexipag es un inhibidor de la agregación plaquetaria in vitro. En un estudio fase 3, placebo controlado en pacientes con HAP, no se observó un incremento en el riesgo de sangrado en los pacientes tratados con selexipag comparado con placebo, incluyendo cuando se administró selexipag junto con anticoagulantes (como la heparina o la cumarina) o bien, con inhibidores de la agregación plaquetaria. En un estudio en sujetos sanos, selexipag (400 microgramos dos veces al día) no altero la exposición a S-warfarina (sustrato CYP2C9) o R-warfarina (sustrato CYP3A4) posterior a una dosis única de 20 mg de warfarina. Selexipag no tuvo influencia en el efecto farmacodinámico de la warfarina en el índice internacional normalizado.
Midazolam: En el estado estacionario, tras alcanzar la dosis máxima de 1,600 microgramos de Selexipag dos veces al día, no se observaron cambios clínicamente relevantes sobre la exposición a midazolam, sustrato sensible de CYP3A4 en el intestino y el hígado, o a su metabolito, 1- hidroximidazolam. La administración concomitante de Selexipag con sustratos de CYP3A4 no requiere ajuste de dosis.
Anticonceptivos Hormonales: No se han realizado estudios específicos de interacción medicamentosa con anticonceptivos hormonales. Dado que selexipag no afecta la exposición al CYP3A4 sustrato midazolam y R-warfarina o al sustrato CYP2C9 S-warfarina. No se espera una disminución en la eficacia de los anticonceptivos hormonales.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Disminución en la Hemoglobina: En un estudio fase 3, place controlado en pacientes con HAP, cambios promedio absolutos en la hemoglobina durante las visitas regulares comparadas con los valores basales fueron de entre -0.34 a -0.02 g/dL en el grupo de Selexipag comparado con -0.05 a 0.25 g/dL en el grupo placebo. Se reportó una disminución desde la basal en la concentración de hemoglobina menor a 1O g/dL en el 8.6% de los pacientes tratados con selexipag y en 5% de los pacientes tratados con placebo.
Pruebas de Función Tiroidea: En un estudio fase 3, placebo controlado en pacientes con HAP, se observó una disminución (de hasta -0.3 MU/L de la mediana en la basal de 2.5 MU/L) del valor promedio de la hormona estimulante de la tiroides (TSH) en casi todas las visitas en el grupo de selexipag. En el grupo placebo, pequeños cambios promedio en los valores fueron aparentes. No se presentaron cambios promedio en ninguno de los grupos para la triyodotironina y la tiroxina.
Incremento en la frecuencia cardiaca: En el estudio fase 3, placebo-controlado en pacientes con HAP, se observó un incremento transitorio en la frecuencia cardiaca media de 3-4 latidos por minuto (Ipm), 2-4 horas posterior a la dosis. La evaluación con electrocardiograma mostró una taquicardia sinusal en 11.3% de los pacientes con selexipag comparado con 8.8% de los pacientes en el grupo placebo.
PRECAUCIONES GENERALES:
Hipotensión: UPTRAVI® posee propiedades de vasodilatación que pueden dar como resultado una disminución en la presión sanguínea. Antes de prescribir UPTRAVI®,.el médico deberá considerar cautelosamente si el paciente con algunas condiciones subyacentes puede" verse afectado de manera adversa con un efecto de vasodilatación (ej., pacientes con tratamiento antihipertensivo o con hipotensión de reposo, hipovolemia, obstrucción severa al flujo ventricular izquierdo o disfunción autonómica).
Hipertiroidismo: La presencia de hipertiroidismo se ha observado con el uso de UPTRAVI®. Pruebas de función tiroidea son recomendadas de acuerdo a sintomatología clínica de hipertiroidismo.
Enfermedad Pulmonar Veno-oclusiva: Casos de Edema pulmonar han sido reportados con el uso de vasodilatadores (principalmente protaciclinas) cuando son usadas en pacientes con enfermedad pulmonar vena-oclusiva. En caso de presentarse síntomas de edema pulmonar, se debe considerar el posible diagnóstico de enfermedad pulmonar vena-oclusiva. En caso de confirmarse, UPTRAVI® deberá ser interrumpido.
lnhibidores moderados de la CYP2CB: La administración concomitante de UPTRAVI® con inhibidores de la CYP2C8 (ej., clopidrogel, deferasirox, teriflunomide), puede incrementar la exposición a selexipag y su metabolito activo. Deberá considerarse realizar algún ajuste de dosis de selexipag cuando un inhibidor de la CYP2C8 se administre de manera concomitante, o bien, descontinuar el tratamiento.
Información en poblciones especiales:
Uso en adultos mayores (>65 años de edad): Existe experiencia clínica limitada del uso de selexipag en pacientes mayores de 75 años de edad, por tal motivo, UPTRAVI® deberá ser empleado con precaución en esta población.
Insuficiencia Hepática: No existe experiencia clínica del uso de selexipag en pacientes con insuficiencia hepática severa (Child-Pugh clase C), por tanto, UPTRAVI® no deberá ser administrado en estos pacientes.
La exposición a selexipag y su metabolito activo se incrementa en sujetos con insuficiencia hepática moderada (Child-Pugh clase B). En los pacientes con insuficiencia hepática moderada, UPTRAVI® deberá de administrarse sólo una vez al día.
Insuficiencia Renal: En pacientes con insuficiencia renal severa (IFG < 30 ml/ min/1.73 m2 se deberá tener especial cuidado en el periodo de titulación. No existe experiencia con el uso de UPTRAVI® en pacientes sometidos a diálisis, por tanto, UPTRAVI® no deberá ser administrado a estos pacientes.
DOSIS Y VÍA DE ADMINISTRACIÓN: El tratamiento sólo puede ser iniciado y deberá ser monitoreado por un médico con experiencia en el tratamiento de la Hipertensión Arterial Pulmonar.
Dosis de titulación individualizada: Cada paciente deberá ser titulado a la dosis más alta tolerada, la cual puede variar de 200 microgramos administrada dos veces al día a 1600 microgramos administrada dos veces al día (dosis individualizada de mantenimiento).
La dosis inicial recomendada de UPTRAVI® es de 200 microgramos administrados dos veces al día en intervalos de aproximadamente 12 horas. La dosis deberá incrementarse en 200 microgramos administrados dos veces al día, usualmente en intervalos semanales. Al inicio del tratamiento y durante cada ajuste de dosis, se recomienda tomar la primera dosis por la noche. Durante el periodo de titulación pueden aparecer algunas reacciones adversas relacionadas con el mecanismo de acción de selexipag (por ejemplo cefalea, diarrea, náusea y vómito, dolor mandibular, mialgias, dolor en las extremidades, artralgias y rubicundez). Estos eventos normalmente son transitorios y manejables con tratamiento sintomático. Sin embargo, si un paciente alcanza una dosis que no es tolerada, la dosis deberá disminuirse a la dosis previa.
En pacientes en los que la titulación se vio limitada por cualquier razón diferente a la aparición de un evento adverso relacionado con el mecanismo de acción de UPTRAVI®, se deberá realizar un segundo intento de titulación a la dosis individualizada de mantenimiento más alta que sea tolerada, siendo la dosis máxima de 1,600 microgramos dos veces al día.
Dosis Individualizada de Mantenimiento: La dosis más alta tolerada que fue alcanzada durante la fase de titulación deberá mantenerse. Si con el tiempo, el tratamiento se torna menos tolerable a la dosis suministrada, se deberá considerar administrar tratamiento sintomático o bien, disminuir la dosis a la dosis inmediata inferior.
Interrupción y descontinuación del tratamiento: Sí una dosis es olvidada, deberá de tomarse a la brevedad posible. La dosis olvidada no deberá tomarse si será administrada prácticamente a la misma hora de la siguiente dosis (dentro de las 6 horas previas a la siguiente toma).
Si por algún motivo el tratamiento se interrumpe por 3 días o más, se deberá re-iniciar el tratamiento con UPTRAVI® en la menor dosis posible y volver a titularse.
Existe experiencia limitada con la descontinuación abrupta de UPTRAVI® en pacientes con HAP. Si se toma la decisión de suspender UPTRAVI®, éste debe de descontinuarse de manera gradual en lo que se administra una terapia alternativa.
Dosis en poblaciones especiales:
Adultos Mayores(> 65 años): No se requiere ajuste de dosis en adultos mayores. Existe experiencia clínica limitada en pacientes mayores de 75 años de edad, por tanto, UPTRAVI® deberá emplearse con precaución en esta población.
Insuficiencia Hepática: UPTRAVI® no debe de administrarse en pacientes con insuficiencia hepática severa (Child-Pugh Clase C). En pacientes con insuficiencia hepática moderada (Child-Pugh Clase B), la dosis de inicio de UPTRAVI® deberá ser de 200 microgramos una vez al día e incrementarlo semanalmente en 200 microgramos una vez al día hasta que un evento adverso relacionado con el mecanismo de acción de selexipag aparezca y no pueda ser tolerado o medicamente manejado. No se requiere ajuste de dosis en pacientes con insuficiencia hepática leve (Child-Pugh clase A).
Insuficiencia renal: No se requiere ajuste de dosis en pacientes con insuficiencia renal leve o moderada. No se requiere modificar la dosis de inicio en pacientes con insuficiencia renal severa (tasa de filtración glomerular estimada <30 ml/ min/1.73 m2); sin embargo, se deberá tener precaución durante el periodo de titulación en estos pacientes.
Población pediátrica(< 18 años): La seguridad y eficacia de UPTRAVI® en niños menores de 18 años de edad no ha sido estudiada. No existe información disponible, por lo tanto, la administración de selexipag no esta recomendada. Estudios animales indican un riesgo incrementado de intususcepción; sin embargo, se desconoce la relevancia clínica de este hallazgo.
Método de administración: Uso oral.
Las tabletas deberán tomarse por vía oral en la mañana y en la noche. Con el fin de mejorar la tolerabilidad, se recomienda tomar UPTRAVI® junto con los alimentos y, con cada nueva dosis durante el periodo de titulación, se recomienda tomar esta nueva dosis por las noches.
Las tabletas no deben partirse, triturarse o masticarse y deberán de tragarse enteras con un poco de agua.
Pacientes con poblemas importantes de visión o ceguera deben ser asistidos por otra persona al tomar UPTRAVI®, especialmente durante el periodo de titulación.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Se reportaron casos aislados de sobredosis de hasta 3200 μg. El único evento reportado fue náusea leve y transitoria. En caso de sobredosis, medidas de soporte general deberán ser administradas según se requieran. Es poco probable que la diálisis resulte efectiva dado que selexipag y su metabolito activo tienen una unión a proteínas muy fuerte.
PRESENTACIONES: Caja de cartón con 60 tabletas. Cada tableta contiene 200, 400, 600, 800, 1000, 1200, 1400 o 1600 microgramos de selexipag.
Caja de cartón con 10 y 140 tabletas de 200 microgramos de Selexipag. (empaque de ajuste de dosis) En envase de burbuja y con instructivoanexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 30ºC.
Consérvese la caja bien cerrada.
No utilizar UPTRAVI® después de la fecha de caducidad que está especificada en la caja de cartón y en el blíster después de la abreviatura "EXP". La fecha de caducidad se refiere al último día de ese mes.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Este producto debe ser prescrito y supervisado por un especialista relacionado con el diagnóstico y tratamiento de HAP. Su venta requiere receta médica. No se utilice durante el embarazo y la lactancia (logo-símbolo). No se deje al alcance de los niños. La eficacia y seguridad de UPTRAVI® no ha sido demostrada en niños.
Reportar las sospechas de reacción adversa al correo electrónico: farmacovigilancia@cofepris.gob.mx y drugsafetymx@actelion.com
Fabricado en Alemania por:
Excella GmbH,
Nürnberger Strasse 12
90537, Feucht,
Alemania
Acondicionado por:
Allpack Group AG,
Pfeffingerstrasse 45
4153 Reinach BL, Suiza
Para:
Actelion Pharmaceuticals Ltd.
Gewerbertrasse 14/16
4123 Allschwil, Switzerland,
Importado y Distribuido por:
ACTELION PHARMACEUTICALS MÉXICO S.A. de C.V.
Calzada Camarones No. 14 lnt. 4
Col. San Salvador Xochimanca,
Deleg. Azcapotzalco,
C.P. 02870, D.F., México
Representante legal:
Actelion Pharmaceuticals México S.A. de C.V.
Diego Rivera No. 40
Col. Altavista, Deleg. Álvaro Obregón
C. P. 01060, D.F., México
®Registered Trademark