
VENTANA PD-L1 (SP142) ASSAY
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
INTERPRETACIÓN DE LAS TINCIONES Y RESULTADOS PREVISTOS:
El procedimiento de inmunotinción automatizada VENTANA hace que un producto de reacción de color marrón DAB se precipite en los sitios del antígeno localizados por el anticuerpo de VENTANA PD-L1 (SP142) ASSAY. El portaobjetos con tinción lo debe interpretar un anatomopatólogo cualificado con microscopía óptica. Un anatomopatólogo cualificado con experiencia en procedimientos de inmunohistoquímica (IHC) debe evaluar los controles tisulares y calificar el producto con tinción antes de interpretar los resultados.
Interpretación del control tisular de amígdala:
El control tisular de amígdala con tinción se debe estudiar para comprobar que la tinción es adecuada. La presencia de tinción de PD-L1 en los macrófagos y los linfocitos de los centros germinales y en el epitelio de la cripta reticulada de la amígdala actúan como elementos de tejido positivos. La ausencia de tinción en el epitelio escamoso superficial y en las células inmunitarias negativas de las regiones interfoliculares de la amígdala actúan como elementos negativos del tejido. Los criterios de aceptabilidad se enumeran en la Tabla 5. (Consulte las guías de interpretación para obtener más información al respecto.)
Si el control tisular no presenta la tinción correspondiente, los resultados de las muestras del paciente se deben considerar no aptos para su evaluación y será preciso repetir la tinción.
Tabla 5. Criterios de evaluación del control tisular de amígdala
|
Aceptable |
Inaceptable |
|
Elementos de tejido positivos: Presencia de tinción de moderada a fuerte de PD-L1 en linfocitos y macrófagos de los centros germinales con tinción difusa en células epiteliales de la cripta reticulada. |
Tinción de fondo no específica excesiva que impide la identificación de las células positivas en PD-L1. |
|
Elementos de tejido negativos: Células inmunitarias negativas en PD-L1 en las regiones interfoliculares con epitelio escamoso superficial negativo. |
Presencia de tinción de moderada a nula de PD-L1 en linfocitos y macrófagos de los centros germinales y en células epiteliales de la cripta reticulada. |
Control de reactivo negativo:
La tinción no específica, de estar presente, tiene un aspecto difuso y se puede evaluar mediante el portaobjetos de control de reactivo negativo teñido con Rabbit Monoclonal Negative Control Ig. Se deben utilizar células intactas para la interpretación de los resultados de tinción, ya que la tinción de las células necróticas o degeneradas suele ser no específica. Si la tinción de fondo es excesiva, los resultados de las muestras de la prueba se deben considerar como no válidos. Los ejemplos de tinción de fondo de este ensayo se pueden encontrar en las guías de interpretación.
Tejido del paciente:
Las células tumorales (TC) se puntúan en función del porcentaje de células tumorales con presencia de tinción de membrana de PD-L1 apreciable de cualquier intensidad. Las células inmunitarias que infiltran el tumor (IC) se puntúan en función de la proporción del área tumoral, incluidos el estroma peritumoral asociado y contiguo, que ocupa la tinción con PD-L1 de IC de cualquier intensidad. Es preciso evaluar el tejido del paciente de acuerdo con el algoritmo de puntuación VENTANA PD-L1 (SP142) ASSAY específico de la indicación que se suministra en la sección Características de rendimiento para esa indicación. Consulte la guía de interpretación específica de la indicación para obtener instrucciones adicionales e imágenes representativas.
ALMACENAMIENTO Y ESTABILIDAD:
Tras la recepción y cuando no se utilice, consérvese de 2-8°C. No lo congele.
Para garantizar una dispensación adecuada del reactivo y la estabilidad del anticuerpo, vuelva a poner el tapón del dispensador después de cada uso y almacene inmediatamente el dispensador en la nevera, en posición vertical.
Todos los dispensadores de anticuerpos tienen una fecha de caducidad. Si se almacena correctamente, el reactivo se mantendrá estable hasta la fecha indicada en la etiqueta. No usar el reactivo después de la fecha de caducidad.
ADVERTENCIAS Y PRECAUCIONES:
1. Para uso diagnóstico in vitro (IVD).
2. Solo para uso profesional.
3. No utilizar por encima del número especificado de ensayos.
4. Los portaobjetos con carga positiva pueden verse afectados por presiones ambientales, dando lugar a una tinción incorrecta. Póngase en contacto con su representante de servicio de Roche para obtener más información sobre el uso de este tipo de portaobjetos.
5. Los materiales de origen animal o humano deben manipularse como materiales biopeligrosos y eliminarse con las precauciones adecuadas. En caso de exposición, deberán seguirse las directivas sanitarias de las autoridades responsables.13,14
6. Evite el contacto de los reactivos con los ojos y las membranas mucosas. Si los reactivos entran en contacto con zonas sensibles, lávelas con agua abundante.
7. Evite la contaminación microbiana de los reactivos, dado que podría dar lugar a resultados incorrectos.
8. Para obtener más información sobre el uso de este dispositivo, consulte el Manual del usuario del instrumento BenchMark IHC/ISH y las instrucciones de uso de todos los componentes necesarios que puede encontrar en navifyportal.roche.com.
9. Consultar a las autoridades locales y/o estatales sobre el método de eliminación recomendado.
10. El etiquetado de seguridad de los productos sigue principalmente las directrices del SGA de la UE. Está disponible bajo petición la hoja de datos de seguridad para los usuarios profesionales.
11. Para comunicar la sospecha de incidentes graves relacionados con este dispositivo, póngase en contacto con su representante local de servicio Roche y con las autoridades competentes del Estado o País Miembro de residencia del usuario.
MATERIAL SUMINISTRADO:
VENTANA PD-L1 (SP142) ASSAY contiene reactivo suficiente para 50 pruebas.
Un dispensador de 5 mL de VENTANA PD-L1 (SP142) ASSAY contiene aproximadamente 36 μg de anticuerpo monoclonal de conejo.
El anticuerpo se diluye en un tampón salino formado por Tris, EDTA y Brij-35 con una proteína transportadora y azida sódica, un conservante.
La concentración del anticuerpo específico es aproximadamente de 7 μg/mL.
VENTANA PD-L1 (SP142) ASSAY contiene un recombinante de anticuerpo monoclonal de conejo producido como sobrenadante de un cultivo celular purificado.
Consulte la guía de interpretación correspondiente para obtener instrucciones detalladas en cuanto a la interpretación de la tinción con VENTANA PD-L1 (SP142) ASSAY para indicaciones específicas:
• VENTANA PD-L1 (SP142) ASSAY Interpretation Guide for Urothelial Carcinoma (P/N 1015704)
• VENTANA PD-L1 (SP142) ASSAY Interpretation Guide for NSCLC ≥ 50% TC or ≥ 10% IC Stepwise Scoring Algorithm (P/N 1015703)
• VENTANA PD-L1 (SP142) ASSAY Interpretation Guide for NSCLC ≥ 1% TC or ≥ 1% IC Stepwise Scoring Algorithm (P/N 1015654)
• VENTANA PD L1 (SP142) ASSAY Interpretation Guide for TNBC (P/N 1018231)
Consulte la hoja de datos del kit de detección VENTANA correspondiente para obtener descripciones detalladas de los siguientes aspectos: Principio del procedimiento, Material y métodos, Recogida y preparación de muestras para análisis, Procedimientos de control de calidad, Resolución de problemas, Interpretación de los resultados y Limitaciones.
MATERIALES NECESARIOS PERO NO SUMINISTRADOS:
No se suministran reactivos de tinción, como kits de detección VENTANA, ni componentes auxiliares, incluyendo portaobjetos de control de tejido negativos y positivos.
Puede que no todos los productos que aparecen en la hoja de datos estén disponibles en todos los lugares. Consulte al representante local de asistencia técnica de Roche.
No se suministran los reactivos y materiales siguientes, pero pueden ser necesarios para la tinción:
1. Tejidos de amígdala humana benignos para su uso como tejido de control
2. Rabbit Monoclonal Negative Control Ig (nº de cat. 790-4795/06683380001)
3. Portaobjetos para microscopio con carga positiva
4. OptiView DAB IHC Detection Kit (nº de cat. 760-700/06396500001)
5. OptiView Amplification Kit (nº cat. 760-099/06396518001 [50 pruebas] o nº cat. 860-099/06718663001 [250 pruebas])
6. EZ Prep Concentrate (10X) (nº cat. 950-102/05279771001)
7. Reaction Buffer Concentrate (10X) (nº cat. 950-300/05353955001)
8. LCS (Predilute) (nº cat. 650-010/05264839001)
9. ULTRA LCS (Predilute) (n.º cat. 650-210/05424534001)
10. Cell Conditioning Solution (CC1) (n.º cat. 950-124/05279801001)
11. ULTRA Cell Conditioning Solution (ULTRA CC1) (nº cat. 950-224/05424569001)
12. Hematoxylin II (nº cat. 790-2208/05277965001)
13. Bluing Reagent (nº de cat. 760-2037/05266769001)
14. Equipo de laboratorio de uso general
15. Instrumento BenchMark IHC/ISH
PROCEDIMIENTOS DE CONTROL DE CALIDAD:
Rabbit Monoclonal Negative Control Ig:
Se debe utilizar el correspondiente portaobjetos de control de reactivo negativo en cada muestra como ayuda para la interpretación de los resultados. Rabbit Monoclonal Negative Control Ig es el anticuerpo de control de reactivo negativo específico para este ensayo y se utiliza en lugar del anticuerpo primario para evaluar una tinción no específica. El procedimiento de tinción del control de reactivo negativo debe ser idéntico al periodo de incubación del anticuerpo primario. El uso de un reactivo de control negativo diferente o no usar el reactivo de control negativo recomendado puede dar lugar a la obtención de resultados erróneos.
Control tisular de amígdala:
Se debe incluir un control tisular en cada sesión de tinción. Es preciso utilizar el tejido de amígdala humano benigno cualificado como control. El tejido de control se debe fijar y procesar con la mayor inmediatez posible de forma idéntica a la que se ha utilizado con los tejidos del paciente. Estos tejidos se utilizan para hacer un seguimiento de todos los pasos que conlleva el proceso, desde la preparación del tejido hasta la tinción. El tejido de amígdala contiene elementos de tinción positivos y negativos para la proteína PD-L1 y, por tanto, es adecuado para su uso como control tisular. Los componentes de tinción positiva y negativa del tejido sirven para comprobar que el ensayo ha funcionado correctamente.
La tinción correcta de los componentes de tejido de amígdala se describe en la Tabla 5 y en la guía de interpretación.
Verificación del ensayo:
Antes de comenzar a utilizar un anticuerpo o un sistema de tinción en un procedimiento diagnóstico, se debe comprobar la especificidad del anticuerpo mediante pruebas en una serie de tejidos que contengan características de rendimiento en inmunohistoquímica (IHC) conocidas y que reflejen tejidos positivos y negativos en PD-L1 (consulte la sección Procedimientos de control de calidad que se ha mencionado anteriormente en esta sección y que se encuentra en la hoja de datos del producto y las recomendaciones sobre control de calidad de College of American Pathologists Laboratory Accreditation Program, Anatomic Pathology Checklist15 o CLSI Approved Guideline16). Estos procedimientos de control de calidad deben repetirse para cada nuevo lote de anticuerpo nuevo o cuando se produzca un cambio en los parámetros del ensayo. Los tejidos de carcinoma urotelial, NSCLC y TNBC con estado de PD-L1 conocido y las muestras de amígdala humana benigna son aptos para la verificación del ensayo.
CARACTERÍSTICAS DE RENDIMIENTO:
Rendimiento de análisis: generalidades:
Se llevaron a cabo pruebas de tinción para evaluar la sensibilidad, la especificidad, el efecto del grosor del tejido, la repetibilidad y la precisión intermedia, así como pruebas de precisión del lector, reproducibilidad entre laboratorios y resultados clínicos, y los resultados de todas ellas se enumeran a continuación.
Comentarios generales sobre el análisis:
A menos que se especifique lo contrario, se ha calculado el intervalo de confianza bilateral del 95% (CI) en torno a las estimaciones de concordancia de todos los estudios (salvo los estudios de eficacia clínica) mediante el método bootstrap percentil de 2000 muestras bootstrap. Si la estimación de punto del porcentaje de concordancia positiva (PPA), del porcentaje de concordancia negativa (NPA) o del porcentaje de concordancia global (OPA) es de 0% o del 100%, se debe calcular el 95% de CI mediante el método de puntuación de Wilson. Si la estimación de punto del promedio de concordancia positiva (APA) y del promedio de concordancia negativa (ANA) es de 0% o del 100%, se debe calcular el 95% de CI mediante el método de puntuación de Wilson por transformación.
Sensibilidad y especificidad:
Se llevó a cabo la tinción con VENTANA PD-L1 (SP142) ASSAY de arrays que contenían una variedad de tejidos normales y neoplásicos y se evaluaron para detectar la presencia de tinción de células inmunitarias (todo tipo de tinción de células inmunitarias, de cualquier intensidad) tal y como se describe en la Tabla 6 y la Tabla 7.
Tabla 6. La sensibilidad/especificidad de VENTANA PD-L1 (SP142) ASSAY se determinó analizando tejidos normales FFPE
|
Tejido |
Nº de casos positivosa/ total |
Tejido |
Nº de casos positivosa/total |
|---|---|---|---|
|
Cerebro |
0/3 |
Timo |
3/3 |
|
Cerebeloc |
0/3 |
Médula ósea |
0/2 |
|
Glándula suprarrenal |
1/3 |
Pulmón |
1/25 |
|
Ovario |
0/3 |
Corazón |
0/3 |
|
Páncreasd |
0/3 |
Esófago |
0/3 |
|
Glándula paratiroidea |
0/2 |
Estómago |
0/3 |
|
Glándula pituitariad |
0/3 |
Intestino delgado |
1/3 |
|
Testículosc |
0/3 |
Colon |
2/3 |
|
Tiroides |
1/3 |
Hígado |
0/3 |
|
Mama |
1/66 |
Glándula salival |
2/4 |
|
Bazo |
3/3 |
Riñón |
2/3 |
|
Amígdalac |
3/3 |
Vejigab |
3/36 |
|
Ganglio linfático |
3/3 |
Próstata |
0/3 |
|
Endometrio |
2/3 |
Cuello del útero |
0/2 |
|
Músculo esquelético |
0/2 |
Piel |
0/3 |
|
Nervio |
0/3 |
Mesotelio |
0/3 |
a Tinción de células inmunitarias de cualquier intensidad
b Tinción focal de células inmunitarias
c Se observó la presencia de puntos DAB focales en 1/3 tejidos de cerebelo, 1/3 tejidos de testículos y en el control de amígdala normal
d Se observó tinción nuclear en 1/3 tejidos de páncreas y en 1/3 tejidos de glándula pituitaria
Tabla 7. La sensibilidad/especificidad de VENTANA PD-L1 (SP142) ASSAY se determinó analizando una variedad de tejidos neoplásicos FFPE
|
Patología |
Nº de casos |
|
|---|---|---|
|
Células inmunitarias |
Células tumorales |
|
|
Glioblastoma (cerebro) |
1/1 |
0/1 |
|
Meningioma (cerebro) |
0/1 |
0/1 |
|
Ependimoma (cerebro) |
0/1 |
0/1 |
|
Oligodendroglioma (cerebro) |
0/1 |
0/1 |
|
Adenocarcinoma seroso (ovario) |
1/1 |
0/1 |
|
Adenocarcinoma (ovario) |
1/1 |
0/1 |
|
Neoplasia neuroendocrina (páncreas) |
0/1 |
0/1 |
|
Adenocarcinoma (páncreas) |
1/1 |
0/1 |
|
Seminoma (testículos) |
1/1 |
0/1 |
|
Carcinoma embrionario (testículos) |
0/1 |
0/1 |
|
Carcinoma medular (tiroides) |
0/1 |
0/1 |
|
Carcinoma papilar (tiroides) |
0/1 |
1/1 |
|
Carcinoma ductal microinvasivo (mama) |
1/1 |
0/1 |
|
Carcinoma ductal invasivo (mama) |
1/2 |
0/2 |
|
Linfoma de linfocitos B, sin especificar (bazo)a |
1/1 |
1/1 |
|
Carcinoma de células pequeñas (pulmón) |
1/1 |
1/1 |
|
Carcinoma de células escamosas (pulmón) |
1/1 |
0/1 |
|
Adenocarcinoma (pulmón) |
0/1 |
0/1 |
|
Carcinoma neuroendocrino (esófago) |
0/1 |
0/1 |
|
Adenocarcinoma (esófago) |
1/1 |
0/1 |
|
Carcinoma de células en anillo de sello (Estómago) |
1/1 |
0/1 |
|
Adenocarcinoma (intestino delgado) |
1/1 |
0/1 |
|
Tumor estromal gastrointestinal (GIST) (intestino delgado) |
1/1 |
0/1 |
|
Tumor estromal gastrointestinal (GIST) (colon) |
0/1 |
0/1 |
|
Adenocarcinoma (colon) |
1/1 |
0/1 |
|
Adenocarcinoma (recto) |
1/1 |
1/1 |
|
Tumor estromal gastrointestinal (GIST) (recto) |
0/1 |
0/1 |
|
Melanoma (recto) |
1/1 |
0/1 |
|
Carcinoma hepatocelular (hígado) |
0/1 |
0/1 |
|
Hepatoblastoma (hígado) |
1/1 |
0/1 |
|
Carcinoma de células claras (riñón) |
1/1 |
0/1 |
|
Adenocarcinoma (próstata) |
0/2 |
0/2 |
|
Leiomioma (útero) |
0/1 |
0/1 |
|
Adenocarcinoma (útero) |
1/1 |
0/1 |
|
Carcinoma de células claras (útero) |
1/1 |
1/1 |
|
Carcinoma de células escamosas (cuello uterino) |
2/2 |
0/2 |
|
Rabdomiosarcoma embrionario (músculo estriado) |
0/1 |
0/1 |
|
Carcinoma de células basales (piel) |
1/1 |
0/1 |
|
Carcinoma de células escamosas (piel) |
1/1 |
0/1 |
|
Neurofibroma (lumbar) |
1/1 |
0/1 |
|
Neuroblastoma (retroperitoneo) |
1/1 |
0/1 |
|
Rabdomiosarcoma de células fusiformes (retroperitoneo) |
0/1 |
0/1 |
|
Mesotelioma (peritoneo) |
1/1 |
0/1 |
|
Linfoma de linfocitos B (ganglio linfático)b |
1/1 |
1/1 |
|
Linfoma de Hodgkin (ganglio linfático) |
1/1 |
1/1 |
|
Linfoma de linfocitos B, sin especificar (mediastino)b |
1/1 |
1/1 |
|
Linfoma anaplásico de células grandes (ganglio linfático)b |
1/1 |
1/1 |
|
Carcinoma urotelial (vejiga) |
1/1 |
0/1 |
|
Leiomiosarcoma (vejiga) |
0/1 |
0/1 |
|
Osteosarcoma (hueso) |
0/1 |
0/1 |
|
Leiomiosarcoma (músculo liso) |
1/1 |
0/1 |
a Tinción de células inmunitarias o de células tumorales de cualquier intensidad.
b No se pudo distinguir la tinción de células tumorales de la de células inmunitarias.
RENDIMIENTO CLÍNICO: CARCINOMA UROTELIAL:
Se investigó el rendimiento clínico del VENTANA PD-L1 (SP142) ASSAY dentro del marco de IMvigor210 (NCT02951767), un ensayo internacional multicéntrico en fase II con dos cohortes y un solo grupo, diseñado para evaluar la eficacia de TECENTRIQ (atezolizumab) en pacientes con carcinoma urotelial localmente avanzado o metastásico. En la cohorte 1 había pacientes que no habían recibido tratamiento previo con carcinoma urotelial metastásico o localmente avanzado que no cumplían los criterios de idoneidad o no eran aptos para recibir tratamiento con quimioterapia con cisplatino. En la Cohorte 1,119 pacientes recibieron tratamiento con 1,200 mg de TECENTRIQ (atezolizumab) por vía intravenosa cada tres semanas hasta que se alcanzara un nivel de toxicidad inaceptable o hasta la progresión de la enfermedad. Las muestras tumorales de todos los pacientes cribados en el estudio IMvigor210 se evaluaron prospectivamente mediante VENTANA PD-L1 (SP142) ASSAY en un laboratorio central y los resultados sirvieron para definir los subgrupos de análisis especificados previamente. La medida primaria de resultado de eficacia fue la tasa de respuesta objetiva (ORR) confirmada según la evaluación de un centro de revisión independiente (IRF) mediante los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST v1.1).
El criterio de valoración de la eficacia primario de la Cohorte 1 de IMvigor210 fue la ORR confirmada según la evaluación de un IRF mediante RECIST v1.1. El análisis primario se llevó a cabo cuando todos los pacientes habían recibido un seguimiento de, al menos, 24 semanas. La duración media del tratamiento fue de 15.0 semanas y la duración media del seguimiento de supervivencia fue de 8.5 meses para todos los participantes. Se observaron ORR evaluadas por un IRF con importancia clínica según RECIST v1.1; sin embargo, cuando se compararon con la tasa de respuesta de control histórica especificada previamente del 10%, no se alcanzó la importancia estadística del criterio de valoración primario. Las ORR confirmadas según RECIST v1.1 del IRF fueron del 21.9% (CI del 95%: 9.3, 40.0) en los pacientes con expresión de PD-L1 ≥ 5%, 18.8% (CI del 95%: 10.9, 29.0) en los pacientes con expresión de PD-L1 ≥ 1% y 19.3% (CI del 95%: 12.7, 27.6) en todos los participantes. La duración de la respuesta (DOR) promedio no se alcanzó en ningún subgrupo con expresión de PD-L1 ni en el total de los participantes. A pesar de que la ORR no alcanzó el punto de importancia clínica, los resultados de la Cohorte 1 de IMvigor210 mostraban respuestas de importancia clínica de la monoterapia con TECENTRIQ (atezolizumab) con independencia de los niveles de expresión de PD-L1, tal y como se detectó mediante el VENTANA PD-L1 (SP142) ASSAY.
Se está estudiando el rendimiento de VENTANA PD-L1 (SP142) ASSAY en IMvigor130 (NCT02807636), un estudio multicéntrico, abierto y aleatorizado que se ha diseñado para evaluar la eficacia de TECENTRIQ (atezolizumab) en pacientes con carcinoma urotelial metastásico a los que no se ha puesto tratamiento previo y que cumplen los criterios de idoneidad para recibir quimioterapia con platino. El estudio está formado por tres grupos: Monoterapia con TECENTRIQ (atezolizumab), TECENTRIQ (atezolizumab) con quimioterapia con platino (como cisplatino o carboplatino con gemcitabina) y únicamente la quimioterapia a base de platino (comparador). Se incorporaron al estudio tanto los pacientes idóneos para recibir cisplatino como los que no eran considerados aptos para recibir cisplatino. Los criterios de valoración de la eficacia coprimarios para la población con intención de tratar (ITT) son la supervivencia libre de progresión (PFS) y la supervivencia global (OS) evaluadas por un investigador según los criterios de investigación RECIST v1.1.
Las muestras de tumores se evaluaron prospectivamente mediante el ensayo VENTANA PD-L1 (SP142) ASSAY en un laboratorio central. El comité de control de datos independiente (iDMC) del estudio llevó a cabo una revisión de los datos de fases tempranas que concluían que, en los pacientes que se habían clasificado por presentar una expresión < 5% de PD-L1 se reducía la supervivencia cuando recibían tratamiento con monoterapia de TECENTRIQ (atezolizumab) en comparación con aquellos que recibían quimioterapia a base de platino.
En un análisis exploratorio de la OS (CCOD de junio de 2020) según el estado de PD-L1 en pacientes que no cumplían los criterios de idoneidad para recibir tratamiento con cisplatino según los criterios de Galsky, se observó una mejora de la OS en pacientes cuyo estado tumoral de PD-L1 rea más elevado en el grupo de la monoterapia con atezolizumab (Atezo Mono) en comparación con el grupo del placebo + quimioterapia (Tabla 13). Entre los pacientes que no cumplían los criterios de idoneidad para recibir el tratamiento con cisplatino y que presentaban expresión de PD-L1 ≥ 5%, el HR sin estratificar de la comparación entre Atezo Mono y placebo y quimioterapia fue del 0.60 (CI del 95%: 0.36, 1.01). La OS media estimada mediante Kaplan-Meier fue de 18.63 meses (CI del 95%: 14.0, NE) en el grupo de Atezo Mono (n= 50) y de 9.95 meses (CI del 95%: 7.36, 18.1) en el grupo del placebo y quimioterapia (n= 43). En el subgrupo de los pacientes que no cumplían con los criterios de idoneidad para recibir el tratamiento con cisplatino y que presentaban expresión de PD-L1 < 5%, el HR sin estratificar de la comparación entre Atezo Mono y placebo y quimioterapia fue del 1.15 (CI del 95%: 0.87, 1.52). La OS media estimada mediante Kaplan-Meier fue de 11.2 meses (CI del 95%: 6.87, 14.65) en el grupo de Atezo Mono (n= 140) y de 11.17 meses (CI del 95%: 9.89, 14.26) en el grupo del placebo y quimioterapia (n= 140).
Tabla 13. Resumen de los análisis exploratorios: supervivencia global mediante el estado de PD-L1 entre los pacientes que no cumplían los criterios de idoneidad para recibir tratamiento con cisplatino (según los criterios de Galsky)
|
Supervivencia global (OS) |
Atezo Mono |
Placebo y quimioterapia |
|
PD-L1 ≥ 5% |
(N = 50) |
(N = 43) |
|
Nº de muertes (%) |
28 (56.0) |
30 (69.8) |
|
Tiempo promedio hasta los eventos, meses (CI del 95%)a,b |
18.63 (14.0, NE) |
9.95 (7.36, 18.1) |
|
Índice de riesgo sin estratificar (CI del 95%)c,b |
0.60 (0.36, 1.01) |
|
|
Valor pd |
0.0509 |
|
|
PD-L1 < 5% |
(N = 140) |
(N = 140) |
|
Nº de muertes (%) |
105 (75.0) |
96 (68.6) |
|
Tiempo promedio hasta los eventos, meses (CI del 95%)a,b |
11.2 (6.87, 14.65) |
11.17 (9.89, 14.26) |
|
Índice de riesgo sin estratificar (CI del 95%)c,b |
1.15 (0.87, 1.52) |
|
|
Valor pd |
0.3229 |
|
a Los resúmenes de la duración (promedio y percentiles) se han calculado mediante Kaplan-Meier.
b Los CI del 95% de la media se computan con el método de Brookmeyer y Crowley.
c Los índices de riesgo se calcularon mediante la regresión de Cox.
d Los resultados se presentan únicamente con fines informativos.
CI = Intervalo de confianza; NE = Imposible de estimar; CCOD=14 de junio de 2020.
El iDMC recomendó cerrar el grupo de monoterapia a la incorporación de pacientes con un nivel de expresión bajo de PD-L1; sin embargo, no se recomendó ningún otro cambio en el estudio; ni siquiera cambios de tratamiento para los pacientes a los que ya se había asignado de forma aleatoria al grupo de monoterapia y estaban recibiendo tratamiento en él.
El estudio IMvigor130 sigue actualmente en curso y se espera disponer de los resultados de la eficacia de TECENTRIQ (atezolizumab) en comparación con la quimioterapia en la población con expresión de PD-L1 en ≥ 5% de IC en la lectura final del estudio. La actual designación de las pruebas diagnósticas con fines terapéuticos se debe al mencionado hallazgo del iDMC, que señala una menor supervivencia de los pacientes con una expresión de PD-L1 < 5% en CI que reciben tratamiento con TECENTRIQ (atezolizumab). Se evaluarán los resultados en la lectura final de IMvigor130 para determinar si se observa una mayor eficacia de TECENTRIQ (atezolizumab) en la población con expresión de PD-L1 ≥ 5% de IC y se actualizará en consecuencia la justificación de las pruebas diagnósticas con fines terapéuticos.
RENDIMIENTO CLÍNICO: NSCLC ≥ 50% TC 0 ≥ 10% IC:
Se estudió el rendimiento del ensayo VENTANA PD-L1 (SP142) ASSAY en IMpower110 (NCT02409342), un ensayo abierto en fase III, multicéntrico, internacional y aleatorizado con 572 pacientes que padecen NSCLC en estadio IV, entre otros, pacientes con anomalías tumorales genómicas de EGFR o ALK, que no habían recibido anteriormente tratamiento con quimioterapia para enfermedades metastásicas. El estudio se diseñó para evaluar la seguridad y eficacia de TECENTRIQ (atezolizumab) en relación con la quimioterapia consistente en un agente platino (cisplatino o carboplatino a criterio del investigador) en combinación con pemetrexed (enfermedad no escamosa) o gemcitabina (enfermedad escamosa).
El estudio IMpower110 se llevó a cabo con el fin de evaluar la eficacia y la seguridad de TECENTRIQ (atezolizumab) en pacientes que no habían recibido nunca quimioterapia y que padecían NSCLC metastásico. En los pacientes se presentaba una expresión de PD-L1 ≥ 1% de TC (tinción de PD-L1 en ≥ 1% de las células tumorales) o ≥ 1% de IC (tinción de PD-L1 de células inmunitarias que infiltran el tumor que ocupaban ≥ 1% del área del tumor), según el ensayo VENTANA PD-L1 (SP142) ASSAY.
Se aleatorizaron un total de 572 pacientes en una proporción 1:1 para recibir un tratamiento con TECENTRIQ (atezolizumab) (Grupo A) o quimioterapia (Grupo B). Se administró una dosis fija de TECENTRIQ (atezolizumab) de 1200 mg mediante infusión intravenosa cada tres semanas hasta la pérdida del beneficio clínico según la evaluación del investigador o hasta alcanzar una toxicidad inaceptable. La aleatorización se clasificó por género, estado de rendimiento ECOG, histología y expresión tumoral de PD-L1 en TC o en IC.
Se equilibraron adecuadamente entre los grupos del tratamiento los datos demográficos y las características iniciales de la enfermedad de los pacientes con expresión de PD-L1 ≥ 1% de TC o ≥ 1% de IC que no presentaban mutaciones de EGFR o reorganizaciones de ALK (n= 554). La media de edad era de 64.5 años (intervalo de entre 30 y 87 años) y el 70% de los pacientes eran hombres. La mayoría de los pacientes eran blancos (84%) y asiáticos (14%). La mayor parte de los pacientes eran fumadores o lo habían sido con anterioridad (87%) y el estado de rendimiento ECOG inicial de los pacientes era 0 (36%) y 1 (64%). En general, el 69% de los pacientes padecía una enfermedad no escamosa y el 31% de ellos, escamosa. Se equilibraron adecuadamente entre los grupos del tratamiento los datos demográficos y las características iniciales de la enfermedad de los pacientes con elevada expresión de PD-L1 (PD-L1 ≥ 50% TC o ≥ 10% de IC) que no presentaban mutaciones de EGFR o reorganizaciones de ALK (n= 205) y se consideraron representativos de la población más amplia del estudio.
El criterio de valoración primario era la supervivencia global (OS). En el momento del análisis de OS provisional, los pacientes con un elevado nivel de expresión de PD-L1, salvo los que padecían mutaciones de EGFR o reorganizaciones de ALK (n= 205), presentaban una mejoría significativa estadísticamente en la OS en aquellos pacientes a los que se había aplicado aleatoriamente el tratamiento con TECENTRIQ (atezolizumab) (Grupo A) en comparación con aquellos a los que se había administrado quimioterapia (grupo B) (HR de 0.59, CI del 95%: 0.40, 0.89; OS media de 20.2 meses frente a 13.1 meses) con un valor p bilateral de 0.0106. El tiempo de seguimiento de la supervivencia promedio en pacientes con niveles altos de expresión de PD-L1 fue de 15.7 meses. Los resultados clave del análisis provisional se resumen en la Tabla 26.
Tabla 26. Resumen de la eficacia en pacientes con nivel elevado de expresión de PD-L1 ≥ 50% en TC o ≥ 10% en IC (IMpower110)
|
Supervivencia global (OS) |
Atezolizumab |
Quimioterapia |
|
Nº de muertes (%) |
44 (41.1%) |
57 (58.2%) |
|
Tiempo promedio hasta los eventos, meses (CI del 95%) |
20.2 (16.5, NE) |
13.1 (7.4, 16.5) |
|
Índice de riesgo estratificadoa (CI del 95%) |
0.59 (0.40, 0.89) |
|
|
Valor pa |
0.0106 |
|
|
OS de 12 meses (%) |
64.9 |
50.6 |
a Estratificación por género y estado de rendimiento ECOG (0 frente a 1).
CI = Intervalo de confianza; NE = Imposible de estimar.
CCOD = 10 de septiembre de 2018.
Se estudió el rendimiento del VENTANA PD-L1 (SP142) ASSAY en OAK (NCT02008227), un ensayo abierto en fase III, multicéntrico, internacional y aleatorizado que se había diseñado para evaluar la eficacia y la seguridad del tratamiento con TECENTRIQ (atezolizumab) en pacientes con NSCLC localmente avanzado o metastásico con progresión durante el régimen de tratamiento con platino o tras haberlo recibido.
En el estudio OAK participaron 1225 pacientes y la población de análisis primario estuvo formada por los primeros 850 pacientes aleatorizados; los pacientes que cumplían los criterios de idoneidad se estratificaron por el estado de expresión de PD-L1 en CI, por el número de regímenes de quimioterapia previos y por la histología. Los pacientes se aleatorizaron (1:1) para recibir un tratamiento de 1200 mg de TECENTRIQ (atezolizumab) por vía intravenosa cada tres semanas hasta alcanzar una toxicidad inaceptable o hasta la progresión radiográfica o clínica o de 75 mg de docetaxel por vía intravenosa 2 cada tres semanas hasta la pérdida del beneficio clínico evaluado por el investigador. Las muestras tumorales se evaluaron prospectivamente para determinar la expresión de PD-L1 en TC e IC mediante VENTANA PD-L1 (SP142) ASSAY y los resultados sirvieron para definir los subgrupos de expresión de PD-L1 de los análisis con especificación previa que se describen a continuación.
La principal medida de eficacia del estudio OAK fue la supervivencia global (OS) en la población del análisis primario (los primeros 850 pacientes aleatorizados). Los resultados del estudio OAK, con un promedio de seguimiento de 21 meses, se muestran en la Tabla 27.
Las muestras tumorales se evaluaron prospectivamente mediante VENTANA PD-L1 (SP142) ASSAY en un laboratorio central y los resultados sirvieron para definir los subgrupos de expresión de PD-L1 de análisis especificados previamente. De los 850 pacientes, se clasificó al 16% por presentar un elevado nivel de expresión de PD-L1, que se definió como la presencia de expresión de PD-L1 en ≥ 50% TC o ≥ 10% IC. En un análisis exploratorio de subgrupos de eficacia de la OS basado en la expresión de PD-L1, el índice de riesgo era 0.41 (CI del 95%: 0.27, 0.64) en el subgrupo con niveles elevados de expresión de PD-L1 y 0.82 (CI del 95%: 0.68, 0.98) en pacientes en los que no se observaban niveles elevados de la expresión de PD-L1.
Tabla 27. Resultados de eficacia de la población del análisis principal del estudio OAK (NCT02008227)
|
Supervivencia global (OS) |
Atezolizumab (N = 425) |
Docetaxel (N = 425) |
|
Muertes (%) |
271 (64) |
298 (70) |
|
Mediana, meses (CI del 95%) |
13.8 (11.8, 15.7) |
9.6 (8.6, 11.2) |
|
Índice de riesgoa (CI del 95%) |
0.73 (0.62, 0.87) |
|
|
Valor pb |
0.0003 |
|
|
OS de 12 meses (%) |
218 (55) |
151 (41) |
|
OS de 18 meses (%) |
157 (40) |
98 (27) |
a Estratificación en función de la expresión de PD-L1 en las células inmunitarias que infiltran el tumor, de la cantidad de regímenes de quimioterapia recibidos anteriormente y de la histología
b Según la prueba del orden logarítmico estratificado.
CI = Intervalo de confianza
RENDIMIENTO CLÍNICO: TNBC:
Se estudió el rendimiento de VENTANA PD-L1 (SP142) ASSAY en IMpassion130 (NCT02425891), un ensayo en fase III, doble ciego, de dos grupos, multicéntrico, internacional, aleatorizado y controlado con placebo, que se llevó a cabo para evaluar la eficacia y la seguridad de TECENTRIQ (atezolizumab) en combinación con nab-paclitaxel en pacientes que padecían TNBC inoperable y localmente avanzado o metastásico y que no habían recibido tratamiento previo con quimioterapia para enfermedades metastásicas.
En el estudio IMpassion130 se inscribió a 902 pacientes; los pacientes que cumplían los criterios de idoneidad se estratificaron por presencia de metástasis hepáticas, tratamiento previo con taxanos y por estado de la expresión de PD-L1 en IC (IC < 1% del área tumoral frente a ≥ 1% del área tumoral) según la evaluación con el VENTANA PD-L1 (SP142) ASSAY. Los pacientes se aleatorizaron para recibir tratamiento con TECENTRIQ (atezolizumab) (840 mg) o placebo mediante infusiones intravenosas los días 1 y 15 de cada ciclo de 28 días, además de nab-paclitaxel (100 mg/m2) administrado mediante infusión intravenosa los días 1, 8 y 15 de cada ciclo de 28 días. Los pacientes recibieron el tratamiento hasta que se detectó una progresión de la enfermedad radiográfica según RECIST v1.1 o hasta alcanzar una toxicidad inaceptable.
Los datos demográficos y las características iniciales de la enfermedad de la población del estudio estaban bien equilibrados entre los grupos de tratamiento. La mayoría de los pacientes eran mujeres (99.6%); el 67.5% eran blancas y el 17.8% asiáticas. La media de edad fue de 55 años (rango: entre 20 y 86). El estado de rendimiento de ECOG inicial era 0 (58.4%) o 1 (41.3%). En general, el 41% de los pacientes que se incorporó al estudio presentaba expresión de PD-L1 en ≥ 1% de IC, el 27% padecía metástasis en el hígado y el 7% metástasis cerebral asintomática en el inicio. Aproximadamente la mitad de los pacientes habían recibido tratamiento con taxano (51%) o antraciclina (54%) en el contexto (neo)adyuvante. Los datos demográficos y la enfermedad tumoral inicial de los pacientes con expresión de PD-L1 en ≥ 1% de IC se consideraron representativos de la población más amplia del estudio. Los criterios de valoración de la eficacia coprimarios fueron la supervivencia libre de progresión (PFS) en la población ITT y en pacientes que presentaban expresión de PD-L1 en ≥ 1% de IC evaluada por el investigador según RECIST v1.1, así como la supervivencia global (OS) en la población ITT y en pacientes con expresión de PD-L1 en ≥ 1% de IC. Entre los criterios secundarios de valoración de la eficacia se encontraban la tasa de respuesta objetiva (ORR) y la duración de la repuesta (DOR) según RECIST v1.1.
Los resultados de PFS, ORR y DOR de los pacientes del estudio IMpassion130 con expresión de PD-L1 en ≥ 1% de IC en el momento del análisis definitivo de la PFS con un tiempo medio de seguimiento de la supervivencia de 13 meses se resumen en la Tabla 35. Los pacientes que presentaban expresión de PD-L1 en < 1% de IC no mostraron mejoría en la PFS al añadir TECENTRIQ (atezolizumab) al nab-paclitaxel (HR de 0.94, CI del 95% 0.78, 1.13).
Se llevó a cabo un análisis de la OS actualizado con un tiempo medio de seguimiento de 18 meses y los resultados de OS se muestran en la Tabla 35. Los pacientes que presentaban expresión de PD-L1 en < 1% de IC no mostraron mejoría en la OS al añadir TECENTRIQ (atezolizumab) al nab-paclitaxel (HR de 0.97, CI del 95% 0.78, 1.20). En el momento del análisis de OS actualizado, se llevó a cabo un análisis exploratorio de PFS que se muestra en la Tabla 35.
Tabla 35. Resultados de la eficacia de IMpassion130 en pacientes con expresión de PD-L1 en ≥ 1% de IC
|
Criterios clave de valoración de la eficacia |
Atezolizumab + nab-paclitaxel |
Placebo + |
|---|---|---|
|
Criterios de valoración de la eficacia principales |
n= 185 |
n= 184 |
|
PFS evaluada por el investigador (RECIST v1.1)c |
||
|
Nº de eventos (%) |
138 (74.6) |
157 (85.3) |
|
Duración media de PFS (meses) |
7.5 |
5.0 |
|
CI del 95% |
(6.7, 9.2) |
(3.8, 5.6) |
|
Índice de riesgo estratificadoe (CI del 95%) |
0.62 (0.49, 0.78) |
|
|
Valor pa |
< 0.0001 |
|
|
PFS de 12 meses (%) |
29.1 |
16.4 |
|
PFS evaluada por el investigador (RECIST v1.1): análisis exploratorio actualizadod |
||
|
Nº de eventos (%) |
149 (80.5%) |
163 (88.6%) |
|
Duración media de PFS (meses) |
7.5 |
5.3 |
|
CI del 95% |
(6.7, 9.2) |
(3.8, 5.6) |
|
Índice de riesgo estratificadoe (CI del 95%) |
0.63 (0.50-0.80) |
|
|
Valor pa |
< 0.0001 |
|
|
PFS de 12 meses (%) |
30.3 |
17.3 |
|
OSa,b,d |
||
|
Nº de muertes (%) |
94 (50.8) |
110 (59.8) |
|
Tiempo promedio hasta los eventos (meses) |
25.0 |
18.0 |
|
CI del 95% |
(19.55, 30.65) |
(13.63, 20.07) |
|
Índice de riesgo estratificadoe (CI del 95%) |
0.71 (0.54, 0.93) |
|
|
Criterios de valoración secundarios y exploratorios |
||
|
ORR evaluada por el investigador (RECIST v1.1)c |
n= 185 |
n= 183 |
|
Nº de pacientes con respuesta (%) |
109 (58.9) |
78 (42.6) |
|
CI del 95% |
(51.5, 66.1) |
(35.4, 50.1) |
|
Nº de respuesta completa (%) |
19 (10.3) |
2 (1.1%) |
|
Nº de respuesta parcial (%) |
90 (48.6) |
76 (41.5) |
|
N.º de enfermedad estable |
38 (20.5) |
49 (26.8) |
|
DOR evaluada por el investigadorc |
n= 109 |
n= 78 |
|
Media en meses |
8.5 |
5.5 |
|
CI del 95% |
(7.3, 9.7) |
(3.7, 7.1) |
a Según la prueba del orden logarítmico estratificado.
b Las comparaciones de OS entre los grupos de tratamiento en pacientes que presentaban expresión de PD-L1 ≥ 1% no se analizaron oficialmente debido a la jerarquía de análisis especificada previamente.
c Según el análisis definitivo de PFS, ORR y DOR y el primer análisis provisional de OS en la fecha de línea de corte clínica del 17 de abril de 2018.
d Según el segundo análisis provisional de OS y el análisis exploratorio de PFS en la fecha de línea corte clínica del 2 de enero de 2019.
e Estratificación en función de la presencia de metástasis en el hígado y de tratamiento previo con taxano.
PFS = Supervivencia libre de progresión; RECIST = Criterios de Evaluación de Respuesta en Tumores Sólidos v1.1.; CI = Intervalo de confianza; ORR = Tasa de respuesta objetiva; DOR = Duración de la repuesta; OS = Supervivencia global, NE = Imposible de estimar.
RENDIMIENTO DE ANÁLISIS: CARCINOMA UROTELIAL:
Algoritmo de puntuación: carcinoma urotelial:
El tejido del carcinoma urotelial debe evaluarse de acuerdo con el algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para carcinoma urotelial que se facilitan en la Tabla 8. Consulte la guía de interpretación (P/N 1015704) para obtener instrucciones adicionales e imágenes representativas.
Tabla 8. Algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para carcinoma urotelial
|
Evaluación de la tinción de células inmunitarias (IC)a |
Expresión de PD-L1 |
|
Ausencia de cualquier tinción de PD-L1 apreciable O Presencia de tinción de PD-L1 apreciable con cualquier intensidad en células inmunitarias que infiltran el tumor con una cobertura < 5% del área tumoral ocupada por las células tumorales, el estroma peritumoral asociado y el contiguo |
< 5% IC |
|
Presencia de tinción de PD-L1 apreciable con cualquier intensidad en células inmunitarias que infiltran el tumor con una cobertura ≥ 5% del área tumoral ocupada por las células tumorales, el estroma peritumoral asociado y el contiguo |
≥ 5% IC |
a La tinción de PD-L1 en células tumorales no debería tenerse en cuenta en la determinación de la puntuación del tejido de paciente de carcinoma urotelial.
Grosor del tejido: carcinoma urotelial:
Se evaluó el grosor del tejido mediante cinco muestras de carcinoma urotelial únicas (3 PD-L1 ≥ 5% de IC y 2 PD-L1 < 5% de IC). Los tejidos se cortaron y se analizaron por duplicado a 2, 3, 4, 5, 6 y 7 micras. En todos los grosores de tejido se observó una tinción específica adecuada de PD-L1 y niveles de tinción de fondo aceptables de la tinción con VENTANA PD-L1 (SP142) ASSAY. En ninguna de las secciones se presentaron cambios en la expresión de PD-L1 dentro del intervalo de grosores que se analizó. Las muestras deben cortarse a 4 μm para su tinción con VENTANA PD-L1 (SP142) ASSAY.
Repetibilidad y precisión intermedia: carcinoma urotelial:
Se llevaron a cabo estudios con la tinción de muestras de carcinoma urotelial con VENTANA PD-L1 (SP142) ASSAY para demostrar:
• La repetibilidad en el mismo día: se tiñeron 5 portaobjetos replicados de cada una de las 24 muestras únicas de carcinoma urotelial (12 con tinción de PD-L1 ≥ 5% de IC y 12 con tinción de PD-L1 < 5% de IC) con el ensayo VENTANA PD-L1 (SP142) ASSAY en un solo instrumento BenchMark ULTRA en el mismo día.
• Precisión entre días: se tiñeron 10 portaobjetos replicados de cada una de las 24 muestras únicas de carcinoma urotelial (12 con tinción de PD-L1 ≥ 5% de IC y 12 con tinción de PD-L1 < 5% de IC) con el VENTANA PD-L1 (SP142) ASSAY en un solo instrumento BenchMark ULTRA en cinco días no consecutivos.
• Precisión entre instrumentos y entre lotes: se tiñeron 27 portaobjetos de cada una de las 24 muestras únicas de carcinoma urotelial (12 con tinción de PD-L1 ≥ 5% de IC y 12 con tinción de PD-L1 < 5% de IC) con el VENTANA PD-L1 (SP142) ASSAY, tres lotes de anticuerpo VENTANA PD-L1 (SP142) y tres lotes emparejados de OptiView DAB IHC Detection Kit y OptiView Amplification Kit en tres instrumentos BenchMark ULTRA.
• Precisión en la misma plataforma: se tiñeron dos portaobjetos replicados de cada una de las 10 muestras únicas de carcinoma urotelial (3 con tinción de PD-L1 ≥ 5% de IC y 7 con tinción de PD-L1 < 5% de IC) con el VENTANA PD-L1 (SP142) ASSAY en tres instrumentos BenchMark ULTRA y tres instrumentos BenchMark GX. Se calcularon los porcentajes de concordancia relativos al modo de muestra de una plataforma.
Todos los portaobjetos se enmascararon, aleatorizaron y evaluaron a través del algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para el carcinoma urotelial que figura en la Tabla 8. Los resultados se resumen en la Tabla 9.
Tabla 9. Repetibilidad y precisión intermedia de la tinción con VENTANA PD-L1 (SP142) ASSAY en muestras de carcinoma urotelial
|
Parámetro de repetibilidad/precisión intermedia |
Porcentaje de concordancia (CI del 95%)a |
|
Repetibilidad en el mismo día (en un solo día) |
PPA: 98.2 (90.4-99.7) NPA: 100.0 (94.4-100.0) OPA: 99.2 (95.4-99.9) |
|
Precisión entre días (5 días no consecutivos) |
PPA: 91.8 (85.2-95.6) NPA: 100.0 (97.1-100.0) OPA: 96.3 (93.0-98.0) |
|
Precisión entre instrumentos y entre lotes (tres instrumentos, tres lotes de anticuerpos y tres lotes de kits de detección y amplificación) |
PPA: 99.4 (97.8-99.8) NPA: 99.7 (98.3-99.9) OPA: 99.5 (98.6-99.8) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark ULTRA) |
PPA: 83.3 (60.8-94.2) NPA: 100.0 (91.2-100.0) OPA: 94.8 (85.9-98.2) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark GX) |
PPA: 94.4 (74.2-99.0) NPA: 100.0 (91.4-100.0) OPA: 98.3 (91.0-99.7) |
a CI bilateral del método de puntuación de Wilson.
Concordancia entre plataformas: carcinoma urotelial:
Se tiñeron portaobjetos únicos de cada una de las 44 muestras únicas de carcinoma urotelial (22 con tinción de PD-L1 ≥ 5% de IC y 22 con tinción de PD-L1 < 5% de IC) con el VENTANA PD-L1 (SP142) ASSAY en un instrumento BenchMark ULTRA (de referencia) y un instrumento BenchMark GX. Todos los portaobjetos se enmascararon, aleatorizaron y evaluaron a través del algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para el carcinoma urotelial que figura en la Tabla 8. Los resultados se resumen en la Tabla 10.
Tabla 10. Concordancia entre plataformas de la tinción con VENTANA PD-L1 (SP142) ASSAY en muestras de carcinoma urotelial
|
Concordancia entre plataformas |
Porcentaje de concordancia (CI del 95%)a |
|
BenchMark ULTRA: BenchMark GX |
PPA: 95.2 (77.3-99.2) NPA: 100.0 (85.1-100.0) OPA: 97.7 (87.9-99.6) |
a CI bilateral del método de puntuación de Wilson.
Precisión del lector: carcinoma urotelial:
Para evaluar la precisión del lector y entre lectores, tres anatomopatólogos evaluaron 60 muestras únicas de carcinoma urotelial (30 con tinción de PD-L1 ≥ 5% de IC y 30 con tinción de PD-L1 < 5% de IC) que se tiñeron con el ensayo VENTANA PD-L1 (SP142) ASSAY. Todas las muestras se enmascararon, aleatorizaron y evaluaron para determinar el estado de PD-L1 a través del algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para el carcinoma urotelial que figura en la Tabla 8. Los lectores puntuaron todas las muestras dos veces, con un mínimo de dos semanas entre lecturas. Los porcentajes de concordancia entre los lectores y entre las lecturas de cada uno de los anatomopatólogos se resumen en la Tabla 11.
Tabla 11. Precisión del lector y entre lectores de la tinción con VENTANA PD-L1 (SP142) ASSAY en muestras de carcinoma urotelial
|
Precisión del lector |
Porcentaje de concordancia (CI del 95%) |
|
Precisión del lector (promedio de las comparaciones entre pares de lectores desde la primera lectura) |
APA: 92.7 (85.9-97.6) ANA: 93.9 (88.1-98.1) OPA: 93.3 (87.8-97.8) |
|
Precisión entre lectores (media de los porcentajes de concordancia de los tres lectores entre la primera y la segunda lectura) |
APA: 93.4 (87.3-97.7) ANA: 94.2 (88.9-98.1) OPA: 93.9 (88.8-97.8) |
Estudio de reproducibilidad entre laboratorios: carcinoma urotelial:
Se llevó a cabo un estudio de reproducibilidad entre laboratorios con VENTANA PD-L1 (SP142) ASSAY para demostrar la reproducibilidad del ensayo a la hora de determinar el estado de PD-L1 en muestras de tejido de carcinoma urotelial. Se tiñeron veintiocho muestras únicas de carcinoma urotelial (14 con tinción de PD-L1 ≥ 5% de IC y 14 con tinción de PD-L1 < 5% de IC) en tres laboratorios externos en cinco días no consecutivos durante un periodo de, al menos, 20 días. Antes de la tinción, se habían enmascarado los portaobjetos y se habían aleatorizado. En cada sitio, dos anatomopatólogos (lectores) evaluaron los portaobjetos con tinción por separado. Los resultados se resumen en la Tabla 12.
Tabla 12. Reproducibilidad entre laboratorios de la tinción con VENTANA PD-L1 (SP142) ASSAY en muestras de carcinoma urotelial
|
Reproducibilidad entre laboratorios |
Porcentaje de concordancia (CI del 95%) |
|
Concordancia global (en comparación con la puntuación consensuada, en todos los sitios, días y lectores) |
PPA: 98.3 (96.6-99.2)a NPA: 87.4 (83.8-90.2)a OPA: 92.8 (90.9-94.4)a |
|
Concordancia entre sitios (promedio de las comparaciones entre pares entre sitios) |
APA: 90.7 (81.2-96.3) ANA: 88.3 (78.5-94.9) OPA: 89.6 (82.5-95.5) |
|
Concordancia del lector (promedio de las comparaciones entre pares entre los lectores en cada sitio) |
APA: 89.3 (78.1-96.0) ANA: 86.6 (75.1-94.6) OPA: 88.1 (84.6-90.8)a |
a CI bilateral del método de puntuación de Wilson.
RENDIMIENTO DE ANÁLISIS: NSCLC:
Algoritmo de puntuaciones: NSCLC
El tejido de NSCLC debe evaluarse de acuerdo con el algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para NSCLC que se facilita en las Tabla 14 y Tabla 15. Se considera que la muestra presenta expresión de PD-L1 cuando se observa en ≥ 1% de TC o ≥ 1% de IC y que la muestra presenta una elevada expresión de PD-L1 cuando se observa en ≥ 50% de TC o ≥ 10% de IC. Consulte la guía de interpretación (P/N 1015703 y 1015654) para obtener instrucciones adicionales e imágenes representativas.
Tabla 14. Algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para NSCLC ≥ 50% de TC o ≥ 10% de IC
|
PASO 1 |
Evaluación de la tinción en células tumorales (TC) |
Expresión de PD-L1 |
|
Presencia de tinción de PD-L1 en la membrana apreciable con cualquier intensidad en ≥ 50% de las células tumorales |
≥ 50% de TC |
|
|
Ausencia de cualquier tinción de PD-L1 apreciable O Presencia de tinción de PD-L1 en la membrana apreciable con cualquier intensidad en < 50% de las células tumorales |
Continúe con el Paso 2 |
|
|
PASO 2 |
Evaluación de la tinción en células inmunitarias que infiltran el tumor (IC) |
Expresión de PD-L1 |
|
Presencia de tinción de PD-L1 apreciable con cualquier intensidad en células inmunitarias que infiltran el tumor con una cobertura ≥ 10% del área tumoral ocupada por las células tumorales, el estroma peritumoral asociado y el contiguo |
≥ 10% IC |
|
|
Ausencia de cualquier tinción de PD-L1 apreciable O Presencia de tinción de PD-L1 apreciable con cualquier intensidad en células inmunitarias que infiltran el tumor con una cobertura < 10% del área tumoral ocupada por las células tumorales, el estroma peritumoral asociado y el contiguo |
< 50% de TC y < 10% IC |
|
Tabla 15. Algoritmo de puntuación VENTANA PD-L1 (SP142) ASSAY para NSCLC ≥ 1% de TC o ≥ 1% de IC
|
PASO 1 |
Evaluación de la tinción en células tumorales (TC) |
Expresión de PD-L1 |
|
Presencia de tinción de PD-L1 en la membrana apreciable con cualquier intensidad en ≥ 1% de las células tumorales |
≥ 1% TC |
|
|
Ausencia de cualquier tinción de PD-L1 apreciable O Presencia de tinción de PD-L1 en la membrana apreciable con cualquier intensidad en < 1% de las células tumorales |
Continúe con el Paso 2 |
|
|
PASO 2 |
Evaluación de la tinción en células inmunitarias que infiltran el tumor (IC) |
Expresión de PD-L1 |
|
Presencia de tinción de PD-L1 apreciable con cualquier intensidad en células inmunitarias que infiltran el tumor con una cobertura ≥ 1% del área tumoral ocupada por las células tumorales, el estroma peritumoral asociado y el contiguo |
≥ 1% IC |
|
|
Ausencia de cualquier tinción de PD-L1 apreciable O Presencia de tinción de PD-L1 apreciable con cualquier intensidad en células inmunitarias que infiltran el tumor con una cobertura < 1% del área tumoral ocupada por las células tumorales, el estroma peritumoral asociado y el contiguo |
< 1% TC y < 1% IC |
|
Grosor del tejido: NSCLC:
Se evaluó el grosor del tejido con muestras de NSCLC. Se tiñeron secciones duplicadas de 3, 4, 5, 6 y 7 μm de grosor con VENTANA PD-L1 (SP142) ASSAY y se evaluaron para detectar la expresión de PD-L1 en TC e IC. Se evaluaron un total de 42 muestras de NSCLC con una variedad de expresiones de PD-L1 en cada nivel de IC y TC.
En todos los grosores de tejido se observó una tinción específica adecuada de PD-L1 y niveles de tinción de fondo aceptables de la tinción con VENTANA PD-L1 (SP142) ASSAY. En ninguna de las secciones se presentaron cambios en el nivel de TC o IC de PD-L1 dentro del intervalo de grosores que se analizó. Las muestras de NSCLC deben cortarse a 4 μm para su tinción con VENTANA PD-L1 (SP142) ASSAY.
Repetibilidad y precisión intermedia: NSCLC
Se llevaron a cabo estudios con la tinción de muestras de NSCLC con VENTANA PD-L1 (SP142) ASSAY para demostrar:
• La repetibilidad en el mismo día: se tiñeron 5 portaobjetos replicados de cada una de las muestras de NSCLC con el ensayo VENTANA PD-L1 (SP142) ASSAY en un solo instrumento BenchMark ULTRA en un único día y se evaluaron para detectar la expresión de PD-L1 en TC e IC. Se evaluaron un total de 66 muestras de NSCLC con una variedad de expresiones de PD-L1 en cada nivel de IC y TC.
• La precisión entre días: se tiñeron 10 portaobjetos de cada una de las muestras de NSCLC con el ensayo VENTANA PD-L1 (SP142) ASSAY en un solo instrumento BenchMark ULTRA a lo largo de cinco días no consecutivos. Se evaluaron un total de 66 muestras de NSCLC con una variedad de expresiones de PD-L1 en cada nivel de expresión de IC y TC.
• La precisión entre instrumentos y entre lotes de anticuerpos y de detección: se tiñeron al menos 9 portaobjetos de cada una de las muestras de NSCLC con VENTANA PD-L1 (SP142) ASSAY y tres lotes del anticuerpo VENTANA PD-L1 (SP142) y tres lotes emparejados de OptiView DAB IHC Detection Kit y OptiView Amplification Kit en tres instrumentos BenchMark ULTRA. Se evaluaron un total de 92 muestras de NSCLC con una variedad de expresiones de PD-L1 en cada nivel de IC y TC.
• La precisión en la misma plataforma: se tiñeron 2 portaobjetos replicados de cada una de las muestras de NSCLC con el VENTANA PD-L1 (SP142) ASSAY en tres instrumentos BenchMark ULTRA y tres instrumentos BenchMark GX. Se evaluaron un total de 38 muestras de NSCLC con una variedad de expresiones de PD-L1 en cada nivel de expresión de IC y TC. Se calcularon los porcentajes de concordancia relativos al modo de muestra de cada plataforma.
Todos los portaobjetos se enmascararon, se aleatorizaron y se evaluaron posteriormente para la determinación del nivel de expresión de PD-L1 en TC o IC. Los resultados se resumen en la Tabla 16, la Tabla 17, la Tabla 18 y la Tabla 19.
Tabla 16. Repetibilidad y precisión intermedia de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 50% de TC)
|
Parámetro de repetibilidad/precisión intermedia |
Porcentaje de concordancia |
|
Repetibilidad en el mismo día (en un mismo día), 24 muestras de NSCLC |
PPA: 100.0 (94.4-100.0) NPA: 100.0 (93.5-100.0) OPA: 100.0 (96.9-100.0) |
|
Precisión entre días (cinco días no consecutivos), 24 muestras de NSCLC |
PPA: 100.0 (97.1-100.0) NPA: 100.0 (96.5-100.0) OPA: 100.0 (98.4-100.0) |
|
Precisión entre instrumentos y entre lotes (en comparación con el modo de nivel del caso, en todos los instrumentos y lotes), 18 muestras de NSCLC |
PPA: 99.7 (98.1-99.9) NPA: 95.2 (91.2-97.5) OPA: 97.9 (96.2-98.9) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark ULTRA), 10 muestras de NSCLC |
PPA: 100.0 (88.6-100.0) NPA: 100.0 (88.6-100.0) OPA: 100.0 (94.0-100.0) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark GX), 10 muestras de NSCLC |
PPA: 100.0 (88.6-100.0) NPA: 100.0 (88.6-100.0) OPA: 100.0 (94.0-100.0) |
a CI bilateral del método de puntuación de Wilson.
Tabla 17. Repetibilidad y precisión intermedia de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 10% de IC)
|
Parámetro de repetibilidad/precisión intermedia |
Porcentaje de concordancia |
|
Repetibilidad en el mismo día (en un mismo día), 24 muestras de NSCLC |
PPA: 98.3 (91.1-99.7)a NPA: 100.0 (94.0-100.0)a OPA: 99.2 (95.4-99.9)a |
|
Precisión entre días (cinco días no consecutivos), 24 muestras de NSCLC |
PPA: 96.2 (91.3-98.3)a NPA: 98.2 (93.6-99.5)a OPA: 97.1 (94.1-98.6)a |
|
Concordancia entre anticuerpos y entre kits de detección (comparación entre pares), 28 muestras de NSCLC |
APA: 95.1 (91.1-98.1) ANA: 90.2 (82.3-96.2) OPA: 93.4 (88.7-97.5) |
|
Concordancia entre instrumentos y entre lotes de detección (comparación entre pares), 28 muestras |
APA: 96.3 (93.2-98.8) ANA: 92.7 (86.0-97.7) OPA: 95.1 (91.2-98.4) |
|
Concordancia entre instrumentos y entre anticuerpos (comparación entre pares), 28 muestras de NSCLC |
APA: 96.3 (93.1-98.8) ANA: 92.6 (85.9-97.8) OPA: 95.1 (91.1-98.4) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark ULTRA), 8 muestras de NSCLC |
PPA: 100.0 (94.0-100.0)a NPA: 100.0 (94.0-100.0)a OPA: 100.0 (96.9-100.0)a |
|
Precisión en la misma plataforma (tres instrumentos BenchMark GX), 8 muestras de NSCLC |
PPA: 100.0 (94.0-100.0)a NPA: 100.0 (94.0-100.0)a OPA: 100.0 (96.9-100.0)a |
a CI bilateral del método de puntuación de Wilson.
Tabla 18. Repetibilidad y precisión intermedia de la tinción con VENTANA PD-L1 (SP142) de muestra Assays de NSCLC (expresión de PD-L1 ≥ 1% de TC)
|
Parámetro de repetibilidad/precisión intermedia |
Porcentaje de concordancia (CI del 95%)a |
|---|---|
|
Repetibilidad en el mismo día (en un mismo día), 10 muestras de NSCLC |
PPA: 100.0 (86.7-100.0) NPA: 100.0 (86.7-100.0) OPA: 100.0 (92.9-100.0) |
|
Precisión entre días (cinco días no consecutivos), 10 muestras de NSCLC |
PPA: 100.0 (92.9-100.0) NPA: 100.0 (92.9-100.0) OPA: 100.0 (96.3-100.0) |
|
Precisión entre instrumentos y entre lotes (en comparación con el modo de nivel del caso, en todos los instrumentos y lotes), 18 muestras de NSCLC |
PPA: 99.2 (97.0-99.8) NPA: 100.0 (98.4-100.0) OPA: 99.6 (98.5-99.9) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark ULTRA), 12 muestras de NSCLC |
PPA: 100.0 (86.2-100.0) NPA: 95.8 (86.0-98.8) OPA: 97.2 (90.4-99.2) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark GX), 12 muestras de NSCLC |
PPA: 100.0 (86.2-100.0) NPA: 100.0 (92.6-100.0) OPA: 100.0 (94.0-100.0) |
a CI bilateral del método de puntuación de Wilson.
Tabla 19. Repetibilidad y precisión intermedia de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 1% de IC)
|
Parámetro de repetibilidad/precisión intermedia |
Porcentaje de concordancia (CI del 95%) |
|---|---|
|
Repetibilidad en el mismo día (en un mismo día), 8 muestras de NSCLC |
PPA: 100.0 (91.2-100.0)a NPA: 100.0 (91.2-100.0)a OPA: 100.0 (95.4-100.0)a |
|
Precisión entre días (cinco días no consecutivos), 8 muestras de NSCLC |
PPA: 100.0 (96.3-100.0)a NPA: 97.0 (91.6-99.0)a OPA: 98.5 (95.7-99.5)a |
|
Concordancia entre anticuerpos y entre kits de detección (comparación entre pares), 28 muestras de NSCLC |
APA: 98.4 (95.7-100.0) ANA: 98.3 (95.6-100.0) OPA: 98.4 (95.9-100.0) |
|
Concordancia entre instrumentos y entre kits de detección (comparación entre pares), 28 muestras de NSCLC |
APA: 98.4 (95.8-100.0) ANA: 98.3 (95.4-100.0) OPA: 98.4 (95.8-100.0) |
|
Concordancia entre instrumentos y entre anticuerpos (comparación entre pares), 28 muestras de NSCLC |
APA: 98.4 (95.9-100.0) ANA: 98.3 (95.5-100.0) OPA: 98.4 (95.8-100.0) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark ULTRA), 8 muestras de NSCLC |
PPA: 100.0 (94.0-100.0)a NPA: 100.0 (94.0-100.0)a OPA: 100.0 (96.9-100.0)a |
|
Precisión en la misma plataforma (tres instrumentos BenchMark GX), 8 muestras de NSCLC |
PPA: 100.0 (94.0-100.0)a NPA: 100.0 (94.0-100.0)a OPA: 100.0 (96.9-100.0)a |
a CI bilateral del método de puntuación de Wilson.
Concordancia entre plataformas: NSCLC:
Se tiñeron portaobjetos individuales de cada una de las 44 muestras de NSCLC con VENTANA PD-L1 (SP142) ASSAY para determinar la presencia de la expresión de PD-L1 ≥ 50% de TC o ≥ 10% de IC (21 muestras con expresión de PD-L1 ≥ 50% de TC o ≥ 10% de IC y 23 muestras con expresión de PD-L1 < 50% de TC y < 10% de IC) y 44 muestras de NSCLC para determinar la expresión de PD-L1 ≥ 1% de TC o ≥ 1% de IC (23 muestras con expresión de PD-L1 ≥ 1% de TC o ≥ 1% de IC y 21 muestras de PD- L1 < 1% de TC y < 1% de IC) en un instrumento BenchMark ULTRA (instrumento de referencia) y un instrumento BenchMark GX.
Todos los portaobjetos se enmascararon, se aleatorizaron y se evaluaron con el algoritmo de puntuación del VENTANA PD-L1 (SP142) ASSAY para NSCLC (Tabla 14 para NSCLC ≥ 50% de TC o ≥ 10% de IC y Tabla 15 para NSCLC ≥ 1% de TC o ≥ 1% de IC). Los resultados se resumen en la Tabla 20 y la Tabla 21.
Tabla 20. Concordancia entre plataformas de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 50% de TC o ≥ 10% de IC)
|
Concordancia entre plataformas |
Porcentaje de concordancia |
|
BenchMark ULTRA: BenchMark GX |
PPA: 95.2 (77.3-99.2) NPA: 100.0 (85.1-100.0) OPA: 97.7 (87.9-99.6) |
a CI bilateral del método de puntuación de Wilson.
Tabla 21. Concordancia entre plataformas de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 1% de TC o ≥ 1% de IC)
|
Concordancia entre plataformas |
Porcentaje de concordancia |
|
BenchMark ULTRA: BenchMark GX |
PPA: 100.0 (85.7-100.0) NPA: 100.0 (84.5-100.0) OPA: 100.0 (92.0-100.0) |
a CI bilateral del método de puntuación de Wilson.
Estudios de precisión del lector: NSCLC
Para evaluar la precisión del lector y entre lectores, tres anatomopatólogos evaluaron 80 casos únicos de NSCLC con diferentes niveles de expresión de PD-L1 que se tiñeron con VENTANA PD-L1 (SP142) ASSAY. Las muestras se enmascararon, se aleatorizaron y se evaluó el estado de PD-L1 con el algoritmo de puntuación del VENTANA PD-L1 (SP142) ASSAY para NSCLC (Tabla 14 para NSCLC ≥ 50% de TC o ≥ 10% de IC y Tabla 15 para NSCLC ≥ 1% de TC o ≥ 1% de IC). Los lectores puntuaron todas las muestras dos veces, con un mínimo de dos semanas entre lecturas. Los porcentajes de concordancia entre los lectores y entre las lecturas de cada uno de los anatomopatólogos se resumen en la Tabla 22 y en la Tabla 23.
Tabla 22. Precisión del lector de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 50% de TC o ≥ 10% de IC)
|
Precisión del lector |
Porcentaje de concordancia (CI del 95%) |
|
Precisión del lector (promedio de las comparaciones entre pares de lectores desde la primera lectura) |
APA: 88.8 (82.0-94.1) ANA: 89.0 (82.2-94.4) OPA: 88.9 (82.8-94.1) |
|
Precisión entre lectores (media de los porcentajes de concordancia de los tres lectores entre la primera y la segunda lectura) |
APA: 93.7 (89.9-96.6) ANA: 93.6 (89.8-96.7) OPA: 93.6 (90.3-96.6) |
Tabla 23. Precisión del lector de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 1% de TC o ≥ 1% de IC)
|
Precisión del lector |
Porcentaje de concordancia (CI del 95%) |
|
Precisión del lector (promedio de las comparaciones entre pares de lectores desde la primera lectura) |
APA: 93.7 (88.8-97.3) ANA: 90.7 (83.3-96.2) OPA: 92.5 (87.5-96.7) |
|
Precisión entre lectores (media de los porcentajes de concordancia de los tres lectores entre la primera y la segunda lectura) |
APA: 95.4 (92.0-98.1) ANA: 93.4 (88.3-97.2) OPA: 94.6 (90.8- 97.5) |
Estudio de reproducibilidad entre laboratorios: NSCLC
Se llevó a cabo un estudio de reproducibilidad entre laboratorios con la tinción de VENTANA PD-L1 (SP142) ASSAY para demostrar la reproducibilidad del ensayo a la hora de determinar el estado de PD-L1 en muestras de tejido de NSCLC. Se tiñeron veintiocho muestras únicas de NSCLC que contenían una variedad de niveles de expresión de PD-L1 en tres laboratorios externos en cinco días no consecutivos durante un periodo de, al menos, 20 días. Antes de la tinción, se habían enmascarado los portaobjetos y se habían aleatorizado. En cada sitio, dos anatomopatólogos (lectores) evaluaron los portaobjetos con tinción por separado con el algoritmo de puntuación del VENTANA PD-L1 (SP142) ASSAY para NSCLC (Tabla 14 para NSCLC ≥ 50% de TC o ≥ 10% de IC y Tabla 15 para NSCLC ≥ 1% de TC o ≥ 1% de IC). Los resultados se resumen en la Tabla 24 y la Tabla 25.
Tabla 24. Reproducibilidad entre laboratorios de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 50% de TC o ≥ 10% de IC)
|
Reproducibilidad entre laboratorios |
Porcentaje de concordancia |
|
Concordancia global (en comparación con la puntuación consensuada, en todos los sitios, días y lectores) |
PPA: 86.6 (83.0-89.5)a NPA: 99.8 (98.7-100.0)a OPA: 93.2 (91.3-94.7)a |
|
Concordancia entre sitios (promedio de las comparaciones entre pares entre sitios) |
APA: 89.5 (80.9-95.5) ANA: 92.1 (84.4-97.1) OPA: 91.0 (90.3-91.6)a |
|
Concordancia del lector (promedio de las comparaciones entre pares entre los lectores en cada sitio) |
APA: 93.9 (89.3-97.4) ANA: 95.4 (90.6-98.2) OPA: 94.7 (92.2-96.5)a |
a CI bilateral del método de puntuación de Wilson.
Tabla 25. Reproducibilidad entre laboratorios de la tinción con VENTANA PD-L1 (SP142) ASSAY de muestras de NSCLC (expresión de PD-L1 ≥ 1% de TC o ≥ 1% de IC)
|
Reproducibilidad entre laboratorios |
Porcentaje de concordancia (CI del 95%) |
|
Concordancia global (en comparación con la puntuación consensuada, en todos los sitios, días y lectores) |
PPA: 97.1 (94.9-98.3)a NPA: 89.9 (86.6-92.5)a OPA: 93.5 (91.6-95.0)a |
|
Concordancia entre sitios (promedio de las comparaciones entre pares entre sitios) |
APA: 88.4 (78.9- 94.1) ANA: 86.6 (79.6-92.2) OPA: 87.6 (86.8-88.3)a |
|
Concordancia del lector (promedio de las comparaciones entre pares entre los lectores en cada sitio) |
APA: 89.2 (80.8-94.3) ANA: 87.5 (81.3-92.5) OPA: 88.4 (84.9-91.2)a |
a CI bilateral del método de puntuación de Wilson.
RENDIMIENTO DE ANÁLISIS: TNBC:
Algoritmo de puntuaciones: TNBC
El tejido de TNBC debe evaluarse de acuerdo con el algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para TNBC que se facilita en la Tabla 30. Consulte la guía de interpretación (P/N 1018231) para obtener instrucciones adicionales e imágenes representativas.
Tabla 30. VENTANA PD-L1 (SP142) ASSAY para TNBC
|
Evaluación de la tinción de células inmunitarias (IC)a |
Expresión de PD-L1 |
|
Ausencia de cualquier tinción de PD-L1 apreciable O Presencia de tinción de PD-L1 apreciable con cualquier intensidad en células inmunitarias que infiltran el tumor con una cobertura < 1% del área tumoral ocupada por las células tumorales, el estroma peritumoral asociado y el contiguo |
< 1% IC |
|
Presencia de tinción de PD-L1 apreciable con cualquier intensidad en células inmunitarias que infiltran el tumor con una cobertura ≥ 1% del área tumoral ocupada por las células tumorales, el estroma peritumoral asociado y el contiguo |
≥ 1% IC |
a La tinción de PD-L1 en células tumorales no debería tenerse en cuenta en la determinación de la puntuación del tejido de paciente de TNBC.
Grosor del tejido: TNBC:
Se evaluó el grosor del tejido mediante 10 muestras de TNBC únicas (5 con expresión de PD-L1 en ≥ 1% de IC y 5 con expresión de PD-L1 en < 1% de IC). Los tejidos se cortaron y se analizaron por duplicado a 2, 3, 4, 5, 6 y 7 micras. En todos los grosores de tejido se observó una tinción específica adecuada de PD-L1 y niveles de tinción de fondo aceptables de la tinción con VENTANA PD-L1 (SP142) ASSAY. En ninguna de las secciones se presentaron cambios en el estado de PD-L1 dentro del intervalo de grosores que se analizó. Las muestras deben cortarse a 4 μm para su tinción con VENTANA PD-L1 (SP142) ASSAY.
Repetibilidad y precisión intermedia: TNBC:
Se llevaron a cabo estudios con la tinción de muestras de TNBC con VENTANA PD-L1 ASSAY para demostrar:
• La repetibilidad en el mismo día: se tiñeron 5 portaobjetos replicados de cada una de las 24 muestras únicas de TNBC (12 con tinción de PD-L1 ≥ 1% de IC y 12 con tinción de PD-L1 < 1% de IC) con el ensayo VENTANA PD-L1 (SP142) ASSAY en un solo instrumento BenchMark ULTRA en el mismo día.
• Precisión entre días: se tiñeron 10 portaobjetos replicados de cada una de las 24 muestras únicas de TNBC (12 con tinción de PD-L1 ≥ 1% de IC y 12 con tinción de PD-L1 < 1% de IC) con el ensayo VENTANA PD-L1 (SP142) ASSAY en un solo instrumento BenchMark ULTRA en cinco días no consecutivos.
• Precisión entre instrumentos y entre lotes: se tiñeron 27 portaobjetos de cada una de las 24 muestras únicas de TNBC (12 con tinción de PD-L1 ≥ 1% de IC y 12 con tinción de PD-L1 < 1% de IC) con el VENTANA PD-L1 (SP142) ASSAY, tres lotes de anticuerpo VENTANA PD-L1 (SP142) y tres lotes emparejados de OptiView DAB IHC Detection Kit y OptiView Amplification Kit en tres instrumentos BenchMark ULTRA.
• Precisión en la misma plataforma: se tiñeron dos portaobjetos replicados de cada una de las 10 muestras únicas de TNBC (5 con tinción de PD-L1 ≥ 1% de IC y 5 con tinción de PD-L1 < 1% de IC) con el VENTANA PD-L1 (SP142) ASSAY en tres instrumentos BenchMark ULTRA y tres instrumentos BenchMark GX. Se calcularon los porcentajes de concordancia relativos al modo de muestra de cada plataforma.
Todos los portaobjetos se enmascararon, se aleatorizaron y evaluaron a través del algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para TNBC (Tabla 30). Los resultados se resumen en la Tabla 31.
Tabla 31. Repetibilidad y precisión intermedia de la tinción con VENTANA PD-L1 (SP142) ASSAY en muestras de TNBC
|
Parámetro de repetibilidad/precisión intermedia |
Porcentaje de concordancia (CI del 95%) |
|
Repetibilidad en el mismo día (en un solo día) |
PPA: 100.0 (94.0-100.0) NPA: 95.0 (87.2-100.0) OPA: 97.5 (93.3-100.0) |
|
Precisión entre días (5 días no consecutivos) |
PPA: 100.0 (96.9-100.0) NPA: 96.7 (92.7-100.0) OPA: 98.3 (96.3-100.0) |
|
Precisión entre instrumentos y entre lotes (tres instrumentos, tres lotes de anticuerpos y tres lotes de kits de detección y amplificación) |
PPA: 98.3 (96.0-100.0) NPA: 99.2 (97.2-100.0) OPA: 98.6 (97.1-99.8) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark ULTRA) |
PPA: 100.0 (88.6-100.0) NPA: 96.7 (88.9-100.0) OPA: 98.3 (95.0-100.0) |
|
Precisión en la misma plataforma (tres instrumentos BenchMark GX) |
PPA: 93.3 (83.3-100.0) NPA: 96.7 (88.9-100.0) OPA: 95.0 (90.0-100.0) |
Concordancia entre plataformas: TNBC:
Se tiñeron portaobjetos únicos de cada una de las 44 muestras únicas de TNBC (22 con tinción de PD-L1 ≥ 1% de IC y 22 con tinción de PD-L1 < 1% de IC) con el VENTANA PD-L1 (SP142) ASSAY en un instrumento BenchMark ULTRA (instrumento de referencia) y un instrumento BenchMark GX. Todos los portaobjetos se enmascararon, se aleatorizaron y evaluaron a través del algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para TNBC (Tabla 30). Los resultados se resumen en la Tabla 32.
Tabla 32. Concordancia entre plataformas de la tinción con VENTANA PD-L1 (SP142) ASSAY en muestras de TNBC
|
Concordancia entre plataformas |
Porcentaje de |
|
BenchMark ULTRA: BenchMark GX |
PPA: 100.0 (86.2-100.0) NPA: 85.0 (64.0-94.8) OPA: 93.2 (81.8-97.7) |
a CI bilateral del método de puntuación de Wilson
Precisión del lector: TNBC:
Para evaluar la precisión del lector y entre lectores, tres anatomopatólogos evaluaron 60 muestras únicas de TNBC (30 con tinción de PD-L1 ≥ 1% de IC y 30 con tinción de PD-L1 < 1% de IC) que se tiñeron con el ensayo VENTANA PD-L1 (SP142) ASSAY. Las muestras se enmascararon y aleatorizaron antes de la evaluación para determinar el estado de PD-L1 a través del algoritmo de puntuación de VENTANA PD-L1 (SP142) ASSAY para TNBC (Tabla 30). Los lectores puntuaron todas las muestras dos veces, con un mínimo de dos semanas entre lecturas. Los porcentajes de concordancia entre los lectores y entre las lecturas de cada uno de los anatomopatólogos se resumen en la Tabla 33.
Tabla 33. Precisión del lector y entre lectores de la tinción con VENTANA PD-L1 (SP142) ASSAY en muestras de TNBC
|
Precisión del lector |
Porcentaje de concordancia (CI del 95%) |
|
Precisión del lector (promedio de las comparaciones entre pares de lectores desde la primera lectura) |
APA: 91.1 (84.2-96.6) ANA: 91.1 (84.1-96.7) OPA: 91.1 (85.6-96.7) |
|
Precisión entre lectores (media de los porcentajes de concordancia de los tres lectores entre la primera y la segunda lectura) |
APA: 93.8 (89.5-97.1) ANA: 93.9 (89.2-97.3) OPA: 93.9 (89.9-97.2) |
Estudio de reproducibilidad entre laboratorios: TNBC
Se llevó a cabo un estudio de reproducibilidad entre laboratorios con VENTANA PD-L1 (SP142) ASSAY para demostrar la reproducibilidad del ensayo a la hora de determinar el estado de PD-L1 en muestras de TNBC. Se tiñeron veintiocho muestras únicas de TNBC (14 con tinción de PD-L1 ≥ 1% de IC y 14 con tinción de PD-L1 < 1% de IC) en tres laboratorios externos en cinco días no consecutivos durante un periodo de, al menos, 20 días. Antes de la tinción, se habían enmascarado los portaobjetos y se habían aleatorizado. En cada sitio, dos anatomopatólogos (lectores) evaluaron los portaobjetos con tinción por separado. El porcentaje de aceptabilidad de la tinción de VENTANA PD-L1 (SP142) ASSAY fue del 98.6% en el estudio. Los resultados se resumen en la Tabla 34.
Tabla 34. Reproducibilidad entre laboratorios de la tinción con VENTANA PD-L1 (SP142) ASSAY en muestras de TNBC
|
Reproducibilidad entre laboratorios |
Porcentaje de concordancia (CI del 95%) |
|
Concordancia global (en comparación con la puntuación consensuada, en todos los sitios, días y lectores) |
PPA: 93.2 (90.4-95.2)a NPA: 96.6 (94.4-98.0)a OPA: 94.8 (93.1-96.1)a |
|
Concordancia entre sitios (promedio de las comparaciones entre pares entre sitios) |
APA: 91.5 (84.0-96.6) ANA: 91.3 (83.6-96.4) OPA: 91.4 (90.7-92.0)a |
|
Concordancia del lector (promedio de las comparaciones entre pares entre los lectores en cada sitio) |
APA: 93.6 (88.2-97.0) ANA: 93.3 (87.8-96.7) OPA: 93.4 (90.6-95.4)a |
a CI bilateral del método de puntuación de Wilson.
RENDIMIENTO DE VENTANA PD-L1 (SP142) ASSAY EN EL INSTRUMENTO BENCHMARK ULTRA PLUS:
Concordancia entre los instrumentos BenchMark ULTRA PLUS y BenchMark ULTRA en el carcinoma urotelial, NSCLC y TNBC:
Tres laboratorios de instituciones independientes de Estados Unidos participaron en un estudio de concordancia entre el instrumento BenchMark ULTRA PLUS y el instrumento BenchMark ULTRA. Se evaluaron 135 muestras únicas de carcinoma urotelial, NSCLC y TNBC (45 muestras por cada indicación) que representaban el intervalo de tinción de VENTANA PD-L1 (SP142) ASSAY , con una distribución equitativa aproximada entre las muestras positivas en PD-L1 y negativas en PD-L1 de cada indicación. Los portaobjetos de tejido de todas las muestras se tiñeron con H y E junto con VENTANA PD-L1 (SP142) ASSAY en un instrumento BenchMark ULTRA y el protocolo de tinción recomendado. Los portaobjetos con tejido sin tinción de todas las muestras se aleatorizaron y se distribuyeron de forma equitativa (45 muestras por sitio de forma que cada sitio recibiera una muestra representativa de las muestras del estudio) para su tinción en un instrumento BenchMark ULTRA PLUS con el protocolo de tinción recomendado de VENTANA PD-L1 (SP142). Dos anatomopatólogos por sitio, con los estados de las muestras enmascarados, evaluaron los portaobjetos teñidos en el instrumento BenchMark ULTRA PLUS correspondiente y determinaron el estado de PD-L1. Tras un periodo de dos semanas de reposo, los portaobjetos de las muestras correspondientes, previamente teñidas en Roche con el instrumento BenchMark ULTRA se distribuyeron a los sitios oportunos para su evaluación clínica. Además, otro anatomopatólogo revisó todos los portaobjetos de los casos y se incluyó en la lista de patólogos de cada uno de los sitios. Los resultados se analizaron en Roche. Los porcentajes de OPA, PPA y NPA fueron del 91.6% (359/392), del 90.3% (176/195) y del 92.9% (183/197) respectivamente. Los resultados se resumen en la Tabla 36.
Tabla 36. Concordancia combinada del estado de PD-L1 en muestras de carcinoma urotelial, NSCLC y TNBC teñidas con VENTANA PD-L1 (SP142) ASSAY en el instrumento BenchMark ULTRA PLUS frente al instrumento BenchMark ULTRA
|
Estado de PD-L1 con BenchMark ULTRA PLUS |
Estado de PD-L1 con BenchMark ULTRA |
Total |
|
|
Positiva |
Negativa |
||
|
Positiva |
176 |
14 |
190 |
|
Negativa |
19 |
183 |
202 |
|
Total |
195 |
197 |
392 |
|
n/N |
% (CI del 95%) |
||
|
PPA |
176/195 |
90.3 (85.2, 94.7) |
|
|
NPA |
183/197 |
92.9 (89.0, 96.2) |
|
|
OPA |
359/392 |
91.6 (88.4, 94.6) |
|
Nota: Los intervalos de confianza del 95% bilaterales se calcularon mediante el método bootstrap percentil con 2000 replicados cuya estratificación se llevó a cabo en función del estado de la cualificación clínica de PD-L1 (positivo, negativo o en el límite) y de la indicación.
Reproducibilidad entre laboratorios en el instrumento BenchMark ULTRA PLUS con carcinoma urotelial, NSCLC y TNBC:
Se llevó a cabo un estudio de reproducibilidad entre laboratorios (IRL) para evaluar la reproducibilidad de VENTANA PD-L1 (SP142) ASSAY en la determinación del estado de PD-L1 en tejidos de carcinoma urotelial, NSCLC y TNBC teñidos con el instrumento BenchMark ULTRA PLUS junto con los kits OptiView DAB IHC Detection Kit y OptiView Amplification Kit.
Se utilizaron cuarenta y dos muestras únicas de carcinoma urotelial, NSCLC y TNBC (14 muestras por cada indicación) que representaban el intervalo de tinción de VENTANA PD-L1 (SP142) ASSAY, con una distribución equitativa aproximada entre las muestras positivas en PD-L1y negativas en PD-L1 de cada indicación.
Se cortaron varias secciones de cada muestra y se facilitaron a tres sitios de estudio externos. Las 42 muestras se tiñeron en un instrumento BenchMark ULTRA PLUS en cada uno de los 5 días no consecutivos durante un periodo mínimo de 20 días en cada sitio. Las muestras se aleatorizaron y las evaluaron un total de 6 anatomopatólogos (2 lectores/sitio).
Los resultados se resumen en la Tabla 37 y la Tabla 38. Los datos de análisis para determinar los PPA, NPA y OPA se muestran en la Tabla 37 y los APA, ANA y OPA se presentan en la Tabla 38 de todas las observaciones. De cada muestra, todas las observaciones evaluables (positivas frente a negativas) se compararon con el estado modal de cada muestra. Las comparaciones se combinaron en todos los sitios y días y, posteriormente, se agregaron los resultados de todas las muestras.
Tabla 37. Reproducibilidad entre laboratorios: Índices de concordancia con el estado modal en el instrumento BenchMark ULTRA PLUS del carcinoma urotelial, NSCLC y TNBC
|
Reproducibilidad entre laboratorios |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Global |
PPA |
571/629 |
90.8 |
(85.8, 95.7) |
|
NPA |
595/630 |
94.4 |
(90.7, 98.8) |
|
|
OPA |
1166/1259 |
92.6 |
(89.8, 95.3) |
|
|
En el mismo sitio (tres sitios) |
PPA |
586/629 |
93.2 |
(89.4, 96.6) |
|
NPA |
610/630 |
96.8 |
(95.3, 98.5) |
|
|
OPA |
1196/1259 |
95.0 |
(93.2, 96.6) |
|
|
Mismo lector |
PPA |
589/614 |
95.9 |
(94.3, 97.4) |
|
NPA |
628/645 |
97.4 |
(96.1, 98.7) |
|
|
OPA |
1217/1259 |
96.7 |
(95.5, 97.7) |
|
Nota: Los intervalos de confianza (CI) bilaterales del 95% se calcularon mediante el método bootstrap percentil con 2000 replicados estratificados en función de las tres indicaciones (carcinoma urotelial, NSCLC y TNBC) y de los grupos de cribado (positivo, negativo y en el límite).
Tabla 38. Reproducibilidad entre laboratorios: Índices de concordancia entre pares en el instrumento BenchMark ULTRA PLUS del carcinoma urotelial, NSCLC y TNBC
|
Reproducibilidad entre laboratorios |
Concordancia |
|||
|
Tipo |
n/N |
% |
CI del 95% |
|
|
Entre sitios (tres sitios) |
APA |
10640/12108 |
87.9 |
(82.5, 92.4) |
|
ANA |
11584/13052 |
88.8 |
(85.4, 92.2) |
|
|
OPA |
11112/12580 |
88.3 |
(84.1, 92.2) |
|
|
Entre lectores |
APA |
550/605 |
90.9 |
(86.9, 94.1) |
|
ANA |
598/653 |
91.6 |
(89.1, 93.9) |
|
|
OPA |
574/629 |
91.3 |
(88.2, 94.0) |
|
|
Entre días (5 días no consecutivos) |
APA |
2280/2420 |
94.2 |
(92.0, 96.0) |
|
ANA |
2472/2612 |
94.6 |
(92.9, 96.1) |
|
|
OPA |
2376/2516 |
94.4 |
(92.6, 96.0) |
|
Nota: Los intervalos de confianza (CI) bilaterales del 95% se calcularon mediante el método bootstrap percentil con 2000 replicados estratificados en función de las tres indicaciones (carcinoma urotelial, NSCLC y TNBC) y de los grupos de cribado (positivo, negativo y en el límite).
RENDIMIENTO CLÍNICO: NSCLC ≥ 1% TC 0 ≥ 1% IC:
Se estudiaron el rendimiento de VENTANA PD-L1 (SP142) ASSAY y la eficacia de TECENTRIQ (atezolizumab) en IMpower150 (NCT02366143), un ensayo abierto, multicéntrico, internacional y aleatorizado en pacientes sin quimioterapia que padecían NSCLC no escamoso metastásico. Los objetivos de IMpower150 consistían en determinar si la incorporación de TECENTRIQ (atezolizumab) a la quimioterapia a base de platino [carboplatino más paclitaxel (CP)] con o sin bevacizumab en pacientes que no habían recibido nunca un tratamiento con quimioterapia y con NSCLC no escamoso en estadio IV producía mejorías estadísticamente significativas y clínicamente relevantes en la PFS y la OS.
Se inscribieron 1202 pacientes, estratificados por sexo, presencia de metástasis hepáticas y estado de expresión de PD-L1 en TC e IC. Los pacientes se aleatorizaron (1:1:1) y se asignaron a uno de los siguientes tres grupos de tratamiento.
• TECENTRIQ (atezolizumab) en dosis de 1200 mg con paclitaxel y carboplatino cada tres semanas durante cuatro o seis ciclos y, a continuación, 1200 mg de TECENTRIQ (atezolizumab) cada tres semanas hasta la pérdida de beneficio clínico (Grupo A).
• TECENTRIQ (atezolizumab) en dosis de 1200 mg con bevacizumab, paclitaxel y carboplatino cada tres semanas durante cuatro o seis ciclos y, a continuación, 1200 mg de TECENTRIQ (atezolizumab) hasta la pérdida de beneficio clínico y bevacizumab cada tres semanas hasta detectar una progresión de la enfermedad o alcanzar una toxicidad inaceptable (Grupo B).
• Bevacizumab con paclitaxel y carboplatino cada tres semanas durante cuatro o seis ciclos, seguido de bevacizumab cada tres semanas hasta detectar la progresión de la enfermedad o alcanzar una toxicidad inaceptable (Grupo C).
Los datos demográficos y las características iniciales de la enfermedad estaban bien equilibrados entre los grupos del estudio. La media de edad era de 63 años (intervalo de entre 31 y 90 años) y el 60% de los pacientes eran hombres. La mayoría de los pacientes eran blancos (82%). Aproximadamente el 10% de los pacientes presentaba mutación de EGFR conocida, el 4% presentaba reorganizaciones de ALK, el 14% padecía metástasis en el hígado desde el inicio y la mayoría de los pacientes fumaban o habían fumado previamente (80%). El estado de rendimiento de ECOG inicial era 0 (43%) o 1 (57%). Respecto a los tumores de los pacientes, el 51% presentaba expresión de PD-L1 en ≥ 1% de TC o en ≥ 1% de IC, el 49% presentaba expresión de PD-L1 en < 1% de TC y en < 1% de IC.
En el momento del análisis definitivo para evaluar la PFS, el tiempo medio de seguimiento de los pacientes fue de 15.3 meses. La población con intención de tratar (ITT), entre otros, los pacientes con mutaciones de EGFR o con reorganizaciones de ALK que deberían haber recibido previamente tratamiento con inhibidores de tirosina quinasa, presentó una mejoría clínica importante en la PFS en el Grupo B en comparación con el Grupo C (HR de 0.61, CI del 95%: 0.52, 0.72; PFS media de 8.3 frente a 6.8 meses).
En el momento del análisis provisional para evaluar la OS, el tiempo medio de seguimiento de los pacientes fue de 19.7 meses. Los resultados clave de este análisis, así como los del análisis de PFS actualizados, de la población ITT se resumen en la Tabla 28. Se observaron mejorías con importancia clínica en la OS de la población ITT, con una media de OS de 19.8 meses en el Grupo B (n= 400) pero de 14.9 meses en el Grupo C (n= 400; HR estratificado= 0.76; CI del 95%: 0.63, 0.93).
En la Figura 2, se resumen los resultados de OS de los grupos B y C comparados en la población ITT y los subgrupos de PD-L1. Los resultados de PFS del Grupo B en comparación con los del Grupo C se presentan también en la Figura 3.
Los datos indican que todos los pacientes sin quimioterapia y NSCLC no escamoso localmente avanzado o metastásico, independientemente de la expresión de PD-L1, pueden beneficiarse del tratamiento con TECENTRIQ (atezolizumab) junto con quimioterapia y con bevacizumab y, por consiguiente, cumplen los criterios de idoneidad para recibir el tratamiento. Los datos también sugieren que los beneficios de TECENTRIQ (atezolizumab), en combinación con la quimioterapia con bevacizumab, en esta población pueden ser mayores en pacientes con un nivel de expresión tumoral de PD-L1 de ≥ 1% TC o ≥ 1% IC según lo determinado por el VENTANA PD-L1 (SP142) ASSAY.
El beneficio en la OS que se observó en el Grupo B en comparación con el Grupo C en la población ITT en el siguiente análisis (CCOD de septiembre de 2019, Tabla 29) se mantuvo, pero no se probó formalmente según la jerarquía analítica especificada previamente. Las diferencias en la media de la OD del Grupo B en comparación con el Grupo C de la población ITT en la CCOD de septiembre de 2019 fue de 4.8 meses, con un HR estratificado de 0.80 (CI del 95%: 0.68, 0.95).
El beneficio de OS que se observó en el Grupo B en comparación con el Grupo C en el siguiente análisis (CCOD de septiembre de 2019) era superior en el subgrupo con expresión de PD-L1 en ≥ 1% de TC o ≥ 1% de IC que en el subgrupo con expresión de PD-L1 en < 1% de TC o < 1% de IC (Tabla 29). En el primer subgrupo, la media de supervivencia había aumentado en 8.0 meses en el Grupo B en comparación con el Grupo C, con un HR no estratificado de 0.692 (IC del 95%: 0.547, 0.876). En cambio, la supervivencia media del subgrupo con expresión de PD-L1 en < 1% de TC o < 1% de IC se había incrementado solamente en 2.6 meses, con un HR sin estratificar de 0.95 (CI del 95%: 0.76, 1.19).
Los pacientes seguían tolerando bien el tratamiento con TECENTRIQ (atezolizumab), junto con la quimioterapia con bevacizumab o sin él, y las conclusiones de seguridad eran coherentes con los riesgos conocidos de cada tratamiento del estudio. No se detectaron nuevos indicios de seguridad.
En conjunto, los datos demuestran que la presencia de expresión de PD-L1 en TC o en IC, según la detección de VENTANA PD-L1 (SP142) ASSAY, en NSCLC puede asociarse a un mayor beneficio para el paciente al administrar el tratamiento con TECENTRIQ (atezolizumab), junto con quimioterapia con bevacizumab, en la línea de corte de la expresión de PD-L1 en ≥ 1% de TC o ≥ 1% de IC.
Tabla 28. Resultados de eficacia en la población con intención de tratar de IMpower150
|
Criterio de valoración de la eficacia |
Grupo A |
Grupo B |
Grupo C |
|---|---|---|---|
|
Criterios de valoración secundariosa |
|||
|
PFS evaluada por el investigador (RECIST v1.1)g |
n= 402 |
n= 400 |
n= 400 |
|
Nº de eventos (%) |
330 (82.1) |
291 (72.8) |
355 (88.8) |
|
Duración media de PFS (meses) |
6.7 |
8.4 |
6.8 |
|
CI del 95% |
(5.7, 6.9) |
(8.0, 9.9) |
(6.0, 7.0) |
|
Índice de riesgo estratificadoe,f (CI del 95%) |
0.91 (0.78, 1.06) |
0.59 (0.50, 0.69) |
--- |
|
Valor pb,c |
0.2194 |
< 0.0001 |
|
|
PFS de 12 meses (%) |
24 |
38 |
20 |
|
Análisis provisional de OSg |
n= 402 |
n= 400 |
n= 400 |
|
Nº de muertes (%) |
206 (51.2) |
192 (48.0) |
230 (57.5) |
|
Tiempo promedio hasta los eventos (meses) |
19.5 |
19.8 |
14.9 |
|
CI del 95% |
(16.3, 21.3) |
(17.4, 24.2) |
(13.4, 17.1) |
|
Índice de riesgo estratificadoe,f (CI del 95%) |
0.85 (0.71, 1.03) |
0.76 (0.63, 0.93) |
--- |
|
Valor pb,c |
0.0983 |
0.006 |
|
|
OS de 6 meses (%) |
84 |
85 |
81 |
|
OS de 12 meses (%) |
66 |
68 |
61 |
|
Mejor respuesta general evaluada por el investigadord,g (RECIST v1.1) |
n= 401 |
n= 397 |
n= 393 |
|
Nº de pacientes con respuesta (%) |
163 (40.6) |
224 (56.4) |
158 (40.2) |
|
CI del 95% |
(35.8, 45.6) |
(51.4, 61.4) |
(35.3, 45.2) |
|
Nº de respuesta completa (%) |
8 (2.0) |
11 (2.8) |
3 (0.8) |
|
Nº de respuesta parcial (%) |
155 (38.7) |
213 (53.7) |
155 (39.4) |
|
DOR evaluada por el investigadorg (RECIST v1.1) |
n= 163 |
n= 224 |
n= 158 |
|
Criterios de valoración secundariosa |
|||
|
Media en meses |
8.3 |
11.5 |
6.0 |
|
CI del 95% |
(7.1, 11.8) |
(8.9, 15.7) |
(5.5, 6.9) |
a Los criterios de valoración de la eficacia principales fueron la PFS y la OS y se analizaron en la población ITT de tipo silvestre (WT), es decir, excluyendo a los pacientes que presentaban mutaciones de EGFR o reorganizaciones de ALK.
b Según la prueba del orden logarítmico estratificado.
c Con fines informativos, la población ITT, las comparaciones entre el Grupo B y el Grupo C, así como las del Grupo A y el Grupo C, no se habían analizado oficialmente en ese momento según la jerarquía de análisis que se había definido previamente.
d Mejor respuesta general tanto en respuesta completa como parcial.
e Estratificación por género, presencia de metástasis en el hígado y expresión tumoral de PD-L1 en TC o en IC.
f El Grupo C se considera el grupo de comparación de todos los índices de riesgo.
g Análisis de PFS actualizado y análisis provisional de OS en la línea de corte clínica del 22 de enero de 2018.
PFS = Supervivencia libre de progresión; RECIST = Criterios de Evaluación de Respuesta en Tumores Sólidos v1.1.
CI = Intervalo de confianza; DOR = Duración de la respuesta; OS = Supervivencia global.
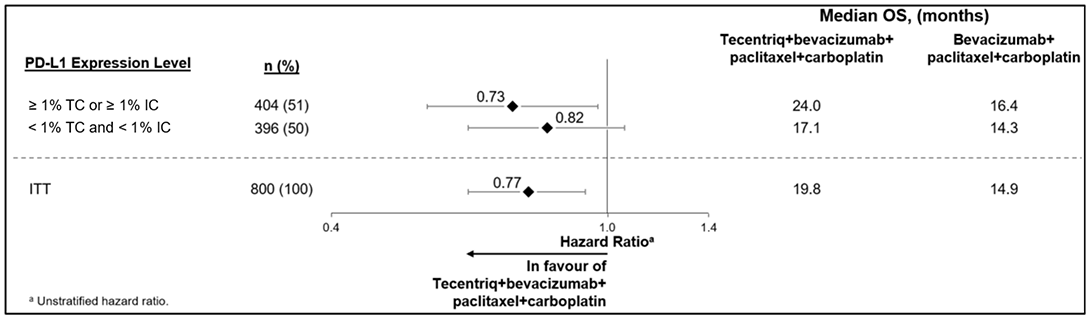
Figura 2. Diagrama de bosque de la supervivencia global en IMpower150, Grupo B frente a C. (CCOD enero de 2018)
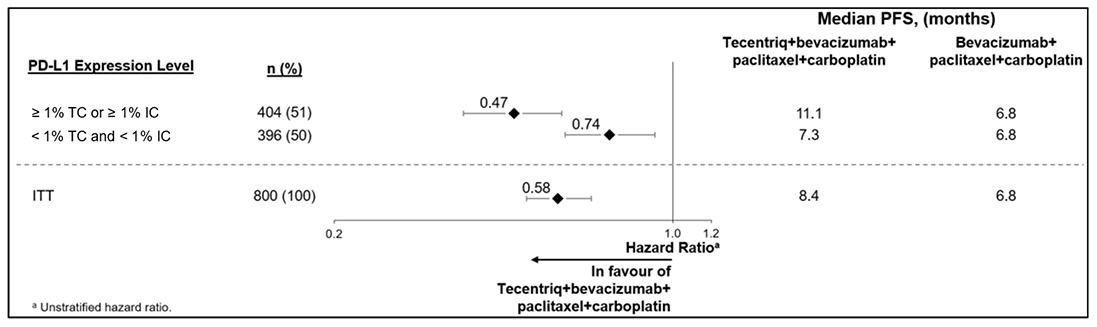
Figura 3. Diagrama de bosque de la supervivencia libre de progresión en IMpower150, Grupo B frente a C. (CCOD enero de 2018)
Tabla 29. Supervivencia global en la población ITT y los subgrupos de PD-L1 (CCOD septiembre de 2019)
|
Supervivencia global (OS): CCOD septiembre de 2019 |
Grupo A |
Grupo B |
Grupo C |
|---|---|---|---|
|
Población ITTa |
n= 402 |
n= 400 |
n= 400 |
|
Muertes, n (%) |
290 (72.1) |
284 (71.0) |
309 (77.3) |
|
Duración media de OS, en meses (CI del 95%)b |
19.0 (16.3, 21.5) |
19.8 (18.2, 22.5) |
15.0 (13.4, 17.1) |
|
Índice de riesgo estratificado (CI del 95%)c,d |
0.86 (0.73, 1.01) |
0.80 (0.68, 0.95) |
--- |
|
Valor p del orden logarítmico estratificadof |
0.0681 |
0.0081 |
--- |
|
Índice de riesgo sin estratificar (CI del 95%)c |
0.84 (0.72, 0.99) |
0.81 (0.69, 0.95) |
--- |
|
Valor p del orden logarítmico sin estratificare |
0.0366 |
0.0084 |
--- |
|
OS de 6 meses, índice de casos sin eventos (%) |
83.5 |
84.8 |
80.8 |
|
OS de 1 año, índice de casos sin eventos (%) |
65.7 |
68.0 |
60.1 |
|
OS de 2 años, índice de casos sin eventos (%) |
41.7 |
43.4 |
33.5 |
|
Subgrupo de ITT con ≥ 1% de TC o ≥ 1% de IC |
n= 213 |
n= 209 |
n= 195 |
|
Muertes, n (%) |
139 (65.3) |
131 (62.7) |
149 (76.4) |
|
Duración media de OS, en mesesb |
24.1 |
24.0 |
16.0 |
|
Índice de riesgo estratificado (CI del 95%)c,d |
0.729 (0.578, 0.92) |
0.697 (0.55, 0.882) |
--- |
|
Valor p del orden logarítmico estratificadoe |
0.0074 |
0.0026 |
--- |
|
Índice de riesgo sin estratificar (CI del 95% Wald)c |
0.72 (0.57, 0.91) |
0.692 (0.547, 0.876) |
--- |
|
Valor p del orden logarítmico sin estratificare |
0.0058 |
0.0021 |
--- |
|
OS de 6 meses, índice de casos sin eventos (%) |
86.3 |
86.3 |
81.5 |
|
OS de 1 año, índice de casos sin eventos (%) |
70.6 |
71.2 |
55.9 |
|
OS de 2 años, índice de casos sin eventos (%) |
50.3 |
50.5 |
37.1 |
|
Subgrupo de ITT con < 1% de TC o < 1% de IC |
n= 188 |
n= 191 |
n= 205 |
|
Muertes, n (%) |
150 (79.8) |
153 (80.1) |
160 (78.0) |
|
Duración media de OS, en meses (CI del 95%)b |
15.3 (13.1, 18.7) |
17.0 (14.1, 19.8) |
14.4 (13.4, 17.1) |
|
Índice de riesgo estratificado (CI del 95%)c,d |
1.00 (0.80, 1.25) |
0.94 (0.75, 1.17) |
--- |
|
Valor p del orden logarítmico estratificadoe |
0.9849 |
0.5670 |
--- |
|
Índice de riesgo sin estratificar (CI del 95%)c |
1.00 (0.80, 1.26) |
0.95 (0.76, 1.19) |
--- |
|
Valor p del orden logarítmico sin estratificare |
0.9739 |
0.6628 |
--- |
|
OS de 6 meses, índice de casos sin eventos (%) |
80.2 |
83.2 |
80.2 |
|
OS de 1 año, índice de casos sin eventos (%) |
60.4 |
64.6 |
64.1 |
|
OS de 2 años, índice de casos sin eventos (%) |
32.2 |
35.8 |
30.0 |
a En la población con intención de tratar (ITT) se encuentran todos los pacientes aleatorizados del estudio GO29436 (hubieran recibido o no el tratamiento del estudio).
b Los resultados de tiempo hasta los eventos (media, percentiles) se han calculado mediante el método de Kaplan-Meier. Los CI del 95% de la media se computan con el método de Brookmeyer y Crowley.
c El Grupo C se considera el grupo de comparación de todos los índices de riesgo
d Estratificación por género, presencia de metástasis en el hígado y expresión tumoral de PD-L1 en TC o en IC.
e Los resultados se presentan únicamente con fines informativos.
CCOD = fecha de línea de corte de los datos clínicos; IIT = Intención de tratar.
USO PREVISTO:
VENTANA PD-L1 (SP142) ASSAY está destinado a su uso en laboratorio para la detección cualitativa inmunohistoquímica del ligando de muerte programada 1 (PD-L1) mediante microscopía óptica en células tumorales y células inmunitarias que infiltran el tumor de los tejidos fijados con formol y embebidos en parafina (FFPE) que se indican a continuación teñidos con OptiView DAB IHC Detection Kit y OptiView Amplification Kit en un instrumento BenchMark IHC/ISH.
La determinación del estado de PD-L1 es específica de la indicación y su evaluación debe sustentarse en la proporción del área del tumor que ocupan las células inmunitarias que infiltran el tumor (% de IC) con expresión de PD-L1 de cualquier intensidad o el porcentaje de células tumorales (% de TC) con expresión de PD-L1 de cualquier intensidad.
Figura 1. PD-L1 en cáncer de pulmón de células no pequeñas.
Consulte las tablas que aparecen a continuación para ver los tipos de tumor específicos y las aplicaciones clínicas. Consulte el etiquetado del correspondiente fármaco para ver las recomendaciones clínicas relativas a la expresión de PD-L1.
Tabla 1. Indicaciones de uso de las pruebas diagnósticas con fines terapéuticos
|
Tipo de tumor |
Valor de corte de la expresión de PD-L1 |
Aplicación clínica |
|
Carcinoma urotelial |
≥ 5% de IC |
La expresión de PD-L1 en las células inmunitarias que infiltran el tumor (IC) que detecta el VENTANA PD-L1 (SP142) ASSAY en el carcinoma urotelial está indicada para ayudar a identificar a los pacientes a los que se podría aplicar el tratamiento con TECENTRIQ (atezolizumab). |
|
Carcinoma de mama triple negativo (TNBC) |
≥ 1% de IC |
La expresión de PD-L1 en las células inmunitarias que infiltran el tumor (IC) que detecta el VENTANA PD-L1 (SP142) ASSAY en el TNBC se indica para ayudar a identificar a los pacientes a los que se podría aplicar el tratamiento con TECENTRIQ (atezolizumab). |
|
Cáncer de pulmón de células no pequeñas (NSCLC) |
≥ 50% TC o ≥ 10% IC |
La expresión de PD-L1 en la membrana de las células tumorales (TC) o en las células inmunitarias que infiltran el tumor (IC) que detecta el VENTANA PD-L1 (SP142) ASSAY en el NSCLC se indica para ayudar a identificar a los pacientes a los que se podría aplicar el tratamiento con TECENTRIQ (atezolizumab). |
Tabla 2. Instrucciones de uso adicionales
|
Tipo de tumor |
Valor de corte de la expresión de PD-L1 |
Aplicación clínica |
|
Cáncer de pulmón de células no pequeñas (NSCLC) |
≥ 50% TC o ≥ 10% IC |
La expresión de PD-L1 en la membrana de las células tumorales (TC) o en las células inmunitarias que infiltran el tumor (IC) que detecta el VENTANA PD-L1 (SP142) ASSAY en el NSCLC puede asociarse a un mayor beneficio para el paciente al que se administra el tratamiento con TECENTRIQ (atezolizumab). |
|
≥ 1% de TC o ≥ 1% IC |
La expresión de PD-L1 en la membrana de las células tumorales (TC) o en las células inmunitarias que infiltran el tumor (IC) que detecta el VENTANA PD-L1 (SP142) ASSAY en el NSCLC puede asociarse a un mayor beneficio para el paciente al que se administra el tratamiento con TECENTRIQ (atezolizumab). |
La interpretación de los resultados de la prueba de este producto debe correr a cargo de un anatomopatólogo cualificado junto con un examen histológico, la información clínica pertinente y los controles adecuados.
Este producto está destinado para uso diagnóstico in vitro (IVD).
LIMITACIONES ESPECÍFICAS:
1. VENTANA PD-L1 (SP142) ASSAY se ha homologado únicamente para los instrumentos BenchMark IHC/ISH con OptiView DAB IHC Detection Kit y OptiView Amplification Kit y no se ha aprobado para ningún otro instrumento o kit de detección.
2. Este ensayo no se ha validado para su uso con muestras de citología o de hueso decalcificadas.
3. El tejido del paciente se debe teñir en un plazo máximo de dos meses a partir del corte del bloque de tejido, en el caso de los tejidos de NSCLC, TNBC y amígdala, y de tres meses en el caso de los tejidos de carcinoma urotelial. Se ha observado pérdida de rendimiento de tinción con la tinción VENTANA PD-L1 (SP142) ASSAY en secciones de tejido que se han conservado a temperatura ambiente durante periodos superiores a los mencionados.
4. Con la presencia de artefactos, como manchas de DAB, manchas en blanco, puntos DAB y/o moteados, será preciso repetir la tinción siempre que interfieran con la interpretación de VENTANA PD-L1 (SP142) ASSAY. Compare siempre el portaobjetos con tinción de PD-L1 con el control de reactivo negativo para garantizar que la tinción de fondo es aceptable. Consulte las guías de interpretación para obtener más información al respecto.
5. Se ha observado la presencia de puntos DAB de forma ocasional en tejido de control de amígdala humana benigna y tejidos de cerebelo y de testículos, así como tinción focal nuclear en tejidos de páncreas normales (células acinosas) y de hipófisis (Tabla 6); no obstante, la tinción nuclear no se incorpora a la puntuación de la tinción con VENTANA PD-L1 (SP142) ASSAY.
6. Es posible que no todos los ensayos estén registrado en todos los instrumentos. Póngase en contacto con el representante local de servicio Roche para obtener más información.
PREPARACIÓN DE MUESTRAS:
Los tejidos FFPE que se procesan de forma habitual resultan adecuados para su uso con este anticuerpo primario cuando se utilizan con los kits de detección de VENTANA y los instrumentos BenchMark IHC/ISH. Se recomienda la fijación de tejido en formol tamponado neutro al 10% (NBF) durante al menos 6 horas y durante un máximo de 72 horas. Los tiempos de fijación inferiores a las 6 horas pueden provocar la pérdida de tinción en PD-L1. La cantidad usada de NBF debe ser de entre 15 y 20 veces el volumen de tejido. Ningún fijador se infiltrará en tejido sólido de más de 2 o 3 mm ni en tejido poroso de 5 mm durante un periodo de 24 horas. La fijación se puede llevar a cabo a temperatura ambiente (de 15 a 25°C).11,12
Con los fijadores como el alcohol-formol-ácido acético (AFA), PREFER y otros que contengan alcohol se ha observado una pérdida de tinción específica de PD-L1 en todos los tiempos de fijación analizados (entre 1 y 72 horas) y no se recomienda su uso con este ensayo. Consulte las guías de interpretación para obtener información más detallada sobre la repercusión de la preparación de muestras en la intensidad de la tinción de PD-L1.
Se deben realizar cortes de unos 4 μm de grosor y colocarse en portaobjetos de vidrio cargados positivamente. Los portaobjetos deben teñirse inmediatamente, ya que la antigenicidad de los cortes de tejido puede disminuir con el tiempo y verse comprometida una vez que han transcurrido tres meses desde el corte del bloque de parafina, en el caso de las muestras de carcinoma urotelial, y dos meses en el caso de las muestras de amígdala, NSCLC y TNBC (consulte las guías de interpretación y la sección sobre Características de rendimiento que aparece a continuación).
Se recomienda analizar controles positivos y negativos a la vez que las muestras desconocidas.
PRINCIPIO DEL PROCEDIMIENTO:
VENTANA PD-L1 (SP142) ASSAY contiene un anticuerpo primario monoclonal de conejo que se une a PD-L1 en secciones de tejido embebido en parafina. El anticuerpo específico se puede visualizar mediante OptiView DAB IHC Detection Kit (nº de cat. 760-700/06396500001) y, a continuación, a través de OptiView Amplification Kit (nº de cat. 760-099/06396518001 [50 pruebas] o 860-099/06718663001 [250 pruebas]).
Consulte la hoja de datos correspondiente para obtener más información.
PROCEDIMIENTO DE TINCIÓN:
Los anticuerpos primarios VENTANA se han desarrollado para su uso en los instrumentos BenchMark IHC/ISH junto con los kits de detección de VENTANA y sus accesorios. Con VENTANA PD-L1 (SP142) ASSAY se debe utilizar un procedimiento de tinción específico del ensayo. Consulte la Tabla 3 y la Tabla 4 para obtener información sobre el protocolo de tinción recomendado y los procedimientos de tinción necesarios. Cualquier diferencia en la forma de llevar a cabo los procedimientos de prueba recomendados pueden invalidar los resultados previstos. Deben emplearse los controles adecuados y documentarlos. Los usuarios que no sigan los procedimientos de prueba recomendados deberán hacerse responsables de la interpretación de los resultados de la paciente.
Este anticuerpo se ha optimizado para tiempos de incubación específicos, pero el usuario debe validar los resultados obtenidos con este reactivo.
Los parámetros de los procedimientos automatizados se pueden visualizar, imprimir y editar según el procedimiento descrito en el Manual del usuario del instrumento. Consulte la hoja de datos del kit de detección VENTANA correspondiente para obtener más detalles sobre los procedimientos de tinción de inmunohistoquímica.
Para obtener más información sobre el uso adecuado de este dispositivo, consulte la hoja de datos del dispensador en línea asociada al N/P 741-4860.
Tabla 3. Utilice los siguientes procedimientos de tinción con VENTANA PD-L1 (SP142) ASSAY en instrumentos BenchMark IHC/ISH
|
Instrumento |
Procedimiento de tinción |
|
BenchMark GX |
GX VENTANA PDL1 (SP142) |
|
BenchMark ULTRA o BenchMark ULTRA PLUS |
ULTRA VENTANA PDL1 (SP142) |
Tabla 4. Protocolo de tinción recomendado para el VENTANA PD-L1 (SP142) ASSAY y Rabbit Monoclonal Negative Control Ig con OptiView DAB IHC Detection Kit y OptiView Amplification Kit en instrumentos BenchMark IHC/ISH
|
Tipo de procedimiento |
Entrada de parámetros |
|
Horneado |
Opcional |
|
Anticuerpo (primario) |
VENTANA PD-L1 (SP142) seleccionado o Control Negativo Seleccionado |
|
Contratinción |
Hematoxylin II, 4 minutos |
|
Post-contratinción |
Bluing, 4 minutos |
RESUMEN Y EXPLICACIÓN:
VENTANA PD-L1 (SP142) ASSAY es un ensayo de inmunohistoquímica con un anticuerpo primario monoclonal de conejo anti-PD-L1 que detecta la proteína PD-L1. Roche/VENTANA Medical Systems, Inc. (VENTANA) y Roche/Genentech han desarrollado conjuntamente este ensayo para identificar a aquellos pacientes que tienen mayor probabilidad de responder al tratamiento con TECENTRIQ® (atezolizumab).
PD-L1 es una proteína transmembrana que reduce las respuestas inmunitarias a través de la unión a sus dos receptores de muerte programada 1 (PD-1) y B7.1.1 PD-1 es un receptor de inhibición cuya expresión se observa en los linfocitos T tras su activación y se mantiene en estados de estimulación crónica, como las infecciones crónicas o el cáncer.1 La unión de PD-L1 con PD-1 inhibe la proliferación de los linfocitos T, la producción de citocinas y la actividad citolítica, dando lugar a la inactivación funcional o al agotamiento de los linfocitos T.1,2 B7.1 es una molécula cuya expresión se observa en las células con antígeno y en los linfocitos T activados.1,2 La unión de PD-L1 a B7.1 en linfocitos T y células con antígeno puede participar en la reducción de las respuestas inmunitarias, como la inhibición de la activación de los linfocitos T y la producción de citocinas.2 La expresión de PD-L1 se ha observado en células inmunitarias y en células tumorales y se ha descrito que la expresión anómala de PD-L1 en células tumorales impide la inmunidad antitumoral, dando como resultado una evasión inmunológica.1,3 Por lo tanto, la interrupción de la vía PD-L1/PD-1 puede resultar una estrategia interesante para revitalizar la inmunidad específica frente a tumores de los linfocitos T que queda suprimida por la expresión de PD-L1 en el microentorno tumoral. Se ha documentado la asociación entre la expresión de PD-L1 en células tumorales (TC) o en células inmunitarias que infiltran el tumor (IC) y el beneficio clínico de los inhibidores de la vía PD-L1/PD-1 en diferentes tipos de cáncer.3-10
Atezolizumab es un anticuerpo monoclonal diseñado mediante fragmentos cristalizables (Fc) y humanizado que se une a PD-L1 y bloquea las interacciones con los receptores de PD-1 y B7.1.3-10
REFERENCIAS:
1. Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56(5):739-745.
2. Butte MJ, Keir ME, Phamduy TB, et al. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111-122.
3. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563-567.
4. Bellmunt J, Mullane SA, Werner L, et al. Association of PD-L1 expression on tumor- infiltrating mononuclear cells and overall survival in patients with urothelial carcinoma. Ann Oncol. 2015;26(4):812-817.
5. Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti- programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167-3175.
6. Fehrenbacher L, Spira A, Ballinger M et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837-1846.
7. Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558-562.
8. Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909–20.
9. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
10. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med. 2018;379(22):2108-2121.
11. Carson FL, Cappellano C. Histotechnology; A Self-Instructional Text, 5th edition. American Society for Clinical Pathology Press; 2020, 2022.
12. Roche PC, Hsi ED. Immunohistochemistry-Principles and Advances. Manual of Clinical Laboratory Immunology, 6th edition. In: NR Rose, ed. ASM Press; 2002.
13. Occupational Safety and Health Standards: Occupational exposure to hazardous chemicals in laboratories. (29 CFR Part 1910.1450). Fed. Register.
14. Directive 2000/54/EC of the European Parliament and Council of 24 June 2020 on the protection of workers from risks related to exposure to biological agents at work.
15. Anatomic Pathology Checklist. College of American Pathologists. Jul 28, 2015.
16. CLSI. Quality assurance for design control and implementation of immunohistochemistry assay: approved guidelines. 2nd edition Wayne, PA, USA: Clinical and Laboratory Standards Institute; 2011.
Nota: En este documento se ha usado el punto como separador decimal para marcar el borde entre la parte entera y la parte fraccionaria de los numerales con decimales. No se han usado separadores para las unidades de millar.
El resumen de los aspectos de seguridad y rendimiento se puede ver a continuación: https://ec.europa.eu/tools/eudamed
Símbolos:
VENTANA usa los siguientes símbolos y signos además de los indicados en la norma ISO 15223-1 (para USA: consulte elabdoc.roche.com/symbols para obtener más información).
|
GTIN |
Número de artículo de Global Trade |
Propiedad intelectual:
VENTANA, BENCHMARK y OPTIVIEW son marcas comerciales de Roche. Todos los demás nombres de productos y marcas comerciales pertenecen a sus respectivos propietarios.
© 2025 Ventana Medical Systems, Inc.
Información de contacto:
ROCHE DIAGNOSTICS GMBH
Sandhofer Strasse 116
68305 Mannheim Germany
+800 5505 6606


