
VEOZA
FEZOLINETANT
Tableta
1 Caja, 1 Envase de burbuja, 30 Tabletas,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA contiene:
Fezolinetant 45 mg
Excipientes cbp 1 tableta
En la sección Leyendas de protección se presenta la lista completa de excipientes.
INDICACIONES TERAPÉUTICAS:
VEOZA® es un antagonista no hormonal selectivo del receptor de neuroquinina 3 (NK3) indicado para el tratamiento de los síntomas vasomotores (SVM) de moderados a severos asociados con la menopausia.
FARMACOCINÉTICA Y FARMACODINAMIA:
Mecanismo de acción:
Fezolinetant es un antagonista no hormonal selectivo del receptor de la NK3 que bloquea a la neuroquinina B (NKB) fijándose a las neuronas kisspeptina/neuroquinina B/dinorfina (KNDy) para modular la actividad neuronal en el centro termorregulador.
El centro termorregulador en el hipotálamo está enervado por las neuronas KNDy, que son inhibidas por el estrógeno y estimuladas el neuropéptido NKB. A lo largo de la transición menopáusica, la disminución del estrógeno altera el equilibrio con el NKB. Sin un antagonista, la señalización del NKB incrementa la actividad neuronal de KNDy conduciendo a la hipertrofia de las neuronas KNDy y a la alteración de la actividad del centro termorregulador, lo que resulta en síntomas vasomotores (SVM), también conocidos como bochornos sudores nocturnos.
Efectos farmacodinámicos:
El tratamiento con fezolinetant proporciona alivio de los SVM por 24 horas. Fezolinetant no es una terapia hormonal y el tratamiento con fezolinetant no mostró una tendencia clara ni cambios clínicamente importantes en las hormonas sexuales medidas (hormona estimulante del folículo, testosterona, estrógeno y sulfato de deshidroepiandrosterona) en mujeres menopáusicas. A las concentraciones pico de fezolinetant se observó la disminución transitoria de los niveles de la hormona luteinizante (LH).
Electrofisiología cardiaca: Se llevó a cabo un método basado en un modelo para evaluar el riesgo de la prolongación del intervalo QT por fezolinetant. Con este modelo no se predijo ninguna prolongación clínicamente importante del intervalo QTc a las concentraciones terapéuticas o supra terapéuticas.
Eficacia y seguridad clínica:
Estudios SKYLIGHT 1 (2693-CL-0301) y SKYLIGHT 2 (2693-CL-0302):
Eficacia:
Efectos en los SVM:
La eficacia de VEOZA® fue evaluada en 1022 mujeres con SVM de moderados a severos asociados con la menopausia en dos estudios fase 3 de 12 semanas de seguimiento, aleatorizados, comparativos con placebo, doble ciegos, seguidos de un periodo de extensión de tratamiento de 40 semanas. La edad promedio fue de 54 años y las mujeres eran caucásicas (81%), negras (17%), asiáticas (1%), e hispana/latinas (24%). En los estudios, se reclutaron mujeres con un promedio mínimo de 7 episodios de SVM de moderados a severos por día.
La población del estudio incluyó mujeres menopáusicas que habían recibido terapia de reemplazo hormonal (TRH) previa (19.9%), con ooforectomía (21.6%) o con histerectomía (32.1%).
Los criterios de valoración de eficacia co-primarios de ambos estudios fueron los cambios con respecto a los valores iniciales en la frecuencia de los SVM de moderados a severos y la intensidad a la semana 4 y 12. Los resultados de los estudios mostraron una reducción estadística y clínicamente significativa (≥ 2 bochornos en 24 horas) con respecto a la línea base en la frecuencia de SVM de moderados a severos a las semanas 4 y 12 con VEOZA® 45 mg en comparación con el placebo. Los resultados de los estudios mostraron una reducción estadísticamente significativa con respecto a la línea base en la intensidad de los SVM de moderados a severos a las semanas 4 y 12 con VEOZA® 45 mg en comparación con el placebo.
Una mayor proporción, estadísticamente significativa, de mujeres alcanzaron una reducción intraindividual clínicamente signiicativa en la frecuencia de los SVM de moderados a severos a las semanas 4 y 12 en el grupo con VEOZA® 45 mg (46.6% a la semana 4 y 47.2% a la semana 12) en comparación con el grupo placebo (24.0% a la semana 4 y 25.7% a la semana 12) con base en un análisis pre-especificado para mujeres que tuvieron cuando menos mejoría moderada en la medición de SVM PGI-C como punto de anclaje primario.
VEOZA® es eficaz en un amplio rango de mujeres, independientemente de la edad, raza, etnicidad, índice de masa corporal (IMC) y tabaquismo. Adicionalmente, la eficacia de VEOZA® se observa sin importar la frecuencia, intensidad y duración de los SVM iniciales, el tiempo transcurrido desde la amenorrea, antecedentes de trastornos del sueño, o antecedentes de hipertensión. Además, VEOZA® es eficaz en mujeres con o sin histerectomía u ooforectomía, con o sin TRH previa, y con o sin uso concurrente de un inhibidor de la recaptación de serotonina (SSRI).
VEOZA® 45 mg redujo la frecuencia y la intensidad de los SVM en una semana. La mejoría en la frecuencia e intensidad de los SVM se mantuvo a lo largo de las 52 semanas de los estudios.
Las mujeres que inicialmente tomaron placebo, y que posteriormente se asignaron aleatoriamente a VEOZA® durante el periodo de extensión, experimentaron una reducción en la frecuencia e intensidad de los SVM consistente con la que experimentaron las mujeres que recibieron VEOZA® a durante todo el estudio.
Los resultados del criterio de valoración co-primario para el cambio a la semana 4 y la semana 12 con respecto a los valores iniciales de la frecuencia media de los SVM de moderados a severos en 24 horas de SKYLIGHT 1 y 2 y de los estudios combinados se presentan en la Tabla 1.
Tabla 1. SKYLIGHT 1 y 2: Valores iniciales promedio y cambio con respecto a los valores iniciales de la frecuencia media de los SVM de moderados a severos en 24 horas a la semana 4 y 12
|
Parámetro |
SKYLIGHT 1 |
SKYLIGHT 2 |
Estudios combinados |
|||
|
VEOZA® 45 mg (n = 174) |
Placebo (n = 175) |
VEOZA® 45 mg (n = 167) |
Placebo (n = 167) |
VEOZA® 45 mg (n = 341) |
Placebo (n = 342) |
|
|
Valores iniciales |
||||||
|
Media (Desv. estd.) |
10.44 (3.92) |
10.51 (3.79) |
11.79 (8.26) |
11.59 (5.02) |
11.10 (6.45) |
11.4 (4.46) |
|
Cambio a la semana 4 con respecto a los valores iniciales |
||||||
|
Media de MC (EE) |
-5.39 (0.30) |
-3.32 (0.29) |
-6.26 (0.33) |
-3.72 (0.33) |
-5.79 (0.23) |
-3.51 (0.22) |
|
Reducción media % |
50.63% |
30.46% |
55.16% |
33.60% |
52.84% |
31.96% |
|
Diferencia vs placebo (EE) |
-2.07 (0.42) |
-- |
-2.55 (0.46) |
-- |
-2.28 (0.32) |
-- |
|
Valor P |
< 0.0011 |
-- |
< 0.0011 |
-- |
< 0.001 |
-- |
|
Cambio a la semana 12 con respecto a los valores iniciales |
||||||
|
Media de MC (EE) |
-6.44 (0.31) |
-3.90 (0.31) |
-7.50 (0.39) |
-4.97 (0.39) |
-6.94 (0.25) |
-4.43 (0.25) |
|
Reducción media % |
61.35% |
34.97% |
64.27% |
45.35% |
62.80% |
40.18% |
|
Diferencia vs placebo (EE) |
-2.55 (0.43) |
-- |
-2.53 (055) |
-- |
-2.51 (0.35) |
-- |
|
Valor P |
< 0.0011 |
-- |
< 0.0011 |
-- |
< 0.001 |
-- |
1 Superior con significancia estadística en comparación con el placebo a un nivel de 0.05 con ajuste por comparaciones múltiples.
Media de MC: media de mínimos cuadrados estimada a partir de un modelo mixto para un análisis de covarianza de mediciones repetidas; Desv. estd.: desviación estándar; EE: error estándar.
Las Figuras 1 y 2 presentan la frecuencia media de los SVM de moderados a severos en 24 horas en SKYLIGHT 1 y 2.
Figura 1. SKYLIGHT 1: Frecuencia media (EE) de los SVM de moderados a severos en 24 horas por semana
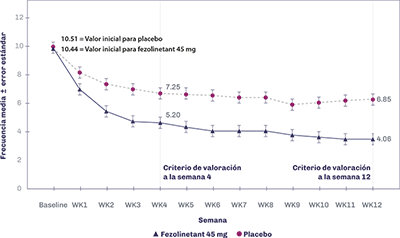
Figura 2. SKYLIGHT 2: Frecuencia media (EE) de los SVM de moderados a severos en 24 horas por semana
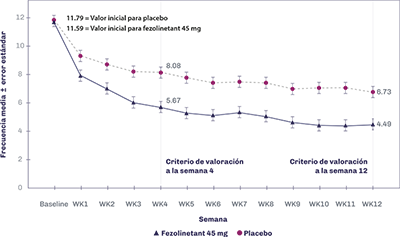
Los resultados del criterio de valoración co-primario para el cambio a la semana 4 y semana 12 con respecto a los valores iniciales en la intensidad media de los SVM de moderados a severos en 24 horas de SKYLIGHT 1 y 2 de los estudios combinados se presentan en la Tabla 2.
Tabla 2. SKYLIGHT 1 y 2: Valores iniciales y cambio con respecto a los valores iniciales de la intensidad media de los SVM de moderados a severos en 24 horas a la semana 4 y 12
|
Parámetro |
SKYLIGHT 1 |
SKYLIGHT 2 |
Estudios combinados |
|||
|
VEOZA® 45 mg (n = 174) |
Placebo (n = 175) |
VEOZA® 45 mg (n = 167) |
Placebo (n = 167) |
VEOZA® 45 mg (n = 341) |
Placebo (n = 342) |
|
|
Valores iniciales |
||||||
|
Media (Desv. Estd.) |
2.40 (0.35) |
2.43 (0.35) |
2.41 (0.34) |
2.41 (0.32) |
2.40 (0.35) |
2.42 (0.34) |
|
Cambio a la semana 4 con respecto a los valores iniciales |
||||||
|
Media de MC (EE) |
-0.46 (0.04) |
-0.27 (0.04) |
-6.61 (0.05) |
-3.32 (0.05) |
-0.53 (0.03) |
-0.30 (0.03) |
|
Diferencia vs placebo (EE) |
-0.19 (0.06) |
-- |
-0.29 (0.06) |
-- |
-0.24 (0.04) |
-- |
|
Valor P |
0.0021 |
-- |
< 0.0011 |
-- |
< 0.001 |
-- |
|
Cambio a la semana 12 con respecto a los valores iniciales |
||||||
|
Media de MC (EE) |
-0.57 (0.05) |
-0.37 (0.05) |
-0.77 (0.06) |
-4.48 (0.06) |
-0.67 (0.04) |
-0.42 (0.04) |
|
Diferencia vs placebo (EE) |
-0.20 (0.08) |
-- |
-0.29 (0.08) |
-- |
-0.24 (0.06) |
-- |
|
Valor P |
0.0071 |
-- |
< 0.0011 |
-- |
< 0.001 |
-- |
1 Superior con significancia estadística en comparación con el placebo con ajuste por comparaciones múltiples.
Media de MC: media de mínimos cuadrados estimada a partir de un modelo mixto para un análisis de covarianza de mediciones repetidas; Desv. Estd.: desviación estándar; EE: error estándar.
Otra eficacia:
Resultados reportados por el paciente:
Trastorno del sueño (PROMIS SD SF 8b):
El análisis de eficacia combinada (SKYLIGHT 1 y 2) del criterio de valoración secundario clave de VEOZA® 45 mg demostró una mejoría a la semana 12 con respecto a los valores iniciales (media de mínimos cuadrados (EE): -1.5 (0.5), 95% CI: (-2.5, -0.5), valor P: 0.004) de alteraciones del sueño en comparación con el placebo, determinados con el Formato 8b abreviado para la Calificación total del trastorno del sueño del Sistema de información de resultados reportados por el paciente (patient-reported outcomes measurement information system/PROMIS). La reducción de la PROMIS SD 8b se mantuvo a lo largo de las 52 semanas. Calidad de vida específico para menopausia (MENQoL).
VEOZA® 45 mg resultó en una mejoría de la calidad de vida en relación con la salud (HRQoL) a la semana 12 con respecto a los valores iniciales en comparación con el placebo, determinados con la calificación total de la MENQoL y la puntuación de dominio de los SVM para MENQoL. La reducción de la MENQoL se mantuvo a lo largo de las 52 semanas. Los resultados se presentan en la Tabla 3.
Tabla 3. SKYLIGHT 1 y 2: Media de los valores iniciales y cambio con respecto a los valores iniciales en la calificación total y puntuación de dominio de los SVM para MENQoL a la semana 12
|
Parámetro |
Estudios combinados (SKYLIGHT 1 y 2) |
|
|---|---|---|
|
VEOZA® 45 mg |
Placebo |
|
|
Calificación total |
||
|
Media de los valores iniciales (Desv. Estd.) |
4.3 (1.4) |
4.3 (1.4) |
|
Media (Desv. Estd.) del cambio a la semana 12 con respecto a los valores iniciales1 |
-1.3 (1.4) |
-0.9 (1.4) |
|
Diferencia de la media de MC (EE) a la semana 122 |
-0.5 (0.1) |
-- |
|
IC al 95% |
-0.7 (0.3) |
-- |
|
Valor P |
< 0.001 |
|
|
Puntuación del dominio de los SVM |
||
|
Media de los valores iniciales (Desv. Estd.) |
6.5 (1.6) |
6.5 (1.4) |
|
Media (Desv. Estd.) del cambio a la semana 12 con respecto a los valores iniciales1 |
-2.4 (2.1) |
-1.6 (2.0) |
|
Diferencia de la media de MC (EE) a la semana 122 |
-0.9 (02) |
-- |
|
IC al 95% |
-1.2, -0.6 |
-- |
|
Valor P |
< 0.001 |
|
1 Un cambio negativo indica una reducción/mejoría con respecto a los valores iniciales.
2 Las diferencias se calculan restando la media de mínimos cuadrados del grupo con placebo de la media de mínimos cuadrados del grupo con fezolinetant.
Media de MC: media de mínimos cuadrados estimados a partir de un modelo mixto para el análisis de covarianza de medidas repetidas; Desv. Estd.: desviación estándar; EE: error estándar; IC: Intervalo de Confianza.
Seguridad:
Seguridad endometrial:
En el estudio de seguridad de largo plazo (SKYLIGHT 4, estudio 2693-CL-0304) se evaluó la seguridad y la tolerabilidad de VEOZA® en un total de 1830 mujeres. La edad promedio fue de 55 años y la etnicidad de las mujeres fue caucásica (80%), negra (17%), asiática (2%), e hispana/latina (20%). La duración promedio de la exposición fue de 296 días. La seguridad endometrial se evaluó mediante ultrasonido transvaginal y biopsia endometrial en mujeres que contaban con una biopsia endometrial inicial y posterior.
En SKYLIGHT 4, VEOZA® demostró no tener efecto en los criterios de valoración primarios de hiperplasia o cáncer endometrial, evaluados mediante biopsia endometrial. VEOZA® no tuvo ningún efecto en el espesor endometrial evaluado mediante ultrasonido transvaginal en comparación con el placebo.
En los grupos con una dosis de VEOZA® de 45 mg en los estudios de fase 3 (SKYLIGHT 1, 2 y 4), las evaluaciones de biopsia endometrial identificaron 1 caso de hiperplasia endometrial, pero ningún caso de cáncer endometrial. La tasa de este tipo de eventos en el grupo de dosis de VEOZA® 45 mg fue ≤ 1%, siendo el límite superior del intervalo de confianza al 95% a una cola ≤ 4%, cumpliendo así con los criterios previamente establecidos para la seguridad endometrial.
Propiedades farmacocinéticas:
En mujeres sanas, la Cmáx y el ABC de fezolinetant se incrementaron proporcionalmente con dosis entre 20 y 60 mg una vez al día.
Después de una dosis de una vez al día, la concentración en plasma de fezolinetant en el estado de equilibrio se alcanzó generalmente al día 2, con una mínima acumulación de fezolinetant. La farmacocinética de fezolinetant no cambia con el tiempo.
Absorción:
El tiempo medio (rango) para alcanzar la Cmáx de fezolinetant es de 1.5 (1 a 4) horas en mujeres sanas.
Efecto del alimento: VEOZA® puede ser administrada con o sin alimento. No se observaron diferencias clínicamente significativas en la farmacocinética de fezolinetant después de la administración de una comida alta en grasas y calorías.
Distribución:
La media del volumen de distribución aparente (Vz/F) de fezolinetant es 189 L. La fijación a las proteínas plasmáticas de fezolinetant es baja (51%). La distribución de fezolinetant en los eritrocitos es casi igual a la del plasma.
Metabolismo:
Fezolinetant se metaboliza primordialmente por el CYP1A2 en humanos para producir el metabolito principal ES259564 oxidado. ES259564 es aproximadamente 20 veces menos potente contra el receptor de NK3 humano sin actividad significativa fuera del objetivo. La proporción metabolito/precursor oscila entre 0.7 y 1.8.
Eliminación y excreción:
La depuración aparente en el estado de equilibrio de fezolinetant es 10.8 L/h. Después de la administración oral, fezolinetant es eliminado principalmente en la orina (76.9%) y en menor grado en heces (14.7%). En orina, una media de 1.1% de la dosis administrada de fezolinetant fue excretada sin cambio y 61.7% de la dosis administrada fue excretada como ES259564. La vida media efectiva (t1/2) de fezolinetant es 9.6 horas en mujeres con síntomas vasomotores.
Poblaciones especiales:
Efecto de la edad, raza y peso corporal: No hay efectos clínicamente relevantes por la edad (18 a 65 años), raza (negra, asiática, otra), peso corporal (42 a 126 kg), o estado de menopausia sobre la farmacocinética de fezolinetant.
Insuficiencia renal: Después de la administración de una sola dosis de 30 mg de fezolinetant, no se presentaron efectos clínicamente relevantes en la exposición de fezolinetant (Cmáx y ABC) en mujeres con insuficiencia renal de leve (eGFR de 60 a menos de 90 mL/min/1.73 m2) a grave (eGFR menos de 30 mL/min/1.73 m2). El ABC de ES259564 no cambió en mujeres con insuficiencia renal leve, pero se incrementó aproximadamente de 1.7 a 4.8 veces con insuficiencia renal moderada (eGFR 30 a menos de 60 mL/min/1.73 m2) y grave. Fezolinetant no se ha estudiado en personas con enfermedad renal en estado terminal (eGFR menor a 15 mL/min/1.73 m2).
Insuficiencia hepática: Después de la administración de una sola dosis de 30 mg de fezolinetant en mujeres con insuficiencia hepática crónica Child-Pugh Clase A (leve), la media de la Cmáx de fezolinetant se incrementó en un 23% y el ABCinf se incrementó en 56%, en comparación con mujeres con función hepática normal. En mujeres con insuficiencia hepática crónica Child-Pugh Clase B (moderada), la media de la Cmáx de fezolinetant disminuyó en un 15% y el ABCinf se incrementó en 96%. La Cmáx de ES259564 disminuyó en ambos grupos con insuficiencia renal crónica de leve a moderada, mientras que el ABCinf y el ABCult se incrementó ligeramente, menos de 15%. Fezolinetant no se ha estudiado en personas con insuficiencia hepática crónica Child-Pugh Clase C (severa).
Menopausia inducida farmacológicamente: Fezolinetant no se ha estudiado en personas con SVM inducidos por tratamiento farmacológico para cáncer (por ejemplo: cáncer de mama).
CONTRAINDICACIONES:
• Cirrosis conocida.
• Insuficiencia hepática crónica.
• Insuficiencia renal severa o enfermedad renal en fase terminal.
• Uso concomitante de inhibidores CYP1A2.
• Hipersensibilidad a la sustancia activa o a cualquiera de los excipientes descritos en la sección Leyendas de protección.
• Embarazo y lactancia. Pacientes menores de 18 años.
REACCIONES SECUNDARIAS Y ADVERSAS:
La seguridad de VEOZA® se evaluó en tres ensayos clínicos de 52 semanas. En los tres ensayos clínicos, un total de 1100 mujeres recibieron VEOZA®. Los ensayos 1 y 2 fueron controlados con placebo durante las primeras 12 semanas, seguidos de una nueva aleatorización de mujeres que previamente habían recibido placebo a VEOZA® (las mujeres que tomaban VEOZA® permanecieron con VEOZA®) durante 40 semanas adicionales de tratamiento no controlado. El ensayo 3 fue un estudio de seguridad doble ciego, aleatorizado, controlado con placebo que evaluó la seguridad de VEOZA® durante 52 semanas.
Las reacciones adversas informadas en al menos el 2% en VEOZA® 45 mg y mayores que en el placebo en el ensayo 3 se presentan en la Tabla 4.
Tabla 4. Reacciones adversas reportadas en al menos 2% con VEOZA® 45 mg y mayores que el placebo en un ensayo doble ciego controlado con placebo de 52 semanas (ensayo 3)
|
Reacción adversa |
VEOZA® 45 mg Total de personas-años = 5042 n(%,EAIR1) |
Placebo (n = 610) Total de personas-años = 475.0 n(%,EAIR1) |
|---|---|---|
|
Dolor abdominal2 |
26 (4.3%, 5.2) |
13 (2.1%, 2.7) |
|
Diarrea |
24 (3.9%, 4.8) |
16 (2.6%, 3.4) |
|
Insomnio |
24 (3.9%, 4.8) |
11 (1.8%, 2.3) |
|
Dolor de espalda |
18 (3.0%, 3.6) |
13 (2.1%, 2.7) |
|
Bochorno |
15 (2.5%, 3.0) |
10 (1.6%, 2.1) |
|
Elevación de trasaminasas hepáticas3 |
14 (2.3%, 2.8) |
5 (0.8%, 1.1) |
1 EAIR = Número de individuos que experimentan un evento de reacción adversa dividido entre el tiempo de exposición (total personas-años) x 100.
2 Dolor abdominal (incluyendo dolor abdominal, dolor abdominal inferior, dolor abdominal superior).
3 Elevación de transaminasas hepáticas (incluyendo alanina aminotransferasa anormal, alanina aminotransferasa elevada, aspartato aminotransferasa anormal, aspartato aminotransferasa elevada).
En los datos de laboratorio combinados de los ensayos 1, 2 y 3, se produjeron elevaciones de las transaminasas hepáticas (más de 3 veces el LSN) en 25 mujeres (2.3%, 2.7 EAIR) expuestas a VEOZA® 45 mg (n = 1100, 912.1 personas en total [años]) en comparación con 8 mujeres (0,9%, 1,5 EAIR) expuestas a placebo (n = 952, 549.1 personas-año en total).
Reporte de sospecha de reacciones adversas:
Es importante reportar la sospecha de reacciones adversas después de la autorización del medicamento, esto permite el monitoreo continuo del balance beneficio/riesgo del medicamento. Se solicita a los profesionales del cuidado de la salud reportar la sospecha de reacciones adversas a los correos electrónicos: farmacovigilancia@cofepris.gob.mx y safety-mx@astellas.com.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
En un estudio de carcinogenicidad a dos años en ratas, se observó un incremento en la incidencia de adenoma folicular de tiroides (186 veces el ABC24 humana a la dosis humana máxima recomendada [DMRH] de 45 mg). El incremento se considera un efecto específico en la rata, secundario a la inducción de las enzimas metabólicas hepatocíticas y, por lo tanto, no constituye un riesgo carcinogénico clínico. En el estudio de carcinogenicidad de 26 semanas en ratones transgénicos rasH2, no se indujeron neoplasmas (47 veces el ABC24 humana a la DMRH).
Fezolinetant y ES259564 no mostraron potencial genotóxico en la prueba de reversión de mutaciones bacterianas, prueba de aberración cromosómica, o prueba de micronúcleo in vivo.
No hay datos del efecto de VEOZA® en la fertilidad humana.
Fezolinetant no mostró efecto alguno en la fertilidad femenina ni en el desarrollo embrionario temprano hasta a 100 mg/kg/día en ratas (143 veces el ABC24 humana a la DMRH).
En los estudios de toxicidad en el desarrollo embriofetal, se observó letalidad embrionaria con un ABC24 128 y 174 veces mayor al ABC24 humana a la DMRH en ratas y conejos, respectivamente. Los conejos también mostraron un incremento en la resorción tardía y se observó peso fetal disminuido con 75 mg/kg/día (28 veces el ABC24 humana a la DMRH). La dosis (máxima) sin efecto adverso observado (no observed adverse effect level/ NOAEL) para el desarrollo embriofetal fue de 50 mg/kg/día en ratas y 45 mg/kg/día en conejos (62 y 16 veces el ABC24 humana a la DMRH en ratas y conejos, respectivamente). Fezolinetant no mostró potencial teratogénico ni en ratas ni en conejos.
En el estudio de desarrollo pre y postnatal en ratas, la NOAEL para la toxicidad materna y fetal fue de 30 mg/kg/día (36 veces el ABC24 humana a la DMRH) con base en el retraso en el parto y letalidad embrionaria con 100 mg/kg/día. La NOAEL para el desarrollo de la generación F1 se determinó en 100 mg/kg/día para las hembras (204 veces el ABC24 humana a la DMRH) y 10 mg/kg/día para machos (11 veces el ABC24 humana a la DMRH). Los machos F1 tuvieron una separación balanoprepucial incompleta, lo que puede retardar la maduración reproductiva o afectar la fertilidad.
En hembras de rata, la administración diaria de fezolinetant por 26 semanas en dosis iguales o mayores a 30 mg/kg/día (56 veces el ABC24 humana a la DMRH) causó atrofia uterina y mucificación epitelial de la vagina y cérvix.
En hembras de monos cynomolgus, la administración diaria por 39 semanas en dosis iguales o mayores a 10 mg/kg/día (19 veces el ABC24 humana a la DMRH) causó la reducción de la actividad ovárica.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Efecto de otros fármacos en fezolinetant:
Inhibidores de CYP1A2: Fezolinetant es un sustrato del CYP1A2. El uso concomitante de VEOZA® con fármacos que son inhibidores moderados o fuertes del CYP1A2 incrementa la Cmáx en plasma y el ABC de fezolinetant.
Evite el uso concomitante de inhibidores moderados o fuertes de CYP1A2 con VEOZA®.
Estudios clínicos:
Inhibidores fuertes de CYP1A2: La coadministración con fluvoxamina, un fuerte inhibidor de CYP1A2, resultó en un incremento general de 1.8 veces de la Cmáx de fezolinetant y un incremento de 9.4 veces del ABC; no se observó ningún cambio en el tmáx.
Inductores moderados de CYP1A2: El tabaquismo (inductor de CYP1A2) disminuyó la Cmáx de fezolinetant a una tasa de la media geométrica de mínimos cuadrados de 71.74%, mientras que el ABC disminuyó a una tasa de la media geométrica de mínimos cuadrados de 48.29%.
Modelos de predicción farmacocinética con base en la fisiología:
Inhibidores de CYP1A2 débiles y moderados: Con base en los modelos farmacocinéticos basados en la fisiología, se puede predecir que un inhibidor débil típico de CYP1A2 (cimetidina) incrementará la Cmáx y el ABC de fezolinetant en 42% y 75%, respectivamente. Se puede predecir que un inhibidor moderado típico de CYP1A2 (mexiletina) incrementará la Cmáx de fezolinetant en 20% y el ABC 3.95 veces.
Estudios in vitro:
Enzimas del complejo del citocromo P450 (CYP): Fezolinetant es principalmente catalizada por CYP1A2 y en menor grado por CYP2C9 y CYP2C19. Fezolinetant y ES259564 no son inhibidores de CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4. Fezolinetant y ES259564 no son inductores de CYP1A2, CYP2B6 y CYP3A4.
Transportadores: ES259564 es un sustrato de la glucoproteína P (P-gp), pero no es inhibidor de ésta. Fezolinetant no es ni sustrato ni inhibidor de la P-gp.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Se presentó la elevación de los niveles séricos de transaminasas en el 2.2% de las mujeres que recibieron VEOZA® 45 mg y en el 0.6% de las mujeres que recibieron placebo en los estudios de fase 3. No se observó la elevación en el total de bilirrubinas (mayor a dos veces el ULN). Las mujeres con elevación de la transaminasa fueron generalmente asintomáticas. Los niveles elevados de transaminasa regresaron a los niveles previos al tratamiento (o cercanos a éste) sin secuelas al continuar la dosis, así como al interrumpir o suspender la dosis. No se estudiaron mujeres con cirrosis.
PRECAUCIONES GENERALES:
Elevación de transaminasas hepáticas:
Se produjeron elevaciones en los niveles de transaminasas séricas [alanina aminotransferasa (ALT) y/o aspartato aminotransferasa (AST)] superiores a tres veces el límite superior normal (LSN) en el 2.3% [tasa de incidencia ajustada por exposición (EAIR) de 2.7 por 100 personas-años] de las mujeres que recibieron VEOZA® y el 0.9% (EAIR de 1.5 por 100 personas-año) de las mujeres que recibieron placebo en tres ensayos clínicos. No se produjeron elevaciones séricas de la bilirrubina total (más de dos veces el LSN). Las mujeres con elevaciones de ALT o AST generalmente eran asintomáticas. Los niveles de transaminasas volvieron a los niveles previos al tratamiento (o cercanos a éstos) sin secuelas con la continuación de la dosis y tras la interrupción o discontinuación de la dosis. No se estudiaron mujeres con cirrosis.
Realice análisis de sangre basal para evaluar la función y daño hepático [incluida la alanina aminotransferasa (ALT) sérica, la aspartato aminotransferasa (AST) sérica y la bilirrubina sérica (total y directa)] antes de iniciar con VEOZA®. No inicie VEOZA® si la concentración de ALT o AST es igual o superior a dos veces el LSN o si la bilirrubina total está elevada (por ejemplo, igual o superior a dos veces el LSN) para las pruebas evaluadas. Si la evaluación inicial de las transaminasas hepáticas es inferior a dos veces el LSN y la bilirrubina total es normal, se puede iniciar VEOZA®. Realice evaluaciones de seguimiento de la concentración de transaminasas hepáticas a los 3 meses, 6 meses y 9 meses después del inicio del tratamiento y cuando los síntomas (como náuseas, vómitos o coloración amarillenta de la piel o los ojos) sugieran lesión hepática.
Efectos en la capacidad para manejar y usar maquinaria: No se han realizado estudios formales sobre los efectos en la capacidad para manejar y usar maquinaria; sin embargo, se considera que VEOZA® tiene una influencia insignificante en la capacidad para manejar y usar maquinaria.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No se recomienda el uso de VEOZA® en mujeres embarazadas. No se cuenta con datos relacionados con el uso de VEOZA® en mujeres embarazadas.
En estudios de toxicidad embriofetal con animales para VEOZA®, se observó letalidad embrionaria con dosis por arriba de la dosis terapéutica humana en ratas y conejos, pero no se observó teratogenicidad (ver sección Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Lactancia: No se recomienda el uso de VEOZA® en mujeres que amamantan. No se cuenta con datos para evaluar los efectos de VEOZA® en el bebé lactante ni los efectos sobre la producción de leche. Se desconoce si VEOZA® está presente en la leche humana.
Después de la administración de fezolinetant radiomarcado a ratas lactantes, la concentración de radiactividad en la leche fue mayor que en plasma en todos los puntos de evaluación, lo que indica que los componentes derivados de fezolinetant se transfieren a los tejidos de las crías de rata a través de la leche.
Toxicología y/o farmacología animal:
Se observó mortalidad con 300 mg/kg/día (197 veces el ABC24 humana a la DMRH DMRH de 45 mg) en un estudio de toxicidad de dosis repetidas con ratas a 4 semanas. Los animales moribundos presentaron letargia, actividad reducida, respiración trabajosa y marcha tambaleante, y pérdida de peso corporal.
No se observó mortalidad en estudios de dosis repetidas de 13 o 26 semanas con dosis de hasta 200 mg/kg/día (148 veces el ABC24 humana a la DMRH). En el mono cynomolgus, la administración de fezolinetant en dosis de 40 mg/kg/día por 39 semanas estuvo asociada con la muerte de un animal (102 veces el ABC24 humana a la DMRH). Los animales moribundos presentaron anemia hemorrágica aguda y trombocitopenia severa. También se observó trombocitopenia en un animal superviviente con una dosis de 40 mg/kg/día, pero no se observó en otros animales.
En un estudio de seguridad farmacológica en rata, se observó la constricción de la pupila con una dosis igual o mayor a 125 mg/kg. Con 250 mg/kg se observó disminución de la actividad, de la respuesta de escape al tacto y de la fuerza de sujeción, lo cual se consideró indicativo de sedación. Estos signos clínicos no fueron evidentes 24 horas después de la dosis. Estos efectos parecidos a la sedación también se confirmaron en los estudios de toxicidad con dosis repetidas en ratas por 4 y 13 semanas. La NOAEL para los efectos parecidos a la sedación fue de 30 mg/kg/día y 60 veces la Cmáx humana a la DMRH.
Por otra parte, no se observaron efectos en el SNC, incluyendo sedación, en monos cynomolgus (estudios de dosis repetidas de 5, 13 y 39 semanas) incluso a la dosis más alta (40 o 50 mg/kg/día, 67 veces la Cmáx humana a la DMRH).
Fezolinetant inhibió la densidad de la corriente hERG con un valor de IC50 de 231.8 μmol/L (83074.8 ng/mL, 371 veces la Cmáx humana a la DMRH). En el estudio de telemetría en monos cynomolgus y en el estudio Langendorff no se observaron efectos en el sistema cardiovascular. Estos resultados indican que fezolinetant tiene poco o ningún efecto cardiovascular.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología: Realice análisis de sangre de referencia para evaluar la función y daño hepático (incluida la alanina aminotransferasa [ALT] sérica, aspartato aminotransferasa [AST] sérica y bilirrubina sérica [total y directa]) antes de iniciar el tratamiento.
Durante el tratamiento se deberán realizar análisis de sangre de seguimiento a los 3 meses, 6 meses y 9 meses después del inicio de la terapia y cuando los síntomas (como náuseas, vómitos o ictericia en piel o los ojos) sugieran lesión hepática.
La dosis recomendada de VEOZA® es de 45 mg una vez al día.
El beneficio del tratamiento de largo plazo debe evaluarse periódicamente, ya que la duración de los SVM puede variar en cada individuo.
Poblaciones especiales:
Adultos mayores: No se han realizado estudios clínicos de seguridad y eficacia en mujeres que inicien el tratamiento con VEOZA® con más de 65 años de edad.
Pediátrica: La seguridad y la eficacia de VEOZA® en esta población no han sido establecidas.
Insuficiencia renal: No se recomienda la modificación de la dosis en personas con insuficiencia renal leve (eGFR de 60 a menos de 90 mL/min/1.73 m2) o moderada (eGFR de 30 a menos de 60 mL/min/1.73 m2) (ver sección Farmacocinética y farmacodinamia). No se recomienda el uso de VEOZA® en personas con insuficiencia renal severa (eGFR menor de 30 mL/min/1.73 m2). VEOZA® no se ha estudiado en personas con enfermedad renal terminal (eGFR menor de 15 mL/min/1.73 m2) y no se recomienda su uso en esta población (ver sección Farmacocinética y farmacodinamia).
Insuficiencia hepática: No se recomienda la modificación de la dosis en personas con insuficiencia hepática crónica Child-Pugh Clase A (leve) (ver sección Farmacocinética y farmacodinamia).
No se recomienda el uso de VEOZA® en personas con insuficiencia hepática crónica Child-Pugh Clase B o C (moderada o severa). VEOZA® no se ha estudiado en personas con insuficiencia hepática crónica Child-Pugh Clase C (severa) (ver sección Farmacocinética y farmacodinamia).
Método de administración: VEOZA® debe administrarse por vía oral una vez al día a aproximadamente la misma hora con o sin alimentos, se debe tomar con líquidos y debe tragarse completa. No la corte, no la triture y no mastique la tableta.
Si se olvida una dosis de VEOZA® o no se toma a la hora usual, administre la dosis que faltó en cuanto sea posible, a menos que falten menos de 12 horas antes de la siguiente dosis. Al siguiente día, vuelva al horario regular.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No hay experiencia con casos de sobredosis inadvertida de fezolinetant. Las dosis de fezolinetant hasta de 900 mg como dosis única y 720 mg una vez al día por 7 días fueron evaluadas en estudios clínicos en mujeres sanas. La dosis máxima tolerada se determinó en 900 mg. Con 900 mg se observó cefalea, náusea y parestesia.
En el caso de sobredosis, la persona debe monitorearse de manera cercana, y se debe considerar tratamiento de apoyo con base en los signos y síntomas. La vida media (t1/2) terminal de fezolinetant es menor a 15 horas.
PRESENTACIÓN:
Caja con 30 tabletas en envase de burbuja e instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Conserve la caja bien cerrada a no más de 30 °C.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. Trague la tableta completa, no la mastique, no la corte y no la triture. No se use durante el embarazo o lactancia. No se use en pacientes menores de 18 años. Mantenga fuera del alcance de los niños. Este medicamento contiene manitol, hidroxipropil celulosa, hidroxipropil celulosa de baja sustitución, celulosa microcristalina, estearato de magnesio, hipromelosa, talco, macrogol, dióxido de titanio, óxido de hierro rojo.
Reporte las sospechas de reacciones adversas a los correos:
farmacovigilancia@cofepris.gob.mx y
safety-mx@astellas.com
Representante Legal e Importador:
ASTELLAS FARMA MÉXICO, S. DE R.L. de C.V.
Avenida Javier Barros Sierra 540, Torre 1 Piso 5,
Col. Santa Fe, C.P. 01210, Álvaro Obregón,
Ciudad de México, México.
Reg. Núm. 191M2024 SSA IV


