
VIDAZA
AZACITIDINA
Suspensión inyectable
1 Frasco(s) ámpula, 100 mg, 25 mg/ml
1 Frasco ámpula de vidrio, 30 ml,
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula con liofilizado contiene:
Azacitidina 100 mg
Manitol 100 mg
Hecha la mezcla, cada ml de la suspensión contiene 25 mg de azacitidina.
INDICACIONES TERAPÉUTICAS: VIDAZA® está indicado como tratamiento de pacientes adultos que no se consideran aptos para el trasplante de células madre hematopoyéticas y que padecen:
• Síndrome mielodisplásico (SMD) intermedio-2 y de alto riesgo, según el sistema internacional de puntuación pronóstica (IPSS por sus siglas en inglés).
• Leucemia mielomonocítica crónica (LMMC) con el 10% al 29% de blastos medulares sin trastorno mieloproliferativo.
• Leucemia mieloide aguda (AML por sus siglas en inglés) con 20-30% de blastos y displasia multilinaje, de acuerdo a la clasificación de la Organización Mundial de la Salud.
• Leucemia mieloide aguda (AML por sus siglas en inglés) con > 30% de blastos en médula ósea según la clasificación de la Organización Mundial de la Salud (OMS) en pacientes de 65 años o más que no son elegibles para trasplante de células madre hematopoyéticas.
• Tratamiento inmunomodulador de mantenimiento para pacientes en estado post trasplante de médula ósea.
FARMACOCINÉTICA Y FARMACODINAMIA:
Farmacodinamia:
Mecanismo de acción: Se cree que azacitidina ejerce sus efectos antineoplásicos mediante diversos mecanismos, que incluyen citotoxicidad sobre las células hematopoyéticas anormales en la médula ósea e hipometilación del ADN. Los efectos citotóxicos de la azacitidina pueden deberse a diversos mecanismos: la inhibición de la síntesis del ADN, ARN y de proteínas, la incorporación en el ARN y en el ADN, y la activación de las vías que causan daño en el ADN. Las células no proliferativas son relativamente insensibles a la azacitidina. La incorporación de azacitidina en el ADN produce la inhibición de las metiltransferasas de ADN, lo que lleva a la hipometilación del ADN. La hipometilación del ADN de genes metilados aberrantemente, que intervienen en las vías de regulación del ciclo celular normal, diferenciación y muerte celular puede producir la re-expresión de genes y el restablecimiento de funciones supresoras en las células cancerosas. La importancia relativa de la hipometilación del ADN frente a la citotoxicidad u otras actividades de azacitidina para los resultados clínicos no ha sido establecida.
Eficacia y seguridad clínica:
Síndromes mielodisplásicos:
Estudio clínico AZA-001: La eficacia y seguridad de VIDAZA® se investigó en un estudio internacional, multicéntrico, controlado, abierto, aleatorizado, con grupos paralelos, comparativo de Fase III (AZA-001) en pacientes con SMD de riesgo intermedio 2 y de alto riesgo de acuerdo con el Sistema Internacional de Puntaje Pronóstico (IPSS), anemia refractaria con exceso de blastos (AREB), anemia refractaria con exceso de blastos en transformación (AREB-T) y leucemia mielomonocítica crónica modificada (mCMML) de acuerdo con el Sistema de Clasificación FAB. Los pacientes con AREB-T (21-30% de blastos) se consideran actualmente pacientes con AML (leucemia mieloide aguda por sus siglas en inglés) bajo el actual Sistema de Clasificación de la OMS. Azacitidina más los mejores cuidados paliativos (BSC) (n = 179) se comparó con los regímenes de cuidados convencionales (CCR). Los CCR consistieron en sólo BSC (n = 105), dosis baja de citarabina más BSC (n = 49) ó quimioterapia de inducción estándar más BSC (n = 25). Los pacientes fueron pre-seleccionados por sus médicos a uno de los tres CCR antes de la aleatorización. Los pacientes recibieron este régimen pre-seleccionado si no eran aleatorizados a VIDAZA®. Como parte de los criterios de inclusión, se requirió que los pacientes tuvieran una categoría de desempeño del ECOG de 0-2. Los pacientes con SMD secundario fueron excluidos del estudio. El objetivo primario del estudio fue la supervivencia global. VIDAZA® se administró a una dosis subcutánea de 75 mg/m2 diarios durante 7 días seguido de un periodo de descanso de 21 días (ciclo de tratamiento de 28 días) por una mediana de 9 ciclos (rango = 1-39) y una media de 10,2 ciclos. Dentro de la población de intención a tratar (ITT), la media de edad fue 69 años (rango = 38-88 años).
En el análisis ITT de 358 pacientes (179 azacitidina y 179 CCR), el tratamiento con VIDAZA® estuvo vinculado a una mediana de supervivencia de 24,46 meses frente a 15,02 meses para aquellos que recibieron el tratamiento CCR, una diferencia de 9,4 meses, con un valor p log-rank estratificado de 0,0001. La proporción de riesgo para el efecto del tratamiento fue de 0,58 (IC 95%: 0,43; 0,77). Las tasas de supervivencia a 2 años fueron 50,8% en los pacientes que recibieron azacitidina frente a 26,2% en pacientes que recibieron CCR (p < 0,0001).
Figura 1. Curva de Kaplan-Meier de tiempo a muerte por cualquier causa (Población con intención de tratar)
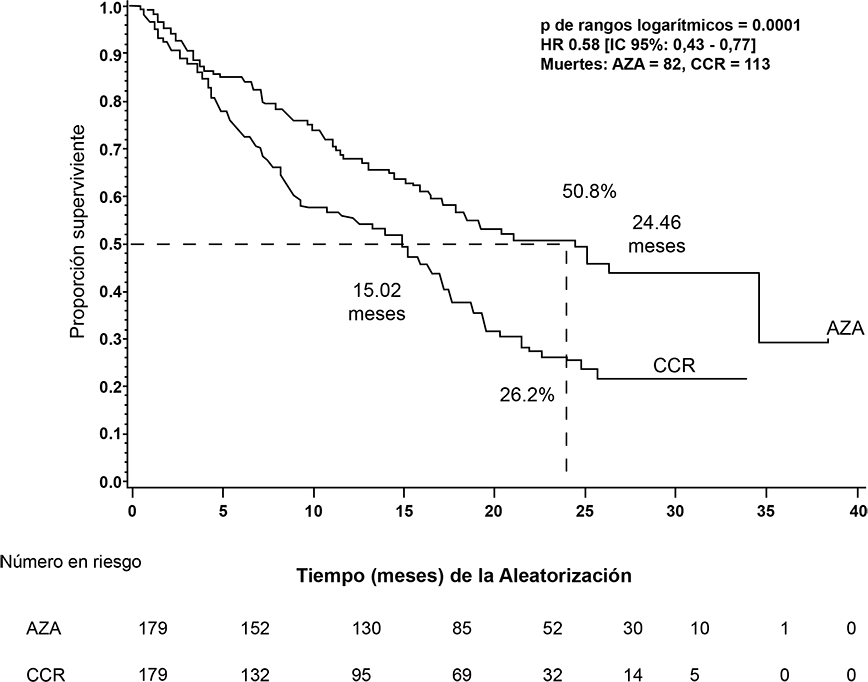
REF: AZA = azacitidina; CCR = régimen de cuidados convencionales; IC = intervalo de confianza, HR = hazard ratio, proporción de riesgo.
Los beneficios de VIDAZA® en supervivencia fueron constantes independientemente de la opción de tratamiento de CCR (BSC solo, dosis baja de citarabina + BSC ó quimioterapia de inducción estándar + BSC) utilizada en el control.
Cuando se analizaron los subgrupos citogenéticos del IPSS se observaron hallazgos similares en términos de la mediana de supervivencia global en todos los grupos (citogenética buena, intermedia, mala incluyendo monosomía 7).
En los análisis de sub-grupos etarios se observó un aumento en la mediana de supervivencia global para todos los grupos (< 65 años; ≥ 65 años y ≥ 75 años).
El tratamiento con VIDAZA® estuvo vinculado a una mediana de tiempo hasta la muerte o la transformación a AML de 13,0 meses frente a 7,6 meses para aquellos que recibieron el tratamiento de CCR, una mejoría de 5,4 meses con un valor p log-rank estratificado de 0,0025.
El tratamiento con VIDAZA® también estuvo vinculado a una reducción de citopenias y sus síntomas relacionados. El tratamiento con VIDAZA® condujo a una menor necesidad de transfusiones de glóbulos rojos y plaquetas. De los pacientes en el grupo con azacitidina que eran dependientes de transfusión de glóbulos rojos en la basal, el 45,0% dejaron de serlo durante el periodo de tratamiento, en comparación con el 11,4% de los pacientes en los grupos de CCR combinados (una diferencia estadísticamente significativa (p < 0,0001) de 33,6% [IC 95%: 22,4; 44,6]). En los pacientes que eran dependientes de transfusión de glóbulos rojos en la basal y dejaron de serlo, la mediana de duración de independencia de transfusión de glóbulos rojos fue de 13 meses en el grupo con azacitidina.
La respuesta fue evaluada por el investigador o por el Comité Independiente de Revisión (IRC). La respuesta global (remisión completa [CR] más remisión parcial [PR]) según determinó el investigador, fue del 29% en el grupo con azacitidina y del 12% en el grupo con CCR combinado (p = 0,0001). La respuesta global (CR + PR) según determinó el IRC en el AZA-001 fue del 7% (12/179) en el grupo con azacitidina en comparación con el 1% (2/179) en el grupo con CCR combinado (p = 0,0113). Las diferencias entre las evaluaciones del IRC y del investigador con respecto a la respuesta fueron consecuencia de los criterios del Grupo Internacional de Trabajo (IWG) que exigen una mejoría en los recuentos de sangre periférica y un mantenimiento de esta mejoría por un mínimo de 56 días. También se demostró un beneficio en supervivencia en pacientes que no habían alcanzado una respuesta completa/parcial luego de tratamiento con azacitidina. La mejoría hematológica (mayor o menor) según determinó el IRC se alcanzó en el 49% de los pacientes que recibieron azacitidina en comparación con el 29% de los pacientes tratados con CCR combinado (p < 0.0001).
En pacientes con una o más anormalidades citogenéticas en el momento basal, el porcentaje de respuesta citogenética mayor fue similar en los grupos con azacitidina y CCR combinado. La respuesta citogenética menor fue significativamente (p = 0,0015) más elevada en el grupo con azacitidina (34%) en comparación con el grupo con CCR combinado (10%).
Estudios clínicos CALGB 9221, CALGB 8921 y CALGB 8421: CALGB 9221, un estudio aleatorizado, abierto, controlado realizado en 53 centros de EUA comparando la seguridad y eficacia de VIDAZA® SC más tratamiento de soporte con tratamiento de soporte únicamente (“observación”) en pacientes con cualquiera de los 5 subtipos de SMD de la FAB: RA, RARS, RAEB, RAEB-T y CMMoL.
Se incluyeron pacientes con RA y RARS si cumplían con uno o más de los siguientes criterios: requerían transfusiones de paquetes globulares (RBC, por sus siglas en inglés); tenían conteos plaquetarios de < 50.0 x 109/L; requerían transfusiones plaquetarias; o tenían neutropenia (conteo de neutrófilos absolutos o ANC por sus siglas en inglés < 1.0 X 109/L) con infecciones que requerían tratamiento con antibióticos. Los pacientes con AML no se intentaron incluir. El tratamiento de soporte permitido en este estudio incluyó transfusiones de productos sanguíneos, antibióticos, antieméticos, analgésicos y antipiréticos. Se prohibió el uso de factores de crecimiento hematopoyético.
Con la población ITT (intención de tratar) las características basales de los pacientes fueron similares entre los grupos de azacitidina y control. La mediana de edad fue 70 años (rango de 31-92), 90% de raza blanca y 70% de sexo masculino. Por la clasificación FAB 20% tenían RA, 6% y 5% respectivamente tenían RARS, 40% tenían RAEB, 16% y 15% respectivamente tenían RAEB-T, 8% tenían CMML. Al momento basal, 10% tenían AML según la clasificación FAB.
VIDAZA® fue administrado en una dosis SC de 75 mg/m² diarios por 7 días cada 4 semanas. La dosis se incrementó a 100 mg/m² si no se observaba beneficio clínico después de 2 ciclos de tratamiento. La dosis se disminuyó y/o retrasó basándose en eventos hematológicos o evidencia de toxicidad renal. Los pacientes en el brazo de observación fueron por protocolo permitidos a cruzar al brazo de VIDAZA® si presentaban incremento en blastos en médula ósea, disminución de la hemoglobina, incremento en el requerimiento transfusional, o disminución en recuento plaquetario, o desarrollaban necesidad de transfusión plaquetaria o desarrollaban infección clínica que requiriera de tratamiento con antibióticos.
Para propósitos de medición de eficacia, el objetivo primario fue la tasa de respuesta.
Criterios de respuesta
|
RA |
RARS |
RAEB |
RAEB-T |
CMMoL |
||
|
Respuesta Completa (CR) duración > 4 semanas |
Médula ósea |
< 5% blastos |
||||
|
Sangre periférica |
CBC (biometría hemática) normal si basalmente estaba anormal |
|||||
|
Respuesta Parcial (PR) duración > 4 semanas |
Médula ósea |
Sin requerimientos por parte de médula ósea |
> 50% disminución en blastos Mejoría de la dispoyesis en médula ósea |
|||
|
Sangre periférica |
> 50% de restauración en el déficit de los niveles normales de células blancas basales, hemoglobina y plaquetas si estaban anormales basalmente No blastos en la sangre periférica Para CMMoL, si los glóbulos blancos estaban elevados en momento basal, una reducción de > 75% en el exceso sobre el límite superior normal |
|||||
De los 191 pacientes incluidos en el estudio, la revisión independiente (diagnóstico adjudicado) encontró que 19 tenían diagnóstico de AML en el momento basal. Estos pacientes fueron excluidos de un primer análisis de tasas de respuesta, aunque sí fueron incluidos en un análisis por ITT de todos los pacientes aleatorizados. Aproximadamente 55% de los pacientes aleatorizados a observación se entrecruzaron a recibir tratamiento con VIDAZA®.
La tasa de respuesta global (CR+PR) de 15.7% en pacientes tratados con VIDAZA® sin AML (16.2% de todos los pacientes aleatorizados incluyendo AML) fue mayor estadísticamente significativo que la tasa de respuesta de 0% en el grupo de observación (p=0.0001).
Tasas de respuesta
|
VIDAZA® N=89 |
Observación antes del entrecruzamiento N=83 |
||
|
Respuesta |
n (%) |
n (%) |
Valor de p |
|
Global (CR+PR) |
14 (15.7) |
0 (0) |
(< 0.0001) |
|
Completa |
5 (5.6%) |
0 (0) |
(0.06) |
|
Parcial |
9 (10.1) |
0 (0) |
– |
La mayoría de los pacientes que lograron ya sea CR o PR tenían anormalidades en 2 o 3 líneas celulares en el momento basal (81%; 13/16) y tenían una elevada cantidad de blastos en médula ósea o eran dependientes de transfusión en la basal.
Los pacientes que respondieron a VIDAZA® tenían una disminución en el porcentaje de blastos en médula ósea, o un incremento en plaquetas, hemoglobina o glóbulos blancos. Más del 90% de los respondedores demostraron inicialmente estos cambios para el 5º ciclo de tratamiento. Todos los pacientes que habían sido dependientes de transfusión llegaron a ser independientes de transfusión durante la PR o la CR. La media y mediana de duración del efecto positivo fue de 418.9 días (1.1 años) y de 284 días respectivamente. La media y la mediana del total de la duración de la respuesta clínica de PR o mejor para los 16 respondedores fue de 224.8 días y de 165.5 días, respectivamente. Las respuestas ocurrieron en todos los tipos de MDS, así como en pacientes a los que se adjudicó diagnóstico de AML en el momento basal.
Los pacientes en el grupo de observación que se entrecruzaron para recibir el tratamiento con VIDAZA® (47 pacientes) tuvieron una tasa de respuesta del 12.8%.
El CALGB 8921 fue un estudio de EUA multicéntrico, abierto, de brazo único, de 72 pacientes con RAEB, RAEB-T, CMMoL, o AML. El tratamiento con VIDAZA® SC dio lugar a una tasa de respuesta (CR + PR) de 13.9% usando criterios similares a los descritos arriba. La media y la mediana del total de la duración total de la respuesta clínica de PR o mejor para los respondedores fue de 565.2 días (1.5 años) y de 117 días, respectivamente. La media y mediana de la duración del efecto positivo fue de 848.2 días (2.3 años) y de 436 días (1.2 años), respectivamente.
CALGB 8421, otro estudio abierto, de brazo único de 48 pacientes con RAEB, RAEB-T, o AML, en el cual el tratamiento con Vidaza® intravenoso dio lugar a una tasa de respuesta de 18.8%, nuevamente usando criterios similares a aquellos descritos arriba. La media y mediana de duración de respuesta clínica de PR o mejor para los respondedores fue de 105.6 días y de 128 días, respectivamente. La media y mediana de la duración del efecto positivo fue de 299.8 días y de 323 días, respectivamente. La respuesta ocurrió en todos los subtipos de SMD así como en pacientes con diagnostico basal adjudicado de AML en estos dos estudios. Los regímenes de dosis de VIDAZA® en estos 2 estudios fueron similares al régimen utilizado en los estudios controlados.
Se observó beneficio en pacientes que no cumplieron los criterios para PR o mejor, pero fueron considerados con ‘mejoría’. Aproximadamente 23% de los pacientes tratados con VIDAZA® fueron considerados con mejoría.
La tasa de respuesta global (CR + PR) en sujetos con SMD estuvo en el rango entre 13.9% y 18.8% en los tres estudios.
Leucemia mieloide aguda:
Estudio clínico AZA-AML-001:
La eficacia y seguridad de VIDAZA® se analizó en un estudio internacional, multicéntrico, controlado, abierto, de grupos paralelos Fase 3 en pacientes de 65 años o mayores con AML de nuevo diagnóstico de novo o secundaria con > 30% de blastos en médula ósea de acuerdo a la clasificación OMS, quienes no fueron elegibles para trasplante de células madre hematopoyéticas (HSCT por sus siglas en inglés).
VIDAZA® más BSC (n = 241) fue comparado con los regímenes de cuidado convencionales (CCR; n = 247). Los CCR consistieron en mejores cuidados de soporte (BSC) solos (n = 45), citarabina a dosis baja más BSC (n = 158), o quimioterapia intensiva estándar con citarabina y antraciclina más BSC (n = 44). Los pacientes fueron preseleccionados por sus médicos en 1 de 3 CCRs previo a la aleatorización. Los pacientes recibieron este régimen pre-seleccionado si no eran aleatorizados a VIDAZA®. Como parte de los criterios de inclusión, los pacientes debían tener un estado de desempeño de ECOG de 0-2 y anormalidades citogenéticas de riesgo intermedio o pobre. El objetivo primario de este estudio fue la supervivencia global.
VIDAZA® se administró vía subcutánea (SC) a una dosis de 75 mg/m²/día para 7 días, seguido de un periodo de descanso de 21 días (ciclo de tratamiento de 28 días), para una mediana de 6 ciclos, rango: 1 a 28) y una media de 8.8 ciclos. Los BSC solos para pacientes tuvieron una mediana de 3 ciclos (rango: 1 a 20), los pacientes con citarabina a dosis bajas recibieron una mediana de 4 ciclos (rango 1 a 25) y los pacientes con quimioterapia intensiva estándar recibieron una mediana de 2 ciclos (rango de 1 a 3, inducción más 1 o 2 ciclos de consolidación).
Los parámetros individuales basales y los grupos con CCR fueron comparables en cuanto a los parámetros basales. La mediana de edad de los sujetos fue de 75.0 años (rango: 64 a 91 años), 75.2% eran Caucásicos y 59.0% eran hombres. A la basal, el 60.7% fue clasificado como AML no especificada, el 32.4% con AML con cambios relacionados con mielodisplasia, el 4.1% con neoplasias mieloides relacionadas a la terapia y el 2.9% con AML con anormalidades genéticas recurrentes de acuerdo a la clasificación OMS.
En el análisis ITT de 488 pacientes (241 con VIDAZA® y 247 con CCR), el tratamiento con VIDAZA® se asoció con una mediana de supervivencia de 10.4 meses versus 6.5 meses para aquellos que recibieron tratamiento con CCR, una diferencia clínicamente significativa de 3.8 meses, con un valor p estratificado log-rank def 0.1009 (dos colas). La proporción de riesgo para el efecto del tratamiento fue de 0.85 (IC 95% = 0.69, 1.03).
Cuando se censuró para terapia subsecuente para AML, la mediana de SG fue mayor en el grupo de tratamiento con VIDAZA® (12.1 meses) comparado con el grupo de tratamiento con CCR (6.9 meses), con un valor p log Rank estratificado de 0.0190 (dos colas). El HR para el efecto del tratamiento fue de 0.76 (IC 95% = 0.60, 0.96).
Las tasas de supervivencia a un año fueron de 46.5% en pacientes que recibieron VIDAZA® versus 34.2% en pacientes que recibieron CCR.
Figura 2. Curva de Kaplan-Meier de tiempo a muerte por cualquier causa (población por intención de tratar)
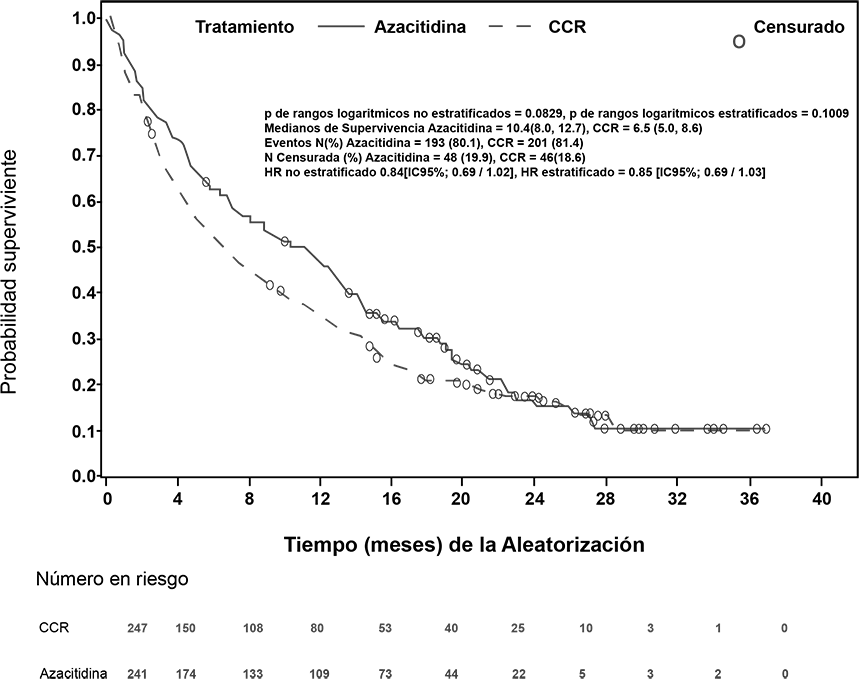
Los beneficios de VIDAZA® en supervivencia fueron mayores cuando se compararon con las opciones de tratamiento de CCR, BSC solo, o dosis baja de citarabina más BSC, y fueron similares cuando se comparó con quimioterapia intensiva estándar más BSC utilizado en el brazo control.
En todos los subgrupos pre-especificados [edad (< 75 años y ≥ 75 años), género, raza, estado de desempeño ECOG (0 a 1 y 2), riesgo citogenético basal (intermedio y pobre), región geográfica, clasificación OMS de AML(incluyendo AML con cambios relacionados a mielodisplasia), cuenta de leucocitos basal (≤ 5 x 109/L y > 5 x 109/L), blastos en médula ósea basales (≤ 50% y > 50%) e historia previa de SMD, sí hubo una tendencia en beneficio en SG a favor de VIDAZA®.
Las respuestas citogenéticas y hematológicas fueron evaluadas por el investigador y por el IRC con resultados similares. La tasa de respuesta global (remisión completa [CR] + remisión completa con recuperación incompleta de la línea roja [CRi]) tal cual fue determinada por el IRC, fue del 27.8% en el grupo con VIDAZA® y del 25.1% en el grupo CCR combinado (p = 0.5384).
En pacientes que lograron CR o CRi, la mediana de duración de la remisión fue de 10.4 meses (IC 95% = 7.2, 15.2) para los sujetos con VIDAZA® y de 12.3 meses (95% IC = 9.0, 17.0) para los pacientes con CCR.
Se demostró también un beneficio en supervivencia en pacientes que no habían logrado una respuesta completa para VIDAZA® comparado con CCR.
El tratamiento con VIDAZA® mejoró las cuentas de sangre periférica y dio lugar a una disminución en la necesidad de células rojas sanguíneas (RBC) y transfusiones plaquetarias. Se consideró a un paciente dependiente de transfusión de RBC o de plaquetas a la basal si el sujeto tenía una o más transfusiones de RBC o de plaquetas sobre los 56 días (8 semanas) o previo a la aleatorización respectivamente. Se consideró a un paciente independiente de transfusión de RBC o plaquetaria durante el periodo de tratamiento si el sujeto no había recibido transfusión de RBC o plaquetas durante cualquiera de los 56 días consecutivos durante el periodo de reporte, respectivamente.
De los pacientes en el grupo VIDAZA® que fueron dependientes de transfusión de RBC a la basal, el 38.5% (IC 95% = 31.1, 46.2) de estos pacientes llegaron a ser independientes de transfusión de RBC durante el periodo de tratamiento comparado con el 27.6% (IC 95% = 20.9, 35.1) de los pacientes en los grupos de CCR combinados. En pacientes que fueron dependientes de transfusión de RBC a la basal y que lograron independencia a la transfusión durante el tratamiento, la mediana de duración de independencia a la transfusión de RBC fue de 13.9 meses en el grupo con Vidaza® y no fue alcanzada en el grupo CCR.
De los pacientes en el grupo de VIDAZA® que eran dependientes de transfusión plaquetaria a la basal, el 40.6% (IC 95% = 30.9, 50.8) de estos pacientes llegaron a ser independientes de transfusión plaquetaria durante el periodo de tratamiento, comparados con el 29.3% (IC 95% = 19.7, 40.4) de los pacientes en los grupos combinados de CCR. En pacientes que fueron dependientes de transfusión plaquetaria en la basal y que lograron independencia transfusional mientras estaban siendo tratados, la mediana de duración de la independencia transfusional plaquetaria fue de 10.8 meses en el grupo de VIDAZA® y de 19.2 meses en el grupo de CCR.
Azacitidina como terapia de mantenimiento en pacientes Post Trasplante de Médula Ósea:
Diversos estudios clínicos han evaluado los resultados del uso de azacitidina en pacientes post trasplante, incluso en aquellos en quienes este recurso terapéutico había fallado en proporcionar una alternativa de remisión de la enfermedad.
Un estudio internacional evaluó el efecto de azacitidina como terapia de rescate en un estudio clínico con 17 pacientes con Leucemia Aguda post Trasplante Alogénico de Médula Ósea, (TAMO) con una dosis diaria de 16 mg/m2 durante 5 días cada 4 semanas, con una mediana de 8 ciclos. De 9 pacientes con enfermedad recurrente, 6 tenían recurrencia medular y 3 pacientes tenían enfermedad extramedular. Cinco de los 9 pacientes que desarrollaron enfermedad recurrente tuvieron respuesta (55%), 3 pacientes alcanzaron una Respuesta Completa (CR) que continuaba más allá de los 4 meses y 17 meses en 2 pacientes, que se perdió después de 4 meses en un paciente.
Dos pacientes presentaron una Respuesta Parcial (PR) que se sostuvo durante 2 y 4 meses. Ocho pacientes que recibieron azacitidina como terapia de sostén permanecieron en CR por una media de 17 meses (rango de 14 a 26 meses). Tres de esos pacientes desarrollaron recurrencia a la enfermedad después de 4 meses, 14 meses y 18 meses de terapia después de que recibieron 3 ciclos, 12 ciclos y 12 ciclos, respectivamente. Después de una mediana de seguimiento y 11 meses después del TAMO y la terapia con azacitidina, 14 pacientes (82%) permanecían vivos, incluyendo 7 pacientes (41%) que tenían CR y 2 pacientes(13%) con PR con una mediana de 20 meses y 12 meses, respectivamente. Los lapsos de supervivencia libre de enfermedad y supervivencia global fueron de 55% y 30%; 90% y 80% a 1 y 2 años respectivamente, demostrándose clínicamente la eficacia de dosis bajas de azacitidina como terapia inmunomoduladora pos trasplante y en pacientes post trasplante de médula ósea (MO) con enfermedad recurrente.
Es importante mencionar que se ha evitado la administración del medicamento ante la presencia de la más mínima manifestación de Enfermedad Injerto contra Huésped (EIH).
Podemos concluir que dosis bajas de azacitidina nos proveen de una herramienta que reduce los rangos de recurrencia a la enfermedad y mejora el quimerismo mixto después de trasplante de MO cuando se da como terapia de mantenimiento o posiblemente como adyuvante a la donación de infusiones de linfocitos para tratar la recurrencia a la enfermedad.
Para determinar la dosis óptima para estos pacientes, De Lima et al., realizó un estudio clínico en 45 pacientes adultos con AML o SMD de alto riesgo, intermedio 2 o alto riesgo con rango de edad de 18 a 75 años (mediana de 60 años), que no fueron candidatos a regímenes de trasplante mieloablativo. Se les proporcionó azacitidina en los días 1 a 4 de ciclos de 30 días por vía subcutánea, iniciando en la sexta semana posterior al TAMO con 5 niveles de dosificación: 8, 16, 24, 32 o 40 mg/m2.
Los diagnósticos de los pacientes fueron los siguientes: AML (n = 37) o SMD (n = 8) el 67% de estos pacientes no presentaban respuesta completa al TAMO. La media de regímenes previos de quimioterapia fue de 2, 39 pacientes recibieron previamente dosis elevadas de quimioterapia basada en Ara-C, y el 18% de los pacientes había fallado al primer TAMO.
La media de seguimiento fue de 20.5 meses. Diecinueve pacientes (42%) había fallecido a la mediana de tiempo de 30.8 meses, 34 pacientes (53%) había desarrollado recurrencia de la enfermedad. Siete recurrencias ocurrieron durante el tratamiento con azacitidina: la primera con 16 mg/m2 durante dos ciclos, una con 24 mg/m2 durante un ciclo, una con 32 mg/m2 por un ciclo, y dos con 40 mg/m2 por dos ciclos.
Veintiocho pacientes (62%) murieron o desarrollaron recurrencia a la enfermedad (mediana de 18.2 meses), con una diferencia significativa en supervivencia libre de enfermedad a favor de los pacientes con CR versus aquellos con enfermedad activa (mediana de 27.2 vs 12 meses).
Se demostró que es posible administrar azacitidina de forma temprana después de un TAMO a la mayoría de los pacientes del grupo de AML/SMD de alto riesgo. Existen razones para pensar que prolongar la duración del tratamiento podría mejorar la Supervivencia Global (SG) y la Supervivencia Libre de Enfermedad y disminuir los riesgos de desarrollar EIH.
Farmacocinética:
Absorción: Azacitidina se absorbe rápidamente después de la administración por vía subcutánea de una dosis única de 75 mg/m²; se produjeron concentraciones plasmáticas máximas de azacitidina de 750 ± 403 ng/ml a las 0,5 horas (primer punto de muestreo) después de la administración de la dosis. La biodisponibilidad absoluta de azacitidina después de la administración por vía subcutánea en relación con la intravenosa fue de aproximadamente el 89%, basado en el área bajo la curva (AUC).
El AUC y Cmax de la administración subcutánea de azacitidina fue aproximadamente un dosis proporcional dentro del rango de dosis de 25 a 100 mg/m².
La administración de dosis múltiples a la dosis-régimen recomendada no da lugar a la acumulación del fármaco.
Distribución: Tras la administración por vía intravenosa, el volumen medio de distribución fue de 76 ± 26 L, y el aclaramiento sistémico fue de 147 ± 47 L/h.
Biotransformación: En base a datos in vitro, el metabolismo de azacitidina no parece estar mediado por las isoenzimas del citrocromo P450 (CYPs), UDP-glucoronosiltransferasas (UGTs), sulfotransferasas (SULTs) y glutatio transferasas (GSTs).
El metabolismo de la azacitidina pasa por hidrólisis espontánea y por desaminación mediada por la citidina desaminasa. En fracciones S9 del hígado humano, la formación de metabolitos fue independiente del NADPH, lo que implica que el metabolismo de azacitidina no fue mediado por las isoenzimas del citocromo P450. Estudios in vitro de hepatocitos humanos cultivados indican que a concentraciones de 1.0 μM a 100 μM (por ejemplo, hasta aproximadamente 30 veces más que las concentraciones clínicamente alcanzables), azacitidina no induce los citocromos CYP 1A2, 2C19 o 3A4 o 3A5. En estudios para evaluar la inhibición de una serie de isoenzimas de P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 y 3A4), la azacitidina hasta 100 μM no produce inhibición. Por lo tanto, la inhibición enzimática mediante azacitidina a concentraciones plasmáticas clínicamente alcanzables es poco probable.
Eliminación: Azacitidina se aclara rápidamente del plasma, con una vida media de eliminación (t½) media de 41 ± 8 minutos, después de la administración por vía subcutánea. No ocurre ninguna acumulación luego de la administración subcutánea de 75 mg/m² de azacitidina una vez por día durante 7 días. La excreción urinaria es la principal ruta de eliminación de la azacitidina y de sus metabolitos. Después de la administración por vía intravenosa y subcutánea de 14C-azacitidina, el 85% y 50% de la radioactividad administrada se recuperó en la orina, respectivamente, mientras que < 1% se recuperó en las heces.
Poblaciones especiales: Los efectos del deterioro hepático, género, edad o raza sobre la farmacocinética de azacitidina no han sido estudiados formalmente.
Farmacocinética en insuficiencia renal: La insuficiencia renal grave no tiene efecto sobre la Farmacocinética de azacitidina, después de la administración subcutánea simple y múltiple. Por lo tanto, la azacitidina puede ser administrada a pacientes con insuficiencia renal.
Azacitidina puede ser administrada a pacientes con insuficiencia renal sin ajuste de dosis inicial.
Farmacogenómica: El efecto de polimorfismos de la citidina deaminasa sobre el metabolismo de azacitidina no ha sido estudiado formalmente.
CONTRAINDICACIONES: Azacitidina está contraindicada en casos de hipersensibilidad conocida al principio activo o a alguno de los excipientes, tumores hepáticos malignos avanzados, durante la lactancia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Mujeres en edad fértil/Anticoncepción en varones y mujeres: Los varones y mujeres en edad fértil deben utilizar un método anticonceptivo efectivo durante y hasta 3 meses después del tratamiento.
Embarazo: No existen datos suficientes sobre la utilización de azacitidina en mujeres embarazadas. Los estudios en ratones han mostrado toxicidad para la reproducción. Se desconoce el riesgo potencial en humanos. En base a los resultados de los estudios en animales y su mecanismo de acción, azacitidina no debe utilizarse durante el embarazo, especialmente durante el primer trimestre, salvo que sea estrictamente necesario. Las ventajas del tratamiento deben considerarse en relación a los posibles riesgos para el feto en cada caso particular.
Lactancia: Se desconoce si azacitidina o sus metabolitos se excretan en la leche humana. Debido a las posibles reacciones adversas graves en el lactante, azacitidina está contraindicada durante este periodo.
Fertilidad: No hay información acerca del efecto de la azacitidina sobre la fertilidad en humanos. En los animales se han documentado reacciones adversas con azacitidina sobre la fertilidad masculina. Se debe aconsejar a los hombres que no conciban un hijo mientras reciben tratamiento, debiendo utilizar un método anticonceptivo eficaz durante el tratamiento y hasta tres meses después del mismo.
Antes de iniciar el tratamiento, debe aconsejarse a los pacientes varones que pidan asesoramiento sobre la conservación de esperma.
REACCIONES SECUNDARIAS Y ADVERSAS:
Información de estudios clínicos: Las reacciones adversas incluidas en esta sección fueron seleccionadas al identificar primeramente aquellos eventos adversos que ocurrieron en una mayor frecuencia en el brazo de VIDAZA comparado con los brazos del comparador en los estudios fundamentales. Estos eventos fueron evaluados clínicamente, con el énfasis en la plausibilidad biológica, relación temporal y el efecto de la retirada y re-introducción del medicamento sospechoso y la enfermedad del paciente y sus condiciones de comorbilidad.
Síndromes mielodisplásicos (AZA-001, CALGB 9221, CALGB 8921, CALGB 8421): En el 97% de los pacientes se han observado reacciones adversas posible o probablemente relacionadas con la administración de VIDAZA®.
Las reacciones adversas informadas con mayor frecuencia con el tratamiento de azacitidina fueron reacciones hematológicas (71,4%) incluyendo anemia, trombocitopenia, neutropenia y leucopenia (usualmente Grado 3-4), eventos gastrointestinales (60,6%) incluyendo náuseas, vómito (usualmente Grado 1-2) o reacciones en el sitio de inyección (77,1%; usualmente Grado 1-2).
Las reacciones adversas serias más comunes (> 2%) observadas en el estudio fundamental (AZA PH GL 2003 CL 001) y también informadas en los estudios de respaldo (CALGB 9221 y CALGB 8921) incluyeron neutropenia febril (8,0%) y anemia (2,3%). Otras reacciones adversas serias informadas incluyeron infecciones como sepsis neutropénica (0.8%) y neumonía (2.5%) (algunas con resultado fatal), trombocitopenia (3.5%), reacciones de hipersensibilidad (0.25%) y eventos hemorrágicos (0.5%) (por ejemplo, hemorragia cerebral) (0.8%), hemorragia gastrointestinal (0.8%) y hemorragia intracraneal (0.5%).
La tabla a continuación contiene las reacciones adversas para las cuales pudo establecerse razonablemente una relación causal con el tratamiento de azacitidina. Las frecuencias mencionadas se basan en las observaciones durante el estudio clínico fundamental, o dos estudios clínicos de respaldo.
Las frecuencias se definen de la siguiente manera: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1,000 a < 1/100); raras (≥ 1/10,000 a < 1/1,000); muy raras (< 1/10,000); desconocidas (no puede calcularse a partir de los datos disponibles). Dentro de cada grupo de frecuencia, los efectos adversos se presentan en orden decreciente de severidad.
Tabla 1. Frecuencia de las reacciones adversas con VIDAZA® en los estudios de Síndrome mielodisplásico (AZA-001, CALGB 9221, CALGB 8921)
|
Clasificación de órganos y sistemas |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Infecciones e infestaciones |
Neumonía, nasofaringitis |
Sepsis neutropénica, infección del tracto respiratorio superior, infección del tracto urinario, celulitis, sinusitis, faringitis, rinitis, herpes simplex |
|
|
Trastornos de la sangre y del sistema linfático |
Neutropenia febril, neutropenia, leucopenia, trombocitopenia, anemia |
Insuficiencia de médula ósea, pancitopenia |
|
|
Trastornos del sistema inmunológico |
Reacciones de hipersensibilidad |
||
|
Trastornos del metabolismo y de la nutrición |
Anorexia |
Hipocalemia |
|
|
Trastornos psiquiátricos |
Confusión, ansiedad, insomnio |
||
|
Trastornos del sistema nervioso |
Mareo, dolor de cabeza |
Hemorragia intracraneal, letargo |
|
|
Trastornos oculares |
Hemorragia ocular, hemorragia conjuntival |
||
|
Trastornos vasculares |
Hipertensión, hipotensión, hematoma |
||
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Disnea de esfuerzo Dolor faringolaríngeo |
|
|
Trastornos gastrointestinales |
Diarrea, vómitos, constipación, náusea, dolores abdominales |
Hemorragia gastrointestinal, hemorragia hemorroidal, estomatitis, sangrado gingival, dispepsia |
|
|
Trastornos de la piel y del tejido subcutáneo |
Petequias, prurito, exantema, equimosis |
Púrpura, alopecia, eritema, exantema macular |
Dermatosis neutrofílica febril aguda |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia |
Mialgia, dolor musculoesquelético |
|
|
Trastornos renales y urinarios |
Insuficiencia renal,hematuria, creatinina sérica elevada |
Acidosis tubular renal |
|
|
Trastornos Generales y alteraciones en el lugar de administración |
Fatiga, pirexia, dolor torácico, eritema en el lugar de inyección, dolor en el lugar de inyección, reacción en el sitio de inyección (no-especificado) |
Sitio de inyección: contusiones, hematoma, induración, exantema, prurito inflamación, decoloración, nódulos y hemorragia, malestar |
|
|
Exploraciones complementarias |
Disminución de peso |
Leucemia mieloide aguda (AZA-AML-001): Dentro del estudio clave de AML (AZA-AML-001), las reacciones adversas reportadas con mayor frecuencia (≥ 30%) con tratamiento de azacitidina fueron estreñimiento (41.9%), náuseas (39.8%), pirexia (37.7%), diarrea. (36.9%), neutropenia febril (32.2%) y neutropenia (30.1%).
Las reacciones adversas al fármaco graves más comunes (≥10%) mencionadas en AZA-AML-001 dentro del grupo de tratamiento con azacitidina, incluyeron neutropenia febril (25.0%), neumonía (20.3%) y pirexia (10.6%). Otras reacciones adversas graves reportadas con menor frecuencia en el grupo de tratamiento con azacitidina incluyeron sepsis (5.1%), anemia (4.2%), sepsis neutropénica (3.0%), infección del tracto urinario (3.0%), trombocitopenia (2.5%), neutropenia (2.1%), celulitis (2.1%), mareos (2.1%) y disnea (2.1%).
La siguiente tabla contiene las reacciones adversas para las que se pudo establecer razonablemente una relación causal con el tratamiento con azacitidina. Las frecuencias que se presentan se basan en las observaciones durante el estudio clave.
Las frecuencias se definen como: muy común (≥ 1/10), común (≥ 1/100 a < 1/10); y poco común (≥ 1/1,000 a < 1/100). Dentro de cada agrupación de frecuencias, los efectos no deseados se presentan en orden decreciente de gravedad.
Tabla 2. Frecuencia de Reacciones Adversas al Fármaco con Azacitidina en el Estudio AML(AZA-AML-001)
|
Clase de Sistema Orgánico |
Muy común |
Común |
Poco común |
|
Infecciones e infestaciones |
Neumonía† (incluyendo bacteriana, viral y fúngica) |
Sepsis† (incluyendo bacteriana, viral y fúngica), sepsis neutropénica†, infección del tracto respiratorio (incluye superior y bronquitis), infección del tracto urinario, celulitis, diverticulitis, infección fúngica oral, nasofaringitis, faringitis, sinusitis, rinitis e infección cutánea |
|
|
Trastornos de la sangre y sistema linfático |
Neutropenia febril†, neutropenia, trombocitopenia, anemia |
Insuficiencia de médula ósea, leucopenia |
Pancitopenia |
|
Trastornos del sistema inmune |
Reacciones de hipersensibilidad |
||
|
Trastornos del metabolismo y nutrición |
Hipocalemia, hiporexia |
Deshidratación |
|
|
Trastornos psiquiátricos |
Insomnio |
Ansiedad |
|
|
Trastornos del sistema nervioso |
Mareo, cefalea |
Hemorragia intracraneal†, síncope, somnolencia |
|
|
Trastornos oculares |
Hemorragia conjuntival |
Hemorragia ocular |
|
|
Trastornos vasculares |
Hipotensión†, hipertensión, hipotensión ortostática, hematoma |
||
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea, epistaxis |
Derrame pleural |
|
|
Trastornos gastrointestinales |
Diarrea, vómito, estreñimiento, náusea, dolor abdominal (incluye molestia superior y abdominal) |
Hemorragia gastrointestinal† (incluye hemorragia bucal), estomatitis, sangrado gingival, dispepsia |
|
|
Trastornos de la piel y tejido subcutáneo |
Exantema, prurito (incluye generalizado) |
Petequia, eritema urticarial |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Artralgia, dolor musculoesquelético (incluye dolor de espalda, óseo y dolor en extremidades) |
Espasmos musculares, mialgia |
|
|
Trastornos generales y condiciones del sitio de administración |
Pirexia†, astenia, fatiga, reacción en el sitio de inyección, eritema en el sitio de inyección |
Malestar, escalofríos, dolor de pecho, hemorragia en el sitio del catéter, sitio de inyección: dolor, equimosis, hematoma, induración, exantema, hemorragia, inflamación y prurito |
|
|
Investigaciones |
Disminución de peso |
† Indica reportes con resultado fatal.
Descripción de reacciones adversas selectas:
Reacciones adversas hematológicas: Las reacciones adversas informadas con mayor frecuencia relacionadas con el tratamiento con azacitidina fueron hematológicas, incluyendo trombocitopenia, neutropenia y leucopenia, y fueron usualmente de Grado 3 ó 4. Existe un mayor riesgo de que ocurran estos eventos durante los primeros dos ciclos, luego de los cuales suceden con menor frecuencia en pacientes con restauración de la función hematológica. La mayoría de las reacciones adversas hematológicas fueron tratadas mediante un monitoreo de rutina de recuentos sanguíneos completos y el retraso de la administración de azacitidina en el siguiente ciclo, antibióticos profilácticos y/o soporte de factor de crecimiento (por ejemplo, G-CSF) para neutropenia y transfusiones para la anemia o trombocitopenia, según fuera necesario.
Infecciones: La mielosupresión puede conducir a neutropenia y a un mayor riesgo de infección. Las infecciones serias, tales como sepsis neutropénica (0,8%) y neumonía (2,5%) fueron informadas en pacientes que recibían azacitidina, algunas con resultado fatal. Las infecciones pueden manejarse con el uso de anti-infecciosos más soporte de factor de crecimiento (por ejemplo, G-CSF) para neutropenia.
Sangrado: Puede observarse sangrado en pacientes que reciben azacitidina. Se han informado reacciones adversas serias tales como hemorragia gastrointestinal (0,8%) y hemorragia intracraneal (0,5%). Se debe monitorear en los pacientes los signos y síntomas de sangrado, especialmente en aquellos con trombocitopenia preexistente o relacionada con el tratamiento.
Hipersensibilidad: Se han informado reacciones serias de hipersensibilidad (0,25%) en pacientes que reciben azacitidina. En caso de una reacción tipo anafiláctica, el tratamiento con azacitidina debe discontinuarse de inmediato e iniciarse un tratamiento sintomático adecuado.
Reacciones adversas de la piel y del tejido subcutáneo: La mayoría de las reacciones adversas de la piel y del tejido subcutáneo estuvieron relacionadas con el sitio de inyección. Ninguna de estas reacciones adversas condujo a la discontinuación temporaria o permanente de azacitidina, o la reducción de la dosis de azacitidina en el estudio fundamental. La mayoría de las reacciones adversas tuvo lugar durante los primeros dos ciclos y tendieron a disminuir con los ciclos subsiguientes. Las reacciones adversas subcutáneas, tales como exantema/inflamación/prurito en el sitio de inyección, exantema, eritema y lesión de la piel pueden requerir el manejo concomitante con productos medicinales, tales como antihistamínicos, corticosteroides y drogas antiinflamatorias no-esteroideas (AINES).
Estas reacciones cutáneas tienen que distinguirse de las infecciones en tejidos blandos, a veces se producen en el sitio de la inyección. Con azacitidina, en el escenario posterior a la comercialización, se han reportado infecciones en tejidos blandos, incluyendo celulitis y fascitis necrotizante que en raras ocasiones pueden llevar a la muerte. Para el manejo clínico de las reacciones adversas infecciosas.
Reacciones adversas gastrointestinales: Las reacciones adversas gastrointestinales informadas más frecuentemente relacionadas con el tratamiento con azacitidina incluyen, constipación, diarrea, náusea y vómito. Estas reacciones adversas fueron tratadas de forma sintomática con antieméticos para la náusea y el vómito; antidiarreicos para la diarrea y laxantes para la constipación.
Reacciones adversas renales: En pacientes tratados con azacitidina se notificaron anomalías renales que abarcaron desde elevación de creatinina sérica y hematuria hasta acidosis tubular renal, insuficiencia renal y muerte.
Reacciones adversas hepáticas: En pacientes con una gran carga tumoral por enfermedad metastásica, se han notificado aparición de insuficiencia hepática, coma hepático progresivo y muerte durante el tratamiento con azacitidina.
Acontecimientos cardiacos: Los datos de un estudio clínico que permitió la inclusión de pacientes con antecedentes conocidos de enfermedad cardiovascular o pulmonar mostraron un aumento estadísticamente significativo de los acontecimientos cardiacos en pacientes con LMA recién diagnosticada tratados con VIDAZA® en combinación con antraciclina más citarabina en dosis alta.
Luego de la comercialización, fueron informados los siguientes eventos adversos:
– Enfermedad pulmonar intersticial.
– Síndrome de lisis tumoral.
– Necrosis en el lugar de inyección.
– Fascitis necrotizante.
– Dermatosis neutrofílica febril aguda.
– Pioderma gangrenosa.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Azacitidina induce tanto mutaciones genéticas como aberraciones cromosómicas en los sistemas celulares bacterianos y mamíferos in vitro. La potencial carcinogenicidad de azacitidina se evaluó en ratones y ratas. Azacitidina indujo tumores del sistema hematopoyético en ratas hembra, cuando fue administrada por vía intraperitoneal 3 veces por semana durante 52 semanas. Se observó una mayor incidencia de tumores en el sistema linforeticular, pulmones, glándulas mamarias y piel en ratones tratados con azacitidina administrada por vía intraperitoneal durante 50 semanas. Un estudio de tumorogenicidad en ratas reveló una mayor incidencia de tumores testiculares.
Estudios de embriotoxicidad temprana en ratones revelaron una frecuencia del 44% de muerte embrional intrauterina (mayor resorción) después de una inyección única intraperitoneal de azacitidina durante la organogénesis. Se han detectado anomalías en el desarrollo en el cerebro en ratones que recibieron azacitidina antes o al momento del cierre del paladar. En ratas, azacitidina no causó eventos adversos al ser administrada antes de la implantación, pero fue evidentemente embriotóxica cuando fue administrada durante la organogénesis. Las anomalías fetales causadas durante la organogénesis incluyeron: anomalías del SNC (exencefalia/encefalocele), anomalías de las extremidades (micromelia, pie martillo, dedos soldados, oligodactilia) y otros (microftalmia, micrognatia, gastrosquisis, edema y anormalidades de costillas).
La administración de azacitidina a ratones machos antes del apareamiento con hembras no tratadas tuvo como resultado una menor fertilidad y la pérdida de crías durante el desarrollo embrionario y el subsiguiente desarrollo post natal. El tratamiento de ratas macho tuvo como resultado una disminución del peso de los testículos y epidídimos, menores recuentos de esperma, menores tasas de preñez, un aumento de embriones anormales y mayores pérdidas de embriones en gestación.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: En base a los datos in vitro, el metabolismo de azacitidina no está mediado por las isoenzimas del citocromo P450 (CYP), las UDP-glucuronosiltransferasas (UGT), sulfotransferasas (SULT) ni glutatión transferasas (GST); por lo tanto, las interacciones relacionadas con estas enzimas metabolizantes in vivo se consideran improbables.
Los efectos inhibitorios o inductores clínicamente significativos de la azacitidina sobre las enzimas del citocromo P450 son improbables.
No se han realizado estudios formales de interacción farmacológica clínica con la azacitidina.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Deben determinarse antes del comienzo de la terapia y antes de cada ciclo de tratamiento pruebas de la función hepática, creatinina y bicarbonato séricos. Debe realizarse biometria hemática completa antes del comienzo de la terapia y, según sea necesario, para monitorear datos de respuesta y toxicidad, por lo menos antes de cada ciclo de tratamiento.
PRECAUCIONES GENERALES:
Toxicidad hematológica: El tratamiento con azacitidina se asocia con anemia, neutropenia y trombocitopenia, especialmente en los dos primeros ciclos. Deben efectuarse recuentos sanguíneos completos cuando sea necesario para vigilar la respuesta y la toxicidad, al menos, antes de cada ciclo de tratamiento. Luego de la administración de la dosis recomendada para el primer ciclo, se debe reducir la dosis para los ciclos subsiguientes o retrasar su administración en base a los recuentos de nadir y la respuesta hematológica. Se debe recomendar a los pacientes que informen inmediatamente episodios de fiebre. Se recomienda especial atención a la presencia de signos y síntomas de hemorragia.
Insuficiencia hepática: No se han realizado estudios formales en pacientes con insuficiencia hepática. En los pacientes con una carga tumoral amplia debido a enfermedad metastásica, se han informado casos de coma hepático progresivo y muerte durante el tratamiento con azacitidina, particularmente en los pacientes con albúmina sérica en línea de base < 30 g/l. Azacitidina está contraindicado en pacientes con tumores hepáticos malignos avanzados.
Insuficiencia renal: En los pacientes tratados con azacitidina intravenosa en combinación con otros agentes quimioterapéuticos, se han informado anomalías renales que abarquen desde niveles de creatinina sérica elevada hasta insuficiencia renal y muerte. Además, la acidosis tubular renal definida como una caída del bicarbonato sérico a < 20 mmol/l en combinación con orina alcalina e hipocalemia (potasio sérico < 3 mmol/l) se manifestó en 5 pacientes con leucemia mieloide crónica (CML) tratados con azacitidina y etopósido. Si se observan reducciones inexplicadas de bicarbonato sérico (< 20 mmol/l) o elevaciones de creatinina sérica o BUN, se debe reducir la dosis o retrasar la administración.
Se les debe recomendar a los pacientes que informen inmediatamente a los profesionales de la salud si presentan oliguria o anuria.
Se debe monitorear la toxicidad en pacientes con insuficiencia renal ya que azacitidina y/o sus metabolitos se excretan principalmente por el riñón.
Cardiopatía y enfermedad pulmonar: Los pacientes con antecedentes de insuficiencia cardiaca congestiva grave, cardiopatía clínicamente inestable o enfermedad pulmonar, fueron excluidos del estudio pivotal para registro (AZA-001), por lo tanto, no se ha establecido la seguridad ni la eficacia de VIDAZA® en estos pacientes. Los datos recientes de un estudio clínico en pacientes con antecedentes conocidos de enfermedad cardiovascular o pulmonar mostraron un aumento significativo de la incidencia de acontecimientos cardiacos con VIDAZA® en combinación con antraciclina más citarabina a dosis alta (ver sección Reacciones secundarias y adversas). Por lo tanto, se aconseja precaución al prescribir VIDAZA® a estos pacientes. Se debe considerar una evaluación cardiopulmonar antes y durante el tratamiento con VIDAZA® .
Fascitis Necrotizante: Se ha reportado fascitis necrotizante en pacientes tratados con VIDAZA® , incluyendo casos fatales. La terapia con VIDAZA® debe interrumpirse en pacientes que desarrollen fascitis necrotizante, y sin demora, debe iniciarse el tratamiento adecuado.
Síndrome de Lisis Tumoral: Los pacientes en riesgo de síndrome de Lisis Tumoral son aquellos con alta carga tumoral, previa al tratamiento. Estos pacientes deben ser monitoreados estrechamente y deben tomarse las precauciones apropiadas.
Efectos sobre la capacidad de conducir cualquier vehículo y utilizar maquinarias: No se han realizado estudios de los efectos sobre la capacidad de conducir autos y utilizar maquinarias. Se debe advertir a los pacientes que pueden experimentar efectos adversos, tales como fatiga durante el tratamiento. Por lo tanto, se debe recomendar cautela al conducir autos u operar maquinarias.
DOSIS Y VÍA DE ADMINISTRACIÓN: El tratamiento con VIDAZA® debe iniciarse y monitorearse bajo la supervisión de un médico con experiencia en el uso de fármacos quimioterapéuticos. Los pacientes deben ser tratados previamente con antieméticos para controlar la náusea y el vómito.
Posología:
Tratamiento de Síndrome Mielodisplásico, la LMMC y AML: La dosis inicial recomendada para el primer ciclo de tratamiento, para todos los pacientes, independientemente de los valores hematológicos iniciales, es de 75 mg/m2 de superficie corporal, inyectada por vía subcutánea, diariamente, durante siete días, seguido de un periodo de reposo de 21 días (ciclo de tratamiento de 28 días).
Se recomienda que los pacientes reciban tratamiento durante un mínimo de seis ciclos. El tratamiento debe continuarse mientras el paciente siga beneficiándose o hasta la progresión de la enfermedad.
Se debe vigilar la respuesta/toxicidad hematológica y la toxicidad renal de los pacientes; puede ser necesario un retraso en el inicio del siguiente ciclo o una disminución de una dosis, como se explica más adelante.
Terapia inmunomoduladora de mantenimiento para pacientes post transplante de médula ósea: La dosis óptima de azacitidina es de 32 mg/m2 de superficie corporal inyectada por vía subcutánea diariamente durante 5 días, seguidos por un periodo de reposo de 25 días (ciclos de tratamiento de 30 días).
Se recomienda el mantenimiento de la terapia mientras el paciente siga beneficiándose o hasta la aparición del primero o temprano dato de Enfermedad Injerto contra Huésped (EIH), en cuyo caso deberá suspenderse de inmediato y revalorar su estado de salud.
Ajuste de la dosis debido a toxicidad hematológica: La toxicidad hematológica se define como el recuento sanguíneo más bajo alcanzado en un ciclo determinado (NADIR), si el recuento de plaquetas ≤ 50,0 x 109/L y/o el recuento absoluto de neutrófilos (RAN) ≤ 1 x 109/L.
La recuperación se define como un aumento de la/s línea/s celular/es en las que se observó una toxicidad hematológica, como mínimo, igual a la mitad de la diferencia entre el NADIR y el recuento inicial, más el recuento NADIR; es decir, recuento sanguíneo en la recuperación ≥ recuento NADIR + (0,5 x [recuento inicial – recuento NADIR]).
Pacientes sin una disminución de los recuentos sanguíneos iniciales (es decir, leucocitos ≥ 3,0 x 109/L y RAN ≥ 1,5 x 109/L, y recuento plaquetario ≥ 75,0 x 109/L) antes del primer tratamiento:
Si se observa toxicidad hematológica después del tratamiento con VIDAZA®, el siguiente ciclo de tratamiento con VIDAZA® debe retrasarse hasta que el recuento plaquetario y el RAN se hayan recuperado. Si la recuperación se alcanza en un plazo de 14 días, no es necesario un ajuste de la dosis. Sin embargo, si la recuperación no se ha alcanzado en un plazo de 14 días, la dosis debe reducirse según la siguiente tabla. Después de las modificaciones de la dosis, la duración del ciclo debe volver a ser de 28 días.
|
Recuentos NADIR |
% de la dosis en el siguiente ciclo, si la recuperación* no se alcanza en un plazo de 14 días |
|
|
RAN (x 109/L) |
Plaquetas (x 109/L) |
|
|
≤ 1,0 |
≤ 50,0 |
50% |
|
> 1,0 |
> 50,0 |
100% |
* Recuperación = recuentos > recuento NADIR + (0,5 x [recuento inicial – recuento NADIR]).
Pacientes con recuentos sanguíneos iniciales reducidos (es decir, leucocitos < 3,0 x 109/L o RAN < 1,5 x 109/L o recuento plaquetario < 75,0 x 109/L) antes del primer tratamiento: Después del tratamiento con VIDAZA®, si la disminución del recuento leucocitario del RAN o del recuento plaquetario con respecto al recuento antes del tratamiento es ≤ 50% o superior al 50%, pero con una mejoría en la diferenciación de cualquier línea celular, el siguiente ciclo no debe retrasarse y no debe efectuarse ningún ajuste de la dosis.
Si la disminución del recuento leucocitario del RAN o del recuento plaquetario es superior al 50% con respecto al recuento antes del tratamiento, y no hay mejoría en la diferenciación de líneas celulares, el siguiente ciclo de tratamiento con VIDAZA® debe retrasarse hasta que el recuento plaquetario y el RAN se hayan recuperado. Si la recuperación se alcanza en un plazo de 14 días, no es necesario un ajuste de la dosis. Sin embargo, si la recuperación no se ha alcanzado en un plazo de 14 días, debe determinarse la producción celular de la médula ósea. Si la producción celular de la médula ósea es > 50%, no debe efectuarse un ajuste de la dosis. Si la producción celular de la médula ósea es ≤ 50%, el tratamiento debe retrasarse y la dosis debe disminuirse, según la siguiente tabla:
|
Producción de la médula ósea |
% de la dosis en el siguiente ciclo, si la recuperación no se alcanza en un plazo de 14 días |
|
|
Recuperación* ≤ 21 días |
Recuperación* > 21 días |
|
|
15-50% |
100% |
50% |
|
< 15% |
100% |
33% |
* Recuperación = recuentos > recuento NADIR + (0,5 x [recuento inicial – recuento NADIR]).
Después de las modificaciones de la dosis, la duración del ciclo debe volver a ser de 28 días.
Poblaciones especiales:
Insuficiencia renal: La azacitidina puede ser administrada a paciente con insuficiencia renal sin ajuste de dosis inicial.
Si ocurren reducciones inexplicables en los niveles séricos de bicarbonato a menos de 20 mmol/l, la dosis se debe reducir en un 50% en el siguiente ciclo. Si ocurren elevaciones inexplicables en la creatinina sérica o el BUN (nitrógeno ureico en sangre por sus siglas en inglés), el siguiente ciclo debe ser retrasado hasta que los valores regresen a estar normales o a nivel basal y la dosis debe ser reducida en un 50% en el siguiente ciclo de tratamiento.
Insuficiencia hepática: No se han realizado estudios formales en pacientes con función hepática disminuida. Se deben vigilar atentamente las reacciones adversas en los pacientes con insuficiencia hepática grave. No se recomienda ninguna modificación específica de la dosis inicial en pacientes con insuficiencia hepática previa al inicio del tratamiento; las modificaciones posteriores deben basarse en los valores hematológicos. VIDAZA® está contraindicado en los pacientes con tumores hepáticos malignos avanzados.
Pacientes de edad avanzada: No se recomienda ningún ajuste específico de la dosis en los pacientes de edad avanzada. Puesto que es más probable que en este tipo de pacientes se presente algún deterioro, es conveniente vigilar la función renal y la cuenta hematológica completa.
Niños y adolescentes: VIDAZA® no está recomendado para uso en menores de 18 años debido a la escasez de datos sobre la seguridad y la eficacia.
VIDAZA® se administrará como una inyección bajo la piel (subcutánea) por un médico o enfermera. Se puede administrar bajo la piel de su muslo, abdomen o parte superior del brazo.
Las nuevas inyecciones deben administrarse como mínimo a 2.5 cm de distancia del lugar anterior y nunca en zonas sensibles, con equimosis, enrojecidas o endurecidas.
Precauciones especiales para la eliminación y manejo:
Recomendaciones para un manejo seguro: VIDAZA® es un medicamento citotóxico y, al igual que otros compuestos potencialmente tóxicos, se debe tener cautela al manejar y preparar suspensiones con azacitidina. Deben emplearse los procedimientos para correcto manejo y eliminación de medicamentos anti-cancerígenos. Si la azacitidina reconstituida entra en contacto con la piel, lavar inmediatamente con agua y jabón en forma meticulosa. Si entra en contacto con las membranas mucosas, enjuagar cuidadosamente con agua.
Después de la reconstitución, no se debe filtrar la suspensión.
Procedimiento de reconstitución:
VIDAZA® se debe reconstituir con agua inyectable. La vida de anaquel del medicamento reconstituido puede prolongarse reconstituyéndolo con agua inyectable refrigerada (entre 2°C y 8°C). A continuación se presenta información sobre la conservación del medicamento reconstituido.
1. Deben reunirse los siguientes suministros:
• Frasco ámpula de azacitidina; frasco ámpula con agua inyectable; guantes quirúrgicos no estériles.
• Torundas de algodón con alcohol; jeringas de 5 ml con agujas.
2. Debe colocarse en la jeringa 4 ml de agua inyectable, asegurándose de eliminar todo aire que pueda quedar dentro de la jeringa.
3. La jeringa con los 4 ml de agua inyectable debe incrustarse en el frasco ámpula de azacitidina y el agua debe pasarse de la jeringa al frasco ámpula.
4. Después de extraer la jeringa y la aguja, el frasco ámpula debe agitarse vigorosamente, hasta obtener una suspensión turbia uniforme.
Después de la reconstitución, cada ml de suspensión contendrá 25 mg de azacitidina (100 mg/4 ml). El producto reconstituido es una suspensión turbia y homogénea, sin aglomerados. La suspensión debe desecharse si contiene partículas grandes o aglomerados. No filtrar la suspensión después de la reconstitución porque esto podría eliminar sustancia activa.
Hay que tener en cuenta que los filtros están presentes en algunos adaptadores, puntas y sistemas cerrados; por lo tanto, tales sistemas no se deben utilizar para la administración del medicamento después de la reconstitución.
5. El tapón de goma debe limpiarse y se introduce una jeringa nueva; a continuación, se extrae la cantidad de medicamento necesario para la dosis correcta, asegurándose de purgar el aire atrapado dentro de la jeringa, posterior a esto, debe extraerse del frasco ámpula la jeringa y la aguja debe desecharse.
6. Seguidamente, debe ajustarse firmemente a la jeringa una aguja subcutánea nueva (se recomienda el calibre 25). La aguja no debe purgarse antes de la inyección, a fin de reducir la incidencia de reacciones locales en el lugar de la aplicación.
7. Si es necesario dosis superiores a 100 mg, deben repetirse todos los pasos anteriores para la preparación de la suspensión. En estos casos la dosis debe dividirse en partes iguales, en dos jeringas (por ejemplo, dosis de 150 mg = 6 ml; dos jeringas con 3 ml en cada jeringa). Inyectar en dos sitios diferentes.
8. El contenido de la jeringa de dosificación debe resuspenderse inmediatamente antes de la administración. La temperatura de la suspensión en el momento de la inyección debe ser de aproximadamente 20 a 25°C. Para volver a suspender, haga rodar vigorosamente la jeringa entre las palmas de las manos, hasta obtener una suspensión uniforme y turbia. La suspensión debe desecharse si contiene partículas grandes o aglomerados.
Conservación del medicamento reconstituido:
Para uso inmediato:
La suspensión de VIDAZA® puede prepararse inmediatamente antes de usarse y la suspensión reconstituida debe administrarse dentro de un lapso de 45 minutos. Si el tiempo transcurrido es mayor a 45 minutos, la suspensión reconstituida debe desecharse apropiadamente y preparar una nueva dosis.
Para uso posterior: Cuando se reconstituye usando agua inyectable no refrigerada, la suspensión reconstituida debe colocarse en un refrigerador (2°C a 8°C) inmediatamente después de la reconstitución y mantenerse en el refrigerador durante un máximo de 8 horas. Si el tiempo transcurrido en el refrigerador es mayor a 8 horas, la suspensión debe desecharse apropiadamente y preparar una nueva dosis.
Cuando se reconstituye usando agua inyectable refrigerada, (2°C a 8°C) la suspensión reconstituida debe colocarse en un refrigerador (2°C a 8°C) inmediatamente después de la reconstitución y mantenerse en el refrigerador durante un máximo de 22 horas. Si el tiempo transcurrido en el refrigerador es mayor a 22 horas, la suspensión debe desecharse apropiadamente y preparar una nueva dosis.
La jeringa cargada con suspensión reconstituida debe dejarse reposar hasta 30 minutos antes de la administración para que alcance una temperatura aproximadamente de 20°C-25°C. Si el tiempo transcurrido es mayor a 30 minutos, la suspensión debe desecharse apropiadamente y preparar una nueva dosis.
Cálculo de una dosis individual: La dosis total, según la superficie corporal (SC), puede calcularse de la siguiente manera:
Dosis total (mg) = dosis (mg/m2) x SC (m2)
La siguiente tabla se presenta sólo como un ejemplo para calcular dosis individuales de azacitidina, basadas en un valor promedio de SC de 1,8 m2.
|
Dosis, mg/m2 (% de la dosis inicial recomendada) |
Dosis total basada en un valor de SC de 1,8 m2 |
Número de viales necesarios |
Volumen total de suspensión reconstituida requerida |
|
75 mg/m2 (100%) |
135 mg |
2 viales |
5,4 ml |
|
37,5 mg/m2 (50%) |
67,5 mg |
1 vial |
2,7 ml |
|
25 mg/m2 (33%) |
45 mg |
1 vial |
1,8 ml |
Forma de administración: VIDAZA® reconstituido debe inyectarse por vía subcutánea (introduzca la aguja con un ángulo de 45° a 90°), con una aguja de calibre 25 en el brazo, el muslo o el abdomen.
Las dosis superiores a 4 ml deben inyectarse en dos lugares separados.
Los lugares de inyección deben someterse a rotación. Las nuevas inyecciones deben administrarse como mínimo a 2,5 cm de distancia del lugar anterior y nunca en zonas sensibles, con equimosis, enrojecidas o endurecidas.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Incompatibilidades: Este medicamento no debe mezclarse con otros medicamentos.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Se notificó un caso de sobredosis con azacitidina durante los ensayos clínicos. Un paciente sufrió diarrea, náusea y vómitos después de recibir una dosis única por vía intravenosa de aproximadamente 290 mg/m2, casi el cuádruple de la dosis inicial recomendada.
En caso de sobredosis, se debe vigilar en el paciente los recuentos sanguíneos adecuados y debe recibir el tratamiento de apoyo que sea necesario. No existe un antídoto específico conocido para la sobredosis de azacitidina.
PRESENTACIONES: 1 frasco ámpula con 100 mg de azacitidina (25 mg/ml).
Frasco ámpula de vidrio de 30 ml tipo I incoloro sellado con tapón elastomérico de butilo y precinto de aluminio con botón plástico de polipropileno.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Los frascos ámpula sin reconstituir deben almacenarse a temperaturas no mayores a 30°C.
Vida media:
Después de la reconstitución: Cuando VIDAZA® se reconstituye con agua para inyectables que no se ha refrigerado, la estabilidad química y física del medicamento reconstituido se ha demostrado a 25ºC durante 45 minutos y entre 2°C a 8°C durante 8 horas.
La vida útil del medicamento reconstituido se puede extender por medio de la reconstitución con agua para inyectables refrigerada (2°C a 8°C). Cuando VIDAZA® se reconstituye con agua para inyectables refrigerada (2°C a 8°C), la estabilidad química y física del medicamento reconstituido se ha demostrado entre 2°C a 8°C durante 22 horas.
Desde el punto de vista microbiológico, el producto reconstituido debe usarse de forma inmediata. Si no se utiliza de inmediato, los tiempos y condiciones de almacenamiento en uso previo al uso son responsabilidad del usuario y no deben superar las 8 horas a 2°C a 8°C cuando se reconstituye con agua para inyectables que no se ha refrigerado o por más de 22 horas cuando se reconstituyó con agua para inyectables refrigerada (2ºC a 8ºC).
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje al alcance de los niños. Este medicamento deberá ser administrado únicamente por médicos especialistas en oncología y con experiencia en quimioterapia antineoplásica. No se use en el embarazo y lactancia ni en menores de 18 años. Literatura exclusiva para el médico.
Reporte las sospechas de reacción adversa al correo: farmacovigilancia@cofepris.gob.mx y
dsmexico@celgene.com o a la línea 01800CELGENE.
Hecho en Alemania por:
Baxter Oncology Gmbh
Kantstrasse 2 33790 Halle/Westfalen
Alemania.
Para:
Celgene Europe LTD.
1 LongWalk Road, Stockley Park
Uxbridge, UB11 1BD
Reino Unido
Representante legal y distribuido en México por:
CELGENE LOGISTICS SARL
CPA Logistics Center Tlalnepantla
Edificio 1, Bodega 7, Almacén 12
Km 12.5 Vía Gustavo Baz,
Col. San Pedro Barrientos,
Tlalnepantla de Baz
C.P. 54010, México.
Reg. Núm. 046M2013, SSA IV


