
XGEVA - Solución inyectable
Sustancia(s):
- Denosumab
Presentaciones:
- 1 Caja, 1 Frasco(s) ámpula, 120/1.7 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
El frasco ámpula contiene:
Denosumab 120 mg
Vehículo cbp 1.7 mL
La jeringa prellenada contiene:
Denosumab 120 mg
Vehículo cbp 1 mL
Anticuerpo monoclonal humano IgG2 de origen ADN recombinante expresado en células de ovario de hámster chino (CHO).
INDICACIONES TERAPÉUTICAS:
XGEVA está indicado para la prevención de eventos relacionados con el esqueleto (fracturas patológicas, radioterapia de hueso, compresión medular o cirugía ósea) en pacientes con mieloma múltiple y en pacientes con metástasis ósea de tumores sólidos.
XGEVA está indicado en adultos y en adolescentes con esqueleto maduro con tumor óseo de células gigantes que es irresecable, o cuando la resección quirúrgica pueda resultar en morbilidad severa.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas:
Mecanismo de acción: Denosumab es un anticuerpo monoclonal humano (IgG2) que se dirige y se une con gran afinidad y especificidad al ligando RANKL impidiendo la activación de su único receptor RANK; el cual, se encuentra en la superficie de los osteoclastos y sus precursores. El ligando RANK existe como una proteína transmembranal o soluble. El ligando RANK es esencial para la formación, función y supervivencia de los osteclastos, el único tipo de células responsable de la resorción ósea. El incremento en la actividad de los osteoclastos, estimulada por el ligando RANK, es un mediador clave en la destrucción del hueso en la enfermedad ósea en tumores metastásicos y en mieloma múltiple. La prevención de la interacción del ligando RANK con el receptor resulta en una reducción en el número y función de los osteoclastos, disminuyendo la resorción y la destrucción ósea inducida por el cáncer.
Una característica de los tumores óseos de células gigantes son las células estromales que expresan el ligando RANK y células gigantes parecidas a osteoclastos que expresan el receptor RANK. En pacientes con tumores óseos de células gigantes, denosumab se une al ligando RANK reduciendo o eliminando significativamente las células gigantes parecidas a osteoclastos. En consecuencia, se reduce la osteolisis y el estroma tumoral proliferativo es reemplazado con tejido óseo denso, nuevo, diferenciado y no proliferativo.
Farmacodinamia: En un estudio fase 2 en pacientes con cáncer de mama y metástasis óseas que no habían recibido previamente terapia con bifosfonatos IV, la administración subcutánea de XGEVA 120 mg cada 4 semanas (Q4W) causó una rápida reducción en los marcadores de resorción ósea (uNTX/creatinina [en orina], y CTx sérico) con una mediana en la reducción de 82% para uNTX/Cr (en orina) en 1 semana. Las reducciones en los marcadores de remodelación ósea se mantuvieron, con una mediana en la reducción de uNTX/Cr de 74% a 82% en las semanas 2 a 25 con la dosis continua de 120 mg cada 4 semanas. En los estudios fase 3 en pacientes con cáncer avanzado, la mediana en la reducción fue aproximadamente de 80% en uNTX/Cr después de 3 meses de tratamiento con XGEVA vs el periodo basal en 2,075 pacientes con cáncer avanzado (mama, próstata, mieloma múltiple y otros tumores sólidos).
De manera similar, en pacientes con cáncer avanzado y metástasis óseas (incluyendo sujetos con mieloma múltiple y enfermedad ósea) quienes estaban recibiendo tratamiento con bifosfonato IV, tuvieron niveles de uNTX/Cr > 50 nM/mM; la administración de XGEVA en dosis SC múltiples, cada 4 semanas o bien cada 12 semanas produjo una reducción de aproximadamente 80% en uNTX/Cr desde el periodo basal y después de 3 y 6 meses de tratamiento. En general, 97% de los pacientes en los grupos con XGEVA tuvieron por lo menos 1 valor de uNTX/Cr < 50 nM/mM a la semana 25 del estudio.
En un estudio de fase 3 de pacientes con mieloma múltiple recién diagnosticado que recibieron dosis SC de XGEVA 120 mg cada 4 semanas (Q4W), se observaron medianas de reducción en uNTx/Cr de aproximadamente 75% en la semana 5. Se mantuvieron las reducciones en los marcadores de recambio óseo, con una mediana de reducciones del 74% al 79% para la uNTx/Cr de las semanas 9 a la 49 de la administración continua de 120 mg cada 4 semanas.
En un estudio fase 2 en pacientes con tumor óseo de células gigantes que recibieron dosis SC de XGEVA 120 mg cada 4 semanas (Q4W) por vía subcutánea; con dosis de carga los días 8 y 15, se observaron reducciones en uNTX/Cr y sCTx mediana de aproximadamente 80% a la semana 9. Se mantuvieron las reducciones en los marcadores de remodelación ósea, con reducciones mediana en uNTX/Cr entre 77% y 87% y en sCTx entre 78% y 83%, de la semana 5 a la 25 con la dosificación continua de 120 mg cada 4 semanas.
lnmunogenicidad:
En estudios clínicos, no se han observado anticuerpos neutralizantes para XGEVA en pacientes con cáncer avanzado o pacientes con tumor de células gigantes de hueso. Utilizando un inmunoanálisis sensible, menos del 1% de los pacientes tratados con denosumab resultaron positivos a anticuerpos de unión no neutralizantes, sin evidencia de alteración en la respuesta farmacocinética, farmacodinámica o toxicidad.
Farmacocinética: Después de la administración subcutánea, la biodisponibilidad fue de 62% y denosumab mostró una farmacocinética no lineal en amplios rangos de dosis; y se observan incrementos proporcionales con exposición a dosis de 60 mg (o 1 mg/kg) y mayores. En pacientes con cáncer avanzado, quienes recibieron dosis múltiples de 120 mg cada 4 semanas se observó una acumulación de aproximadamente 2 veces en las concentraciones séricas de denosumab y el estado de equilibrio se alcanzó a los 6 meses, consistente con la farmacocinética independiente del tiempo. En sujetos con mieloma múltiple que recibieron 120 mg cada 4 semanas, la mediana a través de los niveles más bajos varió en menos del 8% entre los meses 6 y 12. En sujetos con tumor óseo de células gigantes que recibieron 120 mg cada 4 semanas con una dosis adicional de 120 mg en los días 8 y 15, se lograron niveles en estado estable durante el primer mes de tratamiento. Entre las semanas 9 y 49, la mediana en los niveles más bajos varió en menos de 9%. En estado de equilibrio, la concentración media sérica más baja fue 20.6 mcg/mL (rango: 0.456 mcg/mL a 56.9 mcg/mL). En sujetos que descontinuaron la dosis de 120 mg cada 4 semanas, el promedio de vida media fue 28 días (rango: 14 a 55 días).
Se realizó un análisis de farmacocinética poblacional para evaluar los efectos de las características demográficas. Este análisis no mostró diferencia notable en la farmacocinética con relación a la edad (18 a 87 años), raza, peso corporal (36 kg a 174 kg), o entre los pacientes con tumores sólidos, mieloma múltiple y tumores óseos de células gigantes. La farmacocinética y farmacodinamia de denosumab fueron similares en hombres y mujeres y en pacientes que transicionaron de terapia de bifosfonato IV. Los perfiles farmacocinético y farmacodinámico de denosumab no se vieron afectados por la formación de anticuerpos de unión a denosumab.
Poblaciones especiales de pacientes:
Sexo:
No se observaron diferencias en la farmacocinética de denosumab entre hombres y mujeres.
Pacientes geriátricos:
El perfil farmacocinético de denosumab no se vio afectado por el rango de edad entre 18 a 87 años.
Pacientes pediátricos:
En adolescentes con esqueleto maduro (12 a 17 años de edad; N = 10) con tumor de células gigantes de hueso que recibieron 120 mg cada 4 semanas con una dosis de carga los días 8 y 15, la farmacocinética de denosumab fue similar a la observada en sujetos adultos con GCTB (N = 15).
Raza: La farmacocinética de denosumab no fue afectada por la raza.
Insuficiencia renal: En estudios de denosumab (60 mg, N = 55 y 120 mg, N = 32) en pacientes sin cáncer avanzado, pero con grados variables de función renal, incluyendo pacientes en diálisis, el grado de insuficiencia renal no tuvo efecto sobre la farmacocinética y farmacodinamia de denosumab; por lo que no se requiere de ajuste de dosis en caso de insuficiencia renal.
Insuficiencia hepática: No se han realizado estudios clínicos para evaluar el efecto del daño hepático sobre la farmacocinética de denosumab.
Estudios clínicos:
Prevención de complicaciones óseas en adultos con neoplasias malignas avanzadas con afectación ósea:
Se evaluó la eficacia y seguridad de XGEVA en la prevención de Eventos Relacionados con el Esqueleto (ERE) en pacientes con malignidades avanzadas y afectación ósea en tres estudios aleatorizados, doble ciego, controlados con activo (Estudio 1, 2 y 3). Cada estudio evaluó denosumab (120 mg administrado por vía subcutánea) con ácido zoledrónico (4 mg administrado por vía intravenosa, con ajuste de dosis por función renal reducida) una vez cada 4 semanas. Las variables de eficacia primaria y secundaria evaluaron la ocurrencia de uno o más Eventos Relacionados con el Esqueleto (ERE) definidos como cualquiera de los siguientes: fractura patológica, terapia con radiación a hueso, cirugía ósea o compresión medular.
XGEVA redujo o bien fue capaz de prevenir el riesgo de desarrollar un ERE o múltiples ERE (primero o subsecuente) en pacientes con neoplasias malignas avanzadas con afectación ósea. A los pacientes que participaron en los estudios que demostraron superioridad de XGEVA sobre ácido zoledrónico, se les ofreció participar en un estudio abierto en el que se preespecificó una extensión del tratamiento con dos años de duración. Los resultados de eficacia se proporcionan en la Tabla 1.
Tabla 1: Resultados de eficacia de XGEVA en comparación con ácido zoledrónico en pacientes con neoplasias malignas avanzadas con afectación ósea
|
Estudio 1 cáncer de mama avanzado |
Estudio 2 cáncer avanzado (otros tumores sólidos o mieloma múltiple |
Estudio 3 cáncer de próstata avanzado |
Combinación de cáncer avanzado |
|||||
|---|---|---|---|---|---|---|---|---|
|
XGEVA |
Ácido zoledrónico |
XGEVA |
Ácido zoledrónico |
XGEVA |
Ácido zoledrónico |
XGEVA |
Ácido zoledrónico |
|
|
N |
1026 |
1020 |
886 |
890 |
950 |
951 |
2862 |
2861 |
|
Primer ERE† |
||||||||
|
Tiempo en meses (mediana) |
NA |
26.4 |
20.6 |
16.3 |
20.7 |
17.1 |
27.6 |
19.4 |
|
Diferencia en tiempo mediana (meses) |
ND |
4.2 |
3.5 |
8.2 |
||||
|
Tasa de riesgo (IC 95%) |
0.82 (0.71, 0.95) |
0.84 (0.71, 0.98) |
0.82 (0.71, 0.95) |
0.83 (0.76, 0.90) |
||||
|
Reducción del riesgo (%) |
18 |
16 |
18 |
17 |
||||
|
No inferioridad valor-p |
< 0.0001 |
0.0007 |
0.0002 |
< 0.0001 |
||||
|
Superioridad valor-p |
0.0101 |
0.0619 |
0.0085 |
< 0.0001 |
||||
|
Proporción de sujetos (%) |
30.7 |
36.5 |
31.4 |
36.3 |
35.9 |
40.6 |
32.6 |
37.8 |
|
Primer ERE y subsecuente*† |
||||||||
|
Número promedio/paciente |
0.46 |
0.60 |
0.44 |
0.49 |
0.52 |
0.61 |
0.48 |
0.57 |
|
Proporción de la tasa (IC 95%) |
0.77 (0.66, 0.89) |
0.90 (0.77, 1.04) |
0.82 (0.71, 0.94) |
0.82 (0.75, 0.89) |
||||
|
Reducción del riesgo (%) |
23 |
10 |
18 |
18 |
||||
|
Superioridad valor-p |
0.0012 |
0.1447 |
0.0085 |
< 0.0001 |
||||
|
Tasa de morbilidad esquelética por año |
0.45 |
0.58 |
0.86 |
1.04 |
0.79 |
0.83 |
0.69 |
0.81 |
|
Primer ERE o HNM |
||||||||
|
Tiempo en meses (mediana) |
NA |
25.2 |
19.0 |
14.4 |
20.3 |
17.1 |
26.6 |
19.4 |
|
Tasa de riesgo (IC 95%) |
0.82 (0.70, 0.95) |
0.83 (0.71, 0.97) |
0.83 (0.72, 0.96) |
0.83 (0.76, 0.90) |
||||
|
Reducción del riesgo (%) |
18 |
17 |
17 |
17 |
||||
|
Superioridad valor-p |
0.0074 |
0.0215 |
0.0134 |
< 0.0001 |
||||
|
Primera radiación a hueso |
||||||||
|
Tiempo en meses (mediana) |
NA |
NA |
NA |
NA |
NA |
28.6 |
NA |
33.2 |
|
Tasa de riesgo (IC 95%) |
0.74 (0.59, 0.94) |
0.78 (0.63, 0.97) |
0.78 (0.66, 0.94) |
0.77 (0.69, 0.87) |
||||
|
Reducción de riesgo (%) |
26 |
22 |
22 |
23 |
||||
|
Superioridad valor-p |
0.0121 |
0.0256 |
0.0071 |
< 0.0001 |
||||
NA = no se alcanzó; ND = no disponible; HNM = hipercalcemia maligna; SMR = tasa de morbilidad esquelética; ERE = evento relacionado con el esqueleto; IC = intervalo de confianza; † Los valores-p ajustados se presentan para los estudios 1, 2 y 3 (variables: primer ERE, y primer ERE y subsecuente); * Cuenta para todos los eventos esqueléticos en el tiempo; sólo eventos que ocurren ≥ 21 días después de que los eventos previos son contados.
Efecto sobre el Dolor: El análisis incluyó la evaluación de los cambios desde la basal con relación al peor dolor reportado mediante el BPI-SF (Brief Pain Inventory Short Form); también incluyó las evaluaciones en el tiempo, relacionadas con empeoramiento del dolor, el dolor moderado o severo, la mejoría en el dolor, y la proporción de sujetos que cumplieran estos criterios. En un análisis ad-hoc de los datos combinados, la mediana en el tiempo hasta el empeoramiento del dolor (> 4 puntos en la puntuación del peor dolor e incremento de ≥ 2 puntos respecto al valor basal) fue mayor para XGEVA en comparación con el ácido zoledrónico (65 contra 59 días y 181 contra 169 días, respectivamente). En un análisis adicional ad-hoc en un subgrupo de pacientes con dolor leve o sin dolor al inicio del estudio, el tiempo al empeoramiento del dolor (> 4 puntos en la puntuación de peor dolor) se retrasó en el grupo XGEVA en comparación con el grupo de tratamiento con ácido zoledrónico (198 contra 143 días). El tiempo para la mejoría del dolor (es decir, disminución de ≥ 2 puntos respecto al valor basal en la puntuación de BPI-SF) fue similar para denosumab y ácido zoledrónico en cada estudio y en el análisis integrado.
Supervivencia global y progresión de la enfermedad: La progresión de la enfermedad fue similar entre XGEVA y ácido zoledrónico en los tres estudios y en el análisis preespecificado de los tres estudios combinados. En los tres estudios, la supervivencia global estuvo equilibrada entre XGEVA y ácido zoledrónico en pacientes con neoplasias malignas avanzadas con afectación ósea: pacientes con cáncer de mama (tasa de riesgo e IC 95% fue de 0.95 [0.81, 1.11]), pacientes con cáncer de próstata (tasa de riesgo e IC 95% fue de 1.03 [0.91, 1.17]), y pacientes con otros tumores sólidos o mieloma múltiple (tasa de riesgo e IC 95% fue de 0.95 [0.83, 1.08]).
Además, un análisis ad-hoc en el Estudio 2 examinó la supervivencia global para los 3 tipos de tumor utilizado para la estratificación (cáncer de pulmón de células no pequeñas, mieloma múltiple, y otros). La supervivencia fue mayor para el grupo de tratamiento XGEVA en cáncer de pulmón de células no pequeñas (tasa de riesgo [IC 95%] de 0.79 [0.65, 0.95]; n = 702) y mayor para el grupo de tratamiento de ácido zoledrónico en mieloma múltiple (tasa de riesgo [IC 95%] de 2.26 [1.13, 4.50], n = 180) y similar entre los grupos de tratamiento de XGEVA y de ácido zoledrónico en la categoría de otros tipos de tumores (tasa de riesgo [IC 95%] de 1.08 (0.90, 1.30); n = 894). En este estudio no se controlaron los factores de pronóstico ni los tratamientos antineoplásicos para el mieloma múltiple.
Eficacia clínica en pacientes con mieloma múltiple:
En el Estudio 4, XGEVA se evaluó en un estudio internacional, aleatorizado (1:1), doble ciego, controlado con activo, que comparó XGEVA con ácido zoledrónico en pacientes con mieloma múltiple recién diagnosticado.
En este estudio, 1,718 pacientes con mieloma múltiple con al menos 1 lesión ósea fueron aleatorizados para recibir 120 mg de XGEVA por vía subcutánea cada 4 semanas o 4 mg de ácido zoledrónico por vía intravenosa (IV) cada 4 semanas (dosis ajustada para insuficiencia renal y se excluyeron pacientes con depuración de creatinina menor a 30 mL/min de acuerdo a la información para prescribir de Zometa). La medida de resultado primario fue la demostración de la no inferioridad del tiempo hasta el primer evento relacionado con el esqueleto (ERE) en comparación con el ácido zoledrónico. Las medidas del resultado secundario incluyeron la superioridad del tiempo hasta el primer ERE, la superioridad del tiempo hasta el primer ERE y posterior, y la supervivencia global. Se definió un ERE como cualquiera de los siguientes: fractura patológica (vertebral o no vertebral), radioterapia a los huesos (incluido el uso de radioisótopos), cirugía a los huesos o compresión de la médula espinal.
En este estudio, la aleatorización se estratificó por la intención de someterse a un trasplante autólogo de células madre de sangre periférica (PBSC) (sí o no), el agente antimieloma utilizado/planeado para utilizarse en terapia de primera línea (terapia basada en innovador o terapia no-basada en innovador [terapias innovadoras incluyen bortezomib, lenalidomida o talidomida]), etapa en el momento del diagnóstico (Sistema internacional de estadificación [ISS] I o II o III), ERE previo (sí o no) y región (Japón u otros países). A través de ambos brazos del estudio, 54.5% de los pacientes tenían la intención de someterse a un trasplante de PBSC autólogo, el 95.8% de los pacientes utilizaron/planificaron utilizar un nuevo agente antimieloma en el tratamiento de primera línea y el 60.7% de los pacientes tenían un antecedente de ERE previo. El número de pacientes a través de ambos brazos del estudio con ISS estadio I, estadio II y estadio III en el momento del diagnóstico fue 32.4%, 38.2% y 29.3%, respectivamente.
La mediana de edad fue de 63 años, el 82.1% de los pacientes eran blancos y el 45.6% de los pacientes fueron mujeres. La mediana del número de dosis administradas fue 16 para XGEVA y 15 para ácido zoledrónico.
En pacientes con mieloma múltiple recién diagnosticado, XGEVA no fue inferior al ácido zoledrónico para retrasar el tiempo hasta el primer ERE después de la aleatorización (ver Figura 1 y Tabla 2).
Figura 1: Gráfica de Kaplan-Meier para el tiempo hasta el primer ERE en Estudio
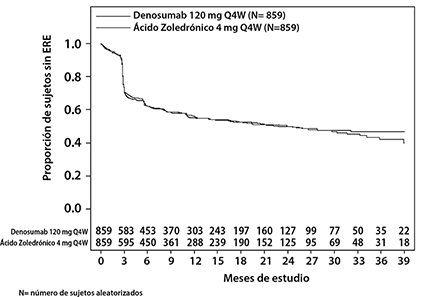
Tabla 2. Resultados de eficacia para XGEVA en comparación con ácido zoledrónico en pacientes con mieloma múltiple recién diagnosticado
|
XGEVA (N = 859) |
Ácido zoledrónico (N = 859) |
|
|---|---|---|
|
Primer ERE |
||
|
Número de pacientes que tuvieron ERE (%) |
376 (43.8) |
383 (44.6) |
|
Mediana de tiempo para ERE (meses) |
22.8 (14.7, NE) |
23.98 (16.56, 33.31) |
|
Tasa de riesgo (95% IC) |
0.98 (0.85, 1.14) |
|
|
No inferioridad valor-p |
0.010 |
|
|
Superioridad valor-p* |
0.84 |
|
|
Componentes del primer ERE |
||
|
Radioterapia a hueso |
47 (5.5) |
62 (7.2) |
|
Fractura patológica |
342 (39.8) |
338 (39.3) |
|
Cirugía de hueso |
37 (4.3) |
48 (5.6) |
|
Compresión de la médula espinal |
6 (0.7) |
4 (0.5) |
|
Primer y subsecuente ERE |
||
|
Número promedio de eventos/paciente |
0.66 |
0.66 |
|
Proporción de tasa (95% IC) |
1.01 (0.89, 1.15) |
|
|
Superioridad valor-p* |
0.84 |
|
|
Tasa de morbilidad esquelética por año |
0.61 |
0.62 |
|
Primer ERE o HNM |
||
|
Tiempo medio (meses) |
22.14 (14.26, NE) |
21.32 (13.86, 29.7) |
|
Tasa de riesgo (IC 95%) |
0.98 (0.85, 1.12) |
|
|
Valor de p |
0.71 |
|
|
Primera radiación a hueso |
||
|
Tiempo medio (meses) |
NE (NE, NE) |
NE (NE, NE) |
|
Tasa de riesgo (IC 95%) |
0.78 (0.53, 1.14) |
|
|
Valor de p |
0.19 |
|
NE = no estimable.
HCM = hipercalcemia maligna.
ERE = evento relacionado con el esqueleto.
IC = intervalo de confianza.
* Valor-p ajustado presentado.
Efecto sobre el dolor: Para las medidas de dolor basadas en BPI-SF, la estimación puntual (IC 95%) del área promedio bajo la curva (ABC) del peor dolor, con respecto a la línea basal, fue -1.04 (-1.32, -0.77) para XGEVA y -0.69 (-0.95, -0.43) para el ácido zoledrónico con una estimación puntual (IC 95%) para la diferencia de tratamiento de -0.35 (-0.73, 0.03) y p = 0.072, y la estimación puntual (IC 95%) del ABC promedio del puntaje de severidad del dolor, con relación al valor basal, fue -0.72 (-0.92, -0.51) para XGEVA y -0.40 (-0.59, -0.20) para el ácido zoledrónico, con una estimación puntual (IC 95%) para la diferencia de tratamiento de -0.32 (-0.60, -0.04) y p = 0.024, otras medidas mostraron resultados similares entre XGEVA y ácido zoledrónico. XGEVA y el ácido zoledrónico mostraron resultados similares en el tiempo para, y en la proporción, por visita para una disminución de ≥ 2 puntos, un aumento de ≥ 2 puntos y > 4 puntos en la puntuación de dolor más alta.
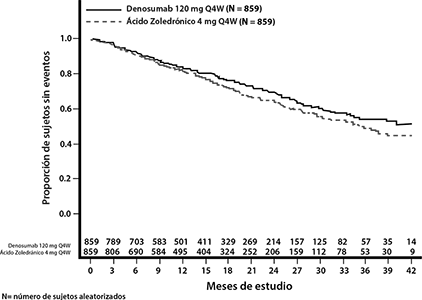
Supervivencia global y supervivencia libre de progresión: La proporción de riesgo entre los grupos de tratamiento con XGEVA y con ácido zoledrónico e IC del 95% para la supervivencia global (SG) fue de 0.90 (0.70, 1.16) (ver Figura 2). La mediana de supervivencia libre de progresión (SLP) (IC 95%) fue 46.1 (34.3, no estimable) meses para el grupo de tratamiento XGEVA y 35.4 (30.2, no estimable) meses para el grupo de ácido zoledrónico (HR [IC 95%] de 0.82 [0.68, 0.99]; valor de p (multiplicidad no ajustada) = 0.036) (ver Figura 3).
Figura 2. Supervivencia global
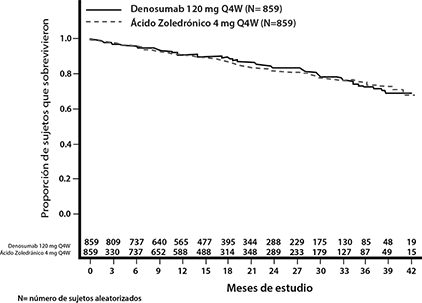
Figura 3. Supervivencia libre de progresión
Tratamiento de tumor óseo de células gigantes en adultos o adolescentes con estructura ósea completamente madura: Se estudió la seguridad y la eficacia de XGEVA en dos estudios fase 2, diseño abierto, de un solo brazo (estudios 5 y 6) que reclutaron a 554 pacientes con tumor óseo de células gigantes no resecable o cuya cirugía estaría asociada con morbilidad grave. Los pacientes recibieron 120 mg vía subcutánea de XGEVA cada 4 semanas con una dosis adicional de 120 mg los días 8 y 15. Los pacientes que suspendieron XGEVA entraron en la fase de seguimiento de seguridad durante un mínimo de 60 meses. Se permitió el retratamiento con XGEVA durante el seguimiento de seguridad para los sujetos que inicialmente demostraron una respuesta a XGEVA (por ejemplo, en caso de enfermedad recurrente).
El estudio 5 reclutó a 37 pacientes adultos con tumor óseo de células gigantes confirmado histológicamente, no resecable o recurrente. El criterio de evaluación principal fue la tasa de respuesta, definida como eliminación de al menos 90% de las células gigantes respecto a la basal (o la eliminación completa de células gigantes en casos donde estas representaran < 5% de las células tumorales), o la falta de progresión de la lesión objetivo mediante mediciones radiográficas en casos donde no estuviera disponible la histopatología. De los 35 pacientes incluidos en el análisis de eficacia, 85.7% (IC 95%: 69.7, 95.2) tuvieron respuesta al tratamiento con XGEVA. Los 20 pacientes (100%) con evaluaciones histológicas cumplieron los criterios de respuesta. De los 15 pacientes restantes, 10 (67%) cumplieron los criterios de respuesta con base en los datos radiológicos.
El estudio 6 reclutó a 535 pacientes adultos o adolescentes con estructura ósea completamente desarrollada con tumor óseo de células gigantes. De estos pacientes, 28 tenían entre 12 a 17 años de edad (ver Dosis y vía de administración: Poblaciones). Los pacientes se asignaron a una de tres cohortes: la cohorte 1 incluyó sujetos con enfermedad irresecable quirúrgicamente (p. ej., en sacro, columna, o lesiones múltiples, incluyendo metástasis pulmonares); la cohorte 2 incluyó sujetos con enfermedad resecable quirúrgicamente cuya cirugía planeada estaba asociada con morbilidad grave (p. ej., resección de articulación, amputación de miembro o hemipelvectomía); y la cohorte 3 incluyó pacientes que participaron en el estudio 5 y posteriormente continuaron en este estudio. Los criterios de evaluación de resultados fueron el tiempo a la progresión de la enfermedad (con base en la evaluación del investigador) para la cohorte 1, y la proporción de sujetos sin cirugía al mes 6 para la cohorte 2.
Un análisis interino retrospectivo incluyó una revisión independiente de los datos de imágenes radiográficas para los pacientes en los estudios 5 y 6. De los 305 pacientes en el momento del análisis interino inscritos en los estudios 5 y 6, 190 tuvieron al menos una respuesta puntual evaluable y fueron incluidos en el análisis (ver tabla 3).
Tabla 3. Respuesta tumoral objetivo en pacientes con tumor óseo de células gigantes
|
Número de pacientes evaluables para el criterio primario de evaluación |
Número de pacientes que cubrió criterio primario de evaluación |
Proporción(%) (IC 95%)a |
KM estimado de la mediana (IC 95%) meses) |
|
|
Proporción de pacientes con respuesta tumoral objetivo (RC, RP) |
||||
|
Basado en mejor respuesta |
190 |
136 |
71.6 (64.6, 77.9) |
– |
|
RECIST 1.1 |
187 |
47 |
25.1 (19.1, 32.0) |
– |
|
EORTC |
26 |
25 |
96.2 (80.4, 99.9) |
– |
|
Densidad/tamaño |
176 |
134 |
76.1 (69.1, 82.2) |
– |
|
Duración de la respuesta tumoral objetivo (tiempo a la progresión desde la primera respuesta tumoral objetivo) |
||||
|
Basado en mejor respuesta |
136 |
1 |
0.7 |
NE (NE, NE)b |
|
RECIST 1.1 |
47 |
3 |
6.4 |
NE (19.94, NE) |
|
EORTC |
25 |
0 |
0.0 |
NE (NE, NE) |
|
Densidad/tamaño |
134 |
1 |
0.7 |
NE (NE, NE) |
|
Tiempo a la primera respuesta tumoral objetivo |
||||
|
Basado en mejor respuesta |
190 |
136 |
71.6 |
3.1 (2.89, 3.65) |
|
RECIST 1.1 |
187 |
47 |
25.1 |
NE (20.93, NE) |
|
EORTC |
26 |
25 |
96.2 |
2.7 (1.64, 2.79) |
|
Densidad/Tamaño |
176 |
134 |
76.1 |
3.0 (2.79, 3.48) |
a Intervalo de confianza exacto.
b NE = no estimable.
Los pacientes fueron evaluados por los siguientes criterios de respuesta para determinar la respuesta objetiva del tumor:
• Criterios de evaluación de respuesta modificada en tumores sólidos (RECIST 1.1) para evaluar la carga tumoral basada en tomografía computarizada (TC)/resonancia magnética (RM).
• Criterios modificados de la Organización Europea para la Investigación y Tratamiento del Cáncer (EORTC) para evaluar la respuesta metabólica mediante tomografía por emisión de positrones de fluorodeoxiglucosa (FDG-PET).
• Criterio de Choi inverso modificado para evaluar el tamaño y la densidad tumoral utilizando unidades de Hounsfield basadas en CT/MRI (densidad/tamaño).
En general, en este análisis provisional retrospectivo, XGEVA logró respuestas objetivas del tumor en el 71.6% (IC 95%: 64.6, 77.9) de los pacientes (consulte la Tabla 3). El tiempo medio de respuesta fue de 3.1 meses (IC 95%: 2.89, 3.65). La duración media de la respuesta no fue estimable, ya que pocos pacientes experimentaron progresión de la enfermedad, con una mediana de seguimiento de 13.4 meses. Los resultados de eficacia en adolescentes esqueléticamente maduros parecían ser similares a los observados en adultos.
En el análisis final, entre las cohortes 1 y 2 combinados, 500 de 501 sujetos evaluables (99.8%) (es decir, aquellos que tuvieron una evaluación del estado de la enfermedad posterior al inicio) tuvieron una mejor respuesta o enfermedad estable reportada por el investigador (RC en 195 sujetos [38.9%], RP en 161 sujetos [32.1%] y enfermedad estable en 144 sujetos [28.7%]).
En la Cohorte 1, al final del análisis, la mediana en el tiempo a la progresión de la enfermedad no se alcanzó, ya que solo 28 de 260 pacientes tratados (10.8%)tuvieron progresión de la enfermedad. En la Cohorte 2, XGEVA prolongó el tiempo a la cirugía, redujo la morbilidad en la cirugía planeada y redujo la proporción de pacientes que requirieron cirugía. 219 de los 238 (92.1%; IC 95%: 87.8%, 95.1%) pacientes evaluables tratados con XGEVA no habían sido sometidos a cirugía para el mes 6. De los 239 sujetos en la cohorte 2 con la localización de la lesión basal o la localización en estudio no en los pulmones o tejidos blandos, un total de 82 sujetos (34,3%) pudieron evitar la cirugía en el estudio. De los 157 sujetos que recibieron cirugía de GCTB en el estudio, 106 (67.5%) se sometieron a un procedimiento de menos morbilidad de lo planificado al inicio del estudio. (ver tabla 4).
Tabla 4. Distribución de la cirugía planificada versus la cirugía real en pacientes con tumor de células gigantes del hueso (cohorte 2)
|
Procedimiento quirúrgico, n |
Planeación basal (N = 239) |
Total actual (N = 239) |
|
Todas las cirugías |
239 |
157 |
|
Cirugías mayores |
109 |
18 |
|
Hemipelvectomía |
11 |
1 |
|
Amputación |
36 |
2 |
|
Reemplazo articular/prótesis |
27 |
11 |
|
Resección articular |
35 |
4 |
|
Escisión marginal, escisión en bloque o resección en bloque |
95 |
42 |
|
Legrado |
29 |
95 |
|
Otro |
6 |
2 |
|
Sin cirugía |
0 |
82 |
N= número de sujetos de la cohorte 2 en el grupo de análisis de eficacia, excluyendo a aquellos con una línea basal de la lesión diana en el pulmón/tejido blando o en la ubicación de la cirugía en el estudio en el pulmón/tejido blando.
En el análisis final, los cohortes 1 y 2 combinados, se informó una reducción clínicamente significativa en el peor dolor (es decir, una disminución de ≥2 puntos con respecto al valor basal) en el 30.8% de los pacientes en riesgo (es decir, aquellos que tuvieron una peor puntuación de dolor de ≥2 al inicio del estudio) dentro de 1 semana de tratamiento, y ≥50% en la semana 5. Estas mejoras de dolor se mantuvieron en todas las evaluaciones posteriores.
Efecto sobre el dolor: En las cohortes 1 y 2 combinadas del estudio 6, se reportó una reducción clínica significativa en el peor dolor (por ejemplo, disminución ≥ 2 puntos desde la basal) en 31.4% de los pacientes en riesgo (por ejemplo, aquellos con una puntuación del peor dolor ≥ 2 en la basal) en 1 semana de tratamiento y ≥ 50% en la semana 5. Esta mejoría en el dolor se mantuvo en todas las evaluaciones subsecuentes. En un análisis post hoc, al menos la mitad de los pacientes evaluables tuvieron una reducción ≥ 30% en la puntuación del peor dolor desde la basal en todos los puntos de tiempo posbasal, iniciando en la semana 9.
CONTRAINDICACIONES: Hipersensibilidad al biofármaco denosumab o a cualquier componente de XGEVA, embarazo y la lactancia, hipocalcemia severa preexistente sin tratamiento, lesiones por procedimientos dentales invasivos sin cicatrizar (extracción dental) o cirugía oral.
XGEVA no está recomendado en niños menores de 18 años excepto en aquellos adolescentes con esqueleto maduro para el tratamiento del tumor óseo de células gigantes que es irresecable.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: No se ha establecido la seguridad ni la eficacia de XGEVA en mujeres embarazadas. XGEVA no está recomendado para su uso en mujeres embarazadas. Se debe aconsejar a las mujeres no embarazarse durante y por al menos 5 meses después del tratamiento con XGEVA.
A exposiciones de ABC hasta 16 veces más altas que la exposición en humanos (120 mg una vez cada 4 semanas), denosumab no mostró evidencia de alteración en la fertilidad en monos Cynomolgus hembras (ver Datos de Seguridad Preclínica).
En un estudio en monos Cynomolgus que recibieron una dosis de denosumab durante el periodo equivalente al primer trimestre, con exposiciones de ABC hasta 10 veces mayores que la dosis en humanos (120 mg cada 4 semanas), no hubo evidencia de daño materno o fetal. En este estudio, no se examinaron los ganglios linfáticos del feto.
En otro estudio, en monos Cynomolgus que recibieron denosumab durante el embarazo a niveles de exposición de ABC 12 veces superior a la dosis utilizada en humanos (120 mg cada 4 semanas), hubo un incremento en mortinatos y en la mortalidad posnatal; crecimiento anormal del hueso resultando en una reducción de la resistencia ósea, reducción en hematopoyesis y mala alineación dental; ausencia de ganglios linfáticos periféricos y disminución del crecimiento del recién nacido. No hubo evidencia de daño materno antes del parto; los efectos adversos maternos fueron poco frecuentes durante el parto. El desarrollo de la glándula mamaria materna fue normal.
Los estudios en ratones knockout sugieren que la ausencia de RANKL podría interferir con la maduración de la glándula mamaria materna provocando alteración de la lactancia posparto.
Lactancia: Se desconoce si denosumab se excreta en la leche humana. Debido a que denosumab tiene el potencial de causar reacciones adversas en los lactantes, debe tomarse una decisión con relación a si se interrumpe la lactancia o se descontinúa el medicamento.
REACCIONES SECUNDARIAS Y ADVERSAS:
Las reacciones adversas identificadas en los ensayos clínicos y en la experiencia poscomercialización con XGEVA se presentan en la tabla a continuación:
Muy común: ≥ 1 en 10.
Común: ≥ 1 en 100 y < 1 en 10.
Poco común: ≥ 1 en 1,000 y < 1 en 100.
Rara: ≥ 1 en 10,000 y < 1 en 1,000.
Muy rara: < 1/10,000.
Dentro de cada grupo de frecuencia y de la clasificación por órganos o sistemas, las reacciones adversas se presentan en orden de gravedad decreciente.
|
Clasificación por órgano o sistema |
Categoría de frecuencia |
Reacción adversa |
|---|---|---|
|
Trastorno del sistema inmune |
Raro |
Hipersensibilidad al fármacoa |
|
Trastornos metabólicos y nutricionales |
Muy común |
Hipocalcemiaa,b |
|
Común |
Hipofosfatemia |
|
|
Poco común |
Hipercalcemia después de la descontinuación del tratamiento en pacientes con tumor de células gigantes de huesob |
|
|
Raro |
Hipercalcemia después de la descontinuación del tratamiento en pacientes con esqueleto en crecimientob |
|
|
Trastornos respiratorios, torácicos y mediastinales |
Muy común |
Disnea |
|
Trastornos músculo esqueléticos y del tejido conectivo |
Muy común |
Dolor musculoesqueléticoa |
|
Común |
Osteonecrosis de la mandíbula (ONM)a,b |
|
|
Poco común |
Fractura femoral atípica (AFF)b |
|
|
Raro |
Múltiples fracturas vertebrales después de la descontinuación del tratamientob |
|
|
Trastornos de la piel y del tejido subcutáneo |
Común |
Alopecia |
|
Poco común |
Dermatitis liquenoidea |
a Ver descripción de reacciones adversas seleccionadas.
b Ver precauciones generales.
Descripción de reacciones adversas seleccionadas:
Eventos de hipersensibilidad al fármaco:
En ensayos clínicos en pacientes con tumores malignos avanzados que involucran hueso, rara vez se informaron eventos de hipersensibilidad al fármaco en sujetos tratados con XGEVA. En el entorno posterior a la comercialización, se ha reportado hipersensibilidad, incluyendo reacciones anafilácticas.
Hipocalcemia:
En el entorno posterior a la comercialización, se ha informado de hipocalcemia sintomática severa (incluidos casos mortales).
Dolor musculoesquelético:
En el entorno posterior a la comercialización, se ha informado dolor musculoesquelético, incluidos casos severos.
Osteonecrosis de la mandíbula (ONM): En tres estudios clínicos fase 3 controlados con activo en pacientes con malignidades avanzadas y afectación ósea, la ONM fue confirmada en 1.8% de los pacientes en el grupo de XGEVA (mediana en exposición de 12.0 meses; rango 0.1 a 40.5) y 1.3% en el grupo de pacientes con ácido zoledrónico. Los ensayos en pacientes con cáncer de mama o próstata incluyeron una fase de extensión del tratamiento con XGEVA (mediana de la exposición general de 14.9 meses; rango 0.1 a 67.2) (ver Estudios clínicos). La incidencia ajustada por paciente-año de ONM confirmada fue de 1.1 por 100 pacientes-año durante el primer año de tratamiento, 3.7 en el segundo año y 4.6 por año posteriormente. La mediana en el tiempo a la presentación de ONM fue de 20.6 meses (rango: 4 a 53).
En un ensayo clínico de fase 3 doble ciego, controlado con activo, en pacientes con mieloma múltiple recién diagnosticado, se confirmó ONM en 4.1% de los pacientes en el grupo XGEVA (mediana de exposición de 15.8 meses; rango de 1 a 49.8) y 2.8% de los pacientes en el grupo de ácido zoledrónico. Al completar la fase de tratamiento doble ciego del ensayo, la incidencia ajustada por año-paciente de ONM confirmada en el grupo XGEVA (mediana de exposición de 19.4 meses, rango de 1 a 52) fue 2.0 para 100 pacientes-año durante el primer año de tratamiento, 5.0 en el segundo año y 4.5 de ahí en adelante. La mediana del tiempo hasta la ONM fue de 18.7 meses (rango: 1 a 44).
En un estudio clínico fase 3 controlado con placebo con una fase de tratamiento de extensión para evaluar XGEVA en la prevención de metástasis óseas en pacientes con cáncer de próstata no metastásico (una población de pacientes para los cuales no está indicado XGEVA), con mayor exposición al tratamiento de hasta 7 años, la incidencia ajustada paciente-año para ONM confirmada fue de 1.1 por 100 paciente-años durante el primer año de tratamiento, 3.0 en el segundo año, y 7.1 de ahí en adelante.
En un estudio clínico abierto fase 2 en pacientes con tumor óseo de células gigantes, se confirmó la ONJ en el 6.8% de los pacientes (mediana del número de 34 dosis; rango 4-116). Al finalizar el estudio, la mediana de tiempo en el estudio, incluida la fase de seguimiento de la seguridad, fue de 60.9 meses (rango: 0-112.6). La incidencia ajustada por paciente-años de ONJ confirmada fue de 1.5 por 100 paciente-años en general (0.2 por 100 paciente-años durante el primer año de tratamiento, 1.5 en el segundo año, 1.8 en el tercer año, 2.1 en el cuarto año, 1.4 en el quinto año, y 2.2 a partir de entonces). La mediana de tiempo hasta ONJ fue de 41 meses (rango: 11-96).
Fractura femoral atípica: En el programa de estudios clínicos, la fractura femoral atípica se ha notificado de manera poco común en pacientes tratados con XGEVA 120 mg y el riesgo aumentó con una mayor duración del tratamiento. Han ocurrido eventos durante el tratamiento y hasta 9 meses después de interrumpirlo.
Dermatitis liquenoide: En la experiencia posterior a la comercialización, se han observado erupciones de medicamentos liquenoides (por ejemplo, reacciones similares a liquen plano).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Datos de seguridad preclínica/toxicología no clínica:
Carcinogenicidad: El potencial carcinogénico de denosumab no se ha evaluado en estudios en animales a largo plazo.
Mutagenicidad: El potencial genotóxico de denosumab no ha sido evaluado.
Toxicología reproductiva:
Fertilidad: Denosumab no tuvo efecto sobre la fertilidad femenina o en los órganos reproductivos masculinos en monos a exposiciones que fueron 9.5 a 16 veces mayores, respectivamente, que la exposición en humanos de 120 mg SC administrados una vez cada 4 semanas.
Farmacología animal: Denosumab ha demostrado ser un potente inhibidor de la resorción ósea por inhibición del ligando RANK.
Debido a que la actividad biológica de denosumab en animales es específica para primates no humanos, la evaluación de ratones con ingeniería genética (knockout) o el uso de otros inhibidores biológicos de la vía RANK/RANKL, tales como OPG-Fc y RANK-Fc, se utilizaron para evaluar las propiedades farmacodinámicas de denosumab en modelos de roedores. En el modelo de ratón de metástasis ósea de cáncer de mama receptor estrogénico positivo o negativo, cáncer de próstata y cáncer pulmonar de células no pequeñas, OPG-Fc redujo las lesiones osteolíticas, osteoblásticas y osteolíticas/osteoblásticas, retardando la formación de metástasis óseas de novo, y con reducción del tamaño del tumor óseo. Se observó una inhibición aditiva del crecimiento del tumor óseo cuando se combinó OPG-Fc con terapia hormonal (tamoxifeno) en los modelos de cáncer de mama o quimioterapia (docetaxel) en cáncer de próstata y de pulmón. En un modelo de ratón de inducción de tumor mamario, RANK-Fc retardó la formación del tumor.
En primates adolescentes dosificados con denosumab a 15 veces (50 mg/kg dosis) y 2.8 veces (10 mg/kg dosis) el área bajo la curva (ABC) de exposición en humanos adultos con dosis subcutáneas de 120 mg cada 4 semanas, se observaron placas de crecimiento anormal, considerado como consistente con la actividad farmacológica de denosumab.
En monos Cynomolgus neonatos expuestos en útero a denosumab a dosis de 50 mg/kg, hubo mortalidad posnatal; el crecimiento anormal del hueso dio como resultado reducción de la resistencia ósea, reducción en hematopoyesis, y mala alineación dental; ausencia de ganglios linfáticos periféricos y disminución del crecimiento del recién nacido. Posterior a un periodo de recuperación del nacimiento hasta los 6 meses de edad, los efectos en hueso regresaron a la normalidad; no hubo efectos adversos durante la erupción dental.
Se observó una mineralización de mínima a moderada en múltiples tejidos en un animal en recuperación. El desarrollo de la glándula mamaria materna fue normal.
Los estudios de distribución en tejidos indican que denosumab no se une a tejidos con expresión conocida de otros miembros de la superfamilia de TNF, incluyendo el ligando inductor de apoptosis relacionado con TNF (TRAIL).
Los ratones knockout con carencia de RANK o RANKL (1) tuvieron ausencia de la lactancia debido a inhibición de la maduración de la glándula mamaria (desarrollo de la glándula lóbulo-alveolar durante el embarazo) (2) mostraron alteración en la formación de nódulos linfáticos (3) exhibieron reducción en el crecimiento óseo, alteración de las placas de crecimiento y falta de erupción de los dientes. La reducción en el crecimiento óseo, la alteración de placas de crecimiento y la alteración en la erupción de los dientes se observaron en estudios con ratas neonatas en quienes se administraron inhibidores de RANKL, y estos cambios fueron parcialmente reversibles cuando se discontinuaron los inhibidores RANKL (ver Restricciones de uso durante el embarazo y la lactancia).
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se han realizado estudios formales de interacción fármaco-fármaco con XGEVA.
En los ensayos clínicos, XGEVA se ha administrado en combinación con el tratamiento estándar contra el cáncer y en pacientes que previamente recibieron bifosfonatos (ver Farmacocinética y Farmacodinamia: Estudios clínicos). La farmacocinética y la farmacodinamia de denosumab no se alteraron por la quimioterapia concomitante y/o la terapia hormonal ni por la exposición previa a bifosfonatos por vía intravenosa.
Incompatibilidades: Debido a que no hay estudios de compatibilidad, XGEVA no debe ser mezclado con otros fármacos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No se conocen.
PRECAUCIONES GENERALES:
Hipocalcemia: La hipocalcemia preexistente debe ser corregida antes de iniciar el tratamiento con XGEVA. La suplementación de calcio y vitamina D es necesaria en todos los pacientes, a menos que exista hipercalcemia. La hipocalcemia puede ocurrir durante el tratamiento con XGEVA. Se recomienda realizar la monitorización de los niveles de calcio durante el tratamiento, especialmente en las primeras semanas de haber iniciado la terapia.
En el periodo poscomercialización se ha reportado hipocalcemia sintomática severa (ver Reacciones secundarias y adversas). Si se produce hipocalcemia, puede ser necesaria una suplementación a corto plazo con calcio (ver Dosis y vía de administración: Poblaciones y Reacciones secundarias y adversas).
Osteonecrosis de la mandíbula (ONM): La ONM ha ocurrido en pacientes tratados con denosumab. En estudios clínicos, la incidencia de ONM fue más alta a mayor duración de la exposición (ver Reacciones secundarias y adversas).
Una higiene bucal deficiente, procedimientos dentales invasivos (por ejemplo, extracción dental), tratamiento con medicamentos antiangiogénicos, infección local de las encías o boca fueron factores para ONM en pacientes que recibían XGEVA en estudios clínicos.
Se recomienda un examen dental con odontología preventiva adecuada antes del tratamiento con XGEVA, especialmente en pacientes con factores de riesgo para ONM. Se deben mantener buenas prácticas de higiene bucal durante el tratamiento con XGEVA.
Evite procesos dentales invasivos durante el tratamiento con XGEVA. Para los pacientes en los que los procedimientos dentales invasivos no pueden ser evitados, el juicio clínico del médico tratante debe guiar el plan de manejo de cada paciente con base en la evaluación beneficio-riesgo individual.
Los pacientes en quienes se sospecha que tienen o que desarrollan osteonecrosis de la mandíbula, mientras están con XGEVA deben recibir atención de un dentista o de un cirujano maxilofacial. En los pacientes que desarrollan ONM durante el tratamiento con XGEVA, se debe considerar la interrupción temporal del tratamiento con base en la evaluación individual del beneficio-riesgo hasta que la condición se resuelva.
Fracturas femorales atípicas: Se han reportado fracturas femorales atípicas con el uso de XGEVA. Las fracturas femorales atípicas pueden ocurrir con poco o ningún trauma en las regiones subtrocantéricas y diafisarias del fémur y pueden ser bilaterales. Los hallazgos radiológicos específicos caracterizan estos eventos. También se han reportado fracturas femorales atípicas en pacientes con ciertas condiciones comórbidas (ejemplo, deficiencia de vitamina D, artritis reumatoide, hipofosfatasia) y con el uso de ciertos agentes farmacéuticos (por ejemplo, bifosfonatos, glucocorticoides, inhibidores de la bomba de protones). Estos eventos también han ocurrido sin tratamiento antiresortivo. Durante el tratamiento con XGEVA los pacientes deben ser advertidos para que reporten cualquier dolor nuevo o inusual en muslo, cadera o ingle. Los pacientes que presenten estos síntomas, deberán ser evaluados para descartar una fractura femoral incompleta y el fémur contralateral también deberá ser examinado.
Hipercalcemia posterior a la descontinuación del tratamiento en pacientes con tumor óseo de células gigantes y en pacientes con esqueleto en crecimiento: XGEVA no está recomendado en pacientes con esqueleto en crecimiento. Se ha reportado hipercalcemia clínicamente significativa que requiere hospitalización y complicada por lesión renal aguda en pacientes tratados con XGEVA con tumor óseo de células gigantes con esqueleto en crecimiento, semanas a meses posteriores a la descontinuación del tratamiento. Después de descontinuar el tratamiento, monitorizar a los pacientes para detectar signos y síntomas de hipercalcemia, considerar la evaluación periódica del calcio sérico según lo indicado clínicamente, y reevaluar los requerimientos de suplementación de calcio y vitamina D de los pacientes. Tratar la hipercalcemia según sea clínicamente apropiado (ver Dosis y vía de Administración: Poblaciones y Reacciones secundarias y adversas).
Fracturas vertebrales múltiples (FVM) después de la descontinuación del tratamiento: Las fracturas vertebrales múltiples (FVM), no debidas a metástasis óseas, pueden ocurrir después de la descontinuación del tratamiento con XGEVA, particularmente en pacientes con factores de riesgo tales como osteoporosis o fracturas previas.
Recomiende a los pacientes no interrumpir el tratamiento con XGEVA sin el consejo de su médico. Cuando el tratamiento con XGEVA es descontinuado, evaluar el riesgo individual del paciente para fracturas vertebrales.
Medicamentos con el mismo ingrediente activo: XGEVA contiene el mismo principio activo que se encuentra en Prolia® (denosumab). Los pacientes que son tratados con XGEVA, no deben recibir Prolia.
Efectos sobre la capacidad para conducir y utilizar maquinaria: No se han realizado estudios del efecto sobre la capacidad para conducir o utilizar maquinaria pesada en pacientes que reciben denosumab.
Pacientes con fenilcetonuria:
Cada 120 mg/1.0 mL de solución de XGEVA en una jeringa prellenada para una dosis única contiene 6.1 mg de fenilalanina. La fenilalanina puede ser dañina para los pacientes con fenilcetonuria (FCU), un trastorno genético poco común en el que se desarrolla fenilalanina porque el cuerpo no la puede eliminar de manera adecuada.
La solución de XGEVA de 120 mg/1.7 mL en un vial de dosis única no contiene fenilalanina. Se debe administrar XGEVA a los pacientes con FCU del vial para dosis única que contiene 120 mg en 1.7 mL de solución.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Administración:
Inyección subcutánea:
• Solución de XGEVA de 120 mg/1.7 mL en un vial para dosis única: la administración con el vial de 120 mg/1.7 mL debe realizarla un profesional de la salud.
• Solución de XGEVA de 120 mg/mL en una jeringa prellenada: la administración con la jeringa prellenada de 120 mg/mL puede realizarla el paciente o el cuidador que haya recibido entrenamiento en las técnicas para aplicar inyecciones por parte de un profesional de la salud. La primera autoadministración con la jeringa prellenada de XGEVA debe ser supervisada por un profesional de la salud.
La administración a pacientes menores de 18 años de edad debe realizarla un profesional de la salud o un cuidador entrenado. Los pacientes deben recibir suplementos adecuados de calcio y vitamina D. No se requieren ajustes en dosis en pacientes con insuficiencia renal.
Dosis:
Eventos relacionados con el esqueleto (EREs): La dosis recomendada de XGEVA es de 120 mg administrada por inyección subcutánea una vez cada 4 semanas en el muslo, abdomen o la parte superior del brazo.
Tumor óseo de células gigantes: La dosis recomendada de XGEVA es de 120 mg administrada mediante inyección subcutánea una vez cada 4 semanas en el muslo, el abdomen o la parte superior del brazo, con una dosis adicional de 120 mg los días 8 y 15 del tratamiento.
Poblaciones:
Pacientes pediátricos: No se ha establecido la seguridad y eficacia de XGEVA en pacientes pediátricos (de 12 a 17 años de edad) que no sean los pacientes con estructura ósea completamente madura con tumor óseo de células gigantes.
XGEVA no está recomendado para su uso en pacientes pediátricos (de 12 a 17 años de edad) que no sean aquellos con esqueleto maduro con tumor óseo de células gigantes. Se ha reportado hipercalcemia clínicamente significativa después de la descontinuación del tratamiento en el entorno posterior a la comercialización en pacientes pediátricos con esqueletos en crecimiento que recibieron denosumab para tumor óseo de células gigantes o para indicaciones no aprobadas (ver Precauciones generales: Hipercalcemia posterior a la descontinuación del tratamiento en pacientes con tumor óseo de células gigantes y en pacientes con esqueleto en crecimiento).
Se estudió XGEVA en un estudio abierto Fase 2 que reclutó a un subgrupo de 28 pacientes pediátricos (entre 12 y 17 años) con tumor óseo de células gigantes con estructura ósea completamente desarrollada definida como al menos 1 hueso largo maduro (por ejemplo, placa de crecimiento epifisiaria cerrada del húmero) y peso corporal ≥ 45 kg (ver Indicaciones terapéuticas y Farmacocinética y farmacodinamia: Estudios clínicos).
Pacientes geriátricos: No se observaron diferencias generales en la seguridad o la eficacia entre los pacientes de más edad y los pacientes más jóvenes. Los estudios clínicos controlados de XGEVA en el tratamiento de mieloma múltiple y metástasis ósea de tumores sólidos en pacientes mayores de 65 años revelaron una eficacia y seguridad similares entre los pacientes mayores y los más jóvenes (ver Farmacocinética y farmacodinamia: Poblaciones Especiales de Pacientes).
Insuficiencia renal: No se requieren ajustes de dosis en pacientes con insuficiencia renal. No es necesario monitorear la función renal con XGEVA (ver Farmacocinética y farmacodinamia: Poblaciones Especiales de Pacientes).
En estudios clínicos de pacientes sin cáncer avanzado con grados variables de función renal (incluyendo pacientes con insuficiencia renal severa [depuración de creatinina < 30 mL/min] o en diálisis), hubo un mayor riesgo de desarrollar hipocalcemia con el incremento en el grado de la insuficiencia renal y en ausencia de suplementos de calcio. La monitorización de los niveles de calcio y la ingesta adecuada de calcio y vitamina D es importante en pacientes con insuficiencia renal grave o que reciben diálisis (ver Precauciones generales).
Insuficiencia hepática: No se realizaron estudios clínicos en sujetos con insuficiencia hepática.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No hay experiencia de sobredosis en los estudios clínicos en humanos.
PRESENTACIÓN: Caja con un frasco ámpula con 120 mg de denosumab en 1.7 mL de solución.
Caja con una jeringa prellenada con 120 mg de denosumab en 1 mL de solución.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Almacenar en refrigeración (2 a 8 °C).
No congelar.
Conservar el frasco ámpula en su caja exterior para protegerlo de la luz directa.
No agitar.
Si se retira de la refrigeración, XGEVA se debe mantener a temperatura ambiente (hasta 25 °C) en la caja de cartón original y debe ser usado en los siguientes 30 días. Si no se utiliza dentro de un plazo de 30 días, XGEVA debe desecharse.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Mantener fuera del alcance de los niños. Su venta requiere receta médica. No se use durante el embarazo y la lactancia. Medicamento de alto riesgo.
Reporte las sospechas de reacción adversa al correo electrónico: farmacovigilancia@cofepris.gob.mx y
farmacovigilanciamx@amgen.com
Titular del Registro:
Amgen Manufacturing Limited.
State Road 31, Km 24.6, Juncos, 00777-4060,
Puerto Rico, EUA.
Representante Legal:
AMGEN MÉXICO, S.A. de C.V.
Av. Vasco de Quiroga No. 3000, piso 4,
Col. Santa Fe, C.P. 01210,
Álvaro Obregón, Ciudad de México, México.
Reg. Núm. 014M2012, SSA IV
®Marca registrada