XULTOPHY - Solución inyectable
Sustancia(s):
- Insulina Degludec, Liraglutida (adn Recombinante)
Presentaciones:
- 1 Caja, 1 Pluma precargada, 3 mL,
- 1 Caja, 3 Pluma precargada, 3 mL,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada mL contiene:
Insulina Degludec* 100 Unidades
Liraglutida** 3.6 mg
Vehículo cbp 1 mL
*Insulina de origen ADN recombinante expresado en Saccharomyces cerviseae.
**Análogo del GLP-1 humano de origen ADN recombinante expresado en Saccharomyces cerviseae.
Una unidad de dosis contiene 1 unidad de insulina degludec y 0.036 mg de liraglutida.
Una pluma precargada contiene 3 mL, equivalentes a 300 unidades de insulina degludec y 10.8 mg de liraglutida.
INDICACIONES TERAPÉUTICAS: XULTOPHY® está indicado para el tratamiento de adultos con diabetes mellitus tipo 2, para mejorar el control glucémico en combinación con medicamentos como metformina o tiazolidinedionas, cuando éstos solos o en combinación con un agonista del receptor GLP-1 o insulina basal, no proporcionan un control glucémico adecuado.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades Farmacocinéticas: En general, la farmacocinética de la insulina degludec y liraglutida no se vieron afectadas de un modo clínicamente relevante al administrarse como XULTOPHY® en comparación con inyecciones independientes de insulina degludec y liraglutida.
A continuación se indican las propiedades farmacocinéticas de XULTOPHY® a menos que se especifique que los datos presentados se refieren a la administración de insulina degludec o liraglutida solas.
Absorción: La exposición total de insulina degludec fue equivalente tras la administración de XULTOPHY® en comparación con insulina degludec sola, mientras la Cmax fue un 12% superior. La exposición total de liraglutida fue equivalente tras la administración de XULTOPHY® en comparación con liraglutida sola, mientras que la Cmax fue inferior un 23%. Las diferencias se consideran carentes de relevancia clínica ya que el tratamiento con XULTOPHY® se inicia y se ajusta de acuerdo con los objetivos de glucosa en sangre de cada paciente.
La exposición a insulina degludec y liraglutida aumentó proporcionalmente con la dosis de XULTOPHY® dentro de todo el rango de dosis, según un análisis farmacocinético de la población.
El perfil farmacocinético de XULTOPHY® es consistente con la administración una vez al día y la concentración en estado estacionario de insulina degludec y liraglutida se alcanza tras de 2-3 días de administración diaria.
Distribución: La insulina degludec y liraglutida se encuentran ampliamente ligadas a proteínas plasmáticas (> 99% y > 98%, respectivamente).
Biotransformación:
Insulina degludec: La degradación de la insulina degludec es similar a la insulina humana. Todos los metabolitos formados son inactivos.
Liraglutida: Durante las 24 horas tras la administración de una única dosis radiomarcada de [3H]-liraglutida a sujetos sanos, el componente mayoritario en el plasma fue liraglutida intacta.
Se detectaron dos metabolitos minoritarios en el plasma (≤ 9% y ≤ 5% de la exposición a radiactividad plasmática total). Liraglutida se metaboliza de un modo similar al de las grandes proteínas sin que se haya identificado un órgano específico como ruta principal de eliminación.
Eliminación: La vida media de la insulina degludec es aproximadamente de 25 horas y la vida media de liraglutida, de aproximadamente 13 horas.
Poblaciones Especiales:
Pacientes de edad avanzada: La edad no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de XULTOPHY® según los resultados de un análisis farmacocinético sobre una población compuesta por pacientes adultos de hasta 83 años tratados con XULTOPHY®.
Genéro: El género no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de XULTOPHY® basado en los resultados de un análisis farmacocinético de la población.
Origen étnico: El origen étnico no tuvo un efecto clínicamente relevante sobre la farmacocinética de XULTOPHY® basado en los resultados de un análisis farmacocinético de la población; incluyendo grupos de población Blanca, Negra, India, Asiática e Hispanoamericana.
Insuficiencia renal:
Insulina degludec: No hay diferencias en la farmacocinética de la insulina degludec entre sujetos sanos y pacientes con insuficiencia renal.
Liraglutida: La exposición a liraglutida disminuyó en sujetos con insuficiencia renal en comparación con los individuos con una función renal normal. La exposición a liraglutida disminuyó un 33%, un 14.%, un 27% y un 26% respectivamente, en sujetos con insuficiencia renal leve (aclaramiento de creatinina, CrCI 50-80 mL/min), moderada (CrCI 30-50 mL/min) y grave (CrCl<30 ml/min) y con enfermedad renal en etapa terminal con necesidad de diálisis, respectivamente.
Del mismo modo, en un ensayo clínico de 26 semanas, los pacientes con diabetes tipo 2 e insuficiencia renal moderada (CrCL 30-59 mL/min) tuvieron una exposición a liraglutida un 26% menor en comparación con un ensayo separado que incluyó pacientes con diabetes tipo 2 con una función renal normal o insuficiencia renal leve.
Insuficiencia hepática:
Insulina degludec: No hay diferencias en la farmacocinética de la insulina degludec entre sujetos sanos y pacientes con insuficiencia hepática.
Liraglutida: Se evaluó la farmacocinética de liraglutida en sujetos con diversos grados de insuficiencia hepática en un ensayo de dosis única. La exposición a liraglutida disminuyó un 13-23% en sujetos con insuficiencia hepática de leve a moderada en comparación con los sujetos sanos.
La exposición fue significativamente menor (44%) en sujetos con insuficiencia hepática grave (puntuación Child Pugh > 9).
Población pediátrica: No se han llevado a cabo estudios con XULTOPHY® en niños y adolescentes menores de 18 años. XULTOPHY® no debe administrarse en menores de 18 años de edad.
Farmacodinamia:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Fármacos utilizados en diabetes. Insulinas y análogos de acción prolongada para inyección.
Código ATC: A10AE56.
Mecanismo de acción: XULTOPHY® es un medicamento combinado que consta de insulina degludec y liraglutida, que tienen mecanismo de acción complementarios para mejorar el control glucémico.
La insulina degludec es una insulina basal que forma multihexámeros solubles cuando se inyecta por vía subcutánea, dando lugar a la formación de un depósito desde el que se absorbe a la circulación de forma continua y lenta, produciendo el efecto hipoglucemiante plano y estable de la insulina degludec con una baja variabilidad día a día en la acción insulínica.
La insulina degludec se une específicamente al receptor de la insulina humana y los resultados son los mismos efectos farmacológicos que la insulina humana.
El efecto hipoglucemiante de insulina degludec se debe a que facilita la absorción de la glucosa al unirse a los receptores de insulina en las células musculares y del tejido graso, y a que inhibe al mismo tiempo la producción hepática de la glucosa.
Liraglutida es un análogo del péptido similar al glucagón tipo 1 (GLP-1) con un 97% de homología de secuencia con el GLP-1 humano que se une al receptor de GLP-1 y lo activa.
Tras la administración subcutánea, el perfil de acción retardada se basa en tres mecanismos: autoasociación, que tiene como resultado una absorción lenta; unión a la albúmina y una estabilidad enzimática superior con respecto a la dipeptidil peptidasa IV (DPP-IV) y a la enzima endopeptidasa neutra (EPN), cuyo resultado es una vida media plásmática prolongada.
La acción de liraglutida es mediada a través de una interacción específica con los receptores de GLP- 1 y mejora el control glucémico al disminuir la glucosa en sangre posprandial y en ayunas. Liraglutida estimula la secreción de insulina y disminuye la secreción de glucagón inadecuadamente elevada, de un modo dependiente de la glucosa. De manera que cuando la glucosa en sangre es elevada, se estimula la secreción de insulina y se inhibe la secreción de glucagón. De forma contraria en la hipoglucemia liraglutida, disminuye la secreción de insulina y no afecta a la secreción de glucagón. El mecanismo hipoglucemiante también implica un retraso leve en el vaciamiento gástrico.
Liraglutida reduce el peso corporal y la masa grasa corporal mediante mecanismo que implican una reducción del apetito y de la ingesta calórica.
El GLP-1 es un regulador fisiológico del apetito y de la ingesta de alimentos, pero el mecanismo exacto de acción no está completamente claro. En estudios llevados a cabo con animales, la administración periférica de liraglutida supuso la absorción en regiones específicas del cerebro implicadas en la regulación del apetito, donde liraglutida, a través de la activación específica de GLP-1R, aumentó las señales de saciedad básicas y redujo las señales de hambre básicas que permitieron perder peso.
Efectos farmacodinámicos: XULTOPHY® tiene un perfil farmacodinámico estable, con una duración de acción efecto que refleja la combinación de los perfiles de acción respectivos de la insulina degludec y liraglutida, que permite la administración de XULTOPHY® una vez al día, a cualquier hora del día, con o sin alimentos. XULTOPHY® mejora el control glucémico gracias a una disminución prolongada de los niveles de glucosa en plasma en ayunas y los niveles de glucosa posprandial después de todas las comidas.
La reducción de la glucosa posprandial se ha confirmado en un subestudio consistente en una prueba estandarizada con comida, de 4 horas de duración, en pacientes no controlados con tratamiento de metformina sola o en combinación con pioglitazona. XULTOPHY® redujo la fluctuación posprandial de la glucosa plasmática (media durante 4 horas) significativamente más que la insulina degludec. Los resultados fueron similares en el caso de XULTOPHY® y liraglutida.
Eficacia y seguridad clínica:
Añadido a medicamentos hipog/ucemiantes orales:
Añadido a metformina sola o en combinación con pioglitazona: La eficacia y seguridad de XULTOPHY® en comparación con insulina degludec y liraglutida, todos ellos administrados una vez al día, se estudió en un ensayo tratar a meta de 26 semanas, con una ampliación de 26 semanas, aleatorizado, controlado y abierto en pacientes con diabetes mellitus tipo 2. La dosis inicial de XULTOPHY® e insulina degludec fue de 10 pasos de dosis (10 unidades de insulina degludec y 0.36 mg de liraglutida) y 10 unidades respectivamente y la dosis fue ajustada dos veces por semana de acuerdo con la Tabla 1, más adelante.
Los pacientes del grupo de liraglutida siguieron un programa de aumento de la dosis fija, con una dosis inicial de 0.6 mg y un aumento de dosis de 0.6 mg semanales hasta alcanzar la dosis de mantenimiento de 1.8 mg. La dosis máxima de XULTOPHY® fue de 50 unidades de dosis, mientras que no hubo dosis máxima en el grupo de insulina degludec.
Tabla 1. Titulación de XULTOPHY® e insulina basal
|
Glucosa plasmática previa la desyuno* |
Ajuste de dosis |
||
|
mmol/L |
mg/dL |
XULTOPHY® (pasos de dosis) |
Insulina basal (unidades) |
|
< 4.0 |
< 72 |
-2 |
-2 |
|
4.0-5.0 |
72-90 |
|
0 |
|
> 5.0 |
> 90 |
+ 2 |
+ 2 |
* Glucosa plasmática medida por el pacientea.
Los principales resultados del ensayo se indican en la figura 1 y en la Tabla 2.
El 60.4% de los pacientes tratados con XULTOPHY® alcanzaron un objetivo de HbA1c < 7% sin episodios hipoglucémicos confirmados tras 26 semanas de tratamiento. La proporción fue significativamente superior a la observada con insulina degludec (40.9%, odds ratio 2.28, p < 0.0001) y similar a la observada con liraglutida (57.7%, odds ratio 1.13, p = 0.3184).
Las tasas de hipoglucemia confirmada fueron inferiores con XULTOPHY® que con insulina degludec, independientemente del control glucémico, ver figura l.

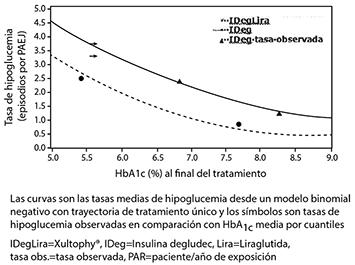
Figura 1. HbA1c media (%) por semana de tratamiento (arriba) y tasa de hipoglucemia confirmada por paciente/año de exposición, frente HbA1c media (%) (abajo) en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con tratamiento de metformlna sola o en combinación con pioglitazona.
La tasa de hipoglucemia grave, definida como episodio que precisa la ayuda de otra persona, por paciente/año de exposición (porcentaje de pacientes) fue de 0.01 (2 pacientes de un total de 825) en el caso de XULTOPHY®, 0.01 (2 pacientes de un total de 412) en el caso de insulina degludec y 0.00 (0 pacientes de un total de 412) en el caso de insulina degludec y 0.00 (0 pacientes de un total de 412) en el caso de liraglutida. La tasa de episodios de hipoglucemia nocturnos fue similar en los casos de tratamiento con XULTOPHY® e insulina degludec.
En general los pacientes tratados con XULTOPHY® experimentaron menos efectos adversos gastrointestinales que los tratados con liraglutida. Esto se puede deber a que cuando se usa XULTOPHY® el aumento de la dosis del componente liraglutida durante el inicio del tratamiento es más lento que cuando se utiliza liraglutida sola.
Datos a largo plazo (52 semanas) en pacíentes no controlados adecuadamente con tratamíento de metformina en monoterapia o en combinación con pioglitazona: La eficacia y seguridad de XULTOPHY® se mantuvieron hasta las 52 semanas de tratamiento.
La reducción de HbA1c desde el inicio hasta las 52 semanas fue del 1.84% con XULTOPHY®, con una diferencia de tratamiento estimada del -0.65% en comparación con liraglutida (p<0.0001) y del -0.46 % en comparación con insulina degludec (p<0.0001). El peso corporal se redujo en 0.4 kg, con una diferencia de tratamiento estimada entre XULTOPHY® e insulina degludec de -2.80 kg (p<0.0001), y la tasa de hipoglucemia confirmada siguió siendo de 1.8 episodios por paciente/año de exposición, manteniendo una reducción significativa en el riesgo total de hipoglucemia confirmada en comparación con insulina degludec.
Añadido a sulfonilurea sola o en combinación con metformina: La eficacia y seguridad de XULTOPHY® como complemento de la sulfonilurea, sola o combinada con metformina, se estudió en un ensayo "tratar a meta" de 26 semanas, aleatorizado, controlado con placebo y doble ciego, en 435 pacientes con diabetes mellitus tipo 2, de los que 289 fueron tratados con XULTOPHY® La dosis inicial de XULTOPHY® fue de 10 pasos de dosis (10 unidades de insulina degludec y 0.36 mg de liraglutida), y la dosis se ajustó dos veces por semana. El ajuste de dosis se realizó según se detalla en la Tabla 1, con un objetivo de titulación de 4-6 mmol/L.
Los principales resultados del ensayo se indican en la figura 2 y en la tabla 2.
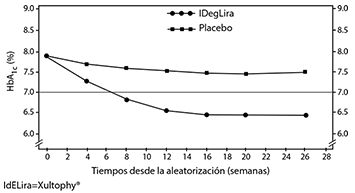
Figura 2. HbA1c media (%) por semana de tratamiento en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con tratamiento de sulfonilurea sola o en combinación con metformina.
La tasa de hipoglucemia grave por paciente/año de exposición (porcentaje de pacientes) fue de 0.02 (2 pacientes de un total de 288) en el caso de XULTOPHY® y 0.00 (0 pacientes de un total de 146) en el caso de placebo.
Tabla 2. Resultados de los estudios de 26 semanas con XULTOPHY® en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con tratamiento de metformina sola o en combinación con pioglitazona (izquierda), o no controlados adecuadamente con tratamiento de sulfonilurea sola o en combinación con metformina (derecha).
|
Tratamiento anterior con metformina + ploglitazona |
Tratamiento anterior con sulfonilurea + metformina |
||||
|
XULTOPHY® |
Insulina degludec |
Liraglutida |
XULTOPHY® |
Placebo |
|
|
N |
833 |
413 |
414 |
289 |
146 |
|
HbA1c (%) |
|||||
|
Valor de referencia → Final del ensayo |
8.3 → 6.4 |
8.3 → 6.9 |
8.3 → 7.0 |
7.9 → 6.4 |
7.9 → 7.4 |
|
Cambio medio |
-1.91 |
-1.44 |
1.28 |
-1.45 |
-1.46 |
|
Diferencia estimada |
-0.47AB [-0.58; -0.36] |
-0.64AB [-0.75; -0.53] |
-1.02AB [-1.18; -0.87] |
||
|
Pacientes (%) que lograron una HbA1c < 7% |
|||||
|
Todos los pacientes |
80.6 |
65.1 |
60.4 |
79.2 |
28.8 |
|
Índice de probabilidad (Odds ratio) estimado |
2.38B [1.78; 3.18] |
8.26B [2.45; 4.33] |
11.95B [7.22; 19.77] |
||
|
Pacientes (%) que lograron una HbA1c ≤ 6.5% |
|||||
|
Todos los pacientes |
69.7 |
47.5 |
41.1 |
64.0 |
12.3 |
|
Índice de probabilidad (Odds ratio) estimado |
2.82B [2.17; 3.67] |
3.98B [3.05; 5.18] |
16.36B [9.05; 29.56] |
||
|
Tasa de hipoglucemia confirmada* por pacientes/año de exposición (porcentaje de pacientes) |
1.80 (31.9%) |
2.57 (38.6%) |
0.22 (6.8%) |
3.52 (41.7%) |
1.35 (17.1%) |
|
Índice estimado |
0.68AC [0.53; 0.87] |
7.61B [5.17; 11.21] |
3.74B [2.28; 6.13] |
||
|
Peso corporal medio (kg) |
|||||
|
Valor de referencia → Final del ensayo |
87.2 → 86.7 |
87.4 → 89.0 |
87.4 → 84.4 |
87.2 → 87.7 |
89.3 → 88.3 |
|
Cambio medio |
-0.5 |
1.06 |
8.0 |
0.5 |
1.0 |
|
Diferencia estimada |
2.22AB [-2.64; -1.80] |
2.44B [2.02; 2.86] |
1.48B [0.90; 2.06] |
||
|
GPA (mmol) |
|||||
|
Valor de referencia → Final del ensayo |
9.2 → 5.6 |
9.4 → 5.8 |
9.0 → 7.3 |
9.1 → 6.5 |
9.1 → 8.8 |
|
Cambio medio |
-3.62 |
-3.61 |
-1.75 |
-2.60 |
-0.31 |
|
Diferencia estimada |
-0.17 [-0.41; 0.07] |
-1.76B [-2.0; -1.53] |
-2.30B [-2.72; 1.89] |
||
|
Dosis al final del ensayo |
|||||
|
Insulina degludec (unidades) |
38 |
53 |
- |
||
|
Liraglutida (mg) |
1.4 |
- |
1.8 |
28 |
- |
|
Diferencia estimada, dosis de insulina degludec |
-14.90AB [-17,14; -12.66] |
1.0 |
- |
||
Los valores de referencia, final del ensayo y variación se determinan mediante el uso de la última observación considerada. El intervalo de confianza del 95% se expresa entre corchetes "[]".
* La hipoglucemia confirmada se define como hipoglucemia grave (episodio que precisa la ayuda de otra persona) y/o hipoglucemia leve (glucosa plasmática < 3.1 mmol, Independientemente de los síntomas).
A Criterios de valoración con superioridad confirmada de XULTOPHY® frente al comparador.
B p<0.0001.
C p<0.05.
Cambio desde tratamiento con agonista del receptor de GLP-1: La eficacia y seguridad de XULTOPHY® (una vez al día) en comparación con el tratamiento con agonista del receptor de GLP-1 sin cambios (dosificado según ficha técnica), se estudió en un ensayo "tratar a meta" de 26 semanas, aleatorizado y abierto, en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con tratamiento de agonista del receptor de GLP-1 y metformina sola (74.2%) o en combinación con pioglitazona (2.5%), sulfonilurea (21.2%) o ambos (2.1%).
La dosis inicial de XULTOPHY® fue de 16 unidades de dosis (16 unidades de insulina degludec y 0.6 mg de liraglutida) y la dosis se ajustó dos veces por semana de acuerdo con la Tabla 1. Los pacientes del brazo del agonista del receptor GLP-1 continuaron con el tratamiento de agonista del receptor de GLP-1 pre-ensayo.
Los principales resultados del ensayo se indican en la Tabla 3 y en la figura 3.
Tabla 3. Resultados de un ensayo de 26 semanas con XULTOPHY® en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con tratamiento de agonistas del receptor de GLP-1.
|
Tratamiento anterior con agonista del receptor de GLP-1 |
||
|
XULTOPHY® |
Agonista del receptor de GLP-I |
|
|
N |
292 |
146 |
|
HbA1c (%) |
||
|
Valor de referencia → Final en ensayo |
7.8 → 6.4 |
7.7 → 7.4 |
|
Cambio medio |
-1.3 |
-0.3 |
|
Diferencia estimada |
-0.94AB [-1.11; -0.78] |
|
|
Pacientes (%) que lograron una HbA1c < 7% |
||
|
Todos los pacientes |
75.3 |
35.6 |
|
Índice de probabilidad (Odds ratio) estimado |
6.84B [4.28; 10.94] |
|
|
Pacientes (%) que lograron una HbA1c ≤ 6.5% |
||
|
Todos los pacientes |
63.0 |
22.6 |
|
Índice de probabilidad (Odds ratio) estimado |
7.53B [4.58; 12.38] |
|
|
Tasa de hipoglucemia confirmada* por pacientes/año de exposición (porcentaje de pacientes) |
2.82 (32.0%) |
0.12 (2.8%) |
|
Índice estimado |
25.36B [10.63; 60.51] |
|
|
Peso corporal medio (kg) |
||
|
Valor de referencia → Final del ensayo |
95.6 → 97.5 |
95.5 → 94.7 |
|
Cambio medio |
2.0 |
-0.8 |
|
Diferencia estimada |
2.89B [2.17; 3.62] |
|
|
GPA (mmol/l) |
||
|
Valor de referencia → Final del ensayo |
9.0 → 6.0 |
9.4 → 8.8 |
|
Cambio medio |
-2.98 |
-0.60 |
|
Diferencia estimada |
-2.64B [-3.03; 2.25] |
|
|
Dosis al final del ensayo |
||
|
Insulina degludec (unidades) |
43 |
La dosis del agonista del receptor de GLP-1 se mantuvo sin cambios desde el valor de referencia |
|
Liraglutida (mg) |
1.6 |
|
|
Diferencia estimada, dosis de insulina degludec |
||
Los valores de referencia, final del ensayo y variación se determinan mediante el uso de la última observación considerada. El intervalo de confianza del 95% se expresa entre corchetes"[]".
* La hipoglucemia confirmada se define como hipoglucemia grave (episodio que precisa la ayuda de otra persona) y/o hipoglucemia leve (glucosa plasmática < 3.1 mmol/L, independientemente de los síntomas).
A Criterios de valoración con superioridad confirmada de XULTOPHY® frente al comparador.
B p<0.0001.

Figura 3. HbA1c media (%) por semana de tratamiento en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con agonistas del receptor de GLP-1.
La tasa de hipoglucemia grave por paciente/año de exposición (porcentaje de pacientes) fue de 0.01 (1 paciente de un total de 291) en el caso de XULTOPHY® y 0.00 (0 pacientes de un total de 199) en el caso de agonistas del receptor de GLP-1.
Cambio desde tratamiento con insulina basal: La eficacia y seguridad de XULTOPHY® en comparación con insulina glargina, ambos una vez al día, se estudió en un ensayo "tratar a meta" de 26 semanas, aleatorizado y abierto, en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con tratamiento de insulina glargina (20-50 unidades) y metformina. La dosis inicial de XULTOPHY® fue de 16 unidades de dosis y la dosis inicial de insulina glargina fue igual a la dosis diaria pre-ensayo. La dosis en ambos grupos se ajustó dos veces por semana de acuerdo con la Tabla l. La dosis máxima permitida fue de 50 unidades de dosis en el caso de XULTOPHY® mientras que para insulina glargina no hubo dosis máxima.
Los principales resultados del ensayo se indican en la Tabla 4 y en la figura 4.
El 54.3% de los pacientes tratados con XULTOPHY® alcanzó el objetivo de HbA1c 7% sin episodios hipoglucémicos confirmados en comparación con el 29.4% de los pacientes tratados con insulina glargina (odds ratio 3.24, p < o.001).

Figura 4. HbA1c media (%) por semana de tratamiento en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con insulina glargina.
La tasa por paciente/año exposición (porcentaje de pacientes) de hipoglucemia grave fue 0.00 (0 pacientes de un total de 278) para XULTOPHY® y 0.01 (1 paciente de un total de 279) para insulina glargina. La tasa de acontecimientos hipoglucémicos nocturnos fue significativamente inferior con XULTOPHY® en comparación con insulina glargina (tasa de tratamiento estimado 0.17, p<0.001).
La eficacia y seguridad de XULTOPHY® en comparación con insulina degludec, ambos una vez al día, se estudió en un ensayo "tratar a meta" de 26 semanas, aleatorizado y doble ciego, en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con tratamiento de insulina basal (20-40 unidades) y metformina sola o en combinación con sulfonilurea/glinidas. Se interrumpió la administración de insulina basal y sulfonilurea/glinidas en la aleatorización.
La dosis inicial de XULTOPHY® e insulina degludec fue de 16 unidades de dosis (16 unidades de insulina degludec y 0.6 mg de liraglutida) y 16 unidades respectivamente y la dosis se ajustó dos veces por semana de acuerdo con la Tabla 1. La dosis máxima permitida fue de 50 unidades de dosis en el caso de XULTOPHY® y 50 unidades en el de insulina degludec.
Los principales resultados del ensayo se indican en la Tabla 4 y en la figura 5.
El 48. 7% de los pacientes alcanzaron un objetivo de HbA1c < 7% sin episodios hipoglucémicos confirmados, lo que supuso una proporción significativamente superior a la observada con insulina degludec (15.6%, odds ratio 5.57, p<0.0001).

Figura 5. HbA1c media (%) por semana de tratamiento en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con insulina basal.
La tasa de hipoglucemia grave por paciente/año de exposición (porcentaje de pacientes) fue de 0.01 (1 paciente de un total de 199) en el caso de XULTOPHY® y 0.00 (0 pacientes de un total de 199) en el caso de insulina degludec. La tasa de episodios de hipoglucemia nocturnos fue similar en los casos de tratamiento con XULTOPHY® e insulina degludec.
Tabla 4. Resultados de dos ensayos de 26 semanas con XULTOPHY® en pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con insulina glargina (izquierda) o con insulina basal (derecha).
|
Tratamiento anterior con insulina glargina |
Tratamiento anterior con insulina basal (NPH, insulina detemir, insulina glargina) |
|||
|
XULTOPHY® |
Insulina glargina ninguna limitación de dosis |
XULTOPHY® |
Insulina degludec, máximo de 50 unidades permitidas |
|
|
N |
278 |
279 |
199 |
199 |
|
HbA1c (%) |
||||
|
Valor de referencia → Final del ensayo |
8.4 → 6.6 |
8.2 → 7.1 |
8.7 → 6.9 |
8.8 → 8.0 |
|
Cambio medio |
-1.81 |
-1.13 |
-1.90 |
-0.89 |
|
Diferencia estimada |
-0.59AB [-0.74; -0.45] |
-1.05AB [-1.25; -0.84] |
||
|
Pacientes (%) que lograron una HbA1c < 7% |
||||
|
Odds ratio de todos los pacientes estimado |
71.6 |
47.0 |
60.3 |
23.1 |
|
3.45B [2.36; 5.05] |
5.44B [3.42; 8.66] |
|||
|
Pacientes (%) que lograron una HbA1c ≤ 6.5% |
||||
|
Todos los pacientes |
55.4 |
30.8 |
45.2 |
13.1 |
|
Índice de probabilidad Odds ratio estimado |
3.29B [2.27; 4.75] |
5.66B [3.37; 9.51] |
||
|
Tasa de hipoglucemia confirmada* por pacientes/año de exposición (porcentaje de pacientes) |
2.23 (28.4%) |
5.05 (49.1%) |
1.53 (24.1%) |
2.63 (24.6%) |
|
Índice estimado |
0.43AB [0.30; 0.61] |
0.66 [0.39; 1.13] |
||
|
Peso corporal (kg) |
||||
|
Valor de referencia → Final del ensayo |
88.3 → 86.9 |
87.3 → 89.1 |
95.4 → 92.7 |
93.5 → 93.5 |
|
Cambio medio |
-1.4 |
1.8 |
-2.7 |
0.0 |
|
Diferencia estimada |
-3.20AB [-3.77; -2.64] |
-2.51B [-3.21; -1.82] |
||
|
GPA (mmol/L) |
||||
|
Valor de referencia →Final del ensayo |
8.9 → 6.1 |
8.9 → 6.1 |
9.7 → 6.2 |
9.6 → 7.0 |
|
Cambio medio |
-2.83 |
-2.77 |
-3.46 |
-2.58 |
|
Diferencia estimada |
-0.01 [-0.35; 0.33] |
-0.73c [-1.19; -0.27] |
||
|
Dosis al final del ensayo |
||||
|
Insulina (unidades) |
41 |
66D |
45 |
45 |
|
Liraglutida (mg) |
1.5 |
- |
1.7 |
- |
|
Diferencia estimada, dosis de insulina degludec |
-25.47B [-28.90; -22.05] |
-0.02 [-1.88; 1.84] |
||
Los valores de referencia, final del ensayo y variacIon se determinan mediante el uso de la última observación considerada. El intervalo de confianza del 95%, se expresa entre corchetes"[]".
* La hipoglucemia confirmada se define como hipoglucemia grave (episodio que precisa la ayuda de otra persona) y/o hipoglucemia leve (glucosa plasmática < 3.1 mmol/I, independientemente de los síntomas).
A Criterios de variación con superioridad confirmada de XULTOPHY® frente al comparador.
B p<0.0001.
C p<0.05.
D La dosis media pre-ensayo de insulina glargina fue de 32 unidades.
El tratamiento con XULTOPHY® en comparación con un régimen de insulina basal en bolo que consiste en insulina basal (insulina glargina 100 unidades/mL) en combinación con insulina en bolo (insulina asparta) se estudió en un ensayo de 26 semanas en pacientes con diabetes mellitus tipo 2 no controlada adecuadamente con insulina glargina y metformina demostraron una reducción similar de HbA1c en los dos grupos (valor promedio de 8.2% a 6.7% en ambos grupos).
En ambos grupos, 66%-67% lograron HbA1c <7%. En comparación con el valor inicial, hubo una reducción en la media en el peso corporal de 0.9 kg para XULTOPHY® y un aumento medio de 2.6 kg para los pacientes tratados con un régimen basal en bolo y la diferencia de tratamiento estimada fue de -3.57 kg [IC 95%: -4.19; -2.95]. El porcentaje de pacientes que experimentaron hipoglucemia sintomática grave o confirmada con glucosa en sangre fue del 19.8% en el grupo de XULTOPHY® y del 52.6% en el grupo de insulina basal-bolo, y el índice de tasa estimado fue de 0.11 [IC 95%: 0.08-0.17]. La dosis diaria total de insulina al final del ensayo fue de 40 unidades para los pacientes tratados con XULTOPHY® y 84 unidades (52 unidades de insulina basal y 32 unidades de insulina en bolo) para los pacientes tratados con un régimen de insulina basal-bolo.
Otros datos clínicos:
Secreción de insulina/función de las células beta: XULTOPHY® mejora la función de las células beta en comparación con la insulina degludec, de acuerdo con el modelo de evaluación de la homeostasia para la función de la célula beta (HOMA-beta). Se demostró una secreción mejorada de insulina en comparación con la insulina degludec, como respuesta a una prueba estandarizada con comida, en 260 pacientes con diabetes tipo2 tras 52 semanas de tratamiento. No se dispone de datos posteriores a las 52 semanas de tratamiento.
Presión arterial: En pacientes no controlados adecuadamente con metformina en monoterapia o en combinación con pioglitazona, XULTOPHY® redujo la presión arterial sistólica media en 1.8 mm Hg en comparación con una reducción de 0.7 mm Hg con insulina degludec y de 2.7 mm Hg con liraglutida. En pacientes no controlados adecuadamente con sulfolinurea en monoterapia o en combinación con metformina, la reducción fue de 3.5 mm Hg con XULTOPHY® y 3.2 mm Hg con placebo. Las diferencias no fueron estadísticamente significativas. En tres ensayos con pacientes no controlados adecuadamente con insulina basal, la presión arterial sistólica se redujo en 5.4 mm Hg con XULTOPHY® y l.7 mm Hg con insulina degludec, con una diferencia de tratamiento estimada estadísticamente significativa de -3.71 mm Hg (p = 0.0028), y se redujo en 3.7 mm Hg con XULTOPHY® vs 0.2 mm Hg con insulina glargina, con una diferencia de tratamiento estimada estadísticamente significativa de -3.57 mm Hg (p< 0.001) y reducido en 4.5 mm Hg con XULTOPHY® vs 1.16 mm Hg con insulina glargina U 100 más insulina asparta, con una diferencia de tratamiento estimada estadísticamente significativa de -3.70 mm Hg (p = 0.0003).
CONTRAINDICACIONES: Hipersensibilidad a cualquiera de los principios activos, a ambos o alguno de los excipientes (Glicerol, Fenal, Acetato de Zinc, Ácido Clorhídrico o Hidróxido de Sodio para ajustar pH, Agua para inyectables). Menores de 18 años de edad, embarazo, lactancia, diabetes mellitus tipo 1, cetoasidosis diabética, insuficiencia cardiaca congestiva clases III y IV.
Insuficiencia renal: Cuando se usa XULTOPHY® en pacientes con insuficiencia renal leve o moderada, es necesario intensificar el control glucémico y ajustar individualmente la dosis. No debe administrarse XULTOPHY® en pacientes con insuficiencia renal grave, incluidos los pacientes con enfermedad renal en etapa terminal (Ver sección 5 Propiedades Farmacodinámicas).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No hay experiencia clínica con el uso de XULTOPHY® insulina degludec o liraglutida en mujeres embarazadas. Se debe interrumpir el tratamiento con XULTOPHY® en caso de que una paciente desee quedarse embarazada o si se produce un embarazo.
Los estudios sobre reproducción animal con insulina degludec no han revelado diferencia alguna entre la insulina degludec y la insulina humana por lo que respecta a la embriotoxicidad y la teratogenicidad. Los estudios en animales con liraglutida han mostrado toxicidad reproductiva, ver sección 12. Precauciones en relación con efectos de carcinogenesis, mutagenesis, teratogenesis y sobre la fertilidad. Se desconoce el riesgo en seres humanos.
XULTOPHY® no debe administrarse durante el embarazo.
Lactancía: No hay experiencia clínica con el uso de XULTOPHY® durante la lactancia. Se desconoce si la insulina degludec o liraglutida se excreta en la leche materna. Dada la falta de experiencia, no se debe usar XULTOPHY® durante el periodo de lactancia.
En ratas, la insulina degludec se excretó en la leche; la concentración en leche fue inferior a la concentración en plasma. Estudios realizados en animales han mostrado que la transferencia a la leche de liraglutida y metabolitos de estrecha relación estructural es baja.
Estudios no clínicos con liraglutida han mostrado una reducción en el crecimiento neonatal relacionada con el tratamiento en crías de rata en período de lactancia (ver sección 12. Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad).
Fertilidad: No hay experiencia clínica con XULTOPHY® en relación con la fertilidad.
Los estudios sobre reproducción animal con insulina degludec no han revelado efectos adversos sobre la fertilidad. Los estudios en animales con liraglutida no han revelado efectos nocivos relacionados con la fertilidad, aparte de una ligera disminución en el número de implantes vivos.
REACCIONES SECUNDARIAS Y ADVERSAS:
Resumen del perfil de seguridad: El programa de desarrollo clínico de XULTOPHY® incluyó aproximadamente a 1,900 pacientes tratados con XULTOPHY®.
La hipoglucemia y las reacciones adversas gastrointestinales fueron las reacciones adversas notificadas más frecuentemente durante el tratamiento con XULTOPHY® (ver sección "Descripción de las reacciones adversas seleccionadas" más adelante).
Lista tabulada de reacciones adversas: A continuación se indican las reacciones adversas asociadas a XULTOPHY®, según el sistema de clasificación de órganos y la frecuencia. Las categorías de frecuencias se definen del siguiente modo: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a <1/10); poco frecuentes (≥ 1/1,000 a < 1/100); raras (≥ 1/10,000 a < 1/1,000); muy raras (< 1/10,000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 5. Reacciones adversas notificadas en estudios controlados de fase 3.
|
Sistema de clasificación de órganos |
Frecuencia |
Reacción Adversa del Medicamento |
|
Trastornos del sistema inmunológico |
Poco frecuente |
Urticaria |
|
Poco frecuente |
Hipersensibilidad |
|
|
Frecuencia no conocida |
Reacción anafiláctica |
|
|
Trastornos del metabolismo y de la nutrición |
Muy frecuente |
Hipoglucemia |
|
Frecuente |
Disminución del apetito |
|
|
Poco frecuente |
Deshidratación |
|
|
Trastornos gastrointestinales |
Frecuente |
Náusea, diarrea, vómito, estreñimiento, dispepsia, gastritis, dolor abdominal, enfermedad de reflujo gastroesofágico, distensión abdominal |
|
Poco frecuente |
Eructos, flatulencia |
|
|
Frecuencia no conocida |
Pancreatitis (incluida pancreatitis necrotizante |
|
|
Desordenes Hepatobiliares |
Poco frecuente |
Colelitiasis |
|
Poco frecuente |
Colecistitis |
|
|
Trastornos de la piel y del tejido subcutáneo |
Poco frecuente |
Sarpullido |
|
Poco frecuente |
Prurito |
|
|
Poco frecuente |
Lipodistrofia adquirida |
|
|
Trastornos generales y alteraciones en el lugar de administración |
Frecuente |
Reacción en la zona de inyección |
|
Frecuencia no conocida |
Edema periférico |
|
|
Investigación |
Frecuente |
Incremento lipasa |
|
Frecuente |
Incremento Amilasa |
|
|
Poco frecuente |
Aumento de la frecuencia cardiaca |
Descripción de las reacciones adversas seleccionadas:
Hipoglucemia: Puede aparecer hipoglucemia si la dosis de XULTOPHY® es superior a la necesaria. Una hipoglucemia grave puede producir un estado de inconsciencia y/o convulsiones y puede dar lugar a una insuficiencia cerebral temporal o permanente o incluso la muerte. Los síntomas de hipoglucemia por lo general aparecen de forma repentina. Pueden incluir sudor frío, piel fría y pálida, fatiga, nerviosismo o temblor, ansiedad, cansancio o debilidad no habitual, confusión, dificultad para concentrarse, somnolencia, apetito excesivo, cambio en la visión, cefalea, náuseas y palpitaciones (Ver sección 5 Propiedades Farmacodinámicas para consultar datos sobre la frecuencia de episodios de hipoglucemia).
Reacciones alérgicas: Se han notificado reacciones alérgicas (manifestadas por signos y síntomas tales como urticaria (0.3% de los pacientes tratados con XULTOPHY®), sarpullido (0. 7%), prurito (0.5%) y/o hinchazón facial (0.2%)) asociadas al uso de XULTOPHY®. Durante la comercialización de liraglutida, se han notificado pocos casos de reacciones anafilácticas con síntomas adicionales tales como hipotensión, palpitaciones, disnea, edema. Las reacciones anafilácticas pueden ser potencialmente mortales.
Reacciones adversas gastrointestinales: Las reacciones adversas gastrointestinales pueden aparecer con mayor frecuencia al comienzo del tratamiento con XULTOPHY® y suelen disminuir al cabo de pocos días o semanas de tratamiento continuado. Se notificaron náuseas en el 7.8% de los pacientes, de carácter transitorio en la mayoría de los casos. La proporción de los pacientes que notificaron náuseas por semana en cualquier momento del tratamiento fue inferior al 4%. Se notificaron diarrea y vómitos en el 7.5% y 3.9% de los pacientes, respectivamente con frecuencia de las náuseas y diarrea fue "frecuente" en el caso de XULTOPHY® y "muy frecuente" en el de liraglutida. Además, se han notificado estreñimiento, dispesia, gastritis, dolor abdominal, enfermedad de reflujo gastroesofágico, distensión abdominal, eructos, flatulencia y disminución del apetito en hasta el 3.6% de los pacientes tratados con XULTOPHY®.
Reacciones en la zona de inyección: Se han notificado reacciones en la zona de inyección (como hematoma, dolor, hemorragia, eritema, nódulos, hinchazón, cambio de color, prurito, calor y abultamiento en el lugar de inyección) en el 2.6% de los pacientes tratados con XULTOPHY®. Estas reacciones fueron por lo general leves y transitorias y normalmente desaparecen durante el tratamiento continuado.
Lipodistrofia: Puede aparecer lipodistrofia (incluidas lipohipertrofia y lipoatrofia) en el lugar de la inyección. La continua rotación del lugar de inyección dentro de un área concreta de inyección puede ayudar a reducir el riesgo de desarrollar estas reacciones.
Aumento de la frecuencia cardiaca: En ensayos clínicos con XULTOPHY® se ha observado un aumento medio de la frecuencia cardiaca desde el nivel de referencias de hasta 2 o 3 latidos más por minutos. No se han determinado los efectos clínicos a largo plazo de este aumento de la frecuencia cardiaca.
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales de la salud a notificar las sospechas de reacciones adversas a través de farmacovigilancia@cofepris.gob.mx.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: El programa de desarrollo no clínico de insulina degludec/liraglutida incluyó estudios pivotales de combinación de toxicidad, de hasta 90 días de duración en una sola especie relevante (ratas Wistar) para sustentar el programa de desarrollo clínico. La tolerancia local se evaluó en conejos y cerdos.
Lo datos de los estudios no clínicos de seguridad no mostraron riesgos de seguridad para los seres humanos según los estudios de toxicidad a dosis repetidas.
Las reacciones del tejido local en los dos estudios con conejos y cerdos, respectivamente, se limitaron a reacciones inflamatorias leves.
No se han llevado a cabo estudios con la combinación de insulina degludec/liraglutida para evaluar la carcinogénesis, mutagénesis o disminución de la fertildad. Los siguientes datos se basan en estudios con insulina degludec y liraglutida por separado.
Insulina degludec: Los datos de los estudios no clínicos no muestran riesgos de seguridad para los seres humanos según los estudios de farmacología de seguridad, toxicidad a dosis repetidas, potencial carcinogénico y toxicidad para la reproducción.
La relación entre la potencia mitógena y la potencia metabólica de la insulina degludec es igual a la de la insulina humana.
Liraglutida: Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas o genotoxicidad. Se observaron tumores no letales en células C de tiroides en estudios de carcinogenicidad de dos años en ratas y ratones. En ratas no se ha observado el nivel sin efecto adverso observado (NOAEL). Estos tumores no se observaron en monos tratados durante 20 meses. Estos hallazgos en roedores están provocados por un mecanismo específico no genotóxico mediado por el receptor GLP-1 al que los roedores son especialmente sensibles. La relevancia en humanos es probablemente baja pero no se puede excluir completamente. No se ha detectado ningún otro tumor relacionado con el tratamiento.
Los estudios en animales no sugieren efectos perjudiciales directos en términos de fertilidad, pero si un leve aumento de las muertes embrionarias tempranas a la dosis más alta. La administración de liraglutida durante el período intermedio de gestación provocó una reducción en el peso de la madre y en el crecimiento del feto con efectos no claros sobre las costillas en ratas y en la variación esquelética en el conejo. El crecimiento neonatal se redujo en el caso de las ratas durante su exposición a liraglutida y continuó durante el periodo de destete en el grupo de dosis elevada. Se desconoce si la disminución en el crecimiento de las crías se debe a una reducción en la ingesta de leche debido a un efecto directo del GLP-1 o a una reducción de la producción de leche materna a causa de una disminución de la ingesta calórica.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones farmacodinámicas: No se han realizado estudios de interacciones con XULTOPHY®.
Hay ciertas sustancias que afectan al metabolismo de la glucosa y pueden requerir un ajuste de la dosis de XULTOPHY®.
Las siguientes sustancias pueden reducir los requerimientos de XULTOPHY®:
Antidiabéticos, inhibidores de la monoaminooxidasa (IMAO), betabloqueadores, inhibidores de la enzima convertidora de angiotensina (IECA), salicilatos, esteroides anabólicos y sulfonamidas.
Las siguientes sustancias pueden aumentar los requerimientos de XULTOPHY®:
Anticonceptivos orales, tiazidas, glucocorticoides, hormonas tiroideas simpaticomiméticos, hormona de crecimiento y danazol.
Los betabloqueadores pueden enmascarar los síntomas de hipoglucemia.
Octreotída y lanreotida pueden aumentar o reducir el requerimiento de XULTOPHY®.
El alcohol puede intensificar o reducir el efecto hipoglucemiante de XULTOPHY®.
Interacciones farmacocinéticas: Los datos procedentes de la investigación in vitro sugieren que la posibilidad de que se produzcan interacciones farmacocinéticas relacionadas con la interacción de CYP y la unión a proteínas plasmáticas es baja, tanto para liraglutida como para la insulina degludec.
El leve retraso en el vaciamiento gástrico asociado a liraglutida puede influir en la absorción de medicamentos orales administrados de forma concomitante. Los estudios de interacción no han demostrado ningún retraso clínicamente significativo en la absorción.
Warfarina v otros derivados de la cumarina: No se han realizado estudios de interacciones. No se puede excluir una interacción clínicamente significativa con principios activos con escasa solubilidad o índice terapéutico estrecho, tales como la warfarina. Al inicio del tratamiento con XULTOPHY® en pacientes tratados con warfarina u otros derivados de la cumarina, se recomienda un control de la INR (Razón Internacional Normalizada) más frecuente.
Paracetamol: Liraglutida no modificó la exposición general del paracetamol tras la administración de una dosis única de 1, 000 mg. Se produjo una disminución del 31% en la Cmax de paracetamol y un retraso en el tmax medio de hasta 15 minutos. No es necesario un ajuste de dosis en el uso concomitante de paracetamol.
Atorvastatina: Liraglutida no modificó la exposición general de atorvastatina hasta un grado clínicamente significativo tras la administración de una dosis única de 40 mg de atorvastatina. Por lo tanto, no es necesario un ajuste de dosis de atorvastatina cuando se administra con liraglutida. Se produjo una disminución del 38% en la Cmax de atorvastatina y el tmax medio se retrasó de 1 h a 3 h con liraglutida.
Griseofulvina: Liraglutida no modificó la exposición general de la griseofulvina tras la administración de una dosis única de 500 mg de griseofulvina. Se produjo un aumento del 37% en la Cmax de griseofulvina y el tmax medio permaneció inalterado. No es necesario un ajuste de dosis de griseofulvina y otros componentes de baja solubilidad y alta permeabilidad.
Digoxina: La administración de una única dosis de 1 mg de digoxina con liraglutida produjo una reducción en la AUC de digoxina de un 16%; la Cmax disminuyó un 31%. Se produjo un retraso en el tiempo medio requerido para alcanzar la concentración máxima (tmax) de digoxina de 1 h a 1.5 h. No es necesario un ajuste de dosis de digoxina en base a estos resultados.
Lisinopril: La administración de una única dosis de 20 mg de lisinopril con liraglutida mostró una reducción en la AUC de lisinopril de un 15%; la Cmax disminuyó un 27%. Se produjo un retraso en el tmax medio del lisinopril que pasó de 6 h a 8h con liraglutida. No es necesario un ajuste de dosis de lisinopril en base a estos resultados.
Anticonceptivos orales: Tras la administración de una única dosis de un medicamento anticonceptivo oral, liraglutida disminuyó la Cmax de etinilestradiol y levonorgestrel en un 12 y 13%, respectivamente. Se produjo un retraso en el tmax de alrededor de 1.5 h con liraglutida para ambos compuestos.
No se observó ningún efecto clínicamente significativo sobre la exposición general ni al estinilestradiol ni a levonorgestrel. Se prevé por lo tanto que el efecto anticonceptivo permanezca inalterado cuando se administran de forma conjunta con liraglutida.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No se identificó ninguna diferencia clínicamente relevante entre los grupos de XULTOPHY® y de los comparadores en relación con los parámetros de laboratorio de hematología, bioquímica (incluyendo calcitonina), análisis de orina o anticuerpos. La respuesta inmunogénica al tratamiento a largo plazo con XULTOPHY® fue baja y no se observó ningún patrón en los AEs reportados, lo que indica un posible efecto clínico de desarrollo de anticuerpos.
Se detectó un aumento de los niveles medios de lipasa en el intervalo de 11-15 U/L con XULTOPHY® y liraglutida, mientras que se observó una disminución en el grupo de Insulina Degludec (en el intervalo de -3 a -7 U/L) después de 26 semanas de tratamiento, con una leve mejoría después de 52 semanas. Se identificó un cambio similar, pero menos pronunciado, en los niveles de amilasa.
PRECAUCIONES GENERALES: No se debe utilizar XULTOPHY® en pacientes con diabetes mellitus tipo 1 o para el tratamiento de la cetoacidosis diabética.
Hipoglucemia: Se puede producir hipoglucemia, si la dosis de XULTOPHY® es superior a la necesaria. La omisión de una comida o el ejercicio físico intenso no previsto, pueden producir hipoglucemia. En combinación con sulfonilurea, es posible disminuir el riesgo de hipoglucemia reduciendo la dosis de sulfonilurea. Las enfermedades concomitantes renales, hepáticas o que afecten a las glándulas suprarrenales, pituitaria o tiroidea pueden requerir un cambio en la dosis de XULTOPHY®. Los pacientes cuyo control glucémico mejora en gran medida (por ejemplo, por medio de terapia intensificada) pueden experimentar un cambio en sus síntomas habituales de aviso de hipoglucemia y deben ser avisados de esta posibilidad. Los síntomas habituales de aviso de hipoglucemia (ver sección 9, Reacciones secundarias y adversas) pueden desaparecer en pacientes con diabetes de larga duración. El efecto prolongado de XULTOPHY® puede retrasar la recuperación de una hipoglucemia.
Hiperqlucemia: La dosificación inadecuada y/o la interrupción del tratamiento antidiabético pueden ocasionar hiperglucemia y potencialmente coma hiperosmolar. En caso de que se interrumpa el tratamiento con XULTOPHY®, asegúrese de que se sigan las instrucciones de inicio de un tratamiento antidiabético alternativo. Asimismo, enfermedades concomitantes, especialmente las infecciones, pueden provocar hiperglucemia y, por tanto, aumentar la necesidad de tratamiento antidiabético. Los primeros síntomas de hiperglucemia generalmente aparecen de forma gradual, a lo largo de un periodo de horas o días. Estos incluyen sed, aumento de la frecuencia de micción, náuseas, vómitos, somnolencia, piel seca y enrojecida, sequedad de boca, pérdida de apetito así como aliento con olor a acetona.
Se debe considerar la administración de insulina de acción rápida en situaciones de hiperglucemia grave. Los acontecimientos hiperglucémicos no tratados pueden acabar dando lugar a un coma hiperosmolar/cetoacidosis diabética, la cual es potencialmente mental.
Combinación de pioglitazona e insulinas: Cuando pioglitazona fue utilizada en combinación con insulinas, se notificaron casos de insuficiencia cardiaca, especialmente en pacientes con factores de riesgo de desarrollar insuficiencia cardiaca. Esto se debe tener en cuenta si se considera el tratamiento combinado de pioglitazona y XULTOPHY®. Si se utiliza esta combinación, se debe vigilar en los pacientes la aparición de signos y síntomas de insuficiencia cardiaca, ganancia de peso y edema. Se debe interrumpir el tratamiento con pioglitazona si tiene lugar cualquier deterioro de los síntomas cardiacos.
Trastornos oculares: La intensificación del tratamiento con insulina, un componente de XULTOPHY®, con una mejora brusca de control glucémico, se puede asociar a un empeoramiento temporal de la retinopatía diabética, mientras que un control glucémico mejorado a lo largo plazo reduce el riesgo del avance de dicha enfermedad.
Formación de anticuerpos: La administración de XULTOPHY® puede provocar la formación de anticuerpos contra la insulina degludec y/o liraglutida. En casos raros, la presencia de estos anticuerpos puede hacer necesario el ajuste de la dosis de XULTOPHY® para corregir una tendencia a la hiper o hipoglucemia. Muy pocos pacientes desarrollaron anticuerpos específicos contra la insulina degludec, anticuerpos con reactividad cruzada a la insulina humana o anticuerpos contra liraglutida tras el tratamiento con XULTOPHY®. La formación de anticuerpos no se ha asociado con una reducción de la eficacia de XULTOPHY®.
Pancreatitis aguda: El uso de agonistas del receptor de GLP-1 como liraglutida, un componente de XULTOPHY®, se ha asociado con un riesgo de desarrollar pancreatitis aguda. Se han notificado pocos casos de pancreatitis aguda. Se debe informar a los pacientes de los síntomas característicos de la pancreatitis aguda. Ante la sospecha de pancreatitis, se debe interrumpir el tratamiento con XULTOPHY®, y este no se debe reanudar si se confirma la pancreatitis aguda. Se debe extremar la precaución en pacientes con antecedentes de pancreatitis.
Acontecimientos adversos tiroideos: Se han notificado acontecimientos adversos tiroideos, que incluyen aumento de calcitonina en sangre, bocio y neoplasia tiroidea, en ensayos clínicos con agonistas del receptor de GLP-1 (como liraglutida, un componente de XULTOPHY®), especialmente en pacientes con enfermedad tiroidea pre-existente y, por lo tanto XULTOPHY® se debe utilizar con precaución en estos pacientes.
Enfermedad inflamatoria intestinal ly gastroparesia diabética: No existe experiencia clínica con XULTOPHY® en pacientes con enfermedad inflamatoria intestinal y gastroparesia diabetica, por lo tanto, no se recomienda el uso de XULTOPHY® en estos pacientes.
Insuficiencia renal:
Insulina degludec: No hay diferencias en la farmacocinética de la insulina degludec entre sujetos sanos y pacientes con insuficiencia renal.
Liraglutida: La exposición a liraglutida disminuyó en sujetos con insuficiencia renal en comparación con los individuos con una función renal normal. La exposición a liraglutida disminuyó un 33%, un 14%, un 27% y un 26% respectivamente, en sujetos con insuficiencia renal leve (aclaramiento de creatinina, CrCI 50-80 mL/min), moderada (CrCI 30-50 mL/min) y grave (CrCl<30 mL/min) y con enfermedad renal en etapa terminal con necesidad de diálisis.
Del mismo modo, en un ensayo clínico de 26 semanas, los pacientes con diabetes tipo 2 e insuficiencia renal moderada (CrCI 30-59 mL/min) tuvieron una exposición a Liraglutida un 26% menor en comparación con un ensayo separado que incluyó pacientes con diabetes tipo 2 con una función renal normal o insuficiencia renal leve.
Deshidratación: Se han notificado signos y síntomas de deshidratación, que incluyen insuficiencia renal y fallo renal agudo, en ensayos clínicos con agonistas del receptor de GLP-1, como liraglutida, un componente de XULTOPHY®. Se debe advertir a los pacientes en tratamiento con XULTOPHY® de que existe un riesgo potencial de deshidratación relacionado con los efectos adversos gastrointestinales y de que tomen precauciones para evitar la pérdida de líquidos.
Cómo evitar errores de medicación: Se debe indicar a los pacientes que comprueben siempre la etiqueta de la pluma antes de cada inyección para evitar confusiones entre XULTOPHY® y otros medicamentos inyectables para la diabetes.
Los pacientes deben verificar visualmente las unidades marcadas en el contador de dosis de la pluma. Por lo tanto, el requisito para que los pacientes se autoinyecten es que pueden leer el contador de dosis en la pluma. Los pacientes que son ciegos o tienen poca visión deben ser instruidos para que siempre obtengan ayuda/asistencia de otra persona que tenga buena visión y esté capacitada para usar el dispositivo de insulina.
Para evitar errores de dosificación y posible sobredosis, los pacientes y los profesionales de la salud nunca deben usar una jeringa para extraer el medicamento del cartucho en la pluma precargada.
En caso de agujas bloqueadas, los pacientes deben seguir las instrucciones descritas en las instrucciones de uso que acompañan al instructivo (ver abajo Precauciones especiales de eliminación y otras manipulaciones).
Poblaciones no estudiadas: No se ha estudiado el cambio de XULTOPHY® desde dosis de insulina basal < 20 y > 50 unidades.
No se ha estudiado XULTOPHY® en combinación con inhibidores de la dipeptidil peptidasa 4 (DPP-4), glinidas o insulina prandial.
La experiencia en pacientes con insuficiencia cardiaca congestiva de clase I y II según la New York Heart Association (NYHA) es limitada y, por lo tanto, XULTOPHY® se debe utilizar con precaución en estos pacientes. No existe experiencia en pacientes con insuficiencia cardiaca congestiva de clase III y IV según la NYHA, y por lo tanto, XULTOPHY® no debe administrarse en estos pacientes.
Excipientes: XULTOPHY® contiene menos de 1 mmol de sodio (23 mg) por dosis, por lo que se considera esencialmente exento de sodio.
Efectos sobre la capacidad para manejar y uso de maquinaria: La capacidad de concentración y de reacción de los pacientes diabéticos se puede ver afectada por una hipoglucemia. Esto puede suponer un riesgo en situaciones en las que estas capacidades sean de especial importancia (por ejemplo conducir o utilizar máquinas).
Se debe advertir a los pacientes que extremen las precauciones para evitar una hipoglucemia mientras conducen. Esto es particularmente importante en aquellos pacientes con reducida o nula capacidad para percibir los síntomas de una hipoglucemia, o que padecen episodios frecuentes de hipoglucemia. Se debe considerar la conveniencia de conducir en estas circunstancias.
Incompatibilidades: Las sustancias añadidas a XULTOPHY® pueden provocar la degradación de los principios activos.
XULTOPHY® no se debe añadir a los fluidos de perfusión.
Este medicamento no debe mezclarse con otros.
Precauciones especiales de eliminación y otras manipulaciones: La pluma precargada está diseñada para utilizarse con agujas Novo Fine® (Reg.No.625C94 SSA) de hasta 8 mm de longitud y un grosor de 32 G.
La pluma precargada está destinada a utilizarse en una sola persona.
XULTOPHY® no se debe utilizar si la solución no tiene un aspecto transparente e incoloro.
Si XULTOPHY® se ha congelado, no se debe utilizar.
Siempre se debe colocar una nueva aguja antes de cada uso. Las agujas no deben reutilizarse. El paciente debe descartar la aguja después de cada inyección.
En caso de agujas bloqueadas, los pacientes deben seguir las instrucciones descritas en las instrucciones de uso que acompañan al instructivo.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normatividad local.
Para ver instrucciones detalladas ver instructivo anexo.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis: XULTOPHY® se administra una vez al día por vía subcutánea. XULTOPHY® se puede administrar en cualquier momento del día, preferentemente a la misma hora.
La dosificación de XULTOPHY® varía en función de las necesidades individuales de cada paciente. Se recomienda optimizar el control glucémico a traves de un ajuste de la dosis basado en el nivel de glucosa en plasma en ayunas.
Puede ser necesario ajustar la dosis si el paciente umenta su actividad física, cambia su dieta habitual o sufre una enfermedad concomitante.
Para los pacientes que olviden administrarse una dosis es aconsejable que, cuando se den cuenta de ello, se le administren y, a continuación, reanuden su esquema de dosificación habitual de una vez al día. Se debe asegurar siempre que pasen un mínimo de 8 horas entre las inyecciones. Esta norma también es aplicable cuando no es posible la administración a la misma hora.
XULTOPHY® se administra en forma de unidades de dosis. Una unidad de dosis contiene 1 unidad de insulina degludec y 0.036 mg de liraglutida. La pluma precargada puede proporcionar desde 1 hasta 50 unidades de dosis en una inyección, en incrementos de 1 unidad de dosis. La dosis máxima diaria de XULTOPHY® es de 50 unidades de dosis (50 unidades de insulina degludec y 1.8 mg de liraglutida). El contador de dosis de la pluma muestra el número de dosis.
Añadido a medicamentos hipoglucemíantes orales:
La dosis inicial recomendada de XULTOPHY® es de 10 unidades (10 unidades de insulina degludec y 0.36 mg de liraglutida).
Es posible añadir XULTOPHY® a un tratamiento existente con antidiabéticos orales. Cuando se añade XULTOPHY® a una terapia con sulfonilurea, se debe considerar la disminución de la dosis de la sulfonilurea (Ver sección 10 Interacciones medicamentosas y de otro género)
Cambio desde agonista del receptor de GLP-1: Antes de iniciar el tratamiento con XULTOPHY®, se debe interrumpir el tratamiento con agonista del receptor de GLP-1. Al cambiar desde un agonista del receptor de GLP-1, la dosis inicial recomendada de XULTOPHY® es de 16 unidades de dosis (16 unidades de insulina degludec y 0.6 mg de liraglutida) (Ver sección 7. Precauciones Generales) No se debe superar la dosis inicial recomendada. Se debe considerar la acción prolongada si se cambia desde un agonista del receptor de GLP-1 de acción prolongada (por ejemplo, de administración una vez a la semana). Se debe iniciar el tratamiento con XULTOPHY® en el momento en el que se debería recibir la siguiente dosis del agonista del receptor de GLP-1 de acción prolongada. Se recomienda un estricto control glucémico durante el cambio y las primeras semanas después de éste.
Cambio desde insulina basal: Antes de iniciar el tratamiento con XULTOPHY®, se debe interrumpir el tratamiento con insulina basal. Al cambiar desde el tratamiento con insulina basal, la dosis inicial recomendada de XULTOPHY® es de 16 unidades de dosis (16 unidades de insulina degludec y 0.6 mg de liraglutida) (Ver sección 7 Precauciones generales y 5 Propiedades Farmacodinámicas). No se debe superar la dosis inicial recomendada. Se recomienda un estricto control glucémico durante el cambio y las primeras semanas después de este.
Titulación Clínica: (Ver sección S. Farmacodinamia. Eficacia y Seguridad Clínica. Titulación de XULTOPHY® e Insulina basal).
Poblaciones especiales:
Pacientes de edad avanzada (> 65 años): XULTOPHY® se puede utilizar en pacientes de edad avanzada. Es necesario monitorear el control glucémico y ajustar individualmente la dosis. La experiencia terapéutica en pacientes ≥ 75 años es limitada.
Insuficiencia renal: Cuando se usa XULTOPHY® en pacientes con insuficiencia renal leve o moderada, es necesario intensificar el control glucémico y ajustar individualmente la dosis. No se puede recomendar el uso de XULTOPHY® en pacientes con insuficiencia renal grave, incluidos los pacientes con enfermedad renal en etapa terminal (Ver sección 5 Propiedades Farmacodinámicas).
Insuficiencia hepática: XULTOPHY® puede usarse en pacientes con insuficiencia hepática leve o moderada. El monitoreo de Glucosa se intensificará y la dosis se ajustará de forma individual. Debido al componente de liraglutida, XULTOPHY® no está recomendado para uso en pacientes con insuficiencia hepática grave (ver SecciónS Propiedades Farmacodinámicas).
Población pediátrica: XULTOPHY® no debe adminisrarse en población pediátrica.
Forma de administración: XULTOPHY® sólo se debe administrar por vía subcutánea. XULTOPHY® no se debe administrar por vía intravenosa o intramuscular.
XULTOPHY® se administra por vía subcutánea mediante inyección en el muslo, la zona superior del brazo o el abdomen. Siempre se debe rotar el punto de inyección dentro de la misma zona para reducir el riesgo de la lipodistrofia. Para más instrucciones sobre la administración ver la sección 7 Precauciones generales.
XULTOPHY® no se debe extraer del cartucho de la pluma precargada en una jeringa (ver sección 7 Precauciones generales).
Los pacientes deben recibir instrucciones de usar siempre una nueva aguja. La reutilización de las agujas de la pluma de insulina aumenta el riesgo de agujas bloqueadas, lo que puede causar una subdosificación o una sobredosificación. En caso de agujas bloqueadas, los pacientes deben seguir las instrucciones descritas en las instrucciones de uso que acompañan al instructivo (ver sección 7 Precauciones generales, Precauciones especiales de eliminación y otras manipulaciones).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: Los datos disponibles relativos a la sobredosis de XULTOPHY® son limitados.
Se puede desarrollar hipoglucemia si el paciente recibe una dosis de XULTOPHY® superior a sus requerimientos:
• Los episodios hipoglucémicos leves se pueden tratar con administración oral de glucosa o productos azucarados. Por consiguiente, se recomienda que los pacientes diabéticos lleven siempre consigo productos azucarados.
• Los episodios hipoglucémicos graves, en los que el paciente no se puede administrar el tratamiento a sí mismo se pueden tratar con inyección intramuscular o subcutánea de glucagón (0.5 a 1 mg) administrada por una persona entrenada, o bien glucosa por vía intravenosa administrada por un profesional de la salud. Se debe administrar glucosa intravenosa, si el paciente no responde al glucagón en 10-15 minutos. Se recomienda la administración de carbohidratos orales al paciente una vez recuperada la conciencia, a fin de prevenir una recaída.
PRESENTACIONES:
Caja de cartón que contiene 1, 3 ó 5* pluma(s) precargada(s) con 3 mL cada una.
Caja de cartón que contiene 2 cajas de cartón, cada una con 5* plumas precargadas con 3 mL (10* plumas precargadas con 3 mL). Todas las presentaciones con instructivo anexo para uso del medicamento e instructivo anexo para uso del dispositivo médico.
No todas las presentaciones pueden estar comercializadas.
*Presentaciones no comercializadas en México.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Antes de la primera apertura: conservar en refrigeración (entre 2°C a 8ºC). No guardar cerca del congelador. No congelar. Conservar la pluma precargada con el capuchón puesto para protegerla de la luz.
Una vez abierto: conservar a un máximo de 30°C o conservar en refrigeración (entre 2ºC y 8ºC). No congelar. Conservar la pluma precargada con el capuchón puesto para protegerla de la luz.
Periodo de caducidad: 24 meses.
Una vez abierto: el producto se puede conservar durante 21 días a una temperatura máxima de 30ºC. El medicamento se debe desechar a los 21 días después de la primera apertura.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. No se deje al alcance de los niños. Su venta requiere receta médica.
Titular del Registro
Novo Nordisk A/S
Novo Alié, Bagsvéerd 2880
Dinamarca
Representante legal:
NOVO NORDISK MÉXICO, S.A. DE C.V.
Homero Núm. 1500, piso 3, Col. Polanco
C.P. 11560, Miguel Hidalgo
Ciudad de México, México.
Reg. Núm. 043M2018, SSA IV
®Marca Registrada