
ZALTRAPZIV
AFLIBERCEPT
Solución inyectable
1 Caja, 1 Frasco(s) ámpula, 100/4 mg/ml
1 Caja, 1 Frasco(s) ámpula, 200/8 mg/ml
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada frasco ámpula contiene:
Aflibercept 100 mg, 200 mg
Vehículo cbp 4 mL, 8 mL
INDICACIONES TERAPÉUTICAS:
ZALTRAPZIV® en combinación con quimioterapia a base de irinotecan-fluoropirimidina está indicado para pacientes con cáncer colorrectal metastásico (CCRM) tratados previamente con un régimen de oxaliplatino.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas:
Mecanismo de acción:
El factor de crecimiento endotelial vascular A y B (VEGF-A, VEGF-B, siglas en inglés) y el factor de crecimiento placentario (PIGF) son miembros de la familia de factores angiogénicos (VEGF) que pueden actuar como potentes factores mitogénicos, quimiotácticos y como factores de permeabilidad vascular en las células endoteliales, VEGF-A actúa a través de dos receptores de tirosina-cinasa, VEGFR-1 y VEGFR-2, presentes en la superficie de las celulas endoteliales. PIGF y VEGF-B sólo se unen a VEGFR-1, que también está presente en la superficie de los leucocitos. La activación excesiva de estos receptores por VEGF-A puede resultar en una neovascularización patológica y una permeabilidad vascular excesiva. El PIGF también está relacionado con la neovascularización patológica y el reclutamiento de células inflamatorias hacia los tumores.
ZALTRAPZIV®, conocido también como VEGF TRAP en la literatura científica, es una proteína de fusión recombinante que consta de las porciones de unión de VEGF de los dominios extracelulares de los receptores VEGF 1 y 2 humanos fusionadas a la porción Fc de la lgG1 humana. Aflibercept actúa como receptor señuelo uniéndose a VEGF-A con una mayor afinidad que los receptores nativos, al igual que a los ligandos relacionados PIGF y VEGF-B. Al actuar como trampa para ligandos, aflibercept previene la unión de ligandos endógenos con sus receptores análogos bloqueando asi la señalización mediada por receptores.
Aflibercept bloquea la activación de los receptores VEGF y la proliferación de las células endoteliales, inhibiendo así el crecimiento de nuevos vasos que suministran oxígeno y nutrientes a los tumores.
ZALTRAPZIV® se une a los factores humanos VEGF-A (constante de disociación en el equilibrio KD = 0.5 pM para VEGF A165 y 0.36 pM para VEGF-A121), PIGF (KD = 39 pM para PIGF-2), y VEGF-B (KD = 1.92 pM) para formar un complejo estable e inerte sin actividad biológica detectable.
Características Farmacodinámicas:
La administración de aflibercept a ratones con xenotrasplantes o alotrasplantes tumorales inhibió el crecimiento de varios tipos de cáncer.
Eficacia Clínica/Estudios Clínicos:
En un estudio aleatorizado, doble ciego, controlado con placebo se evaluaron la eficacia y seguridad de ZALTRAPZIV® en pacientes con cáncer colorrectal metastásico con tratamiento anterior a base de oxaliplatino con o sin bevacizumab previo. Un total de 1,226 pacientes fueron aleatorizados (1:1) para recibir ZALTRAPZIV® (N=612; 4 mg/kg en infusión IV de 1 hora el día 1) o placebo (N=614), en combinación con 5-fluorouracilo más irinotecan [FOLFIRI: irinotecan 180 mg/m2 en infusión IV de 90 minutos y leucovorina (ácido folínico) (dl racémico) 400 mg/m2 en infusión IV de 2 horas a la misma hora del día 1 usando una línea en Y, seguido de 5-FU 400 mg/m2 en bolo IV seguido de 5-FU 2,400 mg/m2 en infusión IV continua de 46 horas. Los ciclos de tratamiento en ambos brazos se repitieron cada 2 semanas. Los pacientes fueron tratados hasta la progresión de la enfermedad o una toxicidad inaceptable. El parámetro primario de eficacia fue la supervivencia global. La asignación del tratamiento se estratificó por el estado funcional ECOG (0 versus 1 versus 2) y la terapia previa con bevacizumab (si o no).
Las características demográficas estuvieron bien balanceadas entre los dos brazos de tratamiento (edad, raza, estado funcional ECOG y bevacizumab previo). De los 1,226 pacientes aleatorizados en el estudio la mediana de edad fue de 61 años, 58.6% fueron varones y 97.8% tuvieron un estado funcional (EF) ECOG basal de 0 ó 1. De los 1,226 pacientes aleatorizados, 89.4% y 90.2% de los pacientes tratados con los regímenes placebo/FOLFIRI y ZALTRAPZIV®/FOLFIRI, respectivamente, recibieron quimioterapia combinada previa a base de oxaliplatino en el estadio metastásico/avanzado. Aproximadamente 10% de los pacientes (10.4% y 9.8% tratados con los regímenes placebo/FOLFIRI y ZALTRAPZIV®/FOLFIRI, respectivamente) recibieron quimioterapia adyuvante previa a base de oxaliplatino y progresaron en un lapso de 6 meses después de completar la quimioterapia adyuvante. Los regímenes a base de oxaliplatino se administraron en combinación con bevacizumab en 373 pacientes (30.4%).
En la Figura 1 y en la Tabla 1 se encuentran resumidos los resultados de eficacia global para el régimen ZALTRAPZIV®/FOLFIRl en comparación con el régimen placebo/FOLFIRI.
Figura 1: Supervivencia Global (SG) (meses) - Curvas Kaplan-Meier por grupo de tratamiento - población IT.
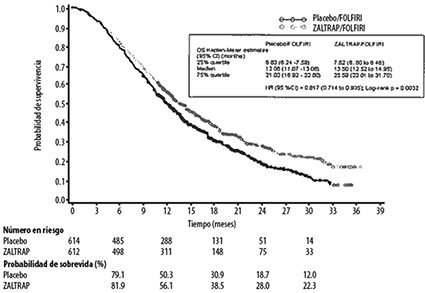
Tabla 1. Principales parámetros de eficaciaa - Población de IT
|
Placebo/FOLFIRI (N=614) |
ZALTRAPZIV®/FOLFIRI (N=612) |
|
|
Supervivencia global |
||
|
Número de eventos de muerte, n (%) |
460 (74.9%) |
403 (65.8%) |
|
Mediana de supervivencia global (IC 95%) (meses) |
12.06 (11.07 a 13.08) |
13.50 (12.52 a 14.95) |
|
Razón de riesgo estratificado (IC 95%) |
0.817 (0.714 a 0.935) |
|
|
Prueba de rango log estratificada, valor p |
0.0032 |
|
|
Supervivencia libre de Progresión (SLP)b 1,2 |
||
|
Número de eventos, n (%) |
454 (73.9%) |
393 (64.2%) |
|
Mediana de la SLP (IC 95%) (meses) |
4.67 (4.21 a 5.36) |
6.90 (6.51 a 7.20) |
|
Razón de riesgo estratificado (IC 95%) |
0.758 (0.661 a 0.869) |
|
|
Prueba de rango log estratificada, valor pb |
0.00007 |
|
|
Tasa de respuesta Global (RC+RP) (IC 95%) (%)c3 |
11.1 (8.5 a 13.8) |
19.8 (16.4 a 23.2) |
|
Prueba Cochran-Mantel-Haenszel estratificada, valor p |
0.0001 |
|
ª Estratificada por Estado Funcional ECOG (0 vs 1 vs 2) y Bevacizumab previo (sí vs no).
b SLP (con base en la evaluación del tumor por un Comité de Revisión Independiente, CRI): El umbral de significancia establecido en 0.0001.
e Tasa de respuesta objetiva global por CRI.
Se realizó un análisis de los factores de estratificación en relación al análisis de supervivencia global (SG) y supervivencia libre de progresión (PFS). Los resultados indicaron que fue observable un efecto consistente del tratamiento en cuanto a beneficios en SG a favor de los pacientes tratados con el esquema ZALTRAPZIV®/FOLFIRI con antecedentes de uso previo de bevacizumab, así como en pacientes sin tratamiento previo con bevacizumab. Los resultados por exposición a bevacizumab se resumen en la Tabla 2.
Tabla 2: SG y PFS por exposición previa a bevacizumaba - Población ITT
|
Placebo/FOLFIRI (N=614) |
ZALTRAPZIV®/FOLFIRI (N=612) |
|
|
Supervivencia global |
||
|
Pacientes con tratamiento previo con bevacizumab (n (%)) |
187(30.5%) |
186 (30.4%) |
|
Mediana SG (95% CI) (meses) |
11.7 (9.96 a 13.77) |
12.5 (10.78 a 15.47) |
|
Razón de riesgo (95% CI) |
0.862 (0.676 a 1.100) |
|
|
Pacientes sin tratamiento previo con bevacizumab (n (%)) |
427 (69.5%) |
426 (69.6%) |
|
Mediana SG (95% CI) (meses) |
12.4 (11.17 a 13.54) |
13.9 (12.72 a 15.64) |
|
Razón de riesgo (95% CI) |
0.788 (0.671 a 0.925) |
|
|
Supervivencia libre de progresión |
||
|
Pacientes con tratamiento previo con bevacizumab (n (%)) |
187 (30.5%) |
186 (30.4%) |
|
Mediana SG (95% CI) (meses) |
3.9 (3.02 a 4.30) |
6.7 (5.75 a 8.21) |
|
Razón de riesgo (95% CI) |
(0.66) (0.512 a 0.80) |
|
|
Pacientes sin tratamiento previo con bevacizumab (n (%)) |
427 (69.5%) |
426 (69.6%) |
|
Mediana SG (95% CI) (meses) |
5.4 (4.53 a 5.68) |
6.0 (6.87 a 7.20) |
|
Razón de riesgo (95% CI) |
0.797 (0.679 a 0.035) |
|
a Como determinante de IVRS
También se realizó un análisis de la SG y la PFS por ECOG PS. La razón de riesgo (95% IC) de supervivencia global fue 0.77 (0.64 a 0.93) para el estado general de ECOG 0 y 0.87 (0.71 a 1.06) para el desempeño ECOG estado 1. La razón de riesgo (95% IC) de la supervivencia libre de progresión fue de 0.76 (0.63 a 0.91) para ECOG estado funcional 0 y 0.75 (0.61 a 0.92) para el estado funcional ECOG 1.
En la tabla 3 se muestran los resultados del análisis post-hoc que excluye a los pacientes que presentaron progresión durante o dentro de los primeros 6 meses de la terapia adyuvante, tanto del subgrupo de pacientes previamente tratados con bevacizumab, como del subgrupo sin uso previo de bevacizumab.
Tabla 3. Análisis Post-hoc excluyendo pacientes con terapia adyuvante a,b
|
Placebo/FOLFIRI (N=550) |
ZALTRAPZIV®/FOLFIRI (N=552) |
|
|
Pacientes con tratamiento previo con bevacizumab excepto aquellos con progresión con la terapia adyuvante (n (%)) |
179 (32.5%) |
177 (32.1%) |
|
Mediana supervivencia global (95% CI) (meses) |
11.7 (9.66 a 13.27) |
13.8 (11.01 a 15.87) |
|
Razón de riesgo (95%) CI |
0.812 (0.498 a 0.835) |
|
|
Mediana PFS (95% CI) (meses) |
3.9 (3.02 a 4.30) |
6.7 (5.72 a 8.21) |
|
Razón de riesgo (95%) CI |
0.645 (0.498 a 0.835) |
|
|
Pacientes sin tratamiento previo con bevacizumab excepto aquellos con progresión con la terapia adyuvante (n (%)) |
371 (67.5%) |
375(67.9%) |
|
Mediana supervivencia global (95% CI) (meses) |
12.4 (11.17 a 13.54) |
13.7 (12.71 a 16.03) |
|
Razón de riesgo (95%) CI |
0.766 (0.645 a 0.908) |
|
|
Mediana PFS (95% CI) (meses) |
5.3 (4.50 a 5.55) |
6.9 (6.24 a 7.20) |
|
Razón de riesgo (95%) CI |
0.777 (0.655 a 0.921) |
|
ª Según determinado por IVRS.
b Supervivencia Global en población de intención de tratamiento (IT) excluyendo población de pacientes cuyo progreso durante o dentro de 6 meses de terapia adyuvante demostraron una RR (95% IC) de 0.78 (0.68 a 0.90) (mediana de SG (95% IC) con Placebo/FOLFIRI 11.9 meses (10.88 a 13.01)y con ZALTRAPZIV®/FOLFIRI 13.8 meses (12.68 a 15.44).
Los análisis por subgrupo para supervivencia global y supervivencia libre de progresión de acuerdo con la edad (< 65 años; ≥ 65 años), sexo, uso previo de bevacizumab, desempeño ECOG 0 y 1, presencia de metástasis hepática únicamente, antecedentes de hipertensión arterial y número de órganos involucrados, demostraron un efecto del tratamiento a favor del régimen ZALTRAPZIV®/FOLFIRI versus placebo/FOLFIRI. En el análisis por subgrupos de la supervivencia global, el beneficio observado fue consistente para la población global tanto < 65 años como ≥ 65 años de edad que recibieron ZALTRAPZIV®/FOLFIRI.
FARMACOCINÉTICA:
Se evaluó la farmacocinética no clínica y clínica de ZALTRAPZIV®
Se realizó un análisis fármaco cinético de poblaciones con los datos de 1,507 pacientes con varios tipos de cáncer avanzado que recibieron aflibercept en monoterapia o en combinación en dosis de 2 a 9 mg/kg administrado cada 2 a 3 semanas en infusión intravenosa de 1 hora. Se midieron las concentraciones plasmáticas del aflibercept libre o unido usando el ensayo de inmuno absorción ligado a enzimas (ELISA).
Absorción:
En los modelos tumorales preclínicos, las dosis biológicamente activas de aflibercept se correlacionaron con las dosis necesarias para producir una concentración de aflibercept libre circulante mayor al aflibercept unido a VEGF. La concentración circulante de aflibercept unido a VEGF aumenta con la dosis hasta que la mayor parte del VEGF disponible se encuentra unido. Un incremento mayor en la dosis de aflibercept da como resultado un incremento de la concentración de aflibercept libre relacionado con la dosis pero sólo un pequeño incremento en la concentración de aflibercept unido a VEGF.
En pacientes, ZALTRAPZIV® se administra en una dosis de 4 mg/kg IV cada dos semanas por lo que hay un exceso de aflibercept libre en comparación con el aflibercept unido a VEGF. En consonancia con la disposición del fármaco mediada por el blanco, el aflibercept libre exhibe una depuración no lineal en dosis menores a 2 mg/kg, posiblemente debido a la alta afinidad de unión del aflibercept al VEGF endógeno. La depuración lineal observada en el rango de dosis de 2 a 9 mg/kg se debe principalmente a los mecanismos biológicos no saturables de eliminación como el catabolismo de proteínas.
La concentración de aflibercept libre con el régimen de dosis recomendado de 4 mg/kg cada dos semanas, casi se alcanzó el estado de equilibrio en el segundo ciclo practicamente sin acumulación (tasa de acumulación 1.2 en el estado de equilibrio en comparación con la primera administración).
Distribución:
El volumen de distribución del aflibercept libre en el estado de equilibrio es de 8 L.
Metabolismo:
No se han realizado estudios de metabolismo con aflibercept ya que es una proteína. Se espera que el aflibercept se degrade hacia péptidos pequeños y aminoácidos individuales.
Eliminación:
El aflibercept libre se depura principalmente al unirse al VEGF endógeno para formar un complejo estable e inerte. Al igual que otras proteínas grandes, se espera que el aflibercept tanto libre como unido se depure más lentamente por mecanismos biológicos como el catabolismo proteolítico. El aflibercept unido a VEGF se elimina sin una disociación reversible apreciable ni la formación de inmunocomplejos de mayor orden.
En dosis de 3 mg/kg, los valores de la vida media, depuración, y del volumen de distribución en estado estacionario (Vss) de aflibercept libre fueron respectivamente de 4.5 días, 18.4 mL/kg/d, y 101 mL/kg.
Las proteínas de alto peso molecular no se eliminan por vía renal, por lo tanto, se espera una eliminación renal mínima de aflibercept.
Poblaciones especiales:
Pacientes pediátricos:
En un estudio Fase I.
Después de la administración intravenosa de 2.0 mg/kg, 2.5 mg/kg o 3.0 mg/kg cada dos semanas de ZALTRAPZIV® a 8 pacientes pediátricos (con edades de 5 a 17 años) con tumores sólidos, la vida media de eliminación de aflibercept libre, determinada después de la primera dosis, fue de aproximadamente 4 días (rango 3-6 días).
Ancianos:
No hubo un efecto de la edad sobre la farmacocinética de ZALTRAPZIV®.
Género:
A pesar de las diferencias en la depuración y el volumen de distribución del aflibercept libre entre varones y mujeres, no se observaron diferencias relacionadas con el género en la exposición al fármaco con una dosis de 4 mg/kg en el estudio fundamental.
Peso:
El peso tuvo un efecto sobre la depuración y el volumen de distribución del aflibercept libre con un incremento de 29% en la exposición al fármaco en pacientes ≥ 100 kg de peso.
Raza:
No hubo un efecto de los grupos étnicos y la raza sobre la farmacocinética de ZALTRAPZIV®.
Insuficiencia hepática:
No se han hecho estudios formales con ZALTRAPZIV® en pacientes con insuficiencia hepática.
En un análisis farmacocinético de poblaciones con datos de 1,507 pacientes con varios tipos de cáncer avanzado en tratamiento con ZALTRAPZIV® con o sin quimioterapia, 63 pacientes tuvieron insuficiencia hepática leve (bilirrubina total >1.0x -1.5x LNS y cualquier valor AST y 5 pacientes insuficiencia hepática moderada (bilirrubina total >1.5x-3x LNS y cualquier valor AST). En los pacientes con insuficiencia hepática leve y moderada no se observó un efecto sobre la depuración de aflibercept. No existen datos disponibles en pacientes con insuficiencia hepática severa (bilirrubina total >3x LNS y cualquier valor AST.
Insuficiencia renal:
No se han realizado estudios formales con ZALTRAPZIV® en pacientes con insuficiencia renal.
Se condujo un análisis farmacocinético de poblaciones con datos de 1,507 pacientes con varios tipos de cáncer avanzado en tratamiento con ZALTRAPZIV® con o sin quimioterapia. Esta población incluyó a 549 pacientes con insuficiencia renal leve (depuración de creatinina entre 50-80 ml/mln), 96 pacientes con insuficiencia renal moderada (depuración de creatinina entre 30-50 ml/min), y 5 pacientes con insuficiencia renal severa (depuración de creatinina < 30 ml/min). Este análisis farmacocinético de poblaciones reveló que no existen diferencias en la exposición sistémica (ABC) de aflibercept libre entre los pacientes con varios grados de insuficiencia renal con la dosis de 4 mg/kg de ZALTRAPZIV®.
CONTRAINDICACIONES:
ZALTRAPZIV® está contraindicado en pacientes con hipersensibilidad severa conocida al aflibercept o a cualquiera de los excipientes de ZALTRAPZIV® (ver sección de Precauciones generales).
Para las contraindicaciones relacionadas con el irinotecan, 5-FU y leucovorina, consultar, información actual del producto.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo:
Los estudios de reproducción en conejos preñados han mostrado que aflibercept es embriotoxico y teratogénico.
No existen datos sobre el uso de aflibercept en mujeres embarazadas. Puesto que la angiogénesis es crítica para el desarrollo fetal, la inhibición de la angiogénesis a consecuencia de la administración de ZALTRAPZIV® podría tener efectos adversos en el embarazo. No se recomienda el uso de ZALTRAPZIV® durante el embarazo o en mujeres que puedan quedar embarazadas. ZALTRAPZIV® debe usarse durante el embarazo sólo si los posibles beneficios justifican el riesgo potencial para el feto.
Se debe aconsejar a las mujeres en edad reproductiva evitar el embarazo durante el tratamiento con ZALTRAPZIV® y se les debe informar sobre el posible daño para el feto.
Con base en estudios en monos, la fertilidad de los varones y las mujeres puede quedar comprometida durante el tratamiento con ZALTRAPZIV® (ver sección de Precauciones en relación con los efectos de carcinogénesis, mutagénesis, teratogénesis y sobre la fertilidad). En los estudios, estos desenlaces fueron reversibles después de 8-18 semanas de discontinuación del tratamiento. Las mujeres en edad reproductiva y los varones fértiles deben usar un método efectivo de anticoncepción durante el tratamiento y durante un mínimo de 6 meses después de la última dosis.
Lactancia:
No se han realizado estudios para evaluar el impacto del ZALTRAPZIV® sobre la producción de leche, su presencia en la leche materna y sus efectos sobre el lactante.
Se desconoce si ZALTRAPZIV® se excreta en la leche humana. Debido a que muchos fármacos se excretan en la leche humana y a las posibles reacciones adversas serias de aflibercept en el lactante, se debe tomar la decisión entre discontinuar la lactancia o discontinuar el fármaco, tomando en cuenta la importancia del fármaco para la madre.
REACCIONES SECUNDARIAS Y ADVERSAS:
La seguridad de ZALTRAPZIV® en combinación con FOLFIRI se evaluó en 1,216 pacientes con cáncer colorrectal metastásico previamente tratados a los que se dio tratamiento con ZALTRAPZIV® 4 mg/kg IV (N=611) o placebo (N=605) cada dos semanas (un ciclo) en un estudio aleatorizado (1:1), doble ciego, controlado con placebo de fase III. Los pacientes recibieron una mediana de 9 ciclos del régimen ZALTRAPZIV®/FOLFIRI y 8 ciclos del régimen placebo/FOLFIRI.
Las reacciones adversas más comunes (todos los grados, incidencia ≥ 20%) reportadas con una incidencia por lo menos 2% mayor para el régimen ZALTRAPZIV®/FOLFIRI en comparación con el régimen placebo/FOLFIRI en orden decreciente de frecuencia fueron: leucopenia, diarrea, neutropenia, proteinuria, incremento de AST, estomatitis, fatiga, trombocitopenia, incremento en ALT, hipertensión, pérdida de peso, pérdida de apetito, epistaxis, dolor abdominal, disfonía, incremento en la creatinina sérica y cefalea (Tabla 4).
Las reacciones adversas grado 3-4 más comunes (≥ 5%) reportadas con una incidencia por lo menos 2% mayor para el régimen ZALTRAPZIV®/FOLFIRI en comparación con el régimen placebo/FOLFIRI en orden decreciente de frecuencia fueron: neutropenia, diarrea, hipertensión, leucopenia, estomatitis, fatiga, proteinuria y astenia (Tabla 4).
La tasa global de discontinuación del tratamiento a causa de reacciones adversas (todos los grados) fue de 26.8% versus 12.1% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI y el régimen placebo/FOLFIRI, respectivamente. Las reacciones adversas más frecuentes que condujeron a la discontinuación permanente en ≥ 1% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI fueron: astenia/fatiga, infecciones, diarrea, deshidratación, hipertensión, estomatitis, eventos de tromboembolismo venoso, neutropenia y proteinuria.
La dosis de ZALTRAPZIV® se modificó (reducción y/u omisión) en 16.7% de los pacientes en comparación 4.8% de los pacientes tratados placebo. El retraso > 7 días de los ciclos se presentó en 59.7% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 42.6% de los pacientes tratados con el régimen placebo/FOLFIRI.
La muerte por causas distintas a la progresión de la enfermedad en los 30 días posteriores a la última administración del tratamiento de estudio se reportó en 16/611 (2.6%) pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI y 6/605 (1.0%) pacientes tratados con el régimen placebo/FOLFIRI. Las causas de muerte en los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRl fueron: infección (incluyendo sepsis por neutropenia) en 4 pacientes, deshidratación en 2 pacientes, hipovolemia en 1 paciente, encefalopatía metabólica en 1 paciente, eventos respiratorios (insuficiencia respiratoria aguda, aspiración, neumonía, y embolia pulmonar) en 3 pacientes, trastornos GI (úlcera duodenal hemorrágica, inflamación GI, y obstrucción del intestino grueso) en 3 pacientes y muerte por causa desconocida en 2 pacientes.
La Tabla 4 presenta las reacciones adversas y las anormalidades de laboratorio (todos los grados) reportadas con una mayor incidencia (≥ 2%) en los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con los pacientes tratados con el régimen de placebo/FOLFIRI en el estudio en pacientes con CCRM.
Tabla 4 - Reacciones adversas y anormalidades de laboratorio (todos los grados) reportados con una mayor incidencia (≥ 2%) en los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con los pacientes tratados con el régimen placebo/FOLFIRl en el estudio en pacientes con CCRM
|
Principales Sistemas Órganos Término Preferente n(%) |
Placebo/FOLFIRI (N=605) |
ZALTRAPZIV®/FOLFIRI (N=611) |
||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Infecciones e infestaciones |
||||
|
Infección del Tracto Urinario |
37 (6.1%) |
5 (0.8%) |
56 (9.2%) |
5 (0.8%) |
|
Nasofaringitis |
15 (2.5%) |
0 |
28 (4.6%) |
0 |
|
Trastornos sanguíneos y del sistema linfático |
||||
|
Leucopenia* |
432 (72.4%) |
73 (12.2%) |
472 (78.3%) |
94 (15.6%) |
|
Neutropenia* |
336 (56.3%) |
176 (29.5%) |
409 (67.8%) |
221 (36.7%) |
|
Trombocitopenia* |
202 (33.8%) |
10(1.7%) |
286(47.4%) |
20 (3.3%) |
|
Neutropenia Febril |
10 (1.7%) |
10 (1.7%) |
26 (4.3%) |
26(4.3%) |
|
Trastornos del metabolismo y la nutrición |
||||
|
Disminución del apetito |
144 (23.8%) |
11 (1.8%) |
195 (31.9%) |
21 (3.4%) |
|
Deshidratación |
18 (3.0%) |
8 (1.3%) |
55 (9.0%) |
26 (4.3%) |
|
Trastornos del sistema nervioso |
||||
|
Cefalea |
53 (8.8%) |
2 (0.3%) |
136 (22.3%) |
10 (1.6%) |
|
Trastornos Vasculares |
||||
|
Hipertensión |
65 (10.7%) |
9 (1.5%) |
252 (41.2%) |
117 (19.1%) |
|
Trastornos Respiratorios, torácicos y del mediastino |
||||
|
Epistaxis |
45 (7.4%) |
0 |
169 (27.7%) |
1 (0.2%) |
|
Disfonía |
20 (3.3%) |
0 |
165 (25.4%) |
3 (0.5%) |
|
Disnea |
52 (8.6%) |
5 (0.8%) |
72 (11.8%) |
5 (0.8%) |
|
Dolor orofaríngeo |
19 (3.1%) |
0 |
48 (5.5%) |
1 (0.2%) |
|
Rinorrea |
11 (1.8%) |
0 |
96 (6.2%) |
0 |
|
Trastornos Gastrointestinales |
||||
|
Diarrea |
342 (56.5%) |
47 (7.8%) |
423 (69.2%) |
118 (19.3%) |
|
Estomatitis |
199 (32.9%) |
28 (4.6%) |
306 (50.1%) |
78 (12.8%) |
|
Dolor Abdominal |
143 (23.6%) |
14 (2.3%) |
164 (26.8%) |
27 (4.4%) |
|
Dolor Abdominal Superior |
48 (7.9%) |
6 (1.0%) |
66 (10.8%) |
7 (1.1%) |
|
Hemorroides |
13 (2.1%) |
0 |
35 (5.7%) |
0 |
|
Hemorragia Rectal |
15 (2.5%) |
3 (0.5%) |
32 (5.2%) |
4 (0.7%) |
|
Proctalgia |
11 (1.8%) |
2 (0.3%) |
32 (5.2%) |
2 (0.3%) |
|
Estomatitis aftosa |
14 (2.3%) |
0 |
30 (4.9%) |
4 (0.7%) |
|
Dolor de dientes |
5 (0.8%) |
0 |
19 (3.1%) |
0 |
|
Trastornos de la piel y tejido subcutáneo |
||||
|
Síndrome de Eritrodisestesia Palmo-Plantar |
26 (4.3%) |
3 (0.5%) |
67 (11.0%) |
17 (2.8%) |
|
Hiperpigmentación de la piel |
17 (2.8%) |
0 |
50 (8.2%) |
0 |
|
Trastornos renales y urinarios |
||||
|
Proteinuria** |
246 (40.7%) |
7 (1.2%) |
380 (62.2%) |
48 (7.9%) |
|
Incremento en la creatinina sérica* |
108 (18.1%) |
3 (0.5%) |
136 (22.6%) |
0 |
|
Trastornos Generales y condiciones en el sitio de administración |
||||
|
Fatiga |
236 (39.0%) |
47 (7.8%) |
292 (47.8%) |
77 (12.6%) |
|
Astenia |
80 (13.2%) |
18 (3.0%) |
221 (18.3%) |
31 (5.1%) |
|
lnvestigaciones |
||||
|
Incremento en AST* |
296 (50.2%) |
10 (1.7%) |
339 (57.5%) |
18 (3.1%) |
|
Incremento en ALP* |
221 (37.1%) |
13 (2.2%) |
284 (47.3%) |
16 (2.7%) |
|
Pérdida de Peso |
87 (14.4%) |
5 (0.8%) |
195 (91.9%) |
16 (2.6%) |
Nota: Las Reacciones Adversas se reportaron usando la versión MEDDRA13.1 y se graduaron con el NCI CIC (versión) 3.0.
* Con base en valores de laboratorio (porcentajes obtenidos con los pacientes con evaluaciones de laboratorio).
** Recopilación de datos clínicos y de laboratorio.
En el estudio fundamental de aflibercept en pacientes con CCRM, los eventos adversos y las anormalidades de laboratorio que se presentaron en ≥ 20% de los pacientes y que fueron comparables entre los grupos (sin una incidencia ≥ 2% en el régimen ZALTRAPZIV®/FOLFIRI) fueron: anemia, náuseas, vómito, estreñimiento, alopecia, incremento en la fosfatasa alcalina e hiperbilirrubinemia.
Hemorragia:
Los pacientes tratados con ZALTRAPZIV® tuvieron un mayor riesgo de hemorragia, incluyendo eventos de hemorragia severa y en ocasiones fatal. En el estudio fundamental en pacientes con CCRM se reportaron episodios de sangrado/hemorragia (todos los grados) en 37.8% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 19.0% de los pacientes tratados con el régimen placebo/FOLFIRI. La forma más común de hemorragia reportada fue epistaxis menor (grado 1-2) en 27.7% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI. La hemorragia grado 3-4, incluyendo hemorragia gastrointestinal, hematuria, y hemorragia posterior al procedimiento se reportó en 2.9% de los pacientes en tratamiento con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 1.7% de los pacientes en tratamiento con el régimen placebo/FOLFIRI. En otros estudios, se observó hemorragia intracraneal severa y hemorragia pulmonar/hemoptisis incluyendo eventos fatales en pacientes en tratamiento con ZALTRAPZIV® (ver sección Precauciones generales).
Insuficiencia cardiaca y disminución de la fracción de eyección:
En pacientes tratados con ZALTRAPZIV® han sido reportados insuficiencia cardiaca y disminución de la fracción de eyección. Los pacientes deben ser monitoreados para detectar signos y síntomas de insuficiencia cardiaca y disminución de la fracción de eyección.
Descontinuar ZALTRAPZIV® en pacientes que sufren de insuficiencia cardiaca y disminución de la fracción de eyección.
Perforación gastrointestinal:
En pacientes tratados con ZALTRAPZIV® se ha reportado perforación gastrointestinal (GI) incluyendo perforación GI fatal. En el estudio fundamental en pacientes con CCRM se reportó perforación GI (todos los grados) en 3 de 611 pacientes (0.5%) tratados con el régimen ZALTRAPZIV®/FOLFIRI y 3 de 605 pacientes (0.5%) tratados con el régimen placebo/FOLFIRI. Se observaron eventos de perforación GI grado 3-4 en 3 pacientes (0.5%) tratados con el régimen ZALTRAPZIV®/FOLFIRI y en 2 pacientes (0.3%) tratados con el régimen placebo/FOLFIRI. En los 3 estudios clínicos controlados con placebo de fase III (poblaciones con cáncer colorrectal, pancreático y pulmonar), la incidencia de perforación GI (todos los grados) fue de 0.8% en los pacientes tratados con ZALTRAPZIV® y 0.3% en los pacientes tratados con placebo. Se observaron eventos de perforación GI grado 3-4 en 0.8% de los pacientes tratados con ZALTRAPZIV® y 0.2% de los pacientes tratados con placebo (ver sección de Precauciones generales).
Formación de fístulas:
En pacientes tratados con ZALTRAPZIV® se ha observado formación de fístulas en sitios gastrointestinales y no gastrointestinales. En el estudio fundamental en pacientes con CCRM se reportaron fístulas (anales, enterovesicales, enterocutáneas, colovaginales, intestinales) en 9 de 611 pacientes (1.5%) tratados con el régimen ZALTRAPZIV®/FOLFIRI y 3 de 605 pacientes (0.5%) tratados con el régimen placebo FOLFIRI. Se observó formación de fístulas GI grado 3 en 2 pacientes tratados con ZALTRAPZIV® (0.3%) y en 1 paciente tratado con placebo (0.2%) (ver sección de Precauciones generales).
Hipertensión:
En pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI se ha observado un incremento en el riesgo de hipertensión grado 3-4 (incluyendo hipertensión y un caso de hipertensión esencial). En el estudio fundamental en pacientes con CCRM, se reportó hipertensión grado 3 (que requiere ajustar la terapia antihipertensiva existente o tratamiento con más de un fármaco) en 1.5% de los pacientes tratados con el régimen placebo/FOLFIRI y 19.1% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI. Se reportó hipertensión (crisis hipertensiva) grado 4 en 1 paciente (0.2%) tratado con el régimen ZALTRAPZIV®/FOLFIRI. En 54% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI que desarrollaron hipertensión grado 3-4, los síntomas iniciaron durante los primeros dos ciclos de tratamiento (ver sección de Precauciones generales.
Eventos tromboembólicos arteriales:
En el estudio fundamental en pacientes con CCRM se reportaron casos de ETA (incluyendo ataque isquémico transitorio, accidente cerebrovascular, angina de pecho, trombo intracardiaco, infarto del miocardio, embolismo arterial, y colitis isquémica) en 2.6% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI y 1.5% de los pacientes tratados con el régimen placebo/FOLFIRI. Los eventos grado 3-4 se presentaron en 11 pacientes (1.8%) tratados con el régimen ZALTRAPZIV®/FOLFIRI y 3 pacientes (0.5%) tratados con el régimen placebo/FOLFIRI (ver sección de Precauciones generales).
Proteinuria:
En el estudio fundamental en pacientes con CCRM se reportó proteinuria (compilada a partir de los datos clínicos y de laboratorio) en 62.2% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 40.7% de los pacientes tratados con el régimen placebo/FOLFIRI. La proteinuria grado 3-4 se presentó en 7.9% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 1.2% de los pacientes tratados con el régimen placebo/FOLFIRI. El Síndrome nefrótico se presentó en 2 pacientes (0.5%) tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con ninguno de los pacientes tratados con el régimen placebo/FOLFIRI. Un paciente tratado con el régimen ZALTRAPZIV®/FOLFIRI que se presentó con proteinuria e hipertensión fue diagnosticado con microangiopatía trombótica (MAT) (ver sección de Precauciones generales).
Neutropenia y complicaciones por neutropenia:
En el estudio fundamental en pacientes con CCRM se observó neutropenia grado 3-4 en 36.7% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 29.5% de los pacientes tratados con el régimen placebo/FOLFIRI. La complicación por neutropenia grado 3-4 más común fue la neutropenia febril con una incidencia de 4.3% en los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 1.7% en los pacientes tratados con el régimen placebo/FOLFIRI. La infección/sepsis por neutropenia grado 3-4 se presentó en 1.5% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI y 1.2% de los pacientes tratados con el régimen placebo/FOLFIRI (ver sección de Precauciones generales).
Diarrea y deshidratación:
En el estudio fundamental en pacientes con CCRM se reportó diarrea grado 3-4 en 19.3% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 7.8% de los pacientes tratados con el régimen placebo/FOLFIRI. Se reportó deshidratación grado 3-4 en 4.3% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en comparación con 1.3% de los pacientes tratados con el régimen placebo/FOLFIRI (ver sección de Precauciones generales).
Reacciones de hipersensibilidad:
En el estudio fundamental en pacientes con CCRM se reportaron reacciones severas de hipersensibilidad en 0.3% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI y 0.5% de los pacientes tratados con el régimen placebo/FOLFIRI (ver sección de Precauciones generales).
Compromiso de la cicatrización de heridas:
El tratamiento con ZALTRAPZIV® está asociado con un posible compromiso de la cicatrización de heridas (dehiscencia de heridas, filtración anastomótica). En el estudio fundamental en pacientes con CCRM se reportó compromiso de la cicatrización de heridas en 3 pacientes (0.5%) tratados con el régimen ZALTRAPZIV®/FOLFIRI y 5 pacientes (0.8%) tratados con el régimen placebo/FOLFIRI. Se reportó compromiso de la cicatrización de heridas grado 3 en 2 pacientes (0.3%) tratados con el régimen ZALTRAPZIV®/FOLFIRI y en ninguno de los pacientes tratados con el régimen placebo/FOLFIRI (ver sección de Precauciones generales).
Síndrome de leucoencefalopatía posterior reversible (SLPR):
En el estudio fundamental de fase III en pacientes con CCR metastásico no se reportaron casos de SLPR. El SLPR se reportó en pacientes tratados con ZALTRAPZIV® en monoterapia (0.5%) y en combinación con otras quimioterapias (ver sección de Precauciones generales).
Infecciones:
Las infecciones se presentaron con mayor frecuencia en pacientes en tratamiento con el régimen ZALTRAPZIV® FOLFIRl (46.2%, todos los grados; 12.3%, grado 3-4) en comparación con los pacientes en tratamiento con el régimen placebo/FOLFIRI (32.7%, todos los grados; 6.9%, grado 3-4), incluyendo infección del tracto urinario, nasofaringitis, infección del tracto respiratorio superior, neumonía, infección en el sitio del catéter e infección dental.
Eventos de tromboembolismo venoso (TEV):
Los términos de eventos adversos agrupados dentro de los eventos de tromboembolismo venoso (TEV) incluyen trombosis venosa profunda y embolia pulmonar. En el estudio fundamental en pacientes con CCRM, el TEV en todos los grados se presentó en 9.3% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI y 7.3% de los pacientes tratados con el régimen placebo/FOLFIRI. El TEV grado 3-4 se presentó en 7.9% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI y en 6.3% de los pacientes tratados con el régimen placebo/FOLFIRI. La embolia pulmonar se presentó en 4.6% de los pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI y 3.5% de los pacientes tratados con el régimen placebo/FOLFIRI.
Poblaciones especiales:
Ancianos:
De los 611 pacientes tratados con el régimen ZALTRAPZIV®/FOLFIRI en el estudio fundamental en pacientes con CCRM, 172 (28.2%) tuvieron ≥ 65 y < 75 años de edad y 33 (5.4%) ≥ 75 años de edad. Los pacientes ancianos (≥ 65 años de edad) pueden ser más propensos a experimentar reacciones adversas. La incidencia de diarrea, mareo, astenia, pérdida de peso y deshidratación fue ≥ 5% más alta en pacientes ancianos en comparación con pacientes más jóvenes. Los pacientes ancianos deben ser monitoreados cuidadosamente en relación con el desarrollo de diarrea y posible deshidratación (ver sección de Precauciones generales).
Insuficiencia renal:
En pacientes en tratamiento con ZALTRAPZIV®, las reacciones adversas en pacientes con insuficiencia renal leve a nivel basal en los estudios de fase III combinados (N=352) fueron comparables con las de los pacientes sin insuficiencia renal (N=642). Un limitado número de pacientes con insuficiencia renal moderada severa a nivel basal (N=49) fueron tratados con ZALTRAPZIV®. En estos pacientes, los eventos no renales fueron en general comparables a los pacientes sin insuficiencia renal, excepto por una incidencia > 10% mayor en la deshidratación (todos los grados).
lnmunogenicidad:
Al igual que con todas las proteínas terapéuticas, existe un potencial de inmunogenicidad con ZALTRAPZIV®.
En todos los estudios clínicos oncológicos se observó una incidencia similar en la respuesta de bajo título de anticuerpos antifármaco (AAF) (a partir del valor basal) en los pacientes tratados con placebo y ZALTRAPZIV® (3.3% y 3.8%, respectivamente). No se detectaron respuestas de alto título de anticuerpo en ningún paciente. Diecisiete pacientes tratados con ZALTRAPZIV® (1.6%) y dos pacientes tratados con placebo (0.2%) también fueron positivos en el ensayo de anticuerpos neutralizantes. En el estudio fundamental en pacientes con CCRM, en el ensayo AAF hubo mayor incidencia de respuestas positivas en los pacientes tratados con el régimen placebo/FOLFIRI [18/526 (3.4%)] en comparación con el régimen ZALTRAPZIV®/FOLFIRI [8/521 (1.5%)]. Los resultados positivos en el ensayo de anticuerpos neutralizantes en el estudio fundamental en pacientes con CCRM también fueron más altos en los pacientes tratados con el régimen placebo/FOLFIRI [2/526 (0.38%)] en comparación con el régimen ZALTRAPZIV®/FOLFIRI [1/521 (0.19%)]. No se observó ningún impacto sobre el pertil farmacocinético de aflibercept en pacientes positivos en los ensayos de inmunogenicidad.
Dada la similitud de los resultados en el ensayo de AAF en pacientes tratados con placebo o ZALTRAPZIV®, es muy probable que se sobreestime la incidencia real de inmunogenicidad con ZALTRAPZIV® con base en estos ensayos.
Los datos de inmunogenicidad son altamente dependientes de la sensibilidad y especificidad del ensayo. Adicionalmente, la incidencia observada de positividad en un ensayo de anticuerpos puede estar influenciada por diversos factores, incluyendo el manejo de la muestra, el momento de la recolección de la muestra, los medicamentos concomitantes y la enfermedad subyacente. Por estas razones la comparación de la incidencia de anticuerpos contra ZALTRAPZIV® con la incidencia de anticuerpos con otros productos puede ser engañosa.
Experiencia post-comercialización:
Las siguientes reacciones adversas se han reportado durante el uso posterior a la aprobación de ZALTRAPZIV®. Las reacciones adversas se derivan de notificaciones espontáneas y, por tanto, la frecuencia es “no conocida” (no puede estimarse a partir de los datos disponibles).
Trastornos musculoesqueléticos y del tejido conjuntivo:
Osteonecrosis de la mandíbula:
Los casos de osteonecrosis de la mandíbula (ONM) han sido reportados en pacientes tratados con aflibercept, primariamente en aquellos pacientes en los que se habían identificado factores de riesgo de osteonecrosis de la mandíbula, incluyendo el uso de bisfosfonatos y/o procedimientos dentales invasivos.
Sobredosis:
No ha habido reportes de sobredosis con ZALTRAPZIV®. No hay información sobre la seguridad de ZALTRAPZIV® administrado en dosis mayores a 7 mg/kg cada 2 semanas o 9 mg/kg cada 3 semanas. Los eventos adversos más comúnmente observados con estas dosis fueron similares a los observados con dosis terapéuticas.
No existe un antídoto específico para la sobredosis de ZALTRAPZIV®. Los casos de sobredosis se deben manejar con medidas de soporte apropiadas, particularmente en relación con el monitoreo y tratamiento de la hipertensión y proteinuria y el paciente debe permanecer bajo estricta supervisión médica para monitorear cualquier reacción farmacológica adversa (ver sección Reacciones secundariasy adversas).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Carcinogenicidad:
No se realizaron estudios para evaluar la carcinogenicidad de ZALTRAPZIV®.
Mutagenicidad:
No se realizaron estudios para evaluar la mutagenicidad de ZALTRAPZIV®.
Genotoxicidad:
No se realizaron estudios para evaluar la genotoxicidad de ZALTRAPZIV®.
Teratogenicidad:
Se ha demostrado que ZALTRAPZIV® es embriotóxico y teratogénico al administarse en conejos preñados cada 3 días durante el periodo organogénesis (días 6 a 18 de gestación) en dosis de aproximadamente 1 a 15 veces la dosis para humanos de 4 mg/kg cada 2 semanas. Los efectos observados incluyeron la disminución del peso corporal materno, incremento en el número de reabsorciones fetales y una mayor incidencia de malformaciones externas (incluyendo anasarca, hernia umbilical, hernia diafragmática, gastrosquisis, paladar hendido, ectrodactilia y atresia), viscerales (en corazón, grandes vasos y arterias) y esqueléticas (incluyendo fusión de vértebras, de esternón y de costillas) del feto.
Alteración de la fertilidad:
No se han realizado estudios específicos con ZALTRAPZIV® en animales para evaluar en efecto sobre la ferlilidad.
Sin embargo, los resultados de estudios de dosis repetida sugieren que existe la posibilidad de que aflibercept altere la función reproductiva y la fertilidad.
En hembras sexualmente maduras de monos cynomolgus tratados intravenosamente (IV) durante 6 meses, fue evidente la inhibición de la función ovárica y del desarrollo folicular por la disminución del peso de los ovarios, la disminución de la cantidad de tejido lúteo, la disminución en el número de folículos maduros, la atrofia del endometrio y el miometrio uterino, la atrofia vaginal, la abrogación de los picos de progesterona y los sangrados menstruales por la administración de 3 mg/kg.
En machos sexualmente maduros de monos cynomolgus tratados intravenosamente (IV) durante 6 meses, se observó una disminución en la movilidad del esperma y un incremento en la incidencia de anormalidades morfológicas en los espermatozoides por la administración de 3 mg/kg.
Los efectos del ZALTRAPZIV® IV sobre la función reproductiva y la fertilidad en monos se presentaron con exposiciones cercanas a la obtenida con la dosis terapéutica recomendada en pacientes. Estos efectos fueron completamente reversibles en un lapso de 8-18 semanas después de la última inyección.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
No se han llevado a cabo estudios formales para evaluar posibles interacciones fármaco-fármaco entre aflibercept y otros medicamentos.
La medición de las concentraciones libre y unida a proteínas de aflibercept en estudios de combinación mostró resultados comparables a aquellos obtenidos con el agente solo, lo cual sugiere que tales combinaciones (con oxaliplatino, cisplatino, 5-fluorouracilo -5-FU-, irinotecan, docetaxel, pemetrexed, gemcitabina y erlotinib) no ejerce impacto sobre la farmacocinética de aflibercept.
Con base en estudios de combinación fase I, y comparando con la información histórica y publicada disponible, aflibercept no afecta la farmacocinética de irinotecan, 5-FU, oxaliplatino, cisplatino, docetaxel, pemetrexed, gemcitabina y erlotinib.
ZALTRAPZIV® es compatible con:
• Equipos de infusión hechos de cloruro de polivinilo (PVC, siglas en inglés) que contenga di(2-etilhexil) ftalato (DEHP, siglas en inglés), PVC libre de DEHP que contenga trioctil-trimellitato (TOTM), PVC cubierto de polietileno, poliuretano, o polipropileno;
• Bolsas de infusión hechas de PVC que contenga DEHP o poliolefin;
• Filtros hechos de polietersulfona.
No use filtros hechos de fluoruro de polivinilideno (PVDF, siglas en inglés) o nailon.
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros productos medicinales o diluyentes excepto con aquellos mencionados en la sección de Dosis y vía de administración inciso D.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
En los estudios no se evaluó la interferencia de ZALTRAPZIV® con las pruebas de laboratorio y/o diagnóstico.
PRECAUCIONES GENERALES:
Hemorragia:
Los pacientes tratados con ZALTRAPZIV® tienen un mayor riesgo de hemorragia, incluyendo eventos de hemorragia severa y en ocasiones fatal (ver sección de Reacciones adversas).
Los pacientes deben ser monitoreados en relación con los signos y síntomas de hemorragía GI y otras hemorragias severas. No administrar ZALTRAPZIV® en pacientes con hemorragia severa (ver sección Dosis y vía de administración).
Insuficiencia cardiaca y disminución de la fracción de eyección:
En pacientes tratados con ZALTRAPZIV® han sido reportados insuficiencia cardiaca y disminución de la fracción de eyección. Los pacientes deben ser monitoreados para detectar signos y síntomas de insuficiencia cardiaca y disminución de la fracción de eyección.
Descontinuar ZALTRAPZIV® en pacientes que sufren de insuficiencia cardiaca y disminución de la fracción de eyección.
Perforación gastrointestinal:
En pacientes tratados con ZALTRAPZIV® se ha reportado perforación gastrointestinal (Gl) incluyendo la perforación GI fatal (ver sección Reacciones secundarias y adversas).
Los pacientes deben ser monitoreados con respecto a los signos y síntomas de perforación GI. Discontinuar la terapia de ZALTRAPZIV® en pacientes que experimente perforación GI (ver sección de Dosis y vía de administración).
Formación de fístulas:
En pacientes tratados con ZALTRAPZIV® se ha observado formación de fístulas en sitios gastrointestinales y no gastrointestinales (ver sección Reacciones secundarias y adversas).
Discontinuar la terapia de ZALTRAPZIV® en pacientes que desarrollen fístulas (ver sección Dosis y vía de administración).
Hipertensión:
En pacientes en tratamiento con un régimen ZALTRAPZIV®/FOLFIRI se ha observado un mayor riesgo de hipertensión grado 3-4 (incluyendo hipertensión y un caso de hipertensión esencial) (ver sección Reacciones secundarias y adversas).
Durante el tratamiento con ZALTRAPZIV® se recomienda monitorear la presión arterial cada dos semanas o según esté clínicamente indicado. En caso de hipertensión, tratar con la terapia antihipertensiva apropiada y monitorear la presión arterial regularmente. Suspender la terapia de ZALTRAPZIV® en pacientes con hipertensión no controlada. En caso de recurrencia de hipertensión severa, suspender hasta su control y reducir la dosis de ZALTRAPZIV® a 2 mg/kg en ciclos subsecuentes. ZALTRAPZIV® se debe discontinuar permanentemente en caso de crisis hipertensiva o encefalopatía hipertensiva (ver sección Dosis y vía de administración).
Actividad física:
Los pacientes en tratamiento con ZALTRAPZIV® y con antecedentes de enfermedad cardiovascular clínicamente importante, como enfermedad de arterias coronarias o insuficiencia cardiaca congestiva, deben tener precaución al llevar a cabo ejercicio físico. No existe experiencia en estudios clínicos con ZALTRAPZIV® en pacientes con insuficiencia cardiaca clase lll o IV NYHA (New York Heart Association).
Eventos tromboembólicos arteriales:
En pacientes que han recibido ZALTRAPZIV® se han observado eventos tromboembólicos arteriales (ETA) (incluyendo ataque isquémico transitorio, accidente cerebrovascular, angina de pecho, trombo intracardiaco, infarto del miocardio, embolismo arterial y colitis isquémica) (ver sección de Reacciones secundarias y adversas).
Discontinuar ZALTRAPZIV® en pacientes que experimenten un ETA (ver sección Dosis y vía de administración).
Proteinuria:
En pacientes tratados con ZALTRAPZIV® se ha observado proteinuria severa, síndrome nefrótico y microangiopatía trombótica (MAT) (ver sección de Reacciones secundarias y adversas).
Durante la terapia con ZALTRAPZIV® monitorear el desarrollo o empeoramiento de la proteinuria por análisis de orina con tira reactiva y/o la proporción proteína-creatinina urinaria (PPCU). En pacientes con una tira reactiva de ≥ 2 + para proteína o una PPCU > 1 o una proporción proteína-creatinina > 100 mg/mmol debe recolectarse orina a lo largo de 24 horas.
Suspender la administración de ZALTRAPZIV® en presencia de proteinuria ≥ 2 gramos/24 horas y reasumir cuando la proteinuria sea < 2 gramos/24 horas. En caso de recurrencia, suspender hasta obtener un valor < 2 gramos/24 horas y reducir la dosis de ZALTRAPZIV® a 2 mg/kg. Discontinuar la terapia de ZALTRAPZIV® en pacientes que desarrollen síndrome nefrótico o MAT (ver sección Dosis y vía de administración).
Neutropenia y complicaciones por neutropenia:
Con el régimen de ZALTRAPZIV®/FOLFIRI se reportó una mayor incidencia de complicaciones por neutropenia (neutropenia febril e infección por neutropenia) (ver sección de Reacciones secundarias y adversas).
Se recomienda el recuento sanguíneo completo con un conteo diferencial a nivel basal y antes del inicio de cada ciclo de ZALTRAPZIV®. La administración de ZALTRAPZIV®/FOLFIRI debe retrasarse hasta que el recuento de neutrófilos sea ≥ 1.5 x 109/L (ver sección Dosis y vía de administración). Se debe considerar el uso terapéutico de G-CSF desde la primera presentación de neutropenia grado ≥ 3 y profilaxis secundaria en pacientes con alto riesgo de complicaciones de neutropenia.
Diarrea y deshidratación:
Con el régimen ZALTRAPZIV®/FOLFIRI hubo una mayor incidencia de diarrea severa (ver sección de Reacciones secundarias y adversas).
Se debe instituir una modificación de la dosis del régimen FOLFIRI (ver sección de Dosis y vía de administración) y la administración de medicamentos antieméticos y rehidratación.
Reacciones de hipersensibilidad:
En el estudio fundamental (VELOUR) en pacientes con CCRM, se reportaron reacciones severas de hipersensibilidad posteriores al régimen ZALTRAPZIV®/FOLFIRI (ver sección de Reacciones secundarias y adversas).
En caso de que se presente una reacción de hipersensibilidad (incluyendo broncoespasmo, disnea angioedema y anafilaxia), se debe discontinuar el tratamiento y administrar la terapia médica apropiada (ver sección de Dosis y vía de administración).
En caso de una reacción de hipersensibilidad leve a moderada (incluyendo enrojecimiento de la piel, erupción cutánea, urticaria y prurito), suspender temporalmente el tratamiento hasta que se reserva la reacción. Tratar con corticosteroides y/o antihistamínicos según esté indicado clínicamente. En los siguientes ciclos se puede considerar el pretratamiento con corticosteroides y/o antihistamínicos (ver sección de Dosis y vía de administración). Se debe tener especial precaución en pacientes con antecedentes de reacciones de hipersensibilidad, ya que se han observado reacciones recurrentes en la readministración de aflibercept aun a pesar del tratamiento profiláctico.
Compromiso de la cicatrización de heridas:
ZALTRAPZIV® afecta la cicatrización de heridas en modelos animales.
El tratamiento con ZALTRAPZIV® está asociado con un posible compromiso de la cicatrización de heridas (dehiscencia de heridas, filtración anastomótica) (ver sección de Reacciones secundarias y adversas).
Suspender ZALTRAPZIV® por lo menos durante 4 semanas antes de la cirugía electiva.
Se recomienda no iniciar ZALTRAPZIV® por lo menos durante 4 semanas después de una cirugía mayor y hasta que la herida quirúrgica haya cicatrizado completamente. Para una cirugía menor como la colocación de un puerto de acceso venoso central, una biopsia o extracción dental, ZALTRAPZIV® puede iniciarse/reasumirse una vez que la herida quirúrgica haya cicatrizado completamente. Discontinuar ZALTRAPZIV® en pacientes con compromiso de la cicatrización de la herida que requieran intervención médica (ver sección de Dosis y vía de administración).
Síndrome de leucoencefalopatía posterior reversible (SLPR):
También conocido como síndrome de encefalopatía reversible posterior. No se reportó ni un solo caso de SLPR en el estudio VELOUR; no obstante, existen reportes de casos de SLPR en pacientes tratados en monoterapia con aflibercept y en combinación con quimioterapia. El SLPR se puede manifestar mediante estado mental alterado, crisis convulsivas, náusea, vómito, cefalea o trastornos visuales. El diagnóstico de SLPR se confirma mediante Resonancia Magnética. En un paciente con sospecha o diagnóstico confirmado de SLPR se debe discontinuar el tratamiento con aflibercept.
Uso parenteral exclusivamente:
ZALTRAPZIV® es una solución hiperosmótica, la cual no está formulada para ser compatible con el medio intraocular; por lo tanto, ZALTRAPZIV® no debe administrarse como inyección intravítrea.
Conducir un vehículo o realizar otras tareas peligrosas:
No se han realizado estudios sobre los efectos de ZALTRAPZIV® en la capacidad para conducir o utilizar máquinas.
El paciente debe ser advertido de no manejar ni utilizar máquinas si experimenta síntomas que afecten su visión o concentración, o su capacidad de reacción, durante el tratamiento.
DOSIS Y VÍA DE ADMINISTRACIÓN:
General:
A) Dosis recomendada y Esquema de Tratamiento:
La dosis recomendada de ZALTRAPZIV®, administrado en infusión intravenosa de 1 hora, es de 4 mg/kg de peso corporal seguida de un régimen FOLFIRI (ver sección de Farmacocinética y Farmacodinamia).
El régimen FOLFIRI usado en el estudio fundamental fue: irinotecan 180 mg/m2 infusión IV de 90 minutos y leucovorina (dl racémico) 400 mg/m2 infusión IV de 2 horas a la misma hora del día 1 usando una línea en Y, seguida de 5-fluorouracilo (5-FU) 400 mg/m2 en Bolo IV, seguido de 5-FU 2400 mg/m2 en infusión IV continua de 46 horas.
Los ciclos de tratamiento se repiten cada 2 semanas.
El tratamiento con ZALTRAPZIV® debe continuarse hasta la progresión de la enfermedad o toxicidad inaceptable.
Modificaciones de la Dosis/Recomendaciones para el Retraso del Tratamiento:
Discontinuar ZALTRAPZIV® por:
• Hemorragia severa (ver sección de Precauciones generales).
• Perforación gastrointestinal (ver sección de Precauciones generales).
• Formación de fístulas (ver sección de Precauciones generales).
• Crisis hipertensiva o encefalopatia hipertensiva (ver sección de Precauciones generales).
• Eventos tromboembólicos arteriales (ver sección de Precauciones generales).
• Síndrome nefrótico o microangiopatía trombótica (MAT) (ver sección de Precauciones generales).
• Reacciones severas de hipersensibilidad (incluyendo broncoespasmo, disnea, angioedema y anafilaxia (ver sección de Precauciones generales y Reacciones secundarias y adversas).
• Compromiso de la cicatrización de heridas que requiera intervención médica (ver sección de Precauciones generales).
• Síndrome de leucoencefalopatía posterior reversible (SLPR).
|
Retraso del Tratamiento de ZALTRAPZIV®/FOLFIRI |
|
|
Neutropenia o Trombocitopenia (ver sección de Precauciones generales y Reacciones secundarias y adversas) |
La administración de ZALTRAPZIV®/FOLFIRI debe retrasarse hasta que el recuento de neutrófilos sea ≥ 1.5 x 109/L o el recuento de plaquetas sea ≥ 75 x 109/L. |
|
Reacción de hipersensibilidad leve a moderada (incluyendo enrojecimienlo, erupción cutánea, urticaria, y prurito) (ver sección de Precauciones generales) |
Suspender temporalmente el tratamiento hasta que la reacción se resuelva. Tratar con corticosteroides y/o antihistamínicos según esté clínicamente indicado. Puede considerarse el pretratamiento con corticosteroides y/o antihistamínicos en ciclos subsecuentes. |
|
Reacción severa de hipersensibilidad (incluyendo broncoespasmo, disnea, angioedema, y anafilaxia). (ver sección de Precauciones generales) |
Discontinuar ZALTRAPZIV® y administrar la terapia médica apropiada. |
|
Hipertensión (ver sección de Precauciones generales) |
Suspender temporalmente ZALTRAPZIV® hasta el control de la hipertensión. En caso de recurrencia de hipertensión severa, suspender hasta el control de la hipertensión y reducir la dosis a 2 mg/kg en los ciclos subsecuentes. |
|
Proteinuria (ver sección de Precauciones generales) |
Suspender ZALTRAPZIV® con proteinuria ≥ 2 gramos por 24 horas y reiniciar cuando la proteinuria sea < 2 gramos por 24 horas. En caso de recurrencia, suspender hasta < 2 gramos por 24 horas y reducir la dosis a 2 mg/kg. |
|
FOLFIRI, Modificación de la Dosis en combinación con ZALTRAPZIV® |
|
|
Estomatitis y Síndrome de Eritrodisestesia Palmo-Plantar Severo |
Reducir la dosis de 5-FU en bolo e infusión 20%. |
|
Diarrea Severa |
Reducir la dosis de irinotecan 15-20%. Si la diarrea severa recurre en el siguiente ciclo, reducir también la dosis de 5-FU en bolo e infusión 20%. Si la diarrea severa persiste con la reducción de ambas dosis, discontinuar FOLFIRI. Tratar con medicamentos antieméticos y rehidratación según se requiera. |
|
Neutropenia Febril o Sepsis par Neutropenia |
Reducir la dosis de irinotecan 15-20% en ciclos subsecuentes. En caso de recurrencia, reducir también la dosis de 5-FU en bolo e infusión 20% en ciclos subsecuentes. Se puede considerar el uso de G-CSF. |
Suspender temporalmente ZALTRAPZIV® por lo menos durante 4 semanas antes de la cirugía electiva (ver sección de Precauciones generales).
Para información adicional sobre la toxicidad relacionada con irinotecan, 5-FU y leucovorina, consultar la información de prescripción respectiva vigente de estos medicamentos.
B) Poblaciones especiales:
Niños:
No se ha establecido la seguridad y eficacia en pacientes pediátricos. Veintiún pacientes con edades de 2 a 21 años (edad media de 12.9 años) con tumores sólidos recibieron ZALTRAPZIV® en un estudio de escalamiento de dosis, seguridad y tolerabilidad, a dosis de entre 2 a 3 mg/Kg IV, cada dos semanas. En 8 de estos pacientes (edades de 5 a 17 años) se evaluó la farmacocinética de aflibercept libre (ver sección de Poblaciones especiales). La dosis máxima tolerada en el estudio fue 2.5 mg/Kg, por debajo de la dosis segura y efectiva conocida en adultos con CCRM. Ancianos:
No se requiere ajustar la dosis de ZALTRAPZIV® en ancianos.
Insuficiencia hepática:
No se han realizado estudios formales con ZALTRAPZIV® en pacientes con insuficiencia hepática. De acuerdo con los datos clínicos, en pacientes con deterioro hepático leve a moderado la exposición a aflibercept fue similar a la observada en pacientes con función hepática normal. Estos mismo datos sugieren que no es necesario un ajuste de dosis en pacientes con deterioro de la función hepática leve a moderado. No existe información disponible respecto a aflibercept en pacientes con insuficiencia hepática grave.
Insuficiencia renal:
No se han realizado estudios formales con ZALTRAPZIV® en pacientes con insuficiencia renal. De acuerdo con los datos clínicos, en pacientes con deterioro renal leve a moderado la exposición a aflibercept fue similar a la observada en pacientes con función renal normal. Estos mismo datos sugieren que no es necesario un ajuste de dosis en pacientes con deterioro de la función renal leve a moderado. No existe información disponible respecto a aflibercept en pacientes con insuficiencia renal grave.
C) Administración:
ZALTRAPZIV® se debe administrar bajo la supervisión de un médico experimentado en el uso de productos medicinales antineoplásicos.
No administrar el concentrado sin diluir. No administrar como bolo intravenoso (IV). No es para inyección intravítrea.
Al igual que con todos los productos medicinales parenterales, la solución diluida de ZALTRAPZIV® se debe inspeccionar visualmente para detectar la posible presencia de partículas y decoloración antes de su administración.
La solución diluida de ZALTRAPZIV® se debe administrar usando equipos de infusión hechos de uno de los siguientes materiales:
• Cloruro de polivinilo (PVC) que contenga bis (2-etilhexil) ftalato (DEHP).
• PVC libre de DEHP que contenga trioctil-trimellitato (TOTM).
• polipropileno.
• PVC cubierto de polietileno.
• poliuretano.
Los equipos de infusión deben contener filtros de polietersulfona de 0.2 micras. No usar filtros hechos de fluoruro de polivinilideno (PVDF) o nailon.
D) Recomendación para el manejo seguro:
• ZALTRAPZIV® solución para infusión debe ser preparado por una profesional de la salud usando una técnica aséptica y procedimientos seguros de manejo.
Preparación de la solución para infusión:
• No use el vial si hay partículas presentes o decoloración.
• Se deben usar bolsas de infusión hechas de PVC que contenga DEHP o Poliolefin (PVC libre DEHP libre).
• Sólo para infusión IV debido a la hiperosmolaridad (1,000 mOsmol/kg) del concentrado de ZALTRAPZIV®.
• No para inyección intravítrea.
• El concentrado de ZALTRAPZIV® debe diluirse. Tome la cantidad necesaria del concentrado de ZALTRAPZIV® y dilúyalo hasta el volumen de administración requerido con solución de cloruro de sodio 0.9% o solución de dextrosa 5% para inyección. La concentración final de la solución de ZALTRAPZIV® para infusión IV debe mantenerse en el rango de 0.6-8 mg/ml de aflibercept. Las soluciones diluidas de ZALTRAPZIV® deben usarse inmediatamente. Si no se usan inmediatamente, las soluciones diluidas de ZALTRAPZIV® pueden almacenarse a 2-8ºC (36-46ºF) hasta 24 horas, o a 25ºC (77ºF) hasta 8 horas ya que las soluciones de ZALTRAPZIV® no contienen conservadores.
Disposición de Residuos:
ZALTRAPZIV® es un vial para una sola aplicación. Si no se utiliza todo el producto, deseche el sobrante, ya que el producto no tiene conservadores. No vuelva a entrar en el vial después de la primera punción.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No se han presentado casos de sobredosis con ZALTRAPZIV®. No hay información sobre la seguridad de ZALTRAPZIV® administrado en dosis mayores a 7 mg/kg cada 2 semanas o 9 mg/kg cada 3 semanas. Los eventos adversos más comúnmente observados con estas dosis fueron similares a los observados con dosis terapéuticas.
No existe un antídoto específico para la sobredosis de ZALTRAPZIV®. Los casos de sobredosis se deben manejar con medidas de soporte apropiadas, particularmente en relación con el monitoreo y tratamiento de la hipertensión y proteinuria y el paciente debe permanecer bajo estricta supervisión médica para monitorear cualquier reacción farmacológica adversa (ver sección Reacciones secundarias y adversas).
PRESENTACIONES:
ZALTRAPZIV® fue formulado específicamente para administración intravenosa y no debe usarse por otras vías de administración.
ZALTRAPZIV® 100 mg/4 ml: Caja con 1 frasco ámpula.
ZALTRAPZIV® 200 mg/8 ml: Caja con 1 frasco ámpula.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese en refrigeración entre 2°C a 8°C.
Protéjase de la luz.
No se congele.
Hecha la mezcla el producto se conserva durante 24 horas en refrigeración entre 2°C y 8°C, sin congelar, o durante 8 horas a no más de 25°C.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para especialistas en el manejo de productos oncológicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo y lactancia. No se administre por vía intravenosa directa. No se mezcle con otros fármacos. Si no se administra todo el producto, deséchese el sobrante. No se administre si la solución no es transparente, si contiene partículas en suspensión o sedimentos. No se administre si el cierre ha sido violado.
SANOFI-AVENTIS DE MÉXICO, S.A. de C.V.
Acueducto del Alto Lerma No. 2
Zona Industrial Ocoyoacac
C.P. 52740 Ocoyoacac, México
Reg. Núm. 099M2014 SSA IV
Clave Interna del documento: MX-(IPP- Zaltrapziv)-(Aflibercept CCDS/v3- v4


