

ZATHELO
ROSUVASTATINA
Tabletas
1 Caja, 10 Tabletas, 10 Miligramos
1 Caja, 20 Tabletas, 10 Miligramos
1 Caja, 30 Tabletas, 10 mg
1 Caja, 10 Tabletas, 20 Miligramos
1 Caja, 20 Tabletas, 20 Miligramos
1 Caja, 30 Tabletas, 20 mg
1 Caja, 10 Tabletas, 40 Miligramos
1 Caja, 20 Tabletas, 40 Miligramos
1 Caja, 30 Tabletas, 40 Miligramos
1 Caja, 10 Tabletas, 10 Miligramos
1 Caja, 20 Tabletas, 10 Miligramos
1 Caja, 30 Tabletas, 10 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Tableta
Cada TABLETA contiene: Rosuvastatina cálcica equivalente a 10 mg, 20 mg, 40 mg de rosuvastatina
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS: ZATHELO® debe ser usado como un complemento de la dieta, cuando la respuesta a la dieta y el ejercicio no es suficiente.
Prevención de eventos cardiovasculares:
ZATHELO® está indicado en pacientes adultos con un incremento de riesgo de enfermedad cardiovascular aterosclerótica basado en la presencia de marcadores de riesgo de enfermedad cardiovascular, tales como nivel elevado de PCRas, edad, hipertensión arterial, nivel bajo de C-HDL, tabaquismo o antecedentes familiares de enfermedad coronaria prematura. ZATHELO® está indicado para reducir la mortalidad total y el riesgo de eventos cardiovasculares mayores (muerte cardiovascular, accidente vascular cerebral, infarto al miocardio, angina inestable o revascularización arterial).
En pacientes adultos con hipercolesterolemia: ZATHELO® reduce el colesterol-LDL (C-LDL), colesterol total y triglicéridos elevados e incrementa el colesterol HDL (C-HDL) en pacientes con hipercolesterolemia primaria (familiar heterocigótica y no familiar) y dislipidemia mixta (incluyendo Fredrickson tipos lla y llb). ZATHELO® disminuye también apoB-colesterol-no HDL, colesterol-VLDL, VLDL-TG, así como los índices C-LDL/C-HDL, colesterol total/C-HDL, C-noHDL/C-HDL, apoB/apoA-I e incrementa la apoA-I en estas poblaciones.
ZATHELO® está indicado en pacientes con disbetalipoproteinemia primaria (hiperlipoproteinemia Fredrickson tipo III).
ZATHELO® está indicado para el tratamiento de hipertrigliceridemia aislada (hiperlipidemia Fredrickson tipo IV).
ZATHELO® reduce el colesterol total y C-LDL en pacientes con hipercolesterolemia familiar homocigótica, ya sea solo o como coadyuvante en la dieta y otros tratamientos para reducción de lípidos (p. ej., aféresis LDL).
ZATHELO® retarda o reduce la progresión de aterosclerosis.
Niños y adolescentes de 10 a 17 años de edad: ZATHELO® está indicado para reducir el colesterol total, C-LDL y apo B en pacientes con hipercolesterolemia familiar heterocigótica (HFHe).
FARMACOCINÉTICA Y FARMACODINAMIA:
PROPIEDADES FARMACOCINÉTICAS:
La rosuvastatina se administra oralmente en su forma activa con niveles plasmáticos máximos observados a las 5 horas después de la dosificación. La exposición aumenta linealmente dependiendo de la dosis. La vida media es de 19 horas y no aumenta con una dosis mayor. La biodisponibilidad absoluta es de 20%. Existe una mínima acumulación con la administración una vez al día. La rosuvastatina tiene un efecto de primer paso en el hígado, el cual es el sitio primario de la síntesis de colesterol y depuración de C-LDL.
La rosuvastatina se une aproximadamente 90% a las proteínas plasmáticas, principalmente a la albúmina. El compuesto original representa más del 90% de la actividad inhibidora de HMG CoA reductasa circulante.
La rosuvastatina sufre un metabolismo muy limitado (aproximadamente 10%), principalmente a la forma N-desmetilada y 90% es eliminada como medicamento inalterado en heces y el resto es excretado en la orina.
FARMACOCINÉTICA EN POBLACIONES ESPECIALES: Edad y sexo: En la farmacocinética con rosuvastatina no se demostró ningún efecto clínicamente relevante relacionado con la edad o sexo en adultos. La farmacocinética de rosuvastatina en niños y adolescentes con hipercolesterolemia familiar heterocigótica fue similar a la de voluntarios adultos.
Raza: Estudios farmacocinéticos muestran una elevación promedio del área bajo la curva (ABC) aproximadamente del doble en sujetos asiáticos en comparación con sujetos caucásicos. Un análisis farmacocinético entre grupos caucásicos, hispanos y de raza negra o afrocaribeños, no reveló diferencias clínicamente importantes.
Insuficiencia renal: Un estudio en sujetos con distintos grados de insuficiencia renal o de enfermedad renal leve a moderada demostró que ellas tienen poca influencia en las concentraciones plasmáticas de rosuvastatina. Sin embargo, los sujetos con daño severo (CrCI < 30 mL/min) tuvieron un aumento de 3 veces en la concentración plasmática en comparación con voluntarios sanos.
Insuficiencia hepática: En un estudio en sujetos con grados variables de insuficiencia hepática, no hubo evidencia de tener mayor exposición a la rosuvastatina a excepción de 2 sujetos con enfermedad hepática severa (puntajes de Child-Pugh de 8 y 9). En estos sujetos, la exposición sistémica fue al menos 2 veces más alta en comparación con sujetos con menores puntajes de Child-Pugh.
Polimorfismo genético: El metabolismo de la HMG-CoA reductasa, incluyendo cuando se administra rosuvastatina, involucra al polipéptido transportador de aniones orgánicos 1B1 (OATP1B1 por sus siglas en inglés) y a las proteínas resistentes al cáncer de mama (BCRP por sus siglas en inglés). En los pacientes con polimorfismo genético de sustancias disolutas de la familia de transportadores de aniones 1 B1 [SLCO1B1 por sus siglas en inglés (OATP1B1)] y/o ABCG2 (BCRP) tienen un mayor riesgo de exposición a rosuvastatina. El polimorfismo individual de SLCO1B10 c.521CC o de ABCG2 c.421AA está asociado con aproximadamente 1.6 veces más a la exposición a rosuvastatina (ABC) o 2.4 veces la exposición más alta, respectivamente, en comparación con los genotipos SLCO1B1 c.521TT o ABCG2 c.421CC.
PROPIEDADES FARMACODINÁMICAS:
MECANISMO DE ACCIÓN: La rosuvastatina es un poderoso inhibidor selectivo y competitivo de la HMG-CoA reductasa, la enzima limitante para la conversión de la coenzima A 3-hidroxi-3-metilglutaril en mevalonato, un precursor de colesterol. Los triglicéridos (TG) y el colesterol en hígado se incorporan con apolipoproteína B (ApoB), formando una lipoproteína de muy baja densidad (VLDL) que se libera en plasma para su distribución a tejidos periféricos. Las partículas de VLDL son ricas en TG. La lipoproteína de baja densidad (LDL) rica en colesterol se forma de VLDL y es depurada principalmente a través del receptor LDL de alta afinidad en el hígado.
La rosuvastatina produce sus efectos modificadores de lípidos en dos formas: aumentando el número de receptores LDL hepáticos en la superficie celular, con lo que aumenta la captación y el catabolismo de LDL e inhibiendo la síntesis hepática de VLDL, con lo cual se reduce el número total de partículas de VLDL y LDL.
La lipoproteína de alta densidad (HDL), la cual contiene apoA-I, está involucrada, entre otras cosas, en el transporte de colesterol de los tejidos regresándolos al hígado (transporte inverso de colesterol). La participación de C-LDL en aterogénesis está bien documentada. Estudios epidemiológicos han establecido que C-LDL y TG altos, así como C-HDL y apoA-I bajos se han ligado a un mayor riesgo de enfermedad cardiovascular. Estudios de intervención han demostrado los efectos benéficos al disminuir el índice de mortalidad y de eventos cardiovasculares (CV) al reducir C-LDL y TG o al aumentar C-HDL. Datos más recientes han vinculado los beneficios de los inhibidores de HMG-CoA reductasa sobre la disminución del colesterol no HDL (es decir, todo el colesterol circulante no HDL) y de apoB o sobre la reducción en el índice apoB/apoA-I.
EFICACIA CLÍNICA:
La rosuvastatina reduce C-LDL, colesterol total y triglicéridos altos y aumenta C-HDL. También disminuye apoB, C-no HDL, C-VLDL, VLDL-TG y aumenta apoA-I (véase tablas 1 y 2). La rosuvastatina también disminuye los índices C-LDL/C-HDL, C-total/C-HDL, C-no HDL/C-HDL y ApoB/ApoA-I.
Tabla 1. Respuesta a la dosis en pacientes con hipercolesterolemia primaria (tipos IIa y IIb) (cambio porcentual promedio ajustado desde la línea basal)
|
Dosis |
N |
C-LDL |
C-Total |
C-HDL |
TG |
C-No HDL |
ApoB |
ApoA-I |
|
Placebo |
13 |
-7 |
-5 |
3 |
-3 |
-7 |
-3 |
0 |
|
5 |
17 |
-45 |
-33 |
13 |
-35 |
-44 |
-38 |
4 |
|
10 |
17 |
-52 |
-36 |
14 |
-10 |
-48 |
-42 |
4 |
|
20 |
17 |
-55 |
-40 |
8 |
-23 |
-51 |
-46 |
5 |
|
40 |
18 |
-63 |
-46 |
10 |
-28 |
-60 |
-54 |
0 |
Tabla 2. Respuesta a la dosis en pacientes con hipertrigliceridemia primaria (tipos IIb o IV) (cambio porcentual promedio desde la línea basal)
|
Dosis |
N |
TG |
C-LDL |
C-Total |
C-HDL |
C-No HDL |
C-VLDL |
VLDL-TG |
|
Placebo |
26 |
1 |
5 |
1 |
-3 |
2 |
2 |
6 |
|
5 |
25 |
-21 |
-28 |
-24 |
3 |
-29 |
-25 |
-24 |
|
10 |
23 |
-37 |
-45 |
-40 |
8 |
-49 |
-48 |
-39 |
|
20 |
27 |
-37 |
-31 |
-34 |
22 |
-43 |
-49 |
-40 |
|
40 |
25 |
-43 |
-43 |
-40 |
17 |
-51 |
-56 |
-48 |
Los datos en las tablas 1 y 2 se confirman por el programa clínico más amplio de más de 5,300 pacientes que recibieron rosuvastatina.
En un estudio con pacientes con hipercolesterolemia familiar heterocigótica, 435 sujetos recibieron rosuvastatina de 20 mg a 80 mg en un diseño de titulación forzada. Todas las dosis de rosuvastatina mostraron un efecto benéfico en los parámetros del perfil de lípidos y en el logro de metas del tratamiento.
Después de la titulación a 40 mg (12 semanas de tratamiento) el C-LDL se redujo en un 53%.
En un estudio abierto con titulación forzada, 42 pacientes con hipercolesterolemia familiar homocigótica fueron evaluados respecto a su respuesta a ZATHELO® de 20-40 mg titulándolos en un intervalo de 6 semanas. En general, la población presentó una reducción promedio de C-LDL de 22%. En los 27 pacientes con al menos un 15% de reducción hacia la semana 12 (considerada como la población que responde a tratamiento), la reducción promedio de C-LDL fue de 26% a la dosis de 20 mg y de 30% a la dosis de 40 mg.
De los 13 pacientes con C-LDL menor a 15%, 3 no tuvieron respuesta o tuvieron un aumento en C-LDL.
En el estudio METEOR, se evaluó el efecto de rosuvastatina 40 mg en la progresión de aterosclerosis mediante ultrasonido Bi-modal de las arterias carótidas. En el estudio clínico controlado con placebo, multicéntrico, doble ciego, 984 sujetos con enfermedad coronaria de bajo riesgo (definida como riesgo Framingham < 10%, después de 10 años), con valores promedio de 154.5 mg/dL de C-LDL, pero con aterosclerosis subclínica detectada midiendo el Engrosamiento Promedio de la Íntima de la Carótida (CIMT), fueron asignados al azar en una proporción 5:2 para el tratamiento, ya fuera con rosuvastatina 40 mg o con placebo durante 2 años. La rosuvastatina retardó significativamente la progresión de aterosclerosis en las carótidas en comparación con el placebo. La diferencia en el porcentaje de cambio en las mediciones en CIMT máximo de los diferentes segmentos valorados en las 12 arterias carótidas entre pacientes tratados con rosuvastatina y pacientes tratados con placebo fue de -0.0145 mm/año (IC 95% -0.0196, -0.0093; p < 0.0001).
El cambio en la línea base para el grupo con rosuvastatina fue -0.0014 mm/año (IC 95% -0.0041, 0.0014), pero no fue significativamente diferente de cero (p = 0.3224). Los beneficios de la rosuvastatina fueron consistentes en los cuatro puntos de medición CIMT. Se presentó progresión significativa en el grupo con placebo (+0.0131 mm/año, 95% IC 0.0087, 0.0174; p < 0.0001). En el grupo de rosuvastatina, 52.1% de los pacientes mostraron ausencia en la progresión de la enfermedad (retroceso) en comparación con el 37.7% de los pacientes en el grupo placebo (p = 0.0002). La rosuvastatina 40 mg fue bien tolerada y los resultados fueron consistentes con el perfil de seguridad establecido para la rosuvastatina.
En un estudio aleatorio, multicéntrico, cruzado, doble ciego, con 32 pacientes (27 con genotipo e2/e2 y 4 con mutación apo E [arg145cys]) con disbetalipoproteinemia (Fredrickson tipo III) que recibieron rosuvastatina 10 o 20 mg diariamente por 6 semanas, este fármaco redujo el C-no-HDL (punto final primario) y los niveles remanentes de lipoproteína en circulación (véase tabla 3).
Tabla 3. Efectos en la modificación de lípidos por rosuvastatina 10 mg y 20 mg en disbetalipoproteinemia (hiperlipoproteinemia Fredrickson tipo III)
después de 6 semanas por el cambio de porcentaje medio (95% IC), referencia (N = 32)
|
Dosis |
C-Total |
TG |
C-No-HDL |
C-VLDL+ C-IDL |
C-LDL |
C-HDL |
C-RLP |
Apo-E |
|
10 |
-43.3 |
-40.1 |
-48.2 |
-46.8 |
-54.4 |
10.2 |
-56.4 |
-42.9 |
|
(-46.9, -37.5) |
(-44.9, -33.6) |
(-56.7, -45.6) |
(-53.7, -39.4) |
(-59.1, -47.3) |
(1.9, 12.3) |
(-67.1, -49.0) |
(-46.3, -33.3) |
|
|
20 |
-47.6 |
-43.0 |
-56.4 |
-56.2 |
-57.3 |
11.2 |
-64.9 |
-42.5 |
|
(-51.6, -42.8) |
(-52.5, -33.1) |
(-61.4, -48.5) |
(-67.7, -43.7) |
(-59.4, -52.1) |
(8.3, 20.5) |
(-74.0, -56.6) |
(-47.1, -35.6) |
El estudio CORONA (Controlled Rosuvastatin Multinational Study in Heart Failure) fue un estudio aleatorio, doble ciego, controlado con placebo, en 5,011 pacientes con síntomas de insuficiencia cardiaca crónica sistólica tratados con 10 mg de rosuvastatina (n = 2514) o placebo (n = 2497) con una duración promedio del tratamiento de 2.5 años. Cuando se añadieron 10 mg de rosuvastatina al tratamiento farmacológico previo en estos sujetos, se observó en el punto de corte primario, una disminución no significativa del 8% de muerte cardiovascular, infarto al miocardio no fatal o accidente cerebrovascular no fatal, con respecto al placebo (HR: 0.92, 95% IC: 0.83 a 1.02, p = 0.12).
El perfil de seguridad para los sujetos que tomaron rosuvastatina 10 mg fue comparable al de los sujetos que recibieron placebo. Del estudio, el 1.8% de los sujetos tratados con rosuvastatina en comparación con el 1.7% de los sujetos tratados con placebo interrumpieron el tratamiento debido a reacciones adversas. Las reacciones adversas más comunes que provocaron la interrupción del tratamiento fueron: mialgia, prurito, exantema y mareos. Las reacciones adversas reportadas en > 2% de los pacientes y en una proporción mayor que o igual al placebo se pueden encontrar en la tabla 4.
Tabla 4. Reacciones Adversas
|
Reacciones adversas |
Rosuvastatina 10 mg N = 2514 |
Placebo N = 2497 |
|
Mareos |
7.8 |
7.6 |
|
Mialgia |
5.4 |
5.2 |
|
Dolor de cabeza |
3.5 |
2.5 |
|
Náuseas |
3.5 |
3.2 |
|
Artralgia |
2.8 |
2.7 |
|
Fatiga |
2.8 |
2.1 |
La rosuvastatina es eficaz en una amplia variedad de poblaciones de pacientes con hipercolesterolemia, con o sin hipertrigliceridemia, independientemente de la raza, sexo, edad y en poblaciones especiales, como diabéticos o pacientes con hipercolesterolemia familiar.
En el estudio de intervención JUPITER (Justification for the Use of Statins in Primary Prevention: An lntervention Trial Evaluating Rosuvastatin), se evaluó el efecto de rosuvastatina cálcica en la incidencia de eventos de enfermedad cardiovascular ateroesclerótica en 17,802 hombres (> 50 años de edad) y mujeres (> 60 años de edad) que no tenían enfermedad cardiovascular establecida y que tenían niveles de C-LDL < 130 mg/dL (3.3 mmol/L) y niveles de hs-CRP (> 2 mg/L). La población de estudio tenía un riesgo estimado de referencia de enfermedad coronaria de 11.3% sobre 10 años con base en los criterios de riesgo de Framingham y se incluyó un alto porcentaje de pacientes con factores de riesgo adicionales como hipertensión arterial (58%), niveles bajos de C-HDL (23%), tabaquismo (16%) o con antecedentes familiares de cardiopatía coronaria prematura (12%). Los participantes del estudio fueron asignados aleatoriamente a placebo (n = 8901) o rosuvastatina 20 mg una vez al día (n = 8901) y se les dio seguimiento en promedio durante dos años. El punto de corte primario fue un punto final compuesto consistente en el tiempo de aparición de cualquiera de los siguientes eventos cardiovasculares mayores: muerte cardiovascular, infarto al miocardio no fatal, accidente cerebrovascular no fatal, angina inestable o un procedimiento de revascularización arterial. La rosuvastatina redujo significativamente el riesgo de eventos cardiovasculares (252 eventos en el grupo placebo frente a 142 eventos en el grupo de rosuvastatina) con una diferencia estadísticamente significativa (p < 0.001) de reducción de riesgo relativo de 44% (Figura 1). El beneficio fue aparente en los primeros 6 meses de tratamiento. La reducción del riesgo fue consistente entre varios subgrupos predefinidos de población basados en evaluaciones de edad, sexo, raza, tabaquismo, antecedentes familiares de cardiopatía coronaria prematura, índice de masa corporal, niveles de C-LDL y C-HDL o PCRas, en el momento de ingreso al estudio. Estadísticamente hubo una diferencia significativa con una reducción del 48% en el punto final combinado con muerte cardiovascular, infarto al miocardio y accidente cerebrovascular (HR:0.52, 95% IC:0.40-0.68, p < 0.001), reducción del 54% de infarto al miocardio mortal o no mortal (HR:0.46, 95% ICl:0.30-0.70) y una reducción del 48% de accidente cerebrovascular mortal o no mortal. La mortalidad total se redujo 20% en el grupo de rosuvastatina (HR:0.80, 95% IC:0.67-0.97, p = 0.02).
Figura 1. Tiempo de ocurrencia de los principales eventos cardiovasculares en JUPITER
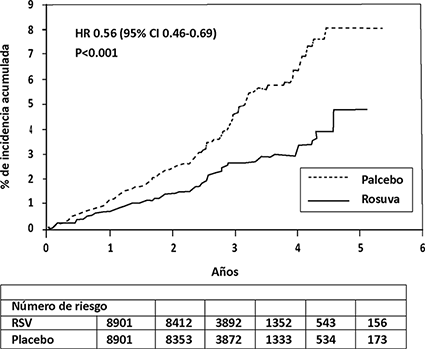
El perfil de seguridad para los sujetos que tomaron rosuvastatina 20 mg fue generalmente similar a la de los sujetos que tomaron placebo. Hubo un 1.6% de sujetos retirados del grupo de rosuvastatina y un 1.8% del grupo de placebo debido a un evento adverso independientemente de la causalidad del tratamiento. Las reacciones adversas más frecuentes que condujeron a la suspensión del tratamiento fueron: mialgia (0.3% rosuvastatina, 0.2% con placebo), dolor abdominal (0.03% de rosuvastatina, 0.02% con placebo) y exantema (0.03% rosuvastatina, 0.03% con placebo). Reacciones adversas reportadas en > 2% de los pacientes y en un porcentaje igual o mayor que el placebo, fueron mialgia (7.6% de rosuvastatina, 6.6% con placebo), estreñimiento (3.3% rosuvastatina, 3.0% con placebo) y náusea (2.4% rosuvastatina, placebo 2.3%). En el estudio JUPITER se produjo un aumento estadísticamente significativo en la frecuencia de diabetes mellitus reportado por los investigadores: 2.8% para los pacientes en el grupo de rosuvastatina y 2.3% en pacientes del grupo placebo (HR:1.27, 95% IC: 1.05-1.53, p = 0.015). La diferencia promedio de HbA1c entre los grupos de tratamiento (rosuvastatina en relación a placebo) fue de aproximadamente 0.1% desde la línea de base.
Un análisis post-hoc de este estudio, sugiere que el riesgo de desarrollar diabetes con el tratamiento con rosuvastatina se limita a los pacientes que ya están en alto riesgo de desarrollarla. Los beneficios cardiovasculares y la mortalidad de una terapia con rosuvastatina supera los riesgos de diabetes en la población del estudio, así como en los participantes con alto riesgo de desarrollarla (véase Precauciones generales y Reacciones secundarias y adversas).
NIÑOS Y ADOLESCENTES CON HIPERCOLESTEROLEMIA: En un estudio doble ciego, aleatorio, multicéntrico, controlado con placebo, de 12 semanas (n = 176, 97 hombres y 79 mujeres), seguido por una fase de titulación de dosis de rosuvastatina, de diseño abierto, de 40 semanas (n = 173, 96 hombres y 77 mujeres), de 10-17 años de edad (Escala de Tanner de la II-V, en mujeres al menos 1 año después de la menarca) con hipercolesterolemia familiar heterocigótica que recibieron rosuvastatina 5, 10 o 20 mg o placebo diariamente durante 12 semanas y que posteriormente recibieron rosuvastatina diariamente durante 40 semanas, 30% de los pacientes estuvieron en el intervalo de edad de 10-13 años y aproximadamente el 17%, 18%, 40% y 25% fueron escala Tanner II, III, IV y V, respectivamente.
La rosuvastatina redujo el C-LDL (punto final primario), el colesterol total y los niveles de apoB (véase tabla 5).
Tabla 5. Efectos de la rosuvastatina sobre la modificación de lípidos en niños y adolescentes con hipercolesterolemia familiar heterocigótica (porcentaje promedio de cambio en mínimos cuadrados a partir de la línea base hasta la semana 12)
|
Dosis (mg) |
N |
C-LDL |
C-HDL |
Colesterol total |
Triglicéridos |
C-No HDL |
ApoB |
ApoA-1 |
|
Placebo |
46 |
-0.7 |
6.9 |
-0.0 |
5.1 |
-0.9 |
-1.7 |
2.8 |
|
5 |
42 |
-38.3 |
4.2 |
-29.9 |
0.3 |
-36.1 |
-31.7 |
1.8 |
|
10 |
44 |
-44.6 |
11.2 |
-34.2 |
-13.6 |
-43.0 |
-38.1 |
5.4 |
|
20 |
44 |
-50.0 |
8.9 |
-38.7 |
-8.1 |
-47.5 |
-40.7 |
4.0 |
Al final de la semana 40 de la fase abierta, se logró el objetivo de C-LDL menor de 110 mg/dL (2.8 mmol/L), con una dosis de hasta un máximo de 20 mg una vez al día en 70 de 173 pacientes (40.5%).
Después de 52 semanas del estudio con el tratamiento no se detectó ningún efecto sobre el crecimiento o la maduración sexual (véase Precauciones generales).
CONTRAINDICACIONES: ZATHELO® está contraindicado en pacientes con hipersensibilidad conocida a cualquier componente de la fórmula, pacientes con enfermedad hepática activa, en mujeres durante el embarazo o lactancia y en mujeres con potencial de embarazarse y que no utilicen medidas anticonceptivas adecuadas.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA: No se ha establecido la seguridad de la rosuvastatina durante el embarazo y la lactancia.
Embarazo: la rosuvastatina está contraindicada durante el embarazo. Las mujeres con potencial de concebir deben utilizar medidas anticonceptivas adecuadas (véase Contraindicaciones).
Lactancia: la rosuvastatina se excreta en la leche en ratas. Se desconoce si la rosuvastatina se excreta en la leche humana. Debido al potencial de reacciones adversas graves de la rosuvastatina en los lactantes, el medicamento está contraindicado en mujeres lactantes.
REACCIONES SECUNDARIAS Y ADVERSAS: La rosuvastatina es generalmente bien tolerada. Los eventos adversos observados con la rosuvastatina son generalmente leves y transitorios. En estudios clínicos controlados, menos del 4% de los pacientes tratados con rosuvastatina fueron retirados de los estudios debido a reacciones adversas. Esta frecuencia de retiro fue comparable a la reportada en pacientes que recibieron placebo.
Eventos/efectos comunes (> 1/100, < 1/10): cefalea, mialgia, astenia, estreñimiento, mareo, náusea, dolor abdominal, diabetes mellitus*.
Poco comunes (>1/1,000, <1/100): prurito, erupción cutánea y urticaria.
Raras (>1/10,000 < 1/1,000): miopatía (incluyendo miositis), reacciones de hipersensibilidad (incluyendo angioedema), rabdomiólisis, pancreatitis.
* Se observó en el estudio JUPITER (frecuencia global reportada 2.8% en el grupo con rosuvastatina y 2.3% en el grupo con placebo) principalmente en pacientes que están en alto riesgo de desarrollar diabetes (véase Precauciones generales y Farmacocinética y farmacodinamia).
Como con otros inhibidores de la HMG-CoA reductasa, la incidencia de reacciones adversas al medicamento tiende a aumentar con dosis altas.
Eventos/efectos musculoesqueléticos: Rara vez se han reportado casos de rabdomiólisis con rosuvastatina, así como con otras estatinas comercializadas; ocasionalmente se asocian con deterioro de la función renal.
Otros eventos/efectos: En un estudio clínico controlado a largo plazo se demostró que la rosuvastatina no tuvo efectos dañinos sobre el cristalino. En pacientes tratados con rosuvastatina no hubo deterioro en la función adrenocortical.
Experiencia durante la comercialización: Además de lo anterior, se han reportado las siguientes reacciones adversas durante la comercialización:
Trastornos hematológicos:
Frecuencia desconocida: trombocitopenia.
Trastornos hepatobiliares:
Frecuencia muy rara: ictericia, hepatitis.
Frecuencia rara: incremento en transaminasas hepáticas.
Trastornos musculoesqueléticos:
Frecuencia desconocida: miopatía necrotizante mediada por el sistema inmune.
Frecuencia muy rara: artralgia.
Al igual que otros inhibidores de HMG-CoA reductasa, la frecuencia de reportes de rabdomiólisis durante la comercialización son mayores con la dosis más alta comercializada.
Trastornos del sistema nervioso:
Frecuencia muy rara: pérdida de la memoria.
Trastornos psiquiátricos:
Frecuencia desconocida: depresión, trastornos del sueño (incluyendo insomnio y pesadillas).
Trastornos del sistema reproductivo y de mama:
Frecuencia desconocida: ginecomastia.
Niños y adolescentes de 10 a 17 años de edad: El perfil de seguridad de la rosuvastatina es similar en niños o adolescentes y adultos, aunque el incremento en los niveles de CPK > 10 x ULN y síntomas musculares después del ejercicio o del aumento en la actividad física, los cuales se resolvieron con el tratamiento continuo, fue observado más frecuentemente en estudios clínicos en niños y adolescentes. Sin embargo, se aplicaron en niños y adolescentes las mismas advertencias y precauciones especiales que para uso en adultos (véase Precauciones generales).
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: Los datos preclínicos no revelaron riesgos especiales para humanos con base en estudios convencionales de farmacología de seguridad, toxicidad con dosis repetidas, toxicidad genética, potencial carcinogénico y toxicidad reproductiva.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Efecto de la coadministración de medicamentos sobre rosuvastatina: in vitro e in vivo, los datos indican que la rosuvastatina no tiene interacciones clínicamente significativas con el citocromo P450 (como un sustrato, como inhibidor o como inductor). La rosuvastatina es un sustrato para ciertas proteínas, incluyendo el transportador de la captación hepática del polipéptido transportador de aniones orgánicos 1B1 (OATP1B1) y el transportador de flujo de salida de proteínas resistentes al cáncer de mama (BCRP). La administración concomitante de ZATHELO® con medicamentos que son inhibidores de estas proteínas transportadoras puede resultar en aumento de las concentraciones plasmáticas de rosuvastatina y un mayor riesgo de miopatía (véase tabla 6, Dosis y vía de administración y Precauciones generales).
Tabla 6. Efecto de la coadministración de los medicamentos sobre la exposición de rosuvastatina (ABC, en orden decreciente de magnitud) de los estudios clínicos publicados
|
Régimen de dosis de medicamentos interactuantes |
Régimen de dosis de rosuvastatina |
Cambio de ABC de rosuvastatina |
|
Ciclosporina 75 mg dos veces al día a 200 mg dos veces al día por 6 meses |
10 mg una vez al día por 10 días |
7.1 veces |
|
Atazanavir 300 mg/ritonavir 100 mg una vez al día por 8 días |
10 mg dosis única |
3.1 veces |
|
Lopinavir 400 mg/ritonavir 100 mg dos veces al día por 17 días |
20 mg una vez al día por 7 días |
2.1 veces |
|
Gemfibrozilo 600 mg dos veces al día por 7 días |
80 mg dosis única |
1.9 veces |
|
Eltrombopag 75 mg una vez al día por 10 días |
10 mg dosis única |
1.6 veces |
|
Darunavir 600 mg/ritonavir 100 mg dos veces al día por 7 días |
10 mg una vez al día por 7 días |
1.5 veces |
|
Tipranavir 500 mg/ritonavir 200 mg dos veces al día por 11 días |
10 mg dosis única |
1.4 veces |
|
Dronedarona 400 mg dos veces al día |
No disponible |
1.4 veces |
|
ltraconazol 200 mg una vez al día por 5 días |
10 u 80 mg, dosis única |
1.4 veces |
|
Ezetimibe 10 mg una vez al día por 14 días |
10 mg una vez al día por 14 días |
1.2 veces |
|
Fosamprenavir 700 mg/ritonavir 100 mg dos veces al día por 8 días |
10 mg dosis única |
« |
|
Aleglitazar 0.3 mg por 7 días |
40 mg por 7 días |
« |
|
Silymarin 140 mg tres veces al día por 5 días |
10 mg dosis única |
« |
|
Fenofibrato 67 mg tres veces al día por 7 días |
10 mg por 7 días |
« |
|
Rifampin 450 mg una vez al día por 7 días |
20 mg dosis única |
« |
|
Ketoconazol 200 mg dos veces al día por 7 días |
80 mg dosis única |
« |
|
Fluconazol 200 mg una vez al día por 11 días |
80 mg dosis única |
« |
|
Eritromicina 500 mg cuatro veces al día por 7 días |
80 mg dosis única |
28% |
|
Baicalin 50 mg tres veces al día por 14 días |
20 mg dosis única |
47% |
Interacciones que requieren ajustes de dosis de rosuvastatina (véase tabla 6): Cuando sea necesario que la coadministración de ZATHELO® con otros medicamentos que aumentan la exposición a rosuvastatina la dosis de ZATHELO® deben ajustarse. Se debe iniciar con una dosis de 5 mg de ZATHELO® una vez al día si el aumento esperado en la exposición área bajo la curva (ABC) es de aproximadamente 2 veces o más. La dosis máxima diaria de ZATHELO® debe ajustarse de manera que la exposición esperada a rosuvastatina no supere una dosis diaria de 40 mg administrada sin que haya interacción con medicamentos, por ejemplo, una dosis de 5 mg de ZATHELO® con ciclosporina (7.1 veces mayor en la exposición), una dosis de 10 mg de ZATHELO® en combinación con ritonavir/atazanavir (3.1 veces mayor) y una dosis de 20 mg de ZATHELO® con gemfibrozilo (1.9 veces mayor).
Otros medicamentos que interactúan:
Antiácidos: La administración simultánea de ZATHELO® con una suspensión antiácida que contenga hidróxido de aluminio y magnesio da como resultado una disminución en la concentración plasmática de rosuvastatina aproximadamente en 50%. Este efecto se mitigó cuando el antiácido se administró 2 horas después en relación a rosuvastatina. La importancia clínica de esta interacción no se ha estudiado.
Efecto de rosuvastatina cuando se administra concomitantemente con otros medicamentos: Warfarina: La farmacocinética de warfarina no se afecta considerablemente después de la coadministración con rosuvastatina. Sin embargo, al igual que otros inhibidores de HMG-CoA reductasa, la coadministración de ZATHELO® y warfarina puede dar como resultado un aumento en INR (International Normalized Ratio) o tiempos de coagulación, en comparación con warfarina sola. En pacientes que reciben antagonistas de la vitamina K se recomienda el monitoreo de INR tanto al inicio como al término de la terapia con ZATHELO® o después de un ajuste de la dosis.
Fenofibratos o derivados del ácido fíbrico: Aunque no se ha observado ninguna interacción farmacocinética entre rosuvastatina y fenofibrato, se puede presentar una interacción farmacodinámica. El gemfibrozil, el fenofibrato y otros ácidos fíbricos, incluyendo el ácido nicotínico, pueden aumentar el riesgo de miopatía cuando se administran de manera concomitante con inhibidores de la HMG-CoA reductasa (véase Precauciones generales).
Ciclosporina: La coadministración de rosuvastatina con ciclosporina dio como resultado cambios no significativos en la concentración plasmática de ciclosporina.
Otros medicamentos: No hubo interacciones clínicamente significativas con anticonceptivos orales, digoxina, ezetimiba o fenofibrato. En estudios clínicos la rosuvastatina se coadministró con agentes antihipertensivos, antidiabéticos y terapia de reemplazo hormonal; estos estudios no produjeron ninguna evidencia de interacciones adversas clínicamente significativas.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: Como con otros inhibidores de HMG-CoA reductasa, se ha observado un aumento relacionado con la dosis en las transaminasas hepáticas y CPK en un pequeño número de pacientes que recibieron rosuvastatina.
También se ha observado un aumento de la HbA1c en pacientes tratados con rosuvastatina (véase Precauciones generales y Farmacocinética y farmacodinamia). Se han observado pruebas de urianálisis anormales (prueba con tira reactiva positiva para proteinuria) en un pequeño número de pacientes que han recibido rosuvastatina y otros inhibidores de HMG-CoA reductasa; la proteína detectada fue principalmente de origen tubular; en la mayoría de los casos, la proteinuria disminuye o desaparece espontáneamente al continuar la terapia y no es predictiva de enfermedad renal aguda o progresiva.
PRECAUCIONES GENERALES:
Hígado: Como con otros inhibidores de la HMG-CoA reductasa, ZATHELO® debe utilizarse con precaución en pacientes que consuman cantidades excesivas de alcohol y/o que tengan antecedentes de enfermedad hepática.
Aparato musculosquelético: Como con otros inhibidores de la HMG-CoA reductasa, se han reportado efectos en el músculo esquelético, por ejemplo, mialgia, miopatía y raramente rabdomiólisis en pacientes tratados con rosuvastatina. Al igual que con otros inhibidores de la HMG-CoA reductasa, la frecuencia de reportes post-comercialización para rabdomiólisis es mayor con la dosis más alta. En aquellos pacientes que desarrollen cualquier signo o síntoma indicativo de miopatía, se deberán determinar los niveles de CPK. La administración de ZATHELO® debe suspenderse si los niveles de CPK son considerablemente elevados (> 10 x ULN) o si se diagnostica o sospecha miopatía. Se han notificado casos muy raros de miopatía necrotizante mediada por el sistema inmune, clínicamente caracterizada por debilidad muscular proximal persistente y elevación en la creatinfosfoquinasa sérica durante el tratamiento o después de la interrupción del tratamiento con estatinas, incluyendo rosuvastatina. Pruebas neuromusculares y serológicas adicionales pueden ser necesarias y también puede requerirse tratamiento con agentes inmunosupresores. En estudios con rosuvastatina no hubo evidencia de aumento de efectos adversos musculosqueléticos cuando se administró rosuvastatina con otra terapia concomitante. Sin embargo, se ha observado mayor incidencia de miositis y miopatía en pacientes que reciben otros inhibidores de la HMG-CoA reductasa junto con ciclosporina, derivados de ácido fíbrico, incluyendo gemfibrozilo, ácido nicotínico, antimicóticos azólicos y antibióticos macrólidos. ZATHELO® debe prescribirse con precaución en pacientes con factores predisponentes a miopatía, por ejemplo, en aquelllos con disfunción renal, edad avanzada e hipotiroidismo o en situaciones donde se pueda tener aumento en los niveles plasmáticos de la rosuvastatina (véase Farmacocinética y farmacodinamia e Interacciones medicamentosas y de otro género). ZATHELO® debe suspenderse temporalmente en cualquier paciente con una condición seria aguda sugerente de miopatía o con predisposición a desarrollar insuficiencia renal secundaria a rabdomiólisis (p. ej. sepsis, hipotensión arterial, cirugía mayor, traumatismos, trastornos metabólicos graves, alteraciones endocrinas y/o trastornos electrolíticos o en status epilepticus).
Diabetes mellitus: Como con otros inhibidores de la HMG-CoA reductasa, se ha observado un incremento en los niveles de HbA1c y glucosa plasmática en pacientes tratados con rosuvastatina y en algunos casos estos aumentos pueden exceder el umbral para el diagnóstico de diabetes mellitus, principalmente en pacientes que ya están en alto riesgo de desarrollar diabetes (véase Farmacocinética y farmacodinamia y Reacciones secundarias y adversas).
Raza: Estudios farmacocinéticos muestran un incremento en la concentración plasmática de rosuvastatina en sujetos asiáticos en comparación con sujetos caucásicos (véase Farmacocinética y farmacodinamia y Dosis y vía de administración).
Niños y adolescentes de 10 a 17 años de edad: La evaluación del crecimiento lineal (altura), peso, IMC (índice de masa corporal y características secundarias de maduración sexual mediante la Clasificación de Tanner en pacientes pediátricos que tomaron rosuvastatina se limitó a un período de un año (véase Farmacocinética y farmacodinamia).
Efectos en la habilidad para conducir u operar maquinaria: Pruebas farmacológicas no revelaron evidencia de efecto sedante con rosuvastatina. Por su perfil de seguridad, no se espera que ZATHELO® afecte la capacidad de manejar u operar maquinaria.
DOSIS Y VÍA DE ADMINISTRACIÓN: Oral.
El rango de la dosis de ZATHELO® es de 10-40 mg por vía oral una vez al día, pudiendo administrarse a cualquier hora y con o sin alimentos. La dosificación de ZATHELO® debe individualizarse de acuerdo con el objetivo terapéutico y la respuesta del paciente. La mayoría de los pacientes se controlan con la dosis inicial.
Sin embargo y, si se requiere, puede hacerse ajuste de la dosis en intervalos de 2 a 4 semanas (véase Farmacocinética y farmacodinamia).
ADULTOS:
Hipercolesterolemia primaria (incluyendo hipercolesterolemia familiar heterocigótica), dislipidemia mixta, disbetalipoproteinemia, hipertrigliceridemia aislada, tratamiento de aterosclerosis y prevención de eventos cardiovasculares: La dosis inicial recomendada de ZATHELO® es de 10 mg una vez al día. Una dosis inicial de 5 mg está disponible para las poblaciones de pacientes especiales, si es requerida. Para pacientes con hipercolesterolemia severa (incluyendo hipercolesterolemia familiar heterocigótica) o aquellos con metas agresivas de reducción de lípidos puede considerarse una dosis inicial de 20 mg.
Hipercolesterolemia familiar homocigótica: Para pacientes con hipercolesterolemia familiar homocigótica, se recomienda una dosis inicial de ZATHELO® de 20 mg una vez al día.
NIÑOS Y ADOLESCENTES DE 10 A 17 AÑOS DE EDAD: En niños y adolescentes con hipercolesterolemia familiar heterocigótica, el rango de dosis habitual de ZATHELO® es de 5-20 mg una vez al día por vía oral. La dosis debe ser titulada adecuadamente para lograr el objetivo del tratamiento. La eficacia y la seguridad de dosis mayores de 20 mg no se ha estudiado en esta población.
En niños y adolescentes con hipercolesterolemia familiar homocigótica, la experiencia está limitada a un pequeño número de niños (8 años de edad en adelante).
POBLACIONES ESPECIALES:
Personas de edad avanzada: Se aplica el rango recomendado de Ia dosis, con precaución (véase Precauciones generales).
Dosificación en pacientes con insuficiencia renal: En pacientes con insuficiencia renal leve a moderada se aplica el rango recomendado de dosis, con precaución (véase Farmacocinética y farmacodinamia). Para pacientes con insuficiencia renal severa, la dosis de ZATHELO® no debe exceder de 10 mg al día (véase Farmacocinética y farmacodinamia).
Dosificación en pacientes con insuficiencia hepática: En pacientes con insuficiencia hepática leve a moderada se aplica el rango recomendado de dosis. Se ha observado un incremento en la concentración plasmática de la rosuvastatina en pacientes con insuficiencia hepática severa, por lo que se debe valorar cuidadosamente el uso de dosis de ZATHELO® mayores a 10 mg (véase Farmacocinética y farmacodinamia).
Raza: Deberá considerarse una dosis inicial de 5 mg de ZATHELO® en pacientes asiáticos. Se ha observado aumento en la concentración plasmática de la rosuvastatina en sujetos asiáticos (véase Farmacocinética y farmacodinamia y Precauciones generales). El incremento en la exposición sistémica se deberá tomar en cuenta al tratar a pacientes asiáticos cuya hipercolesterolemia no esté adecuadamente controlada con dosis de hasta 20 mg/día.
Polimorfismo genético: Se ha demostrado que los genotipos de las sustancias disolutas de la familia de transportadores aniones 1B1 (SLCO1B1) (del polipéptido transportador de aniones orgánicos 1B1 [OATP1B1]) c.521CC y ABCG2 (de las proteínas resistentes al cáncer de mama [BCRP]) c.421AA están asociadas a un incremento en la exposición a la rosuvastatina (ABC) en comparación con SLCO1B1 c.521TT y ABCG2 c.421CC. Para los pacientes que se sabe que tienen el genotipo c.521CC o el c.421AA se recomienda una dosis diaria máxima de ZATHELO® de 20 mg (véase Precauciones generales, Interacciones medicamentosas y de otro género y Farmacocinética y farmacodinamia).
Terapia concomitante: La rosuvastatina es un sustrato de diversas proteínas transportadoras (por ejemplo, OATP1B1 y BCRP). El riesgo de miopatía (incluida la rabdomiólisis) se incrementa cuando se administra ZATHELO® de manera concomitante con ciertos medicamentos que pueden aumentar la concentración plasmática de rosuvastatina debido a interacciones con estas proteínas transportadoras (por ejemplo, ciclosporina y ciertos inhibidores de la proteasa, que incluyen combinaciones de ritonavir con atazanavir, lopinavir y/o tipranavir (véase Precauciones generales e Interacciones medicamentosas y de otro género).
Siempre que sea posible, se deben considerar medicamentos alternativos y, si es necesario, considerar la suspensión temporal de la terapia con ZATHELO®. En situaciones en donde es inevitable la administración conjunta de estos medicamentos con ZATHELO®, el beneficio y el riesgo del tratamiento concomitante y los ajustes de dosis de ZATHELO® se deben considerar cuidadosamente (véase Interacciones medicamentosas y de otro género).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No hay un tratamiento específico en el caso de sobredosis. En este caso, debe tratarse al paciente de manera sintomática y deben instituirse medidas generales de apoyo según se requiera. Es poco probable que la hemodiálisis aporte algún beneficio.
PRESENTACIONES:
Caja con 10, 20 o 30 tabletas de 10 mg.
Caja con 10, 20 o 30 tabletas de 20 mg.
Caja con 10, 20 o 30 tabletas de 40 mg.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 30°C.
LEYENDAS DE PROTECCIÓN:
Literatura exclusiva para médicos. Su venta requiere receta médica. No se deje al alcance de los niños. No se use durante el embarazo y la lactancia. Este medicamento contiene Amarillo no. 6, que puede producir reacciones de hipersensibilidad. Léase instructivo anexo.
Reporte las sospechas de reacciones adversas a los correos: farmacovigilancia@cofepris.gob.mx y
farmacovigilancia@chinoin.com
Hecho en India por:
Hetero Labs Limited Unit V
Sy. No. 439, 440,441 & 458
TSIIC Formulation SEZ,
Polepally Village, Jadcherla, Mandal,
Mahaboobnagar District,
Telangana, India, 509301.
Para y distribuido por:
PRODUCTOS FARMACÉUTICOS, S.A. de C.V.
Km 4.2 Carretera a Pabellón de Hidalgo
C.P. 20420, Rincón de Romos, Aguascalientes, México.
Reg. Núm. 444M2016 SSA IV


