ACTILYSE
ALTEPLASA
Polvo y disolvente para solución inyectable
1 Caja, 2 Frasco ámpula con polvo liofilizado, 50 mL,
COMPOSICIÓN:
Composición cualitativa y cuantitativa:
1 vial de polvo contiene:
50 mg de alteplasa (correspondientes a 29,000,000 UI), respectivamente
La alteplasa es producida mediante la técnica de ADN recombinante, utilizando una línea celular ovárica de hámster chino. La actividad específica de la alteplasa, patrón de referencia interno, es de 580,000 UI/mg. Esto ha sido confirmado por comparación con el segundo patrón internacional de la OMS para t-PA. La especificación para la actividad específica de la alteplasa es de 522,000 a 696,000 UI/mg.
Para consultar la lista completa de excipientes ver sección Lista de excipientes
Forma farmacéutica: Polvo y disolvente para solución inyectable y para perfusión
El polvo se presenta como una pastilla de liofilizado de incolora a amarilla clara. La preparación reconstituida es una solución límpida y de incolora a amarilla clara

INDICACIONES TERAPÉUTICAS:
Datos clínicos:
Tratamiento trombolítico en el infarto agudo de miocardio:
- Régimen de dosificación de 90 minutos (acelerado) (ver sección Posología y forma de administración): para pacientes en los cuales el tratamiento puede iniciarse dentro de las 6 horas después de la presentación de los síntomas.
- Régimen de dosificación de 3 horas (ver sección Posología y forma de administración): para pacientes en los cuales el tratamiento puede iniciarse entre las 6 y 12 horas después de la presentación de los síntomas, siempre que el diagnóstico esté claramente confirmado.
ACTILYSE® ha demostrado reducir la mortalidad al cabo de 30 días en pacientes con infarto agudo de miocardio.
Tratamiento trombolítico en embolia pulmonar masiva aguda con inestabilidad hemodinámica: El diagnóstico deberá ser confirmado, siempre que sea posible, mediante medios objetivos como p. ej. angiografía pulmonar o procedimientos no invasivos como la gammagrafía isotópica pulmonar. No hay evidencia de efectos positivos sobre la mortalidad y morbilidad tardía relacionadas con la embolia pulmonar.
Tratamiento fibrinolítico del ictus isquémico agudo:
El tratamiento debe iniciarse tan pronto como sea posible dentro de las 4.5 horas después de la presentación de los síntomas de ictus y después de la exclusión de hemorragia intracraneal mediante técnicas de imagen apropiadas (p. ej. tomografía computarizada craneal u otros métodos de diagnóstico por imagen sensibles a la presencia de hemorragia). El efecto del tratamiento es dependiente del tiempo; por tanto, un tratamiento temprano aumenta la probabilidad de un desenlace favorable.
PROPIEDADES FARMACOLÓGICAS:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Agentes trombolíticos. Código ATC: B01AD02.
Mecanismo de acción: El principio activo de ACTILYSE® es la alteplasa, un activador recombinante del plasminógeno tisular humano, una glucoproteína que activa directamente el plasminógeno a plasmina. Cuando se administra por vía intravenosa, la alteplasa permanece relativamente inactiva en el sistema circulatorio. Una vez que se conjuga con la fibrina, es activada, induciendo la conversión del plasminógeno en plasmina, lo cual produce la disolución del coágulo de fibrina.
Efectos farmacodinámicos: Debido a su especificidad relativa por la fibrina, la alteplasa a dosis de 100 mg produce una modesta disminución de los niveles de fibrinógeno circulante hasta el 60% aproximadamente a las 4 horas, que generalmente revierte a más del 80% después de 24 horas. El plasminógeno y la α-2-antiplasmina disminuyen hasta aproximadamente el 20% y el 35%, respectivamente, después de 4 horas y aumentan de nuevo a más del 80% a las 24 horas. Sólo en unos pocos pacientes se observa una disminución marcada y prolongada del nivel de fibrinógeno circulante.
Eficacia clínica y seguridad: En un estudio que incluía más de 40,000 pacientes con infarto de miocardio agudo (GUSTO), la administración de 100 mg de alteplasa durante 90 minutos, con perfusión concomitante de heparina intravenosa, redujo la mortalidad a los 30 días (6.3%), en comparación con la administración de estreptoquinasa, 1.5 millones U durante 60 minutos, con heparina subcutánea o intravenosa (7.3%). Los pacientes tratados con ACTILYSE® mostraron una mayor permeabilidad de los vasos relacionados con el infarto a los 60 y 90 minutos de la trombólisis que los pacientes tratados con estreptoquinasa. No se encontraron diferencias en la permeabilidad a los 180 minutos, ni más tarde.
La mortalidad al cabo de 30 días se reduce, en comparación con pacientes que no reciben terapia trombolítica.
La liberación de α-hidroxibutirato-deshidrogenasa (HBDH) se reduce. La función ventricular global, así como la motilidad de la pared regional, resulta menos afectada, en comparación con pacientes que no reciben terapia trombolítica.
Infarto agudo de miocardio: Un estudio controlado con placebo realizado con 100 mg de alteplasa durante 3 horas (LATE), demostró una reducción de la mortalidad al cabo de 30 días, en comparación con el placebo, en pacientes tratados dentro de las 6-12 horas después de la presentación de los síntomas. El tratamiento puede ser beneficioso en los casos en que todavía se observan signos claros de infarto de miocardio, si el tratamiento se inicia hasta 24 horas después de la presentación de los síntomas.
Embolia pulmonar aguda masiva: En pacientes con embolia pulmonar masiva aguda e inestabilidad hemodinámica, el tratamiento trombolítico con ACTILYSE® conduce a una rápida reducción del tamaño del trombo y a una disminución de la presión arterial pulmonar. No se dispone de datos sobre mortalidad.
Pacientes con ictus isquémico agudo: En dos estudios de Estados Unidos (NINDS A/B) una proporción de pacientes significativamente mayor presentó un desenlace favorable con alteplasa en comparación con placebo (sin o con mínima discapacidad). Estos hallazgos se confirmaron en el ensayo ECASS III (ver el siguiente parágrafo), después de que entretanto dos estudios europeos y un estudio adicional realizado en EEUU no hubieran proporcionado la correspondiente evidencia según los requisitos de la información de producto europea vigente.
El ensayo ECASS III fue un ensayo doble ciego controlado con placebo realizado en pacientes con ictus agudo en un intervalo de tiempo de 3 a 4.5 horas en Europa. La administración del tratamiento en el ensayo ECASS III estaba en línea con el Resumen de Características de Producto europeo para ACTILYSE® en la indicación de ictus, excepto el límite superior del intervalo de tiempo de tratamiento, esto es 4.5 horas.
El criterio de valoración principal fue la discapacidad a los 90 días, dividido según desenlace favorable (escala de Rankin modificada [mRS] de 0 a 1) o desfavorable (mRS de 2 a 6). Se aleatorizó un total de 821 pacientes (418 alteplasa/403 placebo). Más pacientes alcanzaron un desenlace favorable con alteplasa (52.4%) que con placebo (45.2%; razón de probabilidad [OR], 1.34; 95% IC 1.02-1.76; P = 0.038). La incidencia de cualquier HIC//HIC sintomática fue superior con alteplasa que con placebo (cualquier HIC 27.0% versus 17.6%, p = 0.0012; HIC sintomática definida en ECASS III 2.4% versus 0.2%, p = 0.008). La mortalidad fue baja y no fue significativamente diferente entre alteplasa (7.7%) y placebo (8.4%; P = 0.681). Los resultados de los subgrupos del ensayo ECASS III confirmaron que un OTT más largo está asociado a un mayor riesgo de mortalidad y de hemorragia intracraneal sintomática. Los resultados del ensayo ECASS III muestran un beneficio clínico neto positivo para ACTILYSE® en el intervalo de tiempo de 3 a 4.5 horas, mientras que los datos combinados demuestran que el beneficio clínico neto ya no es favorable para alteplasa en el intervalo de tiempo por encima de 4.5 horas.
Se ha evaluado la seguridad y la eficacia de ACTILYSE® en el tratamiento del ictus isquémico agudo hasta 4.5 horas de tiempo desde el inicio de la presentación de los síntomas de ictus hasta el tratamiento (OTT) mediante un registro en curso (SITS-ISTR: Registro de la implementación segura de la trombolisis en el ictus). En este estudio observacional se comparan los datos de los resultados de seguridad de 21,566 pacientes tratados en el intervalo de tiempo de 0 a 3 horas con los datos de 2,376 pacientes tratados entre las 3 y las 4.5 horas después del inicio del ictus isquémico agudo. Se observó que la incidencia de hemorragia intracraneal sintomática (de acuerdo con la definición SITS-MOST) era superior en el intervalo de tiempo de 3 a 4.5 horas (2.2%) en comparación con el intervalo de tiempo de hasta 3 horas (1.7%). Las tasas de mortalidad a los 3 meses fueron similares comparando el intervalo de tiempo de 3 a 4.5 horas (12.0%) con el intervalo de tiempo de 0 a 3 horas (12.3%), con un OR no ajustado de 0.97 (95% IC: 0.84-1.13, p = 0.70) y un OR ajustado de 1.26 (95% IC; 1.07-1.49, p = 0.005). Los datos observacionales SITS avalan la evidencia del ensayo clínico de que el OTT es un buen indicador del desenlace tras el tratamiento de un ictus agudo con alteplasa.
Pacientes de edad avanzada (> 80 años): Se usaron los metaanálisis con datos ajustados de pacientes individuales de 6.756 pacientes, incluyendo los de > 80 años en nueve ensayos aleatorizados que comparaban alteplasa con placebo o controles abiertos para evaluar el beneficio-riesgo de alteplasa en pacientes > 80 años. La probabilidad de un buen desenlace del ictus (mRS 0-1 en día 90/180) aumentó y se asoció a un beneficio mayor cuando fueron tratados antes para todos los grupos de edad (valor de p de 0.0203 para la interacción) y fue independiente de la edad.
El efecto del tratamiento de alteplasa fue similar para los pacientes de 80 años o más jóvenes (retraso medio del tratamiento 4,1 horas: 990/2512 (39%) tratados con alteplasa frente a 853/2515 (34%) de control consiguieron un buen desenlace del ictus en día 90/180: OR 1.25, 95% IC 1.10-1.42) y para aquellos mayores de 80 años (retraso medio del tratamiento 3.7 horas: 155/879 (18%) tratados con alteplasa frente a 112/850 (13%) de control consiguieron un buen desenlace del ictus: OR 1.56, 95% IC 1.17-2.08).
En pacientes mayores de 80 años tratados con alteplasa en menos o igual a 3 horas, se consiguió un buen desenlace del ictus en 55/302 (18.2%) frente a 30/264 (11.4%) de control (OR 1.86, 95% IC 1.11-3.13) y en aquellos tratados con alteplasa en 3 horas-4.5 horas 58/342 (17.0%) consiguieron un buen desenlace del ictus frente a 50/364 (13.7%) de control (OR 1.36, 95% IC 0.87-2.14).
231 (6.8%) de 3.391 pacientes asignados a alteplasa sufrieron hemorragia parenquimal de tipo 2 en 7 días frente a 44 (1.3%) de 3.365 asignados al control (OR 5.55, 95% IC 4.01-7.70).
91 pacientes (2.7%) asignados a alteplasa sufrieron hemorragia parenquimal de tipo 2 fatal en 7 días frente a 13 (0.4%) asignados al control (OR 7.14, 95% IC 3.98-12.79).
En pacientes mayores de 80 años tratados con alteplasa 32/879 (3.6%) sufrieron hemorragia intracraneal fatal en 7 días frente a 4/850 (0.5%) en controles (OR 7.95, 95% IC 2.79-22.60).
De un total de 8.658 pacientes > 80 años tratados en < 4.5 horas desde el inicio del ictus en el SITS-ISTR, se compararon los datos de los 2.157 pacientes tratados en > 3 a 4.5 horas desde el inicio del ictus con aquellos de los 6.501 pacientes tratados en < 3 horas.
La independencia funcional a los tres meses (puntuación mRS 0-2) fue de 36% frente al 37% (OR ajustado 0.79, 95% IC 0.68-0.92), la mortalidad fue del 29% frente al 29.6% (OR ajustado 1.10, 95% IC 0.95-1.28), y la HIC sintomática (según la definición SITS-MOST) fue de 2.7% frente al 1.6% (OR ajustado 1.62, 95% IC 1.12-2.34).
Población pediátrica: Se han obtenido datos observacionales no aleatorizados y no comparativos sobre pacientes con ictus de 16-17 años de edad con tratamiento de alteplasa confirmado de SITS-ISTR (Implementación segura de tratamientos en ictus: Registro Internacional de Trombólisis de ictus, un registro internacional independiente). Entre 2003 y finales de 2017, se acumularon en el registro SITS un total de 25 pacientes pediátricos con uso de alteplasa confirmado dentro del grupo de edad de 16 a 17 años. La dosis media de alteplasa utilizada en este grupo de edad fue de 0.9 mg/kg (rango: 0.83 a 0.99 mg/kg). 23 de 25 pacientes iniciaron el tratamiento dentro de las 4.5 h posteriores al inicio de los síntomas del ictus (19 a las 3 h, 4 a las 3-4.5 h, 1 a las 5-5.5 h, 1 caso no informado). El peso varió de 56 a 90 kg. La mayoría de los pacientes presentaron un ictus moderado o de moderado a grave con una NIHSS mediana de 9.0 (rango 1-30) al inicio del estudio.
Las puntuaciones del día 90 mRS estaban disponibles en 21/25 pacientes. En el día 90, 14/21 pacientes tenían una puntuación mRS de 0-1 (sin síntomas o sin discapacidad significativa) y 5 pacientes más tenían mRS = 2 (ligera discapacidad). Esto significa que 19/21 (más del 90%) de los pacientes tuvieron un resultado favorable en el día 90 según mRS. Los 2 pacientes restantes tuvieron un resultado informado de discapacidad moderada grave (mRS = 4, n = 1) o muerte (mRS = 6) dentro de los 7 días (n = 1).
Cuatro pacientes no tenían una puntuación de día 90 mRS informada. La última información disponible mostró que 2/4 pacientes tenían una mRS de 2 al día 7 y 2/4 pacientes informaron una mejoría global clara al día 7.
Los datos de seguridad sobre los eventos adversos de hemorragias y edema también estaban disponibles en el registro. De los 25 pacientes de la categoría de edad de 16-17 años, ninguno tenía hemorragia intracerebral sintomática (ICHH, hemorragia hemorrágica de HCH tipo PH2). 5 casos desarrollaron edema cerebral después del tratamiento con alteplasa. 4/5 pacientes con edema cerebral tenían reportado un día 90 mRS entre 0 y 2 o mostraron una mejoría global el día 7 después del tratamiento. Un paciente tenía una mRS = 4 (discapacidad moderada grave) informada el día 90. Ninguno de los casos experimentó un desenlace fatal.
En resumen, hubo 25 informes del Registro SITS de pacientes entre 16 y 17 años de edad con ictus isquémico agudo que han sido tratados de acuerdo con las recomendaciones de adultos con alteplasa. Aunque el tamaño de muestra pequeño impide un análisis estadístico, los resultados globales muestran una tendencia positiva con la dosis de adultos correspondiente utilizada en estos pacientes. Los datos no parecen mostrar un mayor riesgo de hemorragia intracerebral sintomática o edema en comparación con los adultos.
Propiedades farmacocinéticas:
La alteplasa se elimina rápidamente de la sangre circulante y se metaboliza principalmente a través del hígado (aclaramiento plasmático 550-680 mL/min). En condiciones fisiológicas, la mayor parte de la alteplasa en circulación está unida al inhibidor. La eliminación hepática de alteplasa no se ve obstaculizada por la presencia de otras proteínas, incluidos los inhibidores de alteplasa. Los complejos de alteplasa y su inhibidor se eliminan como alteplasa libre. La vida media plasmática relevante, t½ alfa es de 4-5 minutos. Esto significa que, al cabo de 20 minutos, menos de un 10% del valor inicial está presente en el plasma. Para la cantidad residual que permanece en un compartimento profundo, se determinó una vida media beta de aproximadamente 40 minutos.
DATOS FARMACÉUTICOS:
Lista de excipientes:
Polvo: Arginina. Ácido fosfórico (para el ajuste del pH) Polisorbato 80.
Disolvente: Agua para preparaciones inyectables.
CONTRAINDICACIONES:
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección Lista de excipientes.
Contraindicaciones en caso de infarto agudo de miocardio, embolia pulmonar aguda masiva e ictus isquémico agudo:
ACTILYSE® está contraindicado en casos en los que existe un alto riesgo de hemorragia como p. ej.:
- Trastorno hemorrágico significativo actual o durante los últimos 6 meses.
- Diátesis hemorrágica conocida.
- Pacientes que reciben tratamiento efectivo con anticoagulantes orales (p. ej. warfarina sódica con INR > 1.3) (ver sección Advertencias y precauciones especiales de empleo).
- Hemorragia grave o peligrosa manifiesta o reciente.
- Sospecha o historia conocida de hemorragia intracraneal.
- Sospecha de hemorragia subaracnoidea o trastorno después de una hemorragia subaracnoidea por aneurisma.
- Cualquier historia de lesión del sistema nervioso central (es decir, neoplasia, aneurisma, cirugía intracraneal o espinal).
- Masaje cardíaco externo traumático reciente (menos de 10 días), parto obstétrico reciente, punción reciente de un vaso sanguíneo no comprimible (p. ej. punción de la vena yugular o subclavia).
- Hipertensión arterial grave no controlada.
- Endocarditis bacteriana, pericarditis.
- Pancreatitis aguda.
- Enfermedad gastrointestinal ulcerativa documentada durante los últimos 3 meses, várices esofágicas, aneurismas arteriales, malformaciones venosas/arteriales.
- Neoplasia con riesgo de hemorragia aumentado.
- Enfermedad hepática grave, incluyendo insuficiencia hepática, cirrosis, hipertensión portal (várices esofágicas) y hepatitis activa.
- Cirugía mayor o traumatismo importante en los últimos 3 meses.
Contraindicaciones adicionales en el infarto agudo de miocardio:
- Cualquier historia conocida de ictus hemorrágico o ictus de origen desconocido.
- Historia conocida de ictus isquémico o ataque isquémico transitorio (AIT) en los 6 meses anteriores, excepto ictus isquémico agudo actual durante las 4.5 horas anteriores.
Contraindicaciones adicionales en la embolia pulmonar aguda masiva:
- Cualquier historia conocida de ictus hemorrágico o ictus de origen desconocido.
- Historia conocida de ictus isquémico o ataque isquémico transitorio (AIT) en los 6 meses anteriores, excepto ictus isquémico agudo actual durante las 4.5 horas anteriores.
Contraindicaciones adicionales en el ictus isquémico agudo:
- Síntomas de accidente isquémico que empiezan más de 4.5 horas antes del inicio de la perfusión o síntomas para los cuales se desconoce la hora del inicio y ésta puede ser potencialmente superior a las 4.5 horas (ver sección Propiedades farmacodinámicas).
- Déficit neurológico leve o síntomas de rápida mejora antes del inicio de la perfusión.
- Ictus grave evaluado clínicamente (p. ej. NIHSS > 25) y/o por técnicas de imagen apropiadas.
- Convulsiones al inicio del ictus.
- Evidencia de una hemorragia intracraneal en la TC.
- Síntomas que sugieran hemorragia subaracnoidea, incluso con TC normal.
- Administración de heparina dentro de las 48 horas previas y un tiempo de tromboplastina que exceda el límite superior normal.
- Pacientes con cualquier historia previa de ictus y diabetes concomitante.
- Ictus previo en los últimos 3 meses.
- Recuento plaquetario inferior a 100,000/mm3.
- Presión sanguínea sistólica > 185 mmHg o presión sanguínea diastólica > 110 mmHg, o controles agresivos (farmacoterapia intravenosa) necesarios para reducir la presión sanguínea a estos límites.
- Niveles de glucosa en sangre < 50 mg/dL o > 400 mg/dL (< 2.8 mM o > 22.2 mM).
Uso en niños y adolescentes:
ACTILYSE® no está indicado en el tratamiento del ictus isquémico agudo en niños menores de 16 años (para adolescentes de 16 años o mayores, ver sección Advertencias y precauciones especiales de empleo).
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO:
Trazabilidad:
Con objeto de mejorar la trazabilidad de los medicamentos biológicos, el nombre y el número de lote del medicamento administrado deben estar claramente registrados.
La presentación adecuada del producto alteplasa debe escogerse cuidadosamente y de acuerdo con el uso previsto. El vial de 2 mg de alteplasa no está indicado para utilizarse en infarto agudo de miocardio, embolia pulmonar aguda masiva o ictus isquémico agudo (debido al riesgo de infradosificación masiva). Solamente los viales de 10 mg, 20 mg o 50 mg están indicados para su utilización en estas indicaciones.
El tratamiento trombolítico/fibrinolítico requiere una monitorización adecuada. ACTILYSE® debe ser utilizado sólo bajo la responsabilidad y seguimiento de médicos entrenados y con experiencia en la administración de tratamiento trombolítico y con los medios para monitorizar esta administración. Se recomienda que cuando se administre ACTILYSE® esté disponible en todos los casos un equipo de reanimación estándar y farmacoterapia.
Hipersensibilidad: Las reacciones de hipersensibilidad inmunomediada asociadas a la administración de ACTILYSE® pueden ser provocadas por el principio activo alteplasa o alguno de los excipientes. Tras el tratamiento no se ha observado formación sostenida de anticuerpos para la molécula del activador recombinante de plasminógeno tisular humano. No hay experiencia sistemática con la readministración de ACTILYSE®.
También existe riesgo de reacciones de hipersensibilidad mediadas por un mecanismo no inmunitario.
El angioedema constituye la reacción de hipersensibilidad más frecuente notificada con ACTILYSE®. Este riesgo puede aumentar en la indicación de ictus isquémico agudo y/o por el tratamiento concomitante con inhibidores del ECA (ver sección Interacción con otros medicamentos y otras formas de interacción). Los pacientes tratados para cualquier indicación autorizada se deben vigilar por si presentan angioedema durante o en las 24 h siguientes a la perfusión.
Si se presenta una reacción de hipersensibilidad grave (p. ej. angioedema), se debe suspender la perfusión e iniciar de inmediato un tratamiento adecuado. Esto puede incluir intubación.
Hemorragias: La complicación más común encontrada durante el tratamiento con ACTILYSE® es la hemorragia. El uso concomitante de otras sustancias activas que afectan a la coagulación o la función plaquetaria, puede contribuir a la hemorragia. Como la fibrina se lisa durante el tratamiento con ACTILYSE®, puede producirse hemorragia procedente de lugares de punción recientes. Por lo tanto, el tratamiento trombolítico requiere una atención cuidadosa de todos los posibles lugares de sangrado (incluidos los provocados por la inserción del catéter, el corte de acceso arterial y venoso y la punción con aguja). Debe evitarse el uso de catéteres rígidos, inyecciones intramusculares y el manejo no esencial del paciente durante el tratamiento con ACTILYSE®.
Si se produce una hemorragia potencialmente peligrosa, en particular hemorragia cerebral, debe interrumpirse el tratamiento fibrinolítico y la administración concomitante de heparina se debe interrumpir inmediatamente. Sin embargo, por regla general, no es necesario sustituir los factores de coagulación debido a la corta vida media y al efecto mínimo sobre los factores de la coagulación sistémicos. La mayoría de los pacientes que presentan hemorragia pueden controlarse mediante interrupción del tratamiento trombolítico y anticoagulante, reemplazo del volumen y aplicación de presión manual a un vaso comprimible. Debe considerarse la administración de protamina si se ha administrado heparina dentro de las 4 horas después de la presentación de la hemorragia. Puede indicarse el uso racional de productos de transfusión en los pocos pacientes que no respondan a estas medidas conservadoras. Después de cada administración debe realizarse una reevaluación mediante análisis clínicos y de laboratorio para considerar la necesidad de transfundir crioprecipitado, plasma congelado reciente y plaquetas. Con perfusión de crioprecipitado es deseable alcanzar un nivel de fibrinógeno de 1 g/L. Como última alternativa se dispone de agentes antifibrinolíticos.
El riesgo de hemorragia intracraneal es mayor en pacientes de edad avanzada, por lo tanto en estos pacientes debe valorarse cuidadosamente la relación beneficio/riesgo.
Como con todos los agentes trombolíticos, el beneficio terapéutico esperado debe ponderarse cuidadosamente frente al posible riesgo, especialmente en pacientes con:
- Traumatismos menores recientes, como biopsias, punciones de vasos mayores, inyecciones intramusculares, masaje cardíaco para reanimación.
- Trastornos con un mayor riesgo de hemorragia no mencionados en la sección Contraindicaciones.
Pacientes que reciben tratamiento anticoagulante oral:
Se puede considerar adecuado el uso de ACTILYSE® cuando la dosis o el tiempo transcurrido, desde la última toma del tratamiento anticoagulante, hacen que la eficacia residual sea poco probable, confirmada por las pruebas de actividad anticoagulante apropiadas para los correspondientes medicamentos no muestren actividad clínicamente relevante sobre el sistema de coagulación (p. ej. INR ≤ 1.3 para los antagonistas de la vitamina K u otras pruebas pertinentes para otros anticoagulantes orales que estén dentro del correspondiente límite superior de lo normal).
Advertencias y precauciones especiales adicionales en el infarto agudo de miocardio y en la embolia pulmonar aguda masiva: No debe administrarse una dosis superior a 100 mg de alteplasa debido a que ha sido asociada con un incremento adicional de hemorragias intracraneales. Debe procederse con especial cuidado, para asegurar que la dosis de alteplasa que se perfunde corresponda a la descrita en la sección Posología y forma de administración.
El beneficio terapéutico esperado debe ser ponderado cuidadosamente frente al posible riesgo, especialmente en pacientes con la presión sanguínea sistólica > 160 mmHg (ver la sección Contraindicaciones) y de edad avanzada que pueden aumentar el riesgo de hemorragia intracerebral. Como el beneficio terapéutico también es positivo en pacientes de edad avanzada, la evaluación del beneficio-riesgo se debe realizar cuidadosamente.
Antagonistas GPIIb/IIIa: El uso concomitante de antagonistas GPIIb/IIIa aumenta el riesgo de hemorragia.
Advertencias y precauciones especiales adicionales en el infarto agudo de miocardio:
Arritmias:
La trombólisis coronaria puede provocar arritmia asociada a la reperfusión.
Las arritmias por reperfusión pueden provocar un paro cardíaco, pueden ser potencialmente mortales y pueden requerir el uso de tratamientos antiarrítmicos convencionales.
Tromboembolismo:
El uso de trombolíticos puede aumentar el riesgo de acontecimientos tromboembólicos en pacientes con trombo izquierdo del corazón, por ejemplo, estenosis mitral o fibrilación auricular.
Advertencias y precauciones especiales adicionales en el ictus isquémico agudo:
Precauciones especiales de empleo: El tratamiento sólo debe ser realizado bajo la responsabilidad y supervisión de un médico entrenado y con experiencia en cuidados neurovasculares. Para verificar la indicación a tratar, las medidas de diagnóstico remoto se pueden considerar adecuadas (ver sección Indicaciones terapéuticas).
Advertencias especiales/condiciones con una relación beneficio/riesgo reducida:
La hemorragia intracerebral representa la principal reacción adversa en el tratamiento del ictus isquémico agudo (hasta el 15% de los pacientes sin ningún aumento de la mortalidad global y sin ningún aumento relevante de la mortalidad global y discapacidad grave combinada, por ejemplo, puntuación 5 y 6 en la Escala de Rankin modificada [modified Rankin Scale, mRS]).
Comparado con otras indicaciones, los pacientes con ictus isquémico agudo tratados con ACTILYSE® tienen un riesgo marcadamente aumentado de hemorragia intracraneal, ya que la hemorragia tiene lugar principalmente en el área del infarto. Esto es aplicable en particular a los casos siguientes:
- Todas las situaciones enumeradas en la sección Contraindicaciones y en general todas las situaciones que conlleven alto riesgo de hemorragia.
- A medida que aumenta el tiempo desde la aparición de los síntomas del ictus y el inicio del tratamiento, el beneficio clínico neto disminuye. Por tanto, no se debe retrasar la administración de ACTILYSE®.
- Pacientes pre-tratados con ácido acetilsalicílico (AAS) pueden tener un mayor riesgo de hemorragia intracerebral, sobre todo si se retrasa el tratamiento con ACTILYSE®.
- Comparado con pacientes más jóvenes, los pacientes de edad avanzada (más de 80 años) pueden tener un resultado algo peor independientemente del tratamiento. También son más propensos a sufrir ictus más graves que se asocian con un mayor riesgo absoluto de hemorragia intracerebral cuando se los tromboliza en comparación con ictus más leves cuando se los tromboliza o con pacientes no trombolizados. Aunque los datos disponibles indican que el beneficio neto de ACTILYSE® en pacientes de más de 80 años es menor comparado con pacientes más jóvenes, ACTILYSE® se puede usar en pacientes de más de 80 años con base en el beneficio-riesgo individual (ver la sección Propiedades farmacodinámicas). Los pacientes de edad avanzada se deben seleccionar muy cuidadosamente teniendo en cuenta tanto la salud general como el estado neurológico.
- El beneficio terapéutico se reduce en pacientes con un ictus previo (ver también la sección Contraindicaciones) o en los cuales se conoce una diabetes no controlada, de este modo la relación beneficio/riesgo se considera menos favorable pero todavía positiva para estos pacientes.
- En pacientes con ictus muy leve, los riesgos superan el beneficio esperado (ver la sección Contraindicaciones).
- Los pacientes con ictus muy grave presentan un riesgo mayor de hemorragia intracerebral y muerte y no se deben tratar (ver la sección Contraindicaciones).
- Los pacientes con infartos extensos presentan un mayor riesgo de resultados insatisfactorios, incluyendo hemorragia grave y muerte. En estos pacientes se debe considerar cuidadosamente la relación beneficio/riesgo.
- En los pacientes con ictus la probabilidad de un buen desenlace disminuye con un mayor tiempo desde el inicio de los síntomas hasta el tratamiento, al aumentar la edad, al aumentar la severidad del ictus y con los niveles de glucosa en sangre altos en el momento del ingreso, mientras que la probabilidad de discapacidad grave y muerte o hemorragias intracraneales sintomáticas aumenta, independientemente del tratamiento.
El tratamiento no debe iniciarse más de 4.5 horas después de la aparición de los síntomas debido a una relación beneficio/riesgo desfavorable basada principalmente en lo siguiente:
- Los efectos positivos del tratamiento disminuyen con el tiempo.
- La tasa de mortalidad aumenta particularmente en pacientes con tratamiento con AAA previo.
- Riesgo incrementado de hemorragia sintomática.
Monitorización de la presión sanguínea: La monitorización de la presión sanguínea durante la administración del tratamiento y hasta 24 horas después parece justificada; también se recomienda un tratamiento antihipertensivo por vía intravenosa si la presión sistólica es > 180 mmHg o la presión diastólica es > 105 mmHg.
Otras advertencias especiales: La reperfusión del área isquémica puede inducir a edema cerebral en la zona del infarto.
Debido a un riesgo aumentado de hemorragia, el tratamiento con inhibidores de la agregación plaquetaria no debe iniciarse dentro de las primeras 24 horas después de la trombolisis con alteplasa.
Población pediátrica:
Hasta el momento, sólo se dispone de experiencia limitada sobre el uso de ACTILYSE® en niños y adolescentes.
Cuando se considere el uso de ACTILYSE® para el tratamiento del ictus isquémico agudo en adolescentes de 16 años o mayores, cuidadosamente seleccionados, se debe sopesar la relación beneficio/riesgo de forma individual y discutirse con el paciente y el padre/tutor, según corresponda. Los adolescentes de 16 años o mayores deben ser tratados de acuerdo con las instrucciones de uso del prospecto para la población adulta después de la obtención de imágenes mediante técnicas apropiadas para descartar falsos ictus y confirmar la oclusión arterial correspondiente al déficit neurológico (ver sección Propiedades farmacodinámicas).
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Fertilidad, embarazo y lactancia:
Embarazo: Los datos relativos al uso de alteplasa en mujeres embarazadas son limitados. Los estudios en animales realizados con alteplasa con dosis superiores a las de los humanos mostraron inmadurez fetal y/o embriotoxicidad, secundario a la actividad farmacológica conocida del medicamento. Alteplasa no se considera teratogénica (ver sección Datos preclínicos sobre seguridad).
En caso de una enfermedad aguda que supone un riesgo para la vida del paciente, debe evaluarse el beneficio frente al riesgo potencial.
Lactancia: Se desconoce si alteplasa se excreta en la leche materna y no se dispone de información suficiente relativa a la excreción de alteplasa en la leche de animales.
Se debe tener precaución cuando se usa ACTILYSE® en una mujer en período de lactancia y se debe decidir si es necesario interrumpir la lactancia durante las primeras 24 horas después del uso de ACTILYSE®.
Fertilidad: No se dispone de datos clínicos de fertilidad para ACTILYSE®. Estudios en animales realizados con alteplasa no mostraron ningún efecto adverso en fertilidad (ver sección Datos preclínicos sobre seguridad).
REACCIONES ADVERSAS:
La reacción adversa más frecuente asociada a ACTILYSE® es la hemorragia en diferentes formas con un descenso en los valores de hematocrito y/o hemoglobina.
Las reacciones adversas detalladas a continuación se enumeran según la frecuencia y clasificación por órganos y sistemas. Las frecuencias se definen según las siguientes categorías: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1,000 a < 1/100), raras (≥ 1/10,000 a < 1/1,000), muy raras (< 1/10,000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
A excepción de la hemorragia intracerebral/intracraneal como reacción adversa en la indicación ictus y de las arritmias de reperfusión en la indicación infarto agudo de miocardio, no existe ninguna razón médica para suponer que el perfil cualitativo y cuantitativo de reacciones adversas de ACTILYSE® en las indicaciones embolia pulmonar e ictus isquémico agudo sea diferente del perfil en la indicación infarto agudo de miocardio.
Tabla 1. Reacciones adversas en infarto agudo de miocardio, embolia pulmonar aguda masiva e ictus isquémico agudo
|
Clasificación por órganos y sistemas |
Reacción adversa |
|
Hemorragia |
|
|
Muy frecuentes |
La hemorragia intracerebral representa la reacción adversa principal en el tratamiento del ictus isquémico agudo Todas las hemorragias, incluidas las de esta tabla, por ejemplo, hemorragia intracraneal y no intracraneal |
|
Frecuentes |
Hemorragia intracerebral (como hemorragia cerebral, hematoma cerebral, ictus hemorrágico, transformación hemorrágica del ictus, hematoma intracraneal, hemorragia subaracnoidea) en el tratamiento del infarto agudo de miocardio y embolia pulmonar aguda masiva Hemorragia faríngea Hemorragia gastrointestinal (como hemorragia gástrica, úlcera gástrica sangrante, hemorragia rectal, hematemesis, melena, hemorragia bucal, hemorragia gingival) Equimosis Hemorragia urogenital (como hematuria, hemorragia del tracto urinario) Hemorragia en el lugar de inyección (hemorragia en el lugar de punción) Hematoma en el lugar de inserción de catéter, hemorragia en el lugar de inserción de catéter) |
|
Poco frecuentes |
Hemorragia pulmonar (como hemoptisis, hemotórax, hemorragia del tracto respiratorio) Epistaxis Hemorragia en el oído |
|
Raras |
Hemorragia en el ojo Hemorragia pericardial Hemorragia retroperitoneal (como hematoma retroperitoneal) |
|
Frecuencia no conocida*** |
Hemorragia en los órganos parenquimatosos (como hemorragia hepática) |
|
Trastornos del sistema inmunológico |
|
|
Raras |
Reacciones de hipersensibilidad (p. ej. exantema, urticaria, broncoespasmo, angioedema, hipotensión, shock)* |
|
Muy raras |
Anafilaxia grave |
|
Trastornos del sistema nervioso |
|
|
Muy raros |
Acontecimientos relacionados con el sistema nervioso (p. ej. ataque epiléptico, convulsiones, afasia, trastorno del habla, delirio, síndrome cerebral agudo, agitación, confusión, depresión, psicosis) a menudo asociados a acontecimientos concurrentes de isquemia o hemorragia cerebrovascular |
|
Trastornos cardíacos** |
|
|
Muy frecuentes |
Isquemia/angina de pecho recurrentes, hipotensión e insuficiencia cardíaca/edema pulmonar |
|
Frecuentes |
Shock cardiogénico, paro cardíaco y reinfarto |
|
Poco frecuentes |
Arritmias de reperfusión (como arritmias, extrasístoles, bloqueo aurículo-ventricular (AV) de primer grado a bloqueo auriculoventricular completo, fibrilación/aleteo (flutter) auricular, bradicardia, taquicardia, arritmia ventricular, taquicardia/fibrilación ventricular, disociación electromecánica [DEM]) Regurgitación mitral, embolia pulmonar, otras embolias sistémicas/embolia cerebral, defecto septal ventricular |
|
Trastornos vasculares |
|
|
Raras |
Embolia, la cual puede conducir a las correspondientes consecuencias en los órganos afectados |
|
Trastornos gastrointestinales |
|
|
Raras |
Náuseas |
|
Frecuencia no conocida*** |
Vómitos |
|
Exploraciones complementarias |
|
|
Poco frecuentes |
Descenso de la presión arterial |
|
Frecuencia no conocida*** |
Aumento de la temperatura corporal |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
|
|
Frecuencia no conocida*** |
Embolia grasa (embolización de cristales de colesterol), lo cual puede conducir a las correspondientes consecuencias en los órganos afectados |
|
Procedimientos médicos y quirúrgicos |
|
|
Frecuencia no conocida*** |
Transfusión sanguínea (necesaria) |
* Ver las secciones Advertencias y precauciones especiales de empleo e Interacción con otros medicamentos y otras formas de interacción.
**Trastornos cardíacos
Como ocurre con otros agentes trombolíticos, los acontecimientos descritos anteriormente en la sección respectiva se han notificado como secuelas de infarto de miocardio y/o administración trombolítica. Estos acontecimientos cardíacos pueden suponer un riesgo para la vida, llegando a producir la muerte.
***Cálculo de la frecuencia
Esta reacción adversa se ha observado a partir de la experiencia poscomercialización. Con un 95% de seguridad, la categoría de la frecuencia no es mayor que las “raras”, pero puede ser menor. La estimación precisa de la frecuencia no es posible ya que la reacción adversa no ocurrió en la base de datos de ensayo clínicos de 8,299 pacientes.
En pacientes que han padecido un ictus (incluyendo hemorragia intracraneal) y otros episodios hemorrágicos graves, se han notificado casos de muerte y discapacidad permanente.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: No procede.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacción con otros medicamentos y otras formas de interacción:
No se han realizado estudios formales de interacciones con ACTILYSE® y medicamentos comúnmente administrados en pacientes con infarto agudo de miocardio.
Fármacos que afectan la función de coagulación/plaquetas:
El riesgo de hemorragia aumenta si se administran derivados cumarínicos, anticoagulantes orales, inhibidores de la agregación plaquetaria, heparina no fraccionada o heparina de bajo peso molecular (LMWH) u otras sustancias activas que interfieran con la coagulación (antes, durante o dentro de las primeras 24 horas después del tratamiento con ACTILYSE®). (Ver secciones Posología y forma de administración y Contraindicaciones).
Inhibidores del ECA:
El tratamiento concomitante con inhibidores de la ECA puede aumentar el riesgo de sufrir una reacción de hipersensibilidad (ver sección Advertencias y precauciones especiales de empleo).
El uso concomitante de antagonistas GPIIb/IIIa aumenta el riesgo de hemorragia.
INCOMPATIBILIDADES:
La solución reconstituida puede diluirse con una solución inyectable estéril de cloruro de sodio 9 mg/mL (0.9%) hasta una concentración mínima de 0.2 mg de alteplasa por mL.
Diluciones adicionales, el uso de agua para preparaciones inyectables para dilución o en general el uso de soluciones de carbohidratos para perfusión, como dextrosa, no se recomiendan, debido al aumento de la turbidez de la solución reconstituida.
ACTILYSE® no debe mezclarse con otros medicamentos ni en el mismo vial de perfusión ni en el mismo catéter (ni siquiera con heparina).
HALLAZGOS DE LABORATORIO CLÍNICO:
Datos preclínicos sobre seguridad: En estudios de toxicidad subcrónica en ratas y monos titís no se han observado reacciones adversas inesperadas. En estudios de mutagenicidad, no se observaron indicios de potencial mutagénico.
En animales gestantes no se observaron efectos teratogénicos tras la perfusión intravenosa de dosis farmacológicamente efectivas. En conejos, dosis superiores a 3 mg/kg/día dieron lugar a efectos embriotóxicos (embrioletalidad, retraso en el crecimiento). En ratas, no se observaron efectos en el desarrollo peri-postnatal ni en los parámetros de fertilidad con dosis de hasta 10 mg/kg/día.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología y forma de administración: ACTILYSE® debe administrarse tan pronto como sea posible después de la presentación de los síntomas. Se aplican las siguientes instrucciones de dosificación.
Infarto agudo de miocardio:
Posología:
a) Régimen de dosificación de 90 minutos (acelerado) para pacientes con infarto agudo de miocardio, en los cuales pueda iniciarse el tratamiento dentro de las 6 horas después de la presentación de los síntomas:
En pacientes con un peso corporal ≥ 65 kg:
|
Volumen a administrar en función de la concentración de alteplasa |
||
|
1 mg/mL |
2 mg/mL |
|
|
15 mg en forma de bolo intravenoso, inmediatamente seguido de |
15 mL |
7.5 mL |
|
50 mg en forma de perfusión intravenosa a velocidad constante durante los primeros 30 minutos, inmediatamente seguido de |
50 mL |
25 mL |
|
35 mg en forma de perfusión intravenosa a velocidad constante durante 60 minutos, hasta una dosis máxima total de 100 mg |
35 mL |
17.5 mL |
En pacientes con un peso corporal < 65 kg, la dosis total debe ajustarse al peso de la siguiente forma:
|
Volumen a administrar en función de la concentración de alteplasa |
||
|
1 mg/mL |
2 mg/mL |
|
|
15 mg en forma de bolo intravenoso, inmediatamente seguido de |
15 mL |
7.5 mL |
|
0.75 mg/kg de peso corporal (p.c.) en forma de perfusión intravenosa a velocidad constante durante los primeros 30 minutos, inmediatamente seguido de |
0.75 mL/kg p.c. |
0.375 mL/kg p.c. |
|
0.5 mg/kg de peso corporal (p.c.) en forma de perfusión intravenosa a velocidad constante durante 60 minutos |
0.5 mL/kg p.c. |
0.25 mL/kg p.c. |
b) Régimen de dosificación de 3 horas para pacientes con infarto agudo de miocardio en los cuales pueda iniciarse el tratamiento entre las 6 y 12 horas después de la presentación de los síntomas.
En pacientes con un peso corporal ≥ 65 kg:
|
Volumen a administrar en función de la concentración de alteplasa |
||
|
1 mg/mL |
2 mg/mL |
|
|
10 mg en forma de bolo intravenoso, inmediatamente seguido de |
10 mL |
5 mL |
|
50 mg en forma de perfusión intravenosa a velocidad constante durante la primera hora, inmediatamente seguido de |
50 mL |
25 mL |
|
40 mg en forma de perfusión intravenosa a velocidad constante durante dos horas, hasta una dosis máxima total de 100 mg |
40 mL |
20 mL |
En pacientes con un peso corporal < 65 kg:
|
Volumen a administrar en función de la concentración de alteplasa |
||
|
1 mg/mL |
2 mg/mL |
|
|
10 mg en forma de bolo intravenoso, inmediatamente seguido de |
10 mL |
5 mL |
|
una perfusión intravenosa a velocidad constante durante 3 horas hasta una dosis máxima total de 1.5 mg/kg p.c. |
1.5 mL/kg p.c. |
0.75 mL/kg p.c. |
Tratamiento coadyuvante: Se recomienda tratamiento antitrombótico coadyuvante en cumplimiento con las guías internacionales actuales para el tratamiento de pacientes con infarto de miocardio con elevación del ST.
Forma de administración: La solución reconstituida debe administrarse por vía intravenosa y es para uso inmediato.
Los viales de 2 mg de alteplasa no están indicados para ser usados en esta indicación. Para instrucciones previas a la reconstitución/administración, ver sección Precauciones especiales de eliminación y otras manipulaciones.
Embolia pulmonar aguda masiva:
Posología:
En pacientes con un peso corporal ≥ 65 kg: Debe administrarse una dosis total de 100 mg de alteplasa en 2 horas.
El siguiente régimen de dosificación es con el que se tiene mayor experiencia:
|
Volumen a administrar en función de la concentración de alteplasa |
||
|
1 mg/mL |
2 mg/mL |
|
|
10 mg en forma de bolo intravenoso durante 1-2 minutos, inmediatamente seguido de |
10 mL |
5 mL |
|
90 mg como perfusión intravenosa a velocidad constante durante 2 horas hasta una dosis máxima total de 100 mg |
90 mL |
45 mL |
En pacientes con un peso corporal < 65 kg:
|
Volumen a administrar en función de la concentración de alteplasa |
||
|
1 mg/mL |
2 mg/mL |
|
|
10 mg en forma de bolo intravenoso durante 1-2 minutos, inmediatamente seguido de |
10 mL |
5 mL |
|
una perfusión intravenosa a velocidad constante durante 2 horas hasta una dosis máxima total de 1.5 mg/kg p.c. |
1.5 mL/kg p.c. |
0.75 mL/kg p.c. |
Tratamiento coadyuvante: Después del tratamiento con ACTILYSE® debe iniciarse (o reanudarse) un tratamiento con heparina si los valores aPTT son inferiores al doble del límite superior normal. La perfusión debe ajustarse para mantener los valores de aPTT en el rango de 50-70 segundos (de 1.5 a 2.5 veces el valor de referencia).
Forma de administración: La solución reconstituida debe administrarse por vía intravenosa y es para uso inmediato.
Los viales de 2 mg de alteplasa no están indicados para ser usados en esta indicación. Para instrucciones previas a la reconstitución/administración, ver sección Precauciones especiales de eliminación y otras manipulaciones.
Ictus isquémico agudo: El tratamiento sólo debe ser realizado bajo la responsabilidad y supervisión de un médico entrenado y con experiencia en cuidados neurovasculares (ver secciones Contraindicaciones y Advertencias y precauciones especiales de empleo).
|
El tratamiento con ACTILYSE® debe iniciarse tan pronto como sea posible dentro de las 4.5 horas desde la presentación de los síntomas (ver sección Advertencias y precauciones especiales de empleo). Más allá de las 4.5 horas después de la presentación de los síntomas de ictus hay una relación beneficio/riesgo negativa asociada al tratamiento con ACTILYSE® y no se debe administrar (ver sección Propiedades farmacodinámicas). |
Posología: La dosis recomendada es de 0.9 mg de alteplasa/kg de peso corporal (hasta un máximo de 90 mg) empezando con un 10% de la dosis total en forma de bolo intravenoso inicial, inmediatamente seguido del resto de la dosis total perfundida por vía intravenosa durante 60 minutos.
Tabla de dosificación para ictus isquémico agudo
|
Utilizando la concentración estándar recomendada de 1 mg/mL el volumen (mL) a administrar es igual al valor de dosis recomendado (mg) |
|||
|
Peso (kg) |
Dosis Total (mg) |
Dosis Bolo (mg) |
Dosis Perfusión* (mg) |
|
40 |
36.0 |
3.6 |
32.4 |
|
42 |
37.8 |
3.8 |
34.0 |
|
44 |
39.6 |
4.0 |
35.6 |
|
46 |
41.4 |
4.1 |
37.3 |
|
48 |
43.2 |
4.3 |
38.9 |
|
50 |
45.0 |
4.5 |
40.5 |
|
52 |
46.8 |
4.7 |
42.1 |
|
54 |
48.6 |
4.9 |
43.7 |
|
56 |
50.4 |
5.0 |
45.4 |
|
58 |
52.2 |
5.2 |
47.0 |
|
60 |
54.0 |
5.4 |
48.6 |
|
62 |
55.8 |
5.6 |
50.2 |
|
64 |
57.6 |
5.8 |
51.8 |
|
66 |
59.4 |
5.9 |
53.5 |
|
68 |
61.2 |
6.1 |
55.1 |
|
70 |
63.0 |
6.3 |
56.7 |
|
72 |
64.8 |
6.5 |
58.3 |
|
74 |
66.6 |
6.7 |
59.9 |
|
76 |
68.4 |
6.8 |
61.6 |
|
78 |
70.2 |
7.0 |
63.2 |
|
80 |
72.0 |
7.2 |
64.8 |
|
82 |
73.8 |
7.4 |
66.4 |
|
84 |
75.6 |
7.6 |
68.0 |
|
86 |
77.4 |
7.7 |
69.7 |
|
88 |
79.2 |
7.9 |
71.3 |
|
90 |
81.0 |
8.1 |
72.9 |
|
92 |
82.8 |
8.3 |
74.5 |
|
94 |
84.6 |
8.5 |
76.1 |
|
96 |
86.4 |
8.6 |
77.8 |
|
98 |
88.2 |
8.8 |
79.4 |
|
100+ |
90.0 |
9.0 |
81.0 |
* Administrado a una concentración de 1 mg/mL durante 60 min a una velocidad de perfusión constante.
Tratamiento coadyuvante: La seguridad y eficacia de este régimen con la administración concomitante de heparina o inhibidores de la agregación plaquetaria como el ácido acetilsalicílico durante las primeras 24 horas después de la presentación de los síntomas no han sido suficientemente investigadas. Por lo tanto, la administración de heparina intravenosa o inhibidores de la agregación plaquetaria como el ácido acetilsalicílico debe evitarse en las primeras 24 horas después del tratamiento con ACTILYSE® debido a un riesgo de hemorragia aumentado. Si se requiere heparina para otra indicación (p. ej. prevención de la trombosis venosa profunda) la dosis no debe exceder las 10,000 UI por día, administrada por vía subcutánea.
Forma de administración: La solución reconstituida debe administrarse por vía intravenosa y es para uso inmediato.
Los viales de 2 mg de alteplasa no están indicados para ser usados en esta indicación. Para instrucciones previas a la reconstitución/administración, ver sección Precauciones especiales de eliminación y otras manipulaciones.
Población pediátrica: La experiencia con el uso de ACTILYSE® en niños y adolescentes es limitada. ACTILYSE® está contraindicado en el tratamiento del ictus isquémico agudo en niños y adolescentes menores de 16 años (ver sección Contraindicaciones). La dosis en adolescentes entre 16 y 17 años es la misma que para los adultos (ver sección Advertencias y precauciones especiales de empleo para ver las recomendaciones de las técnicas de imagen previas que se deben utilizar).
SOBREDOSIS:
Síntomas: Si se excede la dosis máxima recomendada, incrementa el riesgo de hemorragia intracraneal.
A pesar de la especificidad relativa por la fibrina, puede producirse una reducción clínicamente significativa del fibrinógeno y otros componentes de la coagulación sanguínea después de una sobredosificación.
Tratamiento: En la mayoría de los casos es suficiente esperar la regeneración fisiológica de estos factores después de haber finalizado el tratamiento con ACTILYSE®. Sin embargo, si se presentan hemorragias graves, se recomienda la perfusión de plasma congelado reciente, y si fuese necesario pueden administrarse antifibrinolíticos sintéticos.
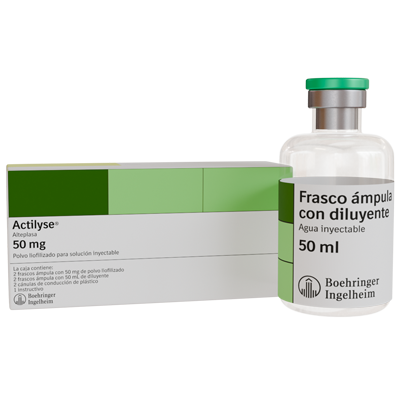
PRESENTACIONES:
Caja contiene:
2 frascos ámpula con 50 mg de polvo liofilizado
2 frascos ámpula con 50 mL de diluyente
2 cánulas de conducción de plástico
1 Instructivo.
Hecho en Alemania por:
Boehringer Ingelheim Pharma GmbH & Co. KG
Birkendorfer Str. 65
88397 Biberach a.d.R., Alemania
Para:
BOEHRINGER INGELHEIM PROMECO S.A. de C.V.
Calle del Maíz No. 49, Col. Barrio Xaltocan,
C.P. 16090, Xochimilco, Ciudad de México, México.
Fecha de revisión: Febrero, 2023
® Marca registrada
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Período de validez:
Viales sin abrir: 3 años.
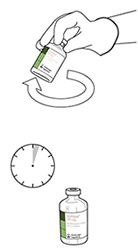
Solución reconstituida: Hecha la mezcla, el producto se conserva en refrigeración entre 2 y 8 ºC por 24 horas, y hasta 8 horas a no más de 30 ºC.
Desde un punto de vista microbiológico, el producto debe ser usado inmediatamente tras su reconstitución. Si no se usa inmediatamente, el período de conservación y las condiciones de uso antes de su utilización son responsabilidad de la persona que lo utilice y normalmente no deberían ser superiores a 24 horas entre 2 y 8 ºC.
Precauciones especiales de conservación: Protéjase de la luz.
Consérvese a no más de 30 °C en el envase original hasta el momento de su preparación. Para las condiciones de conservación tras la reconstitución del medicamento, ver sección Período de validez.
NATURALEZA Y CONTENIDO DEL ENVASE:
Polvo:
Viales de vidrio esterilizados de 50 mL, sellados con tapones de goma butílica, de color gris, siliconados y estériles y con cápsulas "flip-off" de aluminio/plástico.
Disolvente:
En el tamaño de envase de 50 mg, el agua para preparaciones inyectables se envasa en viales de 50 mL. Los viales de agua para preparaciones inyectables se sellan con tapones de goma y cápsulas "flip-off" de aluminio/plástico.
Precauciones especiales de eliminación y otras manipulaciones: Para obtener una concentración final de 1 mg de alteplasa por mL tras la reconstitución, todo el volumen del disolvente proporcionado debe transferirse al vial que contiene el polvo de ACTILYSE®. Con este propósito debe utilizarse la cánula de transferencia que se incluye en la presentación de 50 mg.
Para obtener una concentración final de 2 mg de alteplasa por mL tras la reconstitución, sólo debe utilizarse la mitad del disolvente proporcionado (ver tabla). En estos casos, siempre debe utilizarse una jeringa para transferir la cantidad necesaria de disolvente al vial que contiene el polvo de ACTILYSE®.
El contenido de un vial para inyección de ACTILYSE® (50 mg) debe disolverse bajo condiciones asépticas con agua para preparaciones inyectables, según la tabla siguiente, para obtener una concentración final de alteplasa de 1 mg/mL o de 2 mg/mL:
|
ACTILYSE® sustancia seca |
50 mg |
|
(a) Volumen de agua esterilizada para preparaciones inyectables que se debe añadir a la sustancia seca |
50 mL |
|
Concentración final: |
1 mg alteplasa/mL |
|
(b) Volumen de agua esterilizada para preparaciones inyectables que se debe añadir a la sustancia seca |
25 mL |
|
Concentración final: |
2 mg alteplasa/mL |
La solución reconstituida debe administrarse a continuación por vía intravenosa. La solución reconstituida de 1 mg/mL puede diluirse adicionalmente con una solución inyectable estéril de cloruro de sodio 9 mg/mL (0.9 %) hasta una concentración mínima de 0.2 mg/mL, ya que no puede excluirse la turbidez de la solución reconstituida. No se recomienda la dilución adicional de la solución reconstituida de 1 mg/mL con agua esterilizada para preparaciones inyectables o, en general, el uso de soluciones de carbohidratos para perfusión, p. ej. dextrosa, debido al aumento de formación de turbidez de la solución reconstituida, ACTILYSE® no debe mezclarse con otros medicamentos ni en el mismo vial de perfusión (ni siquiera con heparina).
Para incompatibilidades ver sección Incompatibilidades.
La solución reconstituida es para administración única. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
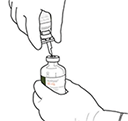
Instrucciones para reconstituir ACTILYSE®
|
1. Reconstituir inmediatamente antes de su administración. |
|
|
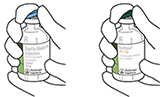
2. Retirar las tapas protectoras de los dos viales que contienen agua estéril y ACTILYSE® sustancia seca, respectivamente, tirándolas hacia arriba con un dedo. |
|
|
3. Limpiar el tapón de goma de cada uno de los viales con una toallita con alcohol. |
|
|
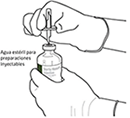
4. Sacar la cánula de transferencia* de su envoltorio. No desinfectar ni esterilizar la cánula de transferencia; es estéril. Quitar la tapa |
|
|
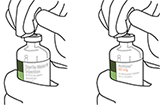
5. Mantener el vial de agua estéril vertical sobre una superficie estable. Directamente desde arriba, perforar el tapón de goma verticalmente en el centro del tapón con la cánula de transferencia, presionando con cuidado pero firmemente, sin girar. |
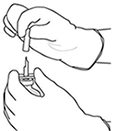
|
|
6. Sujetar el vial de agua estéril y la cánula de transferencia firmemente con una mano utilizando las dos solapas laterales. Retirar la tapa restante de la parte superior de la cánula de transferencia |
|
|
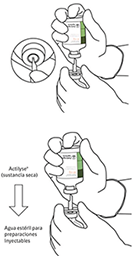
7. Sujetar el vial de agua estéril y la cánula de transferencia firmemente con una mano utilizando las dos solapas laterales. Sujetar el vial con ACTILYSE® sustancia seca verticalmente encima de la cánula de transferencia y posicionar la punta de la cánula de transferencia justo en el centro del tapón. Presionar el vial con la sustancia seca hacia abajo con la cánula de transferencia directamente desde arriba, perforando el tapón de goma verticalmente y con cuidado pero firmemente, sin girar. |
|
|
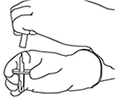
8. Invertir los dos viales y permitir que el agua drene completamente en la sustancia seca. |
|
|
9. Retirar el vial de agua vacío junto con la cánula de transferencia. Se pueden desechar. |
|
|
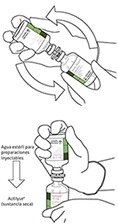
10. Coger el vial con ACTILYSE® reconstituido y girarlo con cuidado para disolver cualquier polvo restante pero no agitar, puesto que esto producirá espuma. Si hay burbujas, mantener la solución inmóvil durante unos minutos para permitir que desaparezcan. |
|
|
11. La solución reconstituida contiene 1 mg/mL de alteplasa. Debe ser límpida y de incolora a amarilla clara y no debe contener ninguna partícula. |
|
|
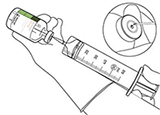
12. Extraer la cantidad requerida solamente utilizando una aguja y una jeringa. No utilizar la zona de punción de la cánula de transferencia para evitar pérdidas |
|
|
13. Utilizar inmediatamente. Desechar la solución no utilizada. |
|
* Sí se incluye en el kit una cánula de transferencia. La reconstitución también se puede realizar con una jeringa y una aguja.