
DETRUSITOL SR
TOLTERODINA
Cápsulas de liberación prolongada
Cápsulas de liberación prolongada, 4 Miligramos
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA:
Ingrediente activo: Tartrato de tolterodina.
Cada CÁPSULA de liberación prolongada para administración oral contiene 4 mg de tartrato de tolterodina que corresponde a 2.74 mg de tolterodina.
INDICACIONES TERAPÉUTICAS: Tolterodina está indicada para el tratamiento de vejiga hiperactiva con síntomas de frecuencia y urgencia urinaria, y/o incontinencia de urgencia.
PARTICULARES FARMACÉUTICAS
Precauciones especiales para almacenamiento: Almacenar a una temperatura menor de 30 °C.
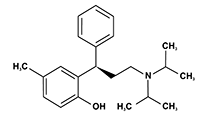
Estructura y nombre químico del principio activo: 2-[(1R)-3-[Bis(1-metiletil)amino]-1-fenilpropil]-4-metilfenol.
Documento de Producto Referencia No.: 841
Fecha: 11 de abril 2008
Reemplaza: 23 de febrero 2007
PFIZER
® Marca registrada
PROPIEDADES FARMACODINÁMICAS: La tolterodina es un antagonista competitivo de los receptores muscarínicos específicos con una selectividad para la vejiga urinaria, y para las glándulas salivales in vivo. Uno de los metabolitos de la tolterodina (el derivado 5-hidroximetilo) muestra un perfil farmacológico similar al del compuesto original. En pacientes que son metabolizadores rápidos, este metabolito contribuye significativamente al efecto terapéutico (ver Propiedades farmacocinéticas, Metabolismo).
El efecto del tratamiento puede esperarse dentro de 4 semanas.
Se estudió un total de 710 pacientes pediátricos (486 con cápsulas de liberación prolongada de tolterodina, 224 con placebo) con edad de 5-10 años con frecuencia urinaria e incontinencia urgente en dos estudios en fase 3, aleatorios, controlados con placebo, doble ciego, con duración de 12 semanas. El porcentaje de pacientes con infecciones de las vías urinarias fue mayor en pacientes tratados con las cápsulas de liberación prolongada de tolterodina (6.6%), en comparación con los pacientes que recibieron placebo (4.5%). Ocurrieron trastornos de la atención y comportamiento agresivo, anormal e hiperactivo en 2.9% de los niños tratados con las cápsulas de liberación prolongada de tolterodina en comparación con 0.9% de los niños tratados con placebo.
En el programa de Fase III, el criterio de valoración primario fue la reducción de los episodios de incontinencia por semana y los criterios de valoración secundarios fueron la reducción de las micturiciones en 24 horas y el incremento del volumen promedio desprovisto por micturición. Estos parámetros se presentan en la siguiente tabla.
|
Tabla 1. Efecto de tratamiento con tolterodina de liberación prolongada de 4 mg una vez al día después de 12 semanas, en comparación con placebo. Cambio absoluto y cambio en el porcentaje relativo a la línea basal. Diferencia de tratamiento de tolterodina de liberación prolongada vs. placebo: Cambio promedio estimado en los cuadrados mínimos e intervalo de confianza del 95%. |
||||
|
Liberación prolongada de tolterodina 4 mg una vez al día (n=507) |
Placebo (n=508) |
Diferencia de tratamiento vs. placebo: Cambio promedio y CI del 95% |
Significancia clínica vs. Placebo (valor p) |
|
|
Número de episodios de incontinencia por semana |
-11.8 (-54%) |
-6.9 (-28%) |
-4.8 (-7.2; -2.5)* |
<0.001 |
|
Número de micciones en 24 horas |
-1.8 (-13%) |
-1.2 (-8%) |
-0.6 (-1.10; -0.2) |
0.005 |
|
Volumen promedio desprovisto por micción (ml) |
+34 (+27%) |
+14 (+12%) |
+20 (14; 26) |
<0.001 |
|
* Intervalo de confianza de 97.5% de acuerdo a Bonferroni |
||||
Después de 12 semanas de tratamiento, 23.8% (121/507) en el grupo de liberación prolongada de tolterodina y 15.7% (80/508) en el grupo de placebo reportaron que subjetivamente no tuvieron problemas con la vejiga, o si los tuvieron fue mínimo.
Se evaluó el efecto de tolterodina en pacientes, que fueron examinados con evaluación urodinámica en la línea basal y, dependiendo del resultado urodinámico, fueron asignados a un grupo positivo urodinámico (urgencia motora) o a un grupo negativo urodinámico (urgencia sensorial). Dentro de cada grupo, los pacientes fueron distribuidos aleatoriamente a recibir ya sea tolterodina o placebo. El estudio no pudo suministrar evidencia convincente de que tolterodina tuviera efecto sobre el placebo en pacientes con urgencia sensorial.
El efecto de 2 mg BID y 4 mg BID de las tabletas de liberación inmediata de tolterodina (tolterodina IR) sobre el intervalo QT se evaluó en un estudio cruzado en 4 sentidos, doble ciego, controlado con placebo y con ingrediente activo (moxifloxacina 400 mg QD) en voluntarios de sexo masculino (N=25) y femenino (N=23) saludables con edad de 18-55 años. Hubo una representación aproximadamente igual de los metabolizadores rápidos de CYP2D6 (EM) y los metabolizadores lentos (PM). Se escogió la dosis de 4 mg BID de tolterodina IR (dos veces la dosis mayor recomendada) debido a que esta dosis resulta en una exposición similar de tolterodina a la observada a partir de la coadministración de tolterodina 2 mg BID con inhibidores potentes de CYP3A4 en pacientes que son metabolizadores lentos de CYP2D6 (ver Precauciones y advertencias especiales para uso y Sobredosis).
La Tabla 2 resume el cambio promedio desde la línea basal hasta el estado estable en el intervalo QT corregido (QTcF Fridericia y QTcP específico de la población) en relación al placebo al momento de las concentraciones pico de tolterodina (1 hora) y moxifloxacina (2 horas). El intervalo QT se midió manualmente y a través de una máquina, y se presentan los datos de ambas mediciones. El motivo para la diferencia entre la lectura manual y de la máquina del intervalo QT no es clara.
|
Tabla 2. |
|||||
|
Fármaco/dosis |
N |
QTcF (mseg) (manual) |
QTcF (mseg) (máquina) |
QTcP (mseg) (manual) |
QTcP (mseg) (máquina) |
|
Tolterodina 2 mg BID1 |
48 |
5.01 |
1.16 |
4.45 |
2.00 |
|
Tolterodina 4 mg BID1 |
48 |
11.84 |
5.63 |
10.31 (5.49, 15.12) |
8.34 (4.53, 12.15) |
|
Moxifloxacina 400 mg QD2 |
45 |
19.263 (15.49, 23.03) |
8.90 (4.77, 13.03) |
19.103 (15.32, 22.89) |
9.29 |
|
1 En Tmáx de 1 h; Intervalo de Confianza de 95%. 2 En Tmáx de 2 h; Intervalo de Confianza de 90%. 3 El efecto sobre el intervalo QT con 4 días de dosificación de moxifloxacina en este estudio de QT puede ser mayor que aquél típicamente observado en estudios QT. |
|||||
El efecto QT de las cápsulas de liberación inmediata de tolterodina parecieron ser mayores para 8 mg/día (dos veces la dosis terapéutica) en comparación a 4 mg/día. El efecto de tolterodina 8 mg/día no fue tan grande como aquel observado después de cuatro días de dosificación terapéutica con el control activo moxifloxacina.
Pareció haber un mayor incremento en el intervalo QTc en PM que en EM después del tratamiento con tolterodina en este estudio (ver Precauciones y advertencias especiales para uso y Sobredosis).
CARACTERÍSTICAS FARMACOCINÉTICAS: Las cápsulas de liberación prolongada de tolterodina suministraron absorción más lenta de tolterodina que las tabletas de liberación inmediata. Como resultado, las concentraciones séricas máximas se observaron a las 4 (2-6) horas después de la administración de las cápsulas. La vida media aparente para tolterodina suministrada como cápsula es aproximadamente 6 horas en metabolizadores rápidos y aproximadamente 10 horas en metabolizadores lentos (CYP2D6 deficiente). Las concentraciones en estado estable se alcanzan dentro de 4 días después de la administración de las cápsulas. No hay efecto de los alimentos sobre la biodisponibilidad de las cápsulas.
Absorción: Después de la administración oral, la tolterodina se somete al metabolismo de primer paso catalizado por CYP2D6 en el hígado, lo cual da como resultado la formación del derivado 5-hidroximetilo, un metabolito principal farmacológicamente equipotente. La biodisponibilidad absoluta de tolterodina es 17% en los metabolizadores rápidos, la mayoría de los pacientes, y 65% en los metabolizadores lentos (con deficiencia de CYP2D6).
Distribución: La tolterodina y el metabolito 5-hidroximetilo se enlazan principalmente a alfa-1-ácido glucoproteína. Las fracciones que no se enlazan son 3.7% y 36%, respectivamente. El volumen de distribución de tolterodina es 113 L.
Metabolismo: La tolterodina se metaboliza extensamente por el hígado tras la dosificación oral. La vía metabólica primaria es mediada por la enzima polimórfica CYP2D6 y conduce a la formación del metabolito 5-hidroximetilo. El metabolismo adicional conduce a la formación de los metabolitos 5-ácido carboxílico y 5-ácido carboxílico N-dealquilatado, lo cual representa 51% y 29% de los metabolitos recuperados en la orina, respectivamente. Un subconjunto (aproximadamente 7%) de la población tiene actividad CYP2D6 deficiente. La vía de metabolismo identificada para estos individuos (metabolizadores lentos) es la vía dealquilación CYP3A4 a tolterodina N-dealquilada, lo cual no contribuye con el efecto clínico. A la población restante se le conoce como metabolizadores rápidos. La depuración sistémica de tolterodina en metabolizadores rápidos es aproximadamente 30 L/h. En metabolizadores lentos, la depuración reducida conduce a concentraciones séricas significativamente mayores de tolterodina (aproximadamente 7 veces) y se observan concentraciones insignificantes del metabolito 5-hidroximetilo.
El metabolito 5-hidroximetilo es farmacológicamente activo y equipotente con la tolterodina. Debido a las diferencias en las características de enlace con proteínas de tolterodina y del metabolito 5-hidroximetilo, la exposición (AUC) de la tolterodina no enlazada en metabolizadores lentos es similar a la exposición combinada de la tolterodina no enlazada y del metabolito 5-hidroximetilo en pacientes con actividad CYP2D6 a los cuales se les administró el mismo régimen de dosificación. La seguridad, tolerabilidad y respuesta clínica son similares independientemente del fenotipo.
Excreción: La excreción de radioactividad después de la administración de [14C]- tolterodina es aproximadamente 77% en orina y 17% en las heces. Se recupera menos del 1% de la dosis como fármaco sin cambio, y aproximadamente 4% como el metabolito 5-hidroximetilo. El metabolito carboxilado y el metabolito dealquilado correspondiente representan aproximadamente 51% y 29% de la recuperación urinaria, respectivamente.
La farmacocinética es lineal en el rango de dosis terapéutica.
Grupos específicos de pacientes
Funcionamiento hepático deteriorado: Se encuentra aproximadamente una exposición 2 veces mayor de tolterodina no enlazada y del metabolito 5-hidroximetilo en sujetos con cirrosis hepática (ver Posología y método de administración, Uso en sujetos con funcionamiento hepático deteriorado, y Precauciones y advertencias especiales para uso).
Funcionamiento renal deteriorado: La exposición promedio de tolterodina no enlazada y de su metabolito 5-hidroximetilo es del doble en pacientes con funcionamiento renal severamente deteriorado (depuración de inulina GFR ≤ 30 ml min). Los niveles plasmáticos de otros metabolitos se incrementaron marcadamente (hasta 12 veces) en estos pacientes. Se desconoce la relevancia clínica de este incremento de exposición de estos metabolitos. No existen datos en sujetos con funcionamiento renal deteriorado de leve a moderado (ver Posología y método de administración – Uso en sujetos con funcionamiento renal deteriorado y Precauciones y advertencias especiales para uso).
CONTRAINDICACIONES:
La tolterodina está contraindicada en pacientes con:
— Hipersensibilidad conocida a tolterodina o a cualquier otro componente del producto.
— Retención urinaria.
— Glaucoma de ángulo estrecho no controlado.
— Colitis ulcerativa grave.
— Megalocolón tóxico.
— Miastemia gravis.
EMBARAZO Y LACTANCIA:
Embarazo: No existen estudios en mujeres embarazadas. Por lo tanto, la tolterodina se debe de utilizar durante el embarazo sólo si el beneficio potencial para la madre justifica el riesgo potencial para el feto.
Lactancia: Se debe de evitar el uso de la tolterodina durante la lactancia, ya que no hay datos disponibles sobre excreción en la leche humana.
EFECTOS SOBRE LA HABILIDAD PARA MANEJAR Y UTILIZAR MÁQUINAS: La habilidad para manejar y utilizar máquinas puede verse afectada negativamente. Se les debe de aconsejar a los pacientes de hacer ejercicio con precaución.
EFECTOS INDESEABLES: La tolterodina puede provocar efectos antimuscarínicos de leves a moderados, como sequedad en la boca, dispepsia, y reducción de lagrimeo.
Estudios clínicos: A continuación se suministran los eventos adversos que se consideran potencialmente relacionados con el fármaco de estudios con cápsulas de tolterodina.
Infecciones e infestaciones: Sinusitis.
Trastornos del sistema inmunológico: Reacciones alérgicas.
Trastornos psiquiátricos: Confusión.
Trastornos del sistema nervioso: Mareo, cefalea, somnolencia.
Trastornos oculares: Visión anormal (incluyendo acomodación anormal), ojos secos.
Trastornos del oído y del laberinto: Vértigo.
Trastornos vasculares: Piel ruborizada.
Trastornos gastrointestinales: Boca seca, dolor abdominal, estreñimiento, dispepsia, flatulencia, reflujo gastroesofágico.
Trastornos renales y urinarios: Disuria, retención urinaria.
Trastornos generales y afecciones en el sitio de administración: Fatiga.
Los siguientes eventos adversos fueron reportados durante vigilancia post-comercialización:
Trastornos del sistema inmunológico: Reacciones anafilactoides.
Trastornos psiquiátricos: Desorientación, alucinaciones.
Trastornos del sistema nervioso: Deterioro de la memoria.
Trastornos cardiacos: Taquicardia, palpitaciones.
Trastornos gastrointestinales: Diarrea.
Trastornos cutáneos y del tejido subcutáneo: Angioedema.
Trastornos generales y afecciones en el sitio de administración: Edema periférico.
Se han reportado casos de agravación de síntomas de demencia (p. ej., confusión, desorientación, delusión) después de que se inició la terapia con tolterodina en pacientes que tomaban inhibidores de colinestarasa para el tratamiento de la demencia.
INTERACCIONES CON OTROS PRODUCTOS MEDICINALES Y OTRAS FORMAS DE INTERACCIÓN: Las interacciones farmacocinéticas son posibles con otros fármacos que son metabolizados por o que inhiben al citocromo P450 2D6 (CYP2D6) o CYP3A4. El tratamiento concomitante con fluoxetina no resulta en una interacción clínicamente significativa.
Ketoconazol, un inhibidor potente de CYP3A4, significativamente incrementa las concentraciones plasmáticas de tolterodina cuando se coadministra a metabolizadores lentos (es decir, personas con deficiencia en la vía metabólica de CYP2D6). Para los pacientes que reciben ketoconazol u otro inhibidor potente de CYP3A4, la dosis diaria total recomendada es 2 mg (ver Posología y método de administración, Uso con inhibidores potentes de CYP3A4, y Precauciones y advertencias especiales para uso – Inhibidores de CYP3A4).
Los estudios clínicos no han mostrado interacciones con warfarina o con anticonceptivos orales combinados (etinil estradiol/levonorgestrel).
Un estudio clínico con fármacos marcadores para las isoenzimas P450 principales no mostró ninguna evidencia de que la actividad de CYP2D6, 2C19, 2C9, 3A4 o 1A2 será inhibida por la tolterodina.
DATOS PRECLÍNICOS DE SEGURIDAD: En estudios de toxicidad, genotoxicidad y carcinogenicidad no se observaron efectos clínicamente relevantes, excepto aquellos relacionados con el efecto farmacológico del fármaco.
Se han realizado estudios de reproducción en ratones y conejos: En ratones, no hubo ningún efecto de tolterodina en la fertilidad o en el funcionamiento reproductivo. La tolterodina produjo muerte del embrión y malformaciones a exposiciones plasmáticas (Cmáx o AUC) 20 o 7 veces mayores a aquellas observadas en humanos tratados.
En conejos, no se observaron efectos de malformación, pero los estudios fueron realizados con exposiciones plasmáticas 20 o 3 veces mayores (Cmáx o AUC) en comparación a aquellas esperadas en los humanos tratados.
Los estudios en ratones preñados han mostrado que altas dosis de tolterodina producen reducción del peso del feto, embrioletalidad e incremento en la incidencia de malformaciones fetales.
La tolterodina, al igual que sus metabolitos activos en humanos, prolongan la duración del potencial de acción (repolarización del 90%) en las fibras de purkinje caninas (23-123 veces los niveles terapéuticos) y bloquea la corriente de K+ en los canales del gen relacionado con el a-go-go humano (hERG) clonado (0.8-14.7 veces los niveles terapéuticos). En perros se ha observado la prolongación del intervalo QT después de la aplicación de la tolterodina en sus metabolitos humanos (5.1-62.7 veces los niveles terapéuticos).
PRECAUCIONES Y ADVERTENCIAS ESPECIALES PARA USO:
Tolterodina debe utilizarse con precaución en los siguientes pacientes:
— En riesgo de retención urinaria.
— En riesgo de disminución de la motilidad gastrointestinal.
— Con funcionamiento renal deteriorado (ver Posología y método de administración, Uso en pacientes con funcionamiento renal deteriorado, y Propiedades farmacocinéticas, Grupos específicos de pacientes).
— Con funcionamiento hepático deteriorado (ver Posología y método de administración, Uso en pacientes con funcionamiento hepático deteriorado, y Propiedades farmacocinéticas, Grupos específicos de pacientes).
— Con miastenia grave.
— Neuropatía autónoma.
— Hernia hiatal.
En un estudio en donde se evaluó el efecto de las tabletas de liberación inmediata de tolterodina sobre el intervalo QT, el efecto sobre el intervalo QT fue mayor para 8 mg/día (dos veces la dosis terapéutica) en comparación con 4 mg/día y fue más pronunciado en los metabolizadores lentos de CYP2D6 (PM) en comparación con los metabolizadores rápidos (EM) (ver Propiedades farmacodinámicas).
El efecto de 8 mg/día de tolterodina no fue tan grande en comparación al observado después de cuatro días de dosificación terapéutica con el control activo moxifloxacina. Sin embargo, los intervalos de confianza se traslaparon.
Estas observaciones deben de considerarse en decisiones clínicas para prescribir las cápsulas de liberación prolongada de tolterodina a pacientes con:
• Prolongación de QT adquirida documentada o congénita.
• Pacientes que toman medicamentos antiarrítmicos clase IA (p. ej., quinidina, procainamida) o clase III (p. ej., amiodarona, sotalol).
Inhibidores de CYP3A4: La dosis diaria total recomendada de tolterodina es 2 mg para pacientes con medicamento concomitante con inhibidores potentes de CYP3A4, tales como antibióticos macrólidos (p. ej., eritromicina y claritromicina) o agentes antifungales de azol (p. ej., ketoconazol, itraconazol y miconazol) (ver Posología y método de administración, Uso con inhibidores potentes de CYP3A4 y Interacciones con otros productos medicinales y otras formas de interacción).
POSOLOGÍA Y MÉTODO DE ADMINISTRACIÓN:
General: Las cápsulas de liberación prolongada pueden tomarse con o sin alimentos y deben de tragarse enteras (ver Propiedades farmacocinéticas, Características farmacocinéticas).
Adultos (incluyendo a adultos mayores): La dosis diaria total recomendada es 4 mg. La dosis con las cápsulas de tolterodina es 4 mg una vez al día. La dosis diaria total puede reducirse a 2 mg, basándose en la tolerabilidad del individuo.
Uso en niños: No se ha establecido la seguridad y la efectividad en niños.
Uso en pacientes con funcionamiento renal deteriorado: La dosis diaria total recomendada es 2 mg (es decir, una cápsula de tolterodina de 2 mg una vez al día) para pacientes con funcionamiento renal deteriorado (ver Precauciones y advertencias especiales para uso).
Uso en pacientes con funcionamiento hepático deteriorado: La dosis diaria total recomendada es 2 mg (es decir, una cápsula de tolterodina de 2 mg una vez al día) para pacientes con funcionamiento hepático deteriorado (ver Precauciones y advertencias especiales para uso).
Uso con inhibidores potentes de CYP3A4: La dosis diaria total recomendada es 2 mg (es decir, una cápsula de tolterodina de 2 mg una vez al día) para los pacientes que reciben ketoconazol concomitante u otro inhibidor potente de CYP3A4 (ver Precauciones y advertencias especiales para uso, inhibidores de CYP3A4, y Interacciones con otros productos medicinales y Otras formas de interacción).
SOBREDOSIS: La dosis más alta de tolterodina suministrada a voluntarios humanos fue 12.8 mg como dosis única. Los eventos adversos más severos observados fueron alteraciones de acomodación y dificultades de micturición.
La sobredosis con tolterodina puede potencialmente resultar en efectos antimuscarínicos centrales severos y deben de tratarse de manera acorde.
En caso de que ocurra sobredosis con tolterodina, se deben de adoptar medidas de apoyo estándar para manejar la prolongación QT (ver Precauciones y advertencias especiales para uso, y Propiedades farmacodinámicas).


