

FIRIALTA
Finerenone
Comprimidos recubiertos
1 Caja, 14 Comprimidos recubiertos,
1 Caja, 28 Comprimidos recubiertos,
FORMA FARMACÉUTICA Y FORMULACIÓN:
FIRIALTA 10 mg:
COMPRIMIDO RECUBIERTO de color rosa, ovalado-oblongo, con una longitud de 10 mm y una anchura de 5 mm, marcado con “10” en una cara y “FI” en la otra.
FIRIALTA 20 mg:
COMPRIMIDO RECUBIERTO de color amarillo, ovalado-oblongo, con una longitud de 10 mm y una anchura de 5 mm, marcado con “20” en una cara y “FI” en la otra.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacológicas:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Diuréticos, antagonistas de la aldosterona.
Código ATC: C03DA05.
Mecanismo de acción: La finerenona es un antagonista selectivo no esteroideo del receptor de mineralocorticoides (RM) activado por la aldosterona y el cortisol que regula la transcripción génica. Su unión al RM da lugar a un complejo receptor-ligando específico que bloquea el reclutamiento de coactivadores transcripcionales implicados en la expresión de mediadores proinflamatorios y profibróticos.
Efectos farmacodinámicos: En FIDELIO-DKD y FIGARO-DKD, dos estudios aleatorizados y multicéntricos de fase III, doble ciego y controlados con placebo en pacientes adultos con ERC y DM2, la reducción relativa en comparación con placebo del cociente albúmina/creatinina en la orina (CACo) en los pacientes aleatorizados a la finerenona fue del 31% y del 32% en el mes 4, respectivamente, y el CACo se mantuvo bajo en ambos estudios.
En ARTS-DN, un estudio aleatorizado y multicéntrico de fase IIb, doble ciego y controlado con placebo, en pacientes adultos con ERC y DM2, la reducción relativa en comparación con placebo del CACo el día 90 fue del 25% y del 38% en los pacientes tratados con finerenona 10 mg y 20 mg una vez al día, respectivamente.
Electrofisiología cardiaca: Un estudio específico sobre el intervalo QT en 57 participantes sanos demostró que la finerenona no tiene ningún efecto sobre la repolarización cardiaca. No hubo indicios de un efecto de prolongación del QT/QTc de la finerenona tras dosis únicas de 20 mg (terapéuticas) u 80 mg (supraterapéuticas).
Eficacia clínica y seguridad: En los estudios FIDELIO-DKD y FIGARO-DKD se investigó el efecto de la finerenona en comparación con el placebo sobre los parámetros renales y cardiovasculares (CV) en pacientes adultos con ERC y DM2.
Los pacientes debían estar recibiendo el tratamiento de referencia, incluyendo la dosis máxima tolerada indicada en la ficha técnica de un inhibidor de la enzima convertidora de la angiotensina (IECA) o un antagonista de los receptores de la angiotensina (ARA). Los pacientes diagnosticados de insuficiencia cardiaca con fracción de eyección reducida y clase II-IV de la New York Heart Association fueron excluidos debido a la recomendación de clase 1A para el tratamiento con antagonistas de los receptores de mineralocorticoides (ARM).
En el estudio FIDELIO-DKD, los pacientes se consideraron aptos si presentaban albuminuria persistente (de > 30 mg/g a 5 000 mg/g), una TFGe de 25 a 75 mL/min/1,73 m2 y potasio sérico ≤ 4,8 mmol/L en la selección.
La variable principal fue una variable compuesta del tiempo hasta la primera aparición de insuficiencia renal (definida como diálisis crónica o trasplante de riñón, o una disminución sostenida de la TFGe hasta < 15 mL/min/1,73 m2 durante al menos 4 semanas), una disminución sostenida de la TFGe del 40% o más en comparación con el valor inicial durante al menos 4 semanas, o la muerte renal. La variable secundaria fundamental fue una variable compuesta del tiempo hasta la primera aparición de muerte CV, infarto de miocardio (IM) no mortal, ictus no mortal u hospitalización por insuficiencia cardiaca.
Un total de 5 674 pacientes fueron aleatorizados para recibir finerenona (N = 2 833), o placebo (N = 2 841) y se incluyeron en los análisis. La mediana de seguimiento fue de 2,6 años. La dosis de la finerenona o del placebo podía ajustarse entre 10 mg y 20 mg una vez al día durante el transcurso del estudio, basándose principalmente en la concentración de potasio sérico. En el mes 24, de los sujetos tratados con finerenona, el 67% estaba recibiendo 20 mg una vez al día, el 30% 10 mg una vez al día y el 3% se encontraba en una interrupción del tratamiento.
Tras la finalización del estudio, se confirmó que el 99,7% de los pacientes seguían vivos. La población del estudio era un 63% blanca, un 25% asiática y un 5% negra. La media de edad en el momento de la inclusión era de 66 años y el 70% de los pacientes eran varones. Al inicio del estudio, la media de la TFGe era de 44,3 mL/min/1,73 m2, con un 55% de pacientes con una TFGe < 45 mL/min/1,73 m2, la mediana del CACo era de 852 mg/g, y la media de HbA1c era del 7,7%, el 46% tenía antecedentes de enfermedad CV aterosclerótica, el 30% antecedentes de arteriopatía coronaria, el 8% antecedentes de insuficiencia cardiaca y la media de la presión arterial era de 138/76 mmHg. La duración media de la DM2 al inicio del estudio era de 16,6 años, y el 47% y el 26% de los pacientes tenían antecedentes de retinopatía y neuropatía diabéticas, respectivamente. En la situación inicial, casi todos los pacientes recibían IECA (34%) o ARA (66%), y el 97% de los pacientes utilizaba uno o más medicamentos antidiabéticos (insulina [64%], biguanidas [44%], agonistas de los receptores del péptido similar al glucagón de tipo 1 [arGLP-1] [7%], inhibidores del cotransportador de sodio y glucosa de tipo 2 [iSGLT2] [5%]). Los demás medicamentos más frecuentes que se tomaban al inicio eran estatinas (74%) y antagonistas del calcio (63%).
Se mostró una diferencia estadísticamente significativa a favor de finerenona para la variable principal y la variable secundaria fundamental (ver la figura 1 y tabla 4 a continuación). El efecto del tratamiento para las variables principal y secundaria fue, en general, uniforme en todos los subgrupos, incluyendo la región, la TFGe, el CACo, la presión arterial sistólica (PAS) y la HbA1c al inicio.
En el estudio FIGARO-DKD, los pacientes se consideraron aptos si presentaban albuminuria persistente con un CACo de ≥ 30 mg/g a < 300 mg/g y una TFGe de 25 a 90 mL/min/1,73 m2 o un CACo ≥ 300 mg/g y una TFGe ≥ 60 mL/min/1,73 m2 en la selección. Los pacientes debían tener una concentración de potasio sérico ≤ 4,8 mmol/L en la selección.
La variable principal fue una variable compuesta del tiempo hasta la primera aparición de muerte CV, IM no mortal, ictus no mortal u hospitalización por insuficiencia cardiaca. La variable secundaria fue una variable compuesta del tiempo hasta la insuficiencia renal, una disminución sostenida de la TFGe del 40% o más en comparación con el valor inicial durante al menos 4 semanas, o la muerte renal.
Un total de 7,352 pacientes fueron aleatorizados para recibir finerenona (N = 3 686) o placebo (N = 3 666) y se incluyeron en los análisis. La mediana de seguimiento fue de 3,4 años. La dosis de finerenona o de placebo podía ajustarse entre 10 mg y 20 mg una vez al día durante el transcurso del estudio, basándose principalmente en la concentración de potasio sérico. En el mes 24, de los sujetos tratados con finerenona, el 82% estaba recibiendo 20 mg una vez al día, el 15% 10 mg una vez al día y el 3% se encontraba en una interrupción del tratamiento. Tras la finalización del estudio, se confirmó que el 99,8% de los pacientes seguían vivos. La población del estudio era un 72% blanca, un 20% asiática y un 4% negra. La media de edad en el momento de la inclusión era de 64 años y el 69% de los pacientes eran varones. Al inicio del estudio, la media de la TFGe era de 67,8 mL/min/1,73 m2, con un 62% de pacientes con una TFGe ≥ 60 mL/min/1,73 m2, la mediana del CACo era de 308 mg/g, y la media de HbA1c era del 7,7%, el 45% de los pacientes tenía antecedentes de enfermedad CV aterosclerótica, el 8% tenía antecedentes de insuficiencia cardiaca y la media de la presión arterial era de 136/77 mmHg. La duración media de la DM2 al inicio del estudio era de 14,5 años, y el 31% y el 28% de los pacientes tenían antecedentes de retinopatía y neuropatía diabéticas, respectivamente. En la situación inicial, casi todos los pacientes recibían IECA (43%) o ARA (57%), y el 98% de los pacientes utilizaba uno o más medicamentos antidiabéticos (insulina [54%], biguanidas [69%], arGLP-1 [7%], iSGLT2 [8%]). Los otros medicamentos más frecuentes que se tomaban al inicio eran estatinas (71%).
Se mostró una diferencia estadísticamente significativa a favor de finerenona para la variable principal compuesta CV (ver la figura 2 y tabla 5 a continuación). El efecto del tratamiento para la variable principal fue uniforme en todos los subgrupos, incluyendo la región, la TFGe, el CACo, la PAS y la HbA1c al inicio.
Se observó una tasa de incidencia menor de la variable secundaria compuesta de insuficiencia renal, disminución sostenida de la TFGe del 40% o más o la muerte renal en el grupo de finerenona frente al grupo placebo; sin embargo, esta diferencia no alcanzó significación estadística (ver tabla 5 a continuación). El efecto del tratamiento para la variable secundaria compuesta renal fue coherente en todos los subgrupos de TFGe al inicio del estudio, pero para el subgrupo de pacientes con CACo < 300 mg/g el HR fue 1,16 (IC del 95% 0,91; 1,47) y para el subgrupo de pacientes con CACo ≥ 300 mg/g el HR fue 0,74 (IC del 95% 0,62; 0,90). Las variables secundarias preespecificadas adicionales del tiempo hasta el acontecimiento se incluyen en la tabla 5.
Tabla 4. Análisis de las variables principal y secundaria del tiempo hasta el acontecimiento (y sus componentes individuales) en el estudio de fase III FIDELIO-DKD
|
FIRIALTA* (N = 2 833) |
Placebo (N = 2 841) |
Efecto del tratamiento |
|||
|
N (%) |
Aconteci mientos/ 100 p-a |
N (%) |
Aconteci mientos/ 100 p-a |
HR (IC del 95%) |
|
|
Variable principal compuesta renal y sus componentes |
|||||
|
Variable compuesta de insuficiencia renal, disminución sostenida de la TFGe ≥ 40% o muerte renal |
504 (17,8) |
7,59 |
600 (21,1) |
9,08 |
0,82 (0,73; 0,93) p = 0,0014 |
|
Insuficiencia renal |
208 (7,3) |
2,99 |
235 (8,3) |
3,39 |
0,87 (0,72; 1,05) |
|
Disminución sostenida de la TFGe de ≥ 40% |
479 (16,9) |
7,21 |
577 (20,3) |
8,73 |
0,81 (0,72; 0,92) |
|
Muerte renal |
2 (< 0,1) |
- |
2 (< 0,1) |
- |
- |
|
Variable secundaria fundamental compuesta CV y sus componentes |
|||||
|
Variable compuesta de muerte CV, IM no mortal, ictus no mortal u hospitalización por insuficiencia cardiaca |
367 (13,0) |
5,11 |
420 (14,8) |
5,92 |
0,86 (0,75; 0,99) p = 0,0339 |
|
Muerte CV |
128 (4,5) |
1,69 |
150 (5,3) |
1,99 |
0,86 (0,68; 1,08) |
|
IM no mortal |
70 (2,5) |
0,94 |
87 (3,1) |
1,17 |
0,80 (0,58; 1,09) |
|
Ictus no mortal |
90 (3,2) |
1,21 |
87 (3,1) |
1,18 |
1,03 (0,76; 1,38) |
|
Hospitalización por insuficiencia cardiaca |
139 (4,9) |
1,89 |
162 (5,7) |
2,21 |
0,86 (0,68; 1,08) |
|
Variables secundarias de la eficacia |
|||||
|
Mortalidad por cualquier causa |
219 (7,7) |
2,90 |
244 (8,6) |
3,23 |
0,90 (0,75; 1,07)** |
|
Hospitalización por cualquier causa |
1 263 (44,6) |
22,56 |
1 321 (46,5) |
23,87 |
0,95 (0,88; 1,02)** |
|
Variable compuesta de insuficiencia renal, disminución sostenida de la TFGe ≥ 57% o muerte renal |
252 (8,9) |
3,64 |
326 (11,5) |
4,74 |
0,76 (0,65; 0,90)** |
* Tratamiento con 10 o 20 mg una vez al día además de las dosis máximas toleradas según ficha técnica de IECA o ARA.
** p = no estadísticamente significativa después del ajuste por multiplicidad.
IC: Intervalo de confianza.
HR: Cociente de riesgos.
p - a: pacientes-años.
Figura 1. Tiempo hasta la primera aparición de insuficiencia renal, disminución sostenida de la TFGe ≥ 40% desde el inicio, o muerte renal en el estudio FIDELIO-DKD
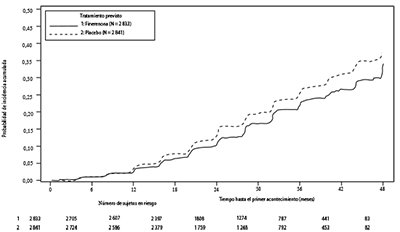
Tabla 5. Análisis de las variables principal y secundaria del tiempo hasta el acontecimiento
(y sus componentes individuales) en el estudio de fase III FIGARO-DKD
|
FIRIALTA* (N = 3 686) |
Placebo (N = 3 666) |
Efecto del tratamiento |
|||
|
N (%) |
Aconteci mientos/100 p-a |
N (%) |
Aconteci mientos/ 100 p-a r |
HR (IC del 95%) |
|
|
Variable principal compuesta CV y sus componentes |
|||||
|
Variable compuesta de muerte CV, IM no mortal, ictus no mortal u hospitalización por insuficiencia cardiaca |
458 (12,4) |
3,87 |
519 (14,2) |
4,45 |
0,87 (0,76; 0,98) p = 0,0264 |
|
Muerte CV |
194 (5,3) |
1,56 |
214 (5,8) |
1,74 |
0,90 (0,74; 1,09) |
|
IM no mortal |
103 (2,8) |
0,85 |
102 (2,8) |
0,85 |
0,99 (0,76; 1,31) |
|
Ictus no mortal |
108 (2,9) |
0,89 |
111 (3,0) |
0,92 |
0,97 (0,74; 1,26) |
|
Hospitalización por insuficiencia cardiaca |
117 (3,2) |
0,96 |
163 (4,4) |
1,36 |
0,71 (0,56; 0,90) |
|
Variable secundaria compuesta renal y sus componentes |
|||||
|
Variable compuesta de insuficiencia renal, disminución sostenida de la TFGe ≥ 40% o muerte renal |
350 (9,5) |
3,15 |
395 (10,8) |
3,58 |
0,87 (0,76; 1,01) p = 0,0689 ** |
|
Insuficiencia renal |
46 (1,2) |
0,40 |
62 (1,7) |
0,54 |
0,72 (0,49; 1,05) |
|
Disminución sostenida de la TFGe ≥ 40% |
338 (9,2) |
3,04 |
385 (10,5) |
3,49 |
0,87 (0,75; > 1,00) |
|
Muerte renal |
0 |
- |
2 (< 0,1) |
- |
- |
|
Variables secundarias de la eficacia |
|||||
|
Mortalidad por cualquier causa |
333 (9,0) |
2,68 |
370 (10,1) |
3,01 |
0,89 (0,77; 1,04) ** |
|
Hospitalización por cualquier causa |
1 573 (42,7) |
16,91 |
1,605 (43,8) |
17,52 |
0,97 (0,90; 1,04) ** |
|
Variable compuesta de insuficiencia renal, disminución sostenida de la TFGe ≥ 57% o muerte renal |
108 (2,9) |
0,95 |
139 (3,8) |
1,23 |
0,77 (0,60; 0,99) ** |
* Tratamiento con 10 o 20 mg una vez al día además de las dosis máximas toleradas según ficha técnica de IECA o ARA.
** No estadísticamente significativa después del ajuste por multiplicidad.
IC: Intervalo de confianza.
HR: Cociente de riesgos.
p-a: Pacientes-años.
Figura 2. Tiempo hasta la primera aparición de muerte CV, infarto de miocardio no mortal, ictus no mortal u hospitalización por insuficiencia cardiaca en el estudio FIGARO-DKD
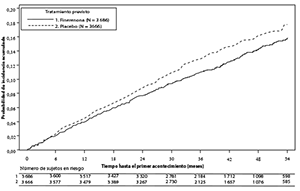
Población pediátrica: La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con FIRIALTA en uno o más grupos de la población pediátrica enel tratamiento de la enfermedad renal crónica (ver sección Dosis y vía de administración; Posología y forma de administración para consultar la información sobre el uso en la población pediátrica).
Propiedades farmacocinéticas:
Absorción: La finerenona se absorbe casi completamente tras su administración oral. La absorción es rápida y las concentraciones plasmáticas máximas (Cmáx) aparecen entre 0,5 y 1,25 horas después de la ingesta del comprimido en ayunas. La biodisponibilidad absoluta de la finerenona es del 43,5% debido al efecto de primer paso en la pared intestinal y en el hígado. La finerenona es un sustrato del transportador de expulsión glucoproteína-P in vitro, que sin embargo no se considera relevante para su absorción in vivo debido a la alta permeabilidad de la finerenona.
Efecto de los alimentos: La ingesta con alimentos ricos en grasas y calorías aumentó el AUC de la finerenona en un 21%, redujo la Cmáx en un 19% y prolongó el tiempo para alcanzar la Cmáx a 2,5 horas. Dado que esto no se considera relevante desde el punto de visto clínico, la finerenona puede tomarse con o sin alimentos.
Distribución: El volumen de distribución en estado estacionario (Vdee) de la finerenona es de 52,6 L. La unión de la finerenona a las proteínas plasmáticas humanas in vitro es del 91,7%, siendo la albúmina sérica la principal proteína de unión.
Biotransformación: Aproximadamente el 90% del metabolismo de finerenona está mediado por el CYP3A4 y el 10% por el CYP2C8. Se encontraron cuatro metabolitos principales en el plasma. Todos los metabolitos son farmacológicamente inactivos.
Eliminación: La eliminación de la finerenona en el plasma es rápida, con una semivida de eliminación (t½) de unas 2 a 3 horas. El aclaramiento sanguíneo sistémico de la finerenona es de unos 25 L/h. Alrededor del 80% de la dosis administrada se excretó por la orina y aproximadamente el 20% de la dosis se excretó por las heces. La excreción fue casi exclusivamente en forma de metabolitos, mientras que la excreción de finerenona inalterada representa una vía menor (< 1% de la dosis en la orina por filtración glomerular, < 0,2% en las heces).
Linealidad: La farmacocinética de la finerenona es lineal en todo el intervalo de dosis investigado, de 1,25 a 80 mg, administrados en comprimidos en dosis única.
Poblaciones especiales:
Pacientes de edad avanzada: De los 2 827 pacientes que recibieron finerenona en el estudio FIDELIO-DKD, el 58% tenían 65 años o más, y el 15% tenían 75 años o más. De los 3 683 pacientes que recibieron finerenona en el estudio FIGARO-DKD, el 52% tenía 65 años o más, y el 13% tenía 75 años o más.
No se observaron diferencias globales de seguridad o eficacia entre estos pacientes y los más jóvenes en ninguno de los estudios.
En un estudio de fase I (N = 48), los participantes sanos de edad avanzada (≥ 65 años) mostraron concentraciones plasmáticas de finerenona más altas que los participantes sanos más jóvenes (≤ 45 años), siendo los valores medios del AUC y la Cmáx un 34% y un 51% más elevados en las personas de edad avanzada (ver sección Dosis y vía de administración; Posología y forma de administración). Los análisis farmacocinéticos poblacionales no identificaron la edad como una covariable para el AUC o la Cmáx de la finerenona.
Insuficiencia renal: La insuficiencia renal leve (aclaramiento de creatinina [CLcr] de 60 a < 90 mL/min) no afectó al AUC ni a la Cmáx de la finerenona.
En comparación con los pacientes con una función renal normal (CLcr ≥ 90 mL/min), el efecto de la insuficiencia renal moderada (CLcr 30 a < 60 mL/min) o grave (CLcr < 30 mL/min) sobre el AUC de la finerenona fue similar, con aumentos del 34-36%. La insuficiencia renal moderada o grave no tuvo efecto sobre la Cmáx (ver sección Dosis y vía de administración; Posología y forma de administración).
Debido a su alta unión a las proteínas plasmáticas, no se espera que la finerenona sea dializable.
Insuficiencia hepática: No hubo cambios en la exposición a la finerenona en pacientes cirróticos con insuficiencia hepática leve (ver sección Dosis y vía de administración; Posología y forma de administración).
En los pacientes cirróticos con insuficiencia hepática moderada, el AUC total y no unida de la finerenona aumentó un 38% y un 55%, respectivamente, mientras que no se observaron cambios en la Cmáx en comparación con los participantes sanos del grupo de referencia (ver sección Dosis y vía de administración; Posología y forma de administración).
No se dispone de datos en pacientes con insuficiencia hepática grave (ver la sección Dosis y vía de administración; Posología y forma de administración y la sección Interacciones medicamentosas y de otro género).
Peso corporal: Los análisis farmacocinéticos poblacionales identificaron el peso corporal como una covariable para la Cmáx de la finerenona. Se estimó que la Cmáx de un sujeto con un peso corporal de 50 kg era entre un 38% y un 51% mayor en comparación con un sujeto de 100 kg. No se justifica la adaptación de la dosis en función del peso corporal (ver sección Dosis y vía de administración; Posología y forma de administración).
Relaciones farmacocinéticas/farmacodinámicas: La relación concentración-efecto a lo largo del tiempo para el CACo se caracterizó por un modelo de efecto máximo que indicaba una saturación a exposiciones altas. El tiempo previsto por el modelo para alcanzar la situación de equilibrio completo (99%) del efecto del fármaco sobre el CACo fue de 138 días. La semivida farmacocinética (FC) fue de 2-3 horas y la situación de equilibrio FC se alcanzó después de 2 días, lo que indica un efecto indirecto y retardado en las respuestas farmacodinámicas.
Estudios clínicos sin interacciones farmacológicas relevantes: El uso concomitante de gemfibrozilo (600 mg dos veces al día), un inhibidor fuerte del CYP2C8, aumentó el AUC y la Cmáx medios de la finerenona 1,1 veces y 1,2 veces, respectivamente. Esto no se considera clínicamente relevante.
El tratamiento previo y el cotratamiento con el inhibidor de la bomba de protones omeprazol (40 mg una vez al día) no tuvieron efecto sobre el AUC media ni sobre la Cmáx media de la finerenona.
El uso concomitante del antiácido hidróxido de aluminio e hidróxido de magnesio (70 mEq) no tuvo ningún efecto sobre el AUC media de la finerenona y redujo su Cmáx media en un 19%. Esto no se considera clínicamente relevante.
In vivo, una pauta multidosis de 20 mg de finerenona administrada una vez al día durante 10 días no tuvo ningún efecto relevante sobre el AUC del sustrato de sonda CYP3A4 utilizado, el midazolam. Por lo tanto, puede excluirse una inhibición o inducción clínicamente relevante del CYP3A4 por parte de la finerenona.
Una dosis única de 20 mg de finerenona tampoco tuvo un efecto clínicamente relevante sobre el AUC y la Cmáx del sustrato de la sonda CYP2C8, la repaglinida. En consecuencia, la finerenona no inhibe el CYP2C8.
Se demostró la ausencia de interacción farmacocinética mutua entre la finerenona y el sustrato del CYP2C9 warfarina, así como entre la finerenona y el sustrato de la gp-P digoxina.
Dosis múltiples de 40 mg de finerenona una vez al día no tuvieron ningún efecto clínicamente relevante sobre la AUC ni la Cmáx de la proteína de resistencia del cáncer de mama (BCRP) y el sustrato del polipéptido transportador de aniones orgánicos (PTAO), la rosuvastatina.
CONTRAINDICACIONES:
- Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección Propiedades farmacéuticas: Lista de excipientes.
- Tratamiento concomitante con inhibidores fuertes del CYP3A4 (ver sección Interacciones medicamentosas y de otro género), por ejemplo.
- Itraconazol.
- Ketoconazol.
- Ritonavir.
- Nelfinavir.
- Cobicistat.
- Claritromicina.
- Telitromicina.
- Nefazodona.
- Enfermedad de Addison.
PRECAUCIONES Y ADVERTENCIAS:
Advertencias y precauciones especiales de empleo:
Hiperpotasemia: Se ha observado hiperpotasemia en pacientes tratados con finerenona (ver sección Reacciones adversas). Algunos pacientes tienen mayor riesgo de desarrollar hiperpotasemia.
Los factores de riesgo son una TFGe baja, potasio sérico elevado y episodios anteriores de hiperpotasemia. En estos pacientes se debe considerar una monitorización más frecuente.
Inicio y continuación del tratamiento (ver sección Dosis y vía de administración; Posología y forma de administración): Si el potasio sérico es > 5,0 mmol/L, no se debe iniciar el tratamiento con finerenona.
Si el potasio sérico es > 4,8 a 5,0 mmol/L, se puede considerar iniciar el tratamiento con finerenona, realizando una monitorización adicional del potasio sérico en las primeras 4 semanas, según las características del paciente y sus niveles de potasio sérico.
Si el potasio sérico es > 5,5 mmol/L, el tratamiento con finerenona debe interrumpirse. Deben seguirse las guías locales para el tratamiento de la hiperpotasemia.
Una vez que el potasio sérico es ≤ 5,0 mmol/L, el tratamiento con finerenona se puede reiniciar con 10 mg una vez al día.
Monitorización: El potasio sérico y la TFGe se deben volver a medir en todos los pacientes 4 semanas después de iniciar, reiniciar o aumentar la dosis de finerenona. A partir de entonces, es preciso volver a evaluar el potasio sérico periódicamente y según sea necesario en función de las características del paciente y de sus niveles de potasio sérico (ver sección Dosis y vía de administración; Posología y forma de administración).
Medicamentos concomitantes: El riesgo de hiperpotasemia también puede aumentar con la ingesta de medicamentos concomitantes que puedan aumentar el potasio sérico (ver sección Interacciones medicamentosas y de otro género). Ver también “Uso concomitante de sustancias que afectan a la exposición a la finerenona”.
La finerenona no se debe administrar de forma concomitante con diuréticos ahorradores de potasio (por ejemplo, amilorida, triamtereno) ni con otros antagonistas de los receptores de mineralocorticoides (ARM), por ejemplo, eplerenona, esaxerenona, espironolactona, canrenona.
La finerenona se debe utilizar con precaución y se debe monitorizar el potasio sérico cuando se tome de forma concomitante con suplementos de potasio, trimetoprim o trimetoprim/sulfametoxazol. Puede ser necesaria la interrupción temporal del tratamiento con finerenona.
Insuficiencia renal: El riesgo de hiperpotasemia aumenta con la disminución de la función renal. Se debe realizar una monitorización continua de la función renal según sea necesario, de acuerdo con la norma habitual (ver sección Dosis y vía de administración; Posología y forma de administración).
Inicio del tratamiento: No se debe iniciar el tratamiento con finerenona en pacientes con una TFGe < 25 mL/min/1,73 m2, ya que los datos clínicos son escasos (ver sección Dosis y vía de administración; Posología y forma de administración y la sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas).
Continuación del tratamiento: Debido a la escasez de datos clínicos, el tratamiento con finerenona se debe interrumpir en los pacientes que hayan progresado a una enfermedad renal terminal (TFGe < 15 mL/min/1,73 m2).
Insuficiencia hepática: No se debe iniciar el tratamiento con finerenona en pacientes con insuficiencia hepática grave (ver sección Dosis y vía de administración; Posología y forma de administración). No se ha estudiado en estos pacientes (ver sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas), pero cabe esperar un aumento significativo de la exposición a la finerenona.
El uso de finerenona en pacientes con insuficiencia hepática moderada puede requerir una monitorización adicional debido al aumento de la exposición a la finerenona. Hay que considerar la monitorización adicional del potasio sérico, así como la adaptación de dicha monitorización según las características del paciente (ver la sección Dosis y vía de administración; Posología y forma de administración y la sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas).
Insuficiencia cardiaca:
Los pacientes diagnosticados con insuficiencia cardiaca con fracción de eyección reducida y clase II-IV de la New York Heart Association fueron excluidos de los ensayos clínicos de fase III (ver sección Farmacocinética y farmacodinamia; Propiedades farmacodinámicas).
Uso concomitante de sustancias que afectan a la exposición a la finerenona:
Inhibidores moderados y débiles del CYP3A4: Se debe monitorizar el potasio sérico durante el uso concomitante de finerenona con inhibidores moderados o débiles del CYP3A4 (ver la sección Dosis y vía de administración; Posología y forma de administración y la sección Interacciones medicamentosas y de otro género).
Inductores fuertes y moderados del CYP3A4: No se debe usar finerenona de forma concomitante con inductores fuertes o moderados del CYP3A4 (ver sección Interacciones medicamentosas y de otro género).
Toronja: No se debe consumir toronja ni jugo de toronja durante el tratamiento con finerenona (ver la sección Dosis y vía de administración; Posología y forma de administración y la sección Interacciones medicamentosas y de otro género).
Toxicidad embriofetal: No se debe utilizar finerenona durante el embarazo, a menos que se haya considerado cuidadosamente el beneficio para la madre y el riesgo para el feto. Si una mujer se queda embarazada mientras está tomando finerenona, debe ser informada de los posibles riesgos para el feto.
Se debe aconsejar a las mujeres en edad fértil que utilicen métodos anticonceptivos efectivos durante el tratamiento con finerenona.
Se debe aconsejar a las mujeres que interrumpan la lactancia durante el tratamiento con finerenona. Para más información, ver la sección Restricciones de uso durante el embarazo y la lactancia y la sección Hallazgos de laboratorio clínico.
Información sobre los excipientes:
FIRIALTA contiene lactosa: Los pacientes con intolerancia hereditaria a galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
FIRIALTA contiene sodio: Este medicamento contiene menos de 1 mmol de sodio (23 mg) por comprimido; esto es, esencialmente “exento de sodio”.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Fertilidad, embarazo y lactancia:
Anticoncepción en mujeres: Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento con finerenona (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Embarazo: No hay datos relativos al uso de finerenona en mujeres embarazadas.
Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver sección Hallazgos de laboratorio clínico).
No se debe utilizar FIRIALTA durante el embarazo a no ser que la situación clínica de la mujer requiera tratamiento con finerenona. Si la mujer se queda embarazada mientras está tomando finerenona, debe ser informada de los posibles riesgos para el feto (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Lactancia: Se desconoce si la finerenona/metabolitos se excretan en la leche materna.
Los datos farmacocinéticos/toxicológicos disponibles en animales muestran que la finerenona y sus metabolitos se excretan en la leche. Las crías de rata expuestas por esta vía mostraron reacciones adversas (ver sección Hallazgos de laboratorio clínico).
No se puede excluir el riesgo en recién nacidos/niños.
Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento de FIRIALTA tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento para la madre (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Fertilidad: No hay datos relativos al efecto de la finerenona en la fertilidad humana.
Los estudios realizados en animales han mostrado una alteración de la fertilidad de las hembras a exposiciones consideradas superiores a la máxima humana, lo que indica una baja relevancia clínica (ver sección Hallazgos de laboratorio clínico).
EFECTO EN LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS:
Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de FIRIALTA sobre la capacidad para conducir y utilizar máquinas es nula.
REACCIONES ADVERSAS:
Resumen del perfil de seguridad: La reacción adversa notificada con más frecuencia con el tratamiento con finerenona fue la hiperpotasemia (14,0%). Consulte la Descripción de reacciones adversas seleccionadas; Hiperpotasemia más adelante y la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo.
Tabla de reacciones adversas: La seguridad de finerenona en pacientes con enfermedad renal crónica (ERC) y diabetes mellitus tipo 2 (DM2) se evaluó en 2 estudios pivotales de fase III: FIDELIO-DKD (enfermedad renal diabética) y FIGARO-DKD. En el estudio FIDELIO-DKD, 2 827 pacientes recibieron finerenona (10 o 20 mg una vez al día) con una duración media del tratamiento de 2,2 años. En el estudio FIGARO-DKD, 3 683 pacientes recibieron finerenona (10 o 20 mg una vez al día) con una duración media del tratamiento de 2,9 años.
Las reacciones adversas observadas se muestran en la tabla 3. Están clasificadas según la base de datos de clasificación por órganos y sistemas de MedDRA y su convención de frecuencia.
Las reacciones adversas se agrupan según su frecuencia en orden de gravedad decreciente. Las frecuencias se definen como sigue:
Muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1 000 a < 1/100), raras (≥ 1/10 000 a < 1/1 000), muy raras (< 1/10 000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 3. Reacciones adversas
|
Clasificación de órganos del sistema MedDRA |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Trastornos del metabolismo y de la nutrición |
Hiperpotasemia |
Hiponatremia, hiperuricemia |
|
|
Trastornos vasculares |
Hipotensión |
||
|
Trastornos de la piel y del tejido subcutáneo |
Prurito |
||
|
Exploraciones complementarias |
Tasa de filtración glomerular disminuida |
Disminución de la hemoglobina |
Descripción de reacciones adversas seleccionadas:
Hiperpotasemia: En los datos agregados de los estudios FIDELIO-DKD y FIGARO-DKD se notificaron acontecimientos de hiperpotasemia en el 14,0% de los pacientes tratados con finerenona, en comparación con el 6,9% de los tratados con placebo. Se observó un aumento desde el inicio en la media del potasio sérico en el primer mes de tratamiento de 0,17 mmol/L en el grupo de finerenona en comparación con el de placebo, que permaneció estable a partir de entonces. La mayoría de los acontecimientos de hiperpotasemia fueron de leves a moderados y se resolvieron en los pacientes tratados con finerenona. Los acontecimientos graves de hiperpotasemia se notificaron con mayor frecuencia con finerenona (1,1%) que con placebo (0,2%). Se notificaron concentraciones de potasio sérico > 5,5 mmol/L y > 6,0 mmol/L en el 16,8% y el 3,3% de los pacientes tratados con finerenona y en el 7,4% y el 1,2% de los pacientes tratados con placebo, respectivamente. La hiperpotasemia que provocó la interrupción definitiva del tratamiento en los pacientes que recibieron finerenona fue del 1,7% frente al 0,6% en el grupo del placebo. La hospitalización por hiperpotasemia en el grupo de la finerenona fue del 0,9% frente al 0,2% en el grupo del placebo.
Para consultar las recomendaciones específicas, ver la sección Dosis y vía de administración; Posología y forma de administración y la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo.
Hipotensión: En los datos agregados de los estudios FIDELIO-DKD y FIGARO-DKD se notificaron acontecimientos de hipotensión en el 4,6% de los pacientes tratados con finerenona, en comparación con el 3,0% de los tratados con placebo. En 3 pacientes (< 0,1%), el tratamiento con finerenona se interrumpió definitivamente debido a la hipotensión. La hospitalización por hipotensión fue la misma en los pacientes que recibieron finerenona o placebo (< 0.1%).
La mayoría de los acontecimientos de hipotensión fueron de leves a moderados y se resolvieron en los pacientes tratados con finerenona.
La presión arterial sistólica media disminuyó en 2-4 mmHg y la presión arterial diastólica media disminuyó en 1-2 mmHg en el mes 1, permaneciendo estable a partir de entonces.
Hiperuricemia: En los datos agregados de los estudios FIDELIO-DKD y FIGARO-DKD se notificaron acontecimientos de hiperuricemia en el 5,1% de los pacientes tratados con finerenona, en comparación con el 3,9% de los tratados con placebo. Todos los acontecimientos fueron no graves y no provocaron la interrupción definitiva en los pacientes que tomaban finerenona. Se observó un aumento desde el inicio en la media del ácido úrico sérico de 0,3 mg/dL en el grupo tratado con finerenona en comparación con el grupo de placebo hasta el mes 16, que se atenuó con el tiempo. No se observó ninguna diferencia entre el grupo de finerenona y el de placebo con respecto a los acontecimientos notificados de gota (3,0%).
Tasa de filtración glomerular (TFG) disminuida: En los datos agregados de los estudios FIDELIO-DKD y FIGARO-DKD se notificaron acontecimientos de TFG disminuida en el 5,3% de los pacientes tratados con finerenona, en comparación con el 4,2% de los pacientes tratados con placebo. Los acontecimientos de TFG disminuida que provocaron la interrupción definitiva del tratamiento fueron los mismos en los pacientes que recibieron finerenona o placebo (0,2%). La hospitalización por TFG disminuida fue la misma en pacientes que recibieron finerenona o placebo (< 0.1%). La mayoría de los acontecimientos de TFG disminuida fueron de leves a moderados y se resolvieron en los pacientes tratados con finerenona. Los pacientes que recibieron finerenona presentaron una disminución inicial de la TFGe (media de 2 mL/min/1,73 m2) que se atenuó con el tiempo en comparación con el placebo. Esta disminución pareció ser reversible durante el tratamiento continuo.
Disminución de la hemoglobina:
En los datos agregados de los estudios FIDELIO-DKD y FIGARO-DKD, finerenona se asoció con una disminución absoluta corregida con placebo de la hemoglobina media de 0,15 g/dL y del hematocrito medio de 0,5% después de 4 meses de tratamiento. La notificación de anemia fue comparable en los pacientes tratados con finerenona (6,5%) y en los pacientes tratados con placebo (6,1%). La frecuencia de acontecimientos graves de anemia fue baja tanto en los pacientes tratados con finerenona como en los pacientes tratados con placebo (0,5%). Los cambios en la hemoglobina y el hematocrito fueron transitorios y alcanzaron niveles comparables a los observados en el grupo tratado con placebo después de aproximadamente 24-32 meses.
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacción con otros medicamentos y otras formas de interacción:
Los estudios de interacciones se han realizado solo en adultos.
La finerenona se elimina casi exclusivamente a través del metabolismo oxidativo mediado por el citocromo P450 (CYP) (principalmente CYP3A4 [90%] con una pequeña contribución del CYP2C8 [10%]).
Uso concomitante contraindicado:
Inhibidores fuertes del CYP3A4: El uso concomitante de FIRIALTA con itraconazol, claritromicina y otros inhibidores fuertes del CYP3A4 (por ejemplo, ketoconazol, ritonavir, nelfinavir, cobicistat, telitromicina o nefazodona) está contraindicado (ver sección Contraindicaciones), ya que se prevé un marcado aumento de la exposición a la finerenona.
No se recomienda el uso concomitante:
Inductores fuertes y moderados del CYP3A4: FIRIALTA no se debe usar de forma concomitante con rifampicina y otros inductores fuertes del CYP3A4 (por ejemplo, carbamazepina, fenitoína, fenobarbital, hierba de San Juan) o con efavirenz y otros inductores moderados del CYP3A4. Se prevé que estos inductores del CYP3A4 disminuyan notablemente la concentración plasmática de finerenona y den lugar a una reducción del efecto terapéutico (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Determinados medicamentos que aumentan el potasio sérico: FIRIALTA no se debe usar de forma concomitante con diuréticos ahorradores de potasio (por ejemplo, amilorida, triamtereno) y otros ARM (por ejemplo, eplerenona, esaxerenona, espironolactona, canrenona). Se prevé que estos medicamentos aumenten el riesgo de hiperpotasemia (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Toronja: No se debe consumir toronja ni jugo de toronja durante el tratamiento con finerenona, ya que se espera que aumente las concentraciones plasmáticas de finerenona debido a la inhibición del CYP3A4 (ver la sección Dosis y vía de administración; Posología y forma de administración y la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Uso concomitante adoptando precauciones:
Inhibidores moderados del CYP3A4: En un estudio clínico, el uso concomitante de eritromicina (500 mg tres veces al día) produjo un aumento de 3,5 veces del AUC de la finerenona y de 1,9 veces de su Cmáx. En otro estudio clínico, el verapamilo (comprimido de liberación controlada de 240 mg una vez al día) ocasionó un aumento de 2,7 y 2,2 veces en el AUC y la Cmáx de la finerenona, respectivamente.
El potasio sérico puede aumentar, por lo que se recomienda monitorizarlo, especialmente durante el inicio o los cambios de la dosis de finerenona o del inhibidor del CYP3A4 (ver la sección Dosis y vía de administración; Posología y forma de administración y la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Inhibidores débiles del CYP3A4: Las simulaciones farmacocinéticas con base fisiológica (PBPK, por sus siglas en inglés) sugieren que la fluvoxamina (100 mg dos veces al día), aumenta el AUC (1,6 veces) y la Cmáx (1,4 veces) de la finerenona.
El potasio sérico puede aumentar, por lo que se recomienda monitorizarlo, especialmente durante el inicio o los cambios de la dosis de finerenona o del inhibidor del CYP3A4 (ver la sección Dosis y vía de administración; Posología y forma de administración y la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Determinados medicamentos que aumentan el potasio sérico (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo): Se prevé que el uso concomitante de FIRIALTA con suplementos de potasio y trimetoprim, o trimetoprim/sulfametoxazol aumente el riesgo de hiperpotasemia. Es necesario monitorizar el potasio sérico.
Puede ser necesaria la interrupción temporal del tratamiento con FIRIALTA durante el tratamiento con trimetoprim o trimetoprim/sulfametoxazol.
Medicamentos antihipertensivos: El riesgo de hipotensión aumenta con el uso concomitante de otros múltiples medicamentos antihipertensivos. En estos pacientes, se recomienda monitorizar la presión arterial.
HALLAZGOS DE LABORATORIO CLÍNICO:
Datos preclínicos sobre seguridad: Los datos de los estudios preclínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad de dosis única, toxicidad a dosis repetidas, genotoxicidad, fototoxicidad, potencial carcinogénico y fertilidad masculina y femenina.
Toxicidad a dosis repetidas: En los perros, se encontró una reducción del peso y del tamaño de la próstata con un AUClibre de unas 10 a 60 veces superior al de los humanos. La dosis sin manifestaciones proporciona un margen de seguridad de aproximadamente 2.
Potencial carcinogénico: En estudios de carcinogénesis de 2 años de duración, la finerenona no mostró potencial carcinogénico en ratas macho y hembra ni en ratones hembra. En ratones macho, la finerenona provocó un aumento de adenomas de células de Leydig a dosis que representaban 26 veces el AUClibre en seres humanos. Una dosis equivalente a 17 veces el AUClibre en seres humanos no causó ningún tumor. Teniendo en cuenta la sensibilidad conocida de los roedores a presentar estos tumores y el mecanismo farmacológico a dosis supraterapéuticas, así como los márgenes de seguridad adecuados, el aumento de los tumores de células de Leydig en ratones macho no es clínicamente relevante.
Toxicidad para el desarrollo: En el estudio de toxicidad embriofetal en ratas, la finerenona dio lugar a una reducción del peso de la placenta y a signos de toxicidad fetal, incluidos la reducción del peso del feto y el retraso en su osificación a la dosis tóxica para la madre de 10 mg/kg/día, lo que corresponde a un AUClibre de 19 veces el de los seres humanos. Con 30 mg/kg/día, aumentó la incidencia de variaciones viscerales y esqueléticas (edema ligero, cordón umbilical acortado, fontanela ligeramente agrandada) y un feto presentó malformaciones complejas, incluida una malformación rara (doble arco aórtico), con un AUClibre de unas 25 veces el de los seres humanos. Las dosis sin manifestaciones (dosis baja en ratas, dosis alta en conejos) proporcionaron márgenes de seguridad de 10 a 13 veces para el AUClibre. Por lo tanto, las manifestaciones en las ratas no apuntan a una mayor inquietud por daño fetal.
Cuando se expuso a las ratas durante la gestación y la lactancia en el estudio de toxicidad para el desarrollo prenatal y posnatal, se observó un aumento de la mortalidad de las crías y otros efectos adversos (menor peso de las crías, retraso en el desdoblamiento del pabellón auditivo) a unas 4 veces la AUClibre esperada en seres humanos. Además, las crías mostraron un ligero aumento de la actividad locomotora, pero ningún otro cambio neuroconductual a partir de unas 4 veces el AUClibre esperado en seres humanos. La dosis sin manifestaciones proporcionó un margen de seguridad de aproximadamente 2 para el AUClibre. El aumento de la actividad locomotora en las camadas puede indicar un posible riesgo para el feto. Además, debido a las manifestaciones en las crías, no se puede excluir un riesgo en recién nacidos/niños lactantes.
Fertilidad femenina: La finerenona provocó una reducción de la fertilidad femenina (disminución en el número de cuerpos lúteos y de lugares de implantación), así como signos de toxicidad embrionaria temprana (aumento de pérdidas postimplantación y disminución del número de fetos viables) a unas 21 veces el AUClibre en seres humanos. Además, se observó una reducción del peso de los ovarios a unas 17 veces el AUClibre en seres humanos. No se hallaron efectos sobre la fertilidad de las hembras ni el desarrollo embrionario temprano a 10 veces el AUClibre en seres humanos. Por lo tanto, los hallazgos en ratas hembras poseen poca relevancia clínica (ver sección Restricciones de uso durante el embarazo y la lactancia).
DOSIS Y VÍA DE ADMINISTRACIÓN:
Forma de administración: Vía oral.
Posología y forma de administración:
Posología: La dosis objetivo recomendada es de 20 mg de finerenona una vez al día.
La dosis máxima recomendada es de 20 mg de finerenona una vez al día.
Inicio del tratamiento: Para determinar si se puede iniciar el tratamiento con finerenona y determinar la dosis inicial, es preciso medir el potasio sérico y la tasa de filtración glomerular estimada (TFGe).
Si el potasio sérico es ≤ 4,8 mmol/L, se puede iniciar el tratamiento con finerenona. Para la monitorización del potasio sérico, ver “Continuación del tratamiento” más adelante.
Si el potasio sérico es > 4,8 a 5,0 mmol/L, se puede considerar iniciar el tratamiento con finerenona, realizando una monitorización adicional del potasio sérico en las primeras 4 semanas, según las características del paciente y de sus niveles de potasio sérico (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo)
Si el potasio sérico es > 5,0 mmol/L, no se debe iniciar el tratamiento con finerenona (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
La dosis inicial recomendada de finerenona se basa en la TFGe y se presenta en la tabla 1.
Tabla 1. Inicio del tratamiento con finerenona y dosis recomendada
|
TFGe (mL/min/1,73 m2) |
Dosis inicial (una vez al día) |
|
≥ 60 |
20 mg |
|
≥ 25 a < 60 |
10 mg |
|
< 25 |
No recomendada |
Continuación del tratamiento:
El potasio sérico y la TFGe se deben volver a medir 4 semanas después de iniciar o reiniciar el tratamiento con finerenona o de aumentar la dosis (ver la tabla 2 para determinar la continuación del tratamiento con finerenona y el ajuste de la dosis).
A partir de entonces, es preciso volver a medir el potasio sérico periódicamente y según sea necesario en función de las características del paciente y de sus niveles de potasio sérico.
Para más información, ver la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo y la sección Interacciones medicamentosas y de otro género.
Tabla 2. Continuación del tratamiento con finerenona y ajuste de la dosis
|
Dosis actual de finerenona (una vez al día) |
|||
|
10 mg |
20 mg |
||
|
Potasio sérico actual (mmol/L) |
≤ 4,8 |
Aumentar a 20 mg de finerenona una vez al día* |
Mantener 20 mg una vez al día |
|
> 4,8 a 5,5 |
Mantener 10 mg una vez al día |
Mantener 20 mg una vez al día |
|
|
> 5,5 |
Interrumpir el tratamiento con finerenona Considerar el reinicio con 10 mg una vez al día cuando el potasio sérico sea ≤ 5,0 mmol/L |
Interrumpir el tratamiento con finerenona. Reiniciar con 10 mg una vez al día cuando el potasio sérico sea ≤ 5,0 mmol/L |
|
* Mantener 10 mg una vez al día si la TFGe ha disminuido > 30% en comparación con la medición anterior
Dosis olvidada: La dosis olvidada se debe tomar tan pronto como el paciente se dé cuenta, pero solo en el mismo día. El paciente no debe tomar 2 dosis para compensar una dosis olvidada.
Poblaciones especiales:
Pacientes de edad avanzada: No es necesario ajustar la dosis en pacientes de edad avanzada (ver sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas).
Insuficiencia renal:
Inicio del tratamiento: En pacientes con una TFGe < 25 mL/min/1,73 m2, no se debe iniciar el tratamiento con finerenona debido a la escasez de datos clínicos (ver la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo y la sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas).
Continuación del tratamiento: En pacientes con una TFGe ≥ 15 mL/min/1,73 m2, el tratamiento con finerenona se puede continuar con un ajuste de la dosis en función del potasio sérico. La TFGe debe medirse 4 semanas después del inicio para establecer si la dosis inicial se puede aumentar hasta la dosis diaria recomendada de 20 mg (ver “Posología, Continuación del tratamiento” y la tabla 2).
Debido a la escasez de datos clínicos, el tratamiento con finerenona debe ser interrumpido en los pacientes que hayan progresado a una enfermedad renal terminal (TFGe < 15 mL/min/1,73 m2) (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo).
Insuficiencia hepática:
Pacientes con:
- Insuficiencia hepática grave: No se debe iniciar finerenona (ver la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo y la sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas). No se dispone de datos.
- Insuficiencia hepática moderada: No se requiere un ajuste inicial de la dosis. Considerar la posibilidad de realizar una monitorización adicional del potasio sérico y adaptar dicha monitorización según las características del paciente (ver la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo y la sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas).
- Insuficiencia hepática leve: No se requiere un ajuste inicial de la dosis.
Medicación concomitante: En los pacientes que toman finerenona de forma concomitante con inhibidores moderados o débiles del CYP3A4, suplementos de potasio, trimetoprim o trimetoprim/sulfametoxazol, se debe considerar la monitorización adicional del potasio sérico, así como la adaptación de dicha monitorización según las características del paciente (ver sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo). Las decisiones sobre el tratamiento con finerenona deben tomarse como se indica en la tabla 2 (Posología, Continuación del tratamiento).
Puede ser necesaria la interrupción temporal del tratamiento con finerenona cuando los pacientes deban tomar trimetoprim o trimetoprim/sulfametoxazol. Para más información, ver la sección Precauciones y advertencias; Advertencias y precauciones especiales de empleo y la sección Interacciones medicamentosas y de otro género.
Peso corporal: No es necesario ajustar la dosis en función del peso corporal (ver sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas).
Población pediátrica: No se ha establecido todavía la seguridad y eficacia de la finerenona en niños y adolescentes menores de 18 años. No se dispone de datos.
Los comprimidos se pueden tomar con un vaso de agua y con o sin alimentos (ver sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas). Los comprimidos no se deben tomar con toronja o jugo de toronja (ver sección Interacciones medicamentosas y de otro género).
Trituración de los comprimidos: Para los pacientes que no puedan tragar comprimidos enteros, los comprimidos de FIRIALTA se pueden triturar y mezclar con agua o alimentos blandos, como compota de manzana, directamente antes de la administración oral (ver sección Farmacocinética y farmacodinamia; Propiedades farmacocinéticas).
PROPIEDADES FARMACÉUTICAS:
Datos farmacéuticos:
Lista de excipientes:
Núcleo del comprimido:
Celulosa microcristalina
Croscarmelosa sódica
Hipromelosa 2910
Lactosa monohidrato
Estearato de magnesio
Laurilsulfato de sodio
Recubrimiento del comprimido:
Hipromelosa 2910
Dióxido de titanio
Talco
FIRIALTA 10 mg comprimidos recubiertos:
Óxido de hierro rojo (E 172)
FIRIALTA 20 mg comprimidos recubiertos:
Óxido de hierro amarillo (E 172)
Incompatibilidades: No procede.
Periodo de validez: 3 años.
Precauciones especiales de conservación:
Consérvese a temperatura no mayor a 30 °C.
Precauciones especiales de eliminación: La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis: Se prevé que la manifestación más probable de sobredosis sea la hiperpotasemia. Si se produce hiperpotasemia, debe iniciarse el tratamiento habitual.
Es poco probable que la finerenona se elimine eficazmente mediante hemodiálisis, ya que su fracción unida a las proteínas plasmáticas es de aproximadamente el 90%.
DESCRIPCIÓN:
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas. Ver la sección Reacciones adversas, en la que se incluye información sobre cómo notificarlas.
Datos clínicos:
Indicaciones terapéuticas:
FIRIALTA está indicado en adultos para el tratamiento de la enfermedad renal crónica (con albuminuria) asociada a diabetes tipo 2.
Para consultar los resultados de los estudios con respecto a los acontecimientos renales y cardiovasculares, ver sección Farmacocinética y farmacodinamia; Propiedades farmacodinámicas.
PRESENTACIONES: Blísteres transparentes con calendario de PVC/PVDC/aluminio con 14 comprimidos recubiertos. Tamaños de envase de 14 o 28 comprimidos recubiertos.
Puede que solamente estén comercializados algunos tamaños de envases.
BAYER, S.A.
Basado en SmPC Ver. 3 (SmpC-Summary of Product Characteristics) aprobado por EMA (European Medicines Agency) el 06 de Febrero de 2023


