
METALYSE
TENECTEPLASA
Polvo y disolvente para solución inyectable
1 Caja, 1 Vial de polvo,
INDICACIONES TERAPÉUTICAS:
Datos clínicos:
METALYSE® está indicado en adultos para el tratamiento trombolítico de sospecha de infarto de miocardio con elevación ST persistente o Bloqueo reciente del Haz de Rama izquierda, en las 6 horas siguientes a la aparición de los síntomas del infarto agudo de miocardio (IAM).
PROPIEDADES FARMACOLÓGICAS:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Agentes antitrombóticos
Código ATC: B01A D11.
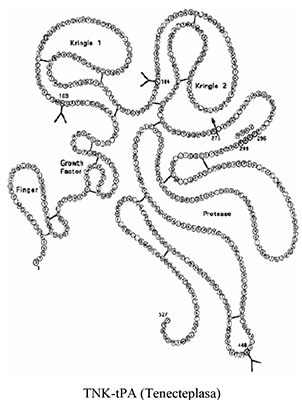
Mecanismo de acción: La Tenecteplasa es un activador recombinante del plasminógeno específico para la fibrina, derivado del t-PA natural por modificación en tres puntos de la estructura proteica. Se une al componente fibrina del trombo (coágulo sanguíneo) y convierte selectivamente el plasminógeno unido al trombo en plasmina, la cual degrada la matriz de fibrina del trombo. La Tenecteplasa posee una mayor especificidad para la fibrina y una mayor resistencia a la inactivación por su inhibidor endógeno (IAP-1), en comparación con t-PA natural.
Efectos farmacodinámicos: Después de la administración de Tenecteplasa, se ha observado un consumo de α2-antiplasmina dosisdependiente (el inhibidor de la plasmina de la fase fluida), con el consiguiente aumento en el nivel de producción de plasmina sistémica. Esta observación es concordante con el pretendido efecto de activación del plasminógeno. En estudios comparativos se observó una reducción del fibrinógeno inferior al 15% y una reducción del plasminógeno inferior al 25%, en sujetos tratados con la dosis máxima de Tenecteplasa (10,000 U, correspondientes a 50 mg), mientras que la Alteplasa ocasionó una disminución de aproximadamente un 50% en los niveles de fibrinógeno y plasminógeno. No se detectó una formación de anticuerpos clínicamente relevante a los 30 días.
Eficacia clínica y seguridad: Los datos de permeabilidad de los estudios angiográficos en Fases I y II sugieren que la Tenecteplasa, administrada como bolo intravenoso único en sujetos con IAM, es eficaz en la disolución de los coágulos sanguíneos de la arteria relacionada con el infarto de forma dosis-dependiente.
ASSENT-2: Un estudio a gran escala de la mortalidad (ASSENT II) en aproximadamente 17.000 pacientes, mostró que la Tenecteplasa es terapéuticamente equivalente a la alteplasa en la reducción de la mortalidad (6.2% para ambos tratamientos, a 30 días, siendo 1,124 el límite superior del intervalo de confianza (IC) del 95% para el riesgo relativo) y que el uso de Tenecteplasa se asocia con una incidencia de hemorragias no intracraneales significativamente inferior (26.4% vs. 28.9%, p = 0,0003). Esto se traduce en una necesidad de transfusiones significativamente inferior (4.3% vs. 5.5%, p = 0.0002). La hemorragia intracraneal se produjo en una proporción del 0.93% vs. 0.94% para Tenecteplasa y Alteplasa, respectivamente.
La permeabilidad coronaria y los datos limitados de los resultados clínicos, mostraron que los pacientes con IAM, después de 6 horas de aparición de los síntomas, han sido tratados satisfactoriamente.
ASSENT-4: El estudio ASSENT-4 PCI se diseñó para poner de manifiesto si en 4,000 pacientes con infarto de miocardio extenso, el pre-tratamiento con dosis completas de Tenecteplasa y un bolo único concomitante de hasta 4,000 UI de heparina no fraccionada, administrados previamente a una intervención coronaria percutánea (ICP) primaria que debe realizarse en los 60-180 minutos posteriores, se obtienen mejores resultados que mediante la ICP primaria solamente. El estudio se detuvo prematuramente con 1,667 pacientes aleatorizados, debido a una mortalidad numérica mayor en el grupo de la ICP facilitada que recibía Tenecteplasa. La incidencia de la variable principal, siendo ésta la combinación de muerte o shock cardiogénico o insuficiencia cardíaca congestiva en 90 días, fue significativamente mayor en el grupo que recibía el tratamiento exploratorio de Tenecteplasa seguido de ICP inmediata de rutina: 18.6% (151/810) en comparación con 13.4% (110/819) en el grupo que únicamente recibió ICP, p = 0.0045. Esta diferencia significativa entre grupos, en cuanto al criterio de valoración primario a los 90 días, ya apareció a nivel intra-hospitalario y a los 30 días.
Numéricamente, todos los componentes de la variable clínica principal combinada eran favorables al tratamiento con ICP únicamente: muerte: 6.7% vs. 4.9% p = 0.14; shock cardiogénico: 6.3% vs. 4.8% p = 0.19; insuficiencia cardíaca congestiva: 12.0% vs. 9.2% p = 0.06, respectivamente. Las variables secundarias, reinfarto y revascularización repetida de los vasos diana, aumentaron significativamente en el grupo pre-tratado con Tenecteplasa: re-infarto: 6.1% vs. 3.7% p = 0.0279; revascularización repetida de los vasos diana: 6.6% vs. 3.4% p = 0.0041. Las siguientes reacciones adversas se presentaron con mayor frecuencia con el uso de Tenecteplasa previamente a la ICP: hemorragia intracraneal: 1% vs. 0% p = 0.0037; ictus: 1.8% vs. 0% p < 0.0001; hemorragias mayores: 5.6% vs. 4.4% p = 0.3118; hemorragias menores: 25.3% vs. 19.0% p = 0.0021; transfusiones de sangre: 6.2% vs. 4.2% p = 0.0873; cierre brusco del vaso: 1.9% vs. 0.1% p = 0.0001.
Estudio STREAM: El estudio STREAM fue diseñado para evaluar la eficacia y seguridad de una estrategia fármaco-invasiva frente a una estrategia de ICP primaria estándar en pacientes con infarto agudo de miocardio con elevación ST en las 3 horas siguientes al inicio de los síntomas y en los que era imposible realizar una ICP primaria en el plazo de una hora desde el primer contacto médico. La estrategia fármaco-invasiva consistió en un tratamiento fibrinolítico precoz con un bolo de Tenecteplasa y tratamiento adicional con medicamentos antiagregantes plaquetarios y antitrombóticos seguido de angiografía en las siguientes 6-24 horas o intervención coronaria de rescate.
La población en estudio consistió en 1,892 pacientes aleatorizados por medio de un sistema de respuesta de voz interactivo. La variable principal, combinación de muerte o shock cardiogénico o insuficiencia cardíaca congestiva o reinfarto en 30 días, se observó en un 12.4% (116/939) en el brazo fármaco-invasivo frente a un 14.3% (135/943) en el brazo de ICP primaria (riesgo relativo 0.86 (0.68-1.09).
Los componentes individuales de la variable principal compuesta para la estrategia fármaco-invasiva frente a la ICP primaria se observaron con las siguientes frecuencias:
|
Fármaco-invasivo (n = 944) |
ICP primaria (n = 948) |
p |
|
|
Combinación de muerte, shock, insuficiencia cardíaca congestiva, reinfarto |
116/939 (12.4%) |
135/943 (14.3%) |
0.21 |
|
Mortalidad por cualquier causa |
43/939 (4.6%) |
42/946 (4.4%) |
0.88 |
|
Shock cardiogénico |
41/939 (4.4%) |
56/944 (5.9%) |
0.13 |
|
Insuficiencia cardíaca congestiva |
57/939 (6.1%) |
72/943 (7.6%) |
0.18 |
|
Reinfarto |
23/938 (2.5%) |
21/944 (2.2%) |
0.74 |
|
Mortalidad cardíaca |
31/939 (3.3%) |
32/946 (3.4%) |
0.92 |
La incidencia observada de hemorragias no-HIC mayor y menor fue similar en los dos grupos:
|
Fármaco-invasivo (n = 944) |
ICP primaria (n = 948) |
p |
|
|
Hemorragia no-HIC mayor |
61/939 (6.5%) |
45/944 (4.8%) |
0.11 |
|
Hemorragia no-HIC menor |
205/939 (21.8%) |
191/944 (20.2%) |
0.40 |
Incidencia de ictus totales y hemorragia intracraneal:
|
Fármaco-invasivo (n = 944) |
ICP primaria (n= 948) |
p |
|
|
Ictus totales (de todo tipo) |
15/939 (1.6%) |
5/946 (0.5%) |
0.03* |
|
Hemorragia intracraneal |
9/939 (0.96%) |
2/946 (0.21%) |
0.04** |
|
Hemorragia intracraneal después de modificar el protocolo reduciendo la dosis a la mitad en pacientes ≥ 75 años: |
4/747 (0.5%) |
2/758 (0.3%) |
0.45 |
* Las incidencias en ambos grupos son las esperadas en pacientes STEMI tratados con fibrinolíticos o ICP primaria (como se observó en estudios previos).
** La incidencia en el grupo fármaco-invasivo es la esperada para fibrinolisis con Tenecteplasa (como se observó en estudios previos).
Después de la reducción a la mitad de la dosis de Tenecteplasa en pacientes ≥ 75 años no hubo más hemorragias intracraneales (0 de 97 pacientes) (95% IC: 0.0-3.7) frente a 8.1% (3 de 37 pacientes) (95% IC: 1.7-21.9) antes de la reducción de la dosis. Los límites del intervalo de confianza de las incidencias observadas antes y después de la reducción de la dosis se superponen.
En pacientes ≥ 75 años la incidencia observada de la eficacia en la variable principal combinada para la estrategia fármaco-invasiva y para la ICP primaria fue la siguiente: antes de la reducción de la dosis 11/37 (29.7%) (95% IC: 15.9-47,0) frente a 10/32 (31.3%) (95% IC: 16.1-50.0), después de la reducción de dosis: 25/97 (25.8%) (95% IC: 17.4-35.7) frente a 25/88 (24.8%) (95% IC: 19.3-39.0). En ambos grupos los límites del intervalo de confianza de las incidencias observadas antes y después de la reducción de la dosis se superponen.
Propiedades farmacocinéticas:
Absorción y distribución: La Tenecteplasa es una proteína recombinante activadora del plasminógeno, que se administra por vía intravenosa. Después de la administración intravenosa de un bolo de 30 mg de Tenecteplasa en pacientes con infarto agudo de miocardio, la concentración plasmática inicialmente estimada fue de 6.45 ± 3.60 μg/mL (media ± DE). La fase de distribución representa del 31% ± 22% al 69% ± 15% (media ± DE) del AUC total después de la administración de dosis en el rango de 5 a 50 mg.
En estudios en ratas con Tenecteplasa marcada radioactivamente, se obtuvieron datos sobre la distribución tisular. El principal órgano en el que se distribuyó la Tenecteplasa fue el hígado. Se desconoce si la Tenecteplasa se une a las proteínas plasmáticas humanas y en qué medida. El tiempo medio de residencia (TMR) en el cuerpo es aproximadamente 1 h y el volumen medio (± DE) de distribución en el estado estacionario (Vss) es de 6.3 ± 2 L a 15 ± 7 L.
Biotransformación: La Tenecteplasa se elimina de la circulación por unión a receptores específicos en el hígado, seguida de su catabolismo a péptidos pequeños. Sin embargo, la unión a receptores hepáticos es reducida si se compara con t-PA natural, dando como resultado una vida media prolongada.
Eliminación: Después de la inyección de un bolo intravenoso único de Tenecteplasa, en pacientes con infarto agudo de miocardio, el antígeno Tenecteplasa muestra una eliminación bifásica del plasma. En el rango de dosis terapéutica, en el aclaramiento de Tenecteplasa no hay dependencia de dosis. La vida media dominante inicial es de 24 ± 5.5 (media ± DE) min, la cual es cinco veces más prolongada que la del t-PA natural. La vida media terminal es de 129 ± 87 min y el aclaramiento plasmático es de 119 ± 49 ml/min.
Un incremento del peso corporal tuvo como consecuencia un aumento moderado del aclaramiento de Tenecteplasa y el aumento de edad tuvo como consecuencia una ligera reducción del aclaramiento. Por lo general, las mujeres presentan un aclaramiento menor que los hombres, pero esto puede explicarse por el peso corporal, que es generalmente inferior en las mujeres.
Linealidad/No-linealidad: El análisis de linealidad de dosis basado en el AUC sugirió que Tenecteplasa muestra una farmacocinética no lineal en el rango de dosis estudiado, es decir, de 5 a 50 mg.
Insuficiencia renal y hepática: Debido a que la Tenecteplasa se elimina a través del hígado, no es de esperar que la insuficiencia renal afecte a su farmacocinética. Esto está también sustentado por los datos en animales. Sin embargo, el efecto de la insuficiencia renal y hepática en la farmacocinética de Tenecteplasa en humanos no ha sido específicamente investigado. En consecuencia, no hay ninguna guía para el ajuste de dosis de Tenecteplasa en pacientes con insuficiencia renal y hepática.
DATOS FARMACÉUTICOS:
Lista de excipientes:
Polvo:
• L-arginina.
• Ácido fosfórico.
• Polisorbato 20.
Disolvente:
• Agua para inyectables.
Incompatibilidades:
METALYSE® es incompatible con soluciones de dextrosa para perfusión intravenosa.
CONTRAINDICACIONES:
METALYSE® no debe ser administrado a pacientes con historia de reacción anafiláctica (i.e. con peligro para la vida) a cualquier de los componentes (i.e. Tenecteplasa o cualquier excipiente) o a la gentamicina (una sustancia residual del proceso de fabricación). Si de todos modos el tratamiento con METALYSE® se considera necesario, se debe disponer inmediatamente de mecanismos de reanimación por si fuera necesario.
Además, como el tratamiento trombolítico se asocia a un mayor riesgo de hemorragia, METALYSE®, está contraindicado en las siguientes situaciones:
• Trastorno hemorrágico significativo actual o durante los últimos 6 meses.
• Pacientes que reciben tratamiento efectivo con anticoagulantes orales, p. ej., warfarina sódica (INR > 1.3) (ver sección Advertencias y precauciones especiales de empleo, subsección Hemorragia).
• Historia de lesión del sistema nervioso central (por ej., neoplasma, aneurisma, cirugía intracraneal o espinal).
• Diátesis hemorrágica conocida.
• Hipertensión no controlada grave.
• Cirugía mayor, biopsia de un órgano parenquimatoso o traumatismo significativo durante los últimos 2 meses (incluyendo cualquier traumatismo asociado con el IAM actual).
• Traumatismo reciente de la cabeza o el cráneo.
• Reanimación cardiopulmonar prolongada (> 2 minutos) durante las últimas 2 semanas.
• Pericarditis aguda y/o endocarditis bacteriana subaguda.
• Pancreatitis aguda.
• Disfunción hepática grave, incluyendo fallo hepático, cirrosis, hipertensión portal (várices esofágicas) y hepatitis activa.
• Úlcera péptica activa.
• Aneurisma arterial y malformación arterial/venosa conocida.
• Neoplasma con riesgo aumentado de hemorragia.
• Cualquier historia conocida de ictus hemorrágico o ictus de origen desconocido.
• Historia conocida de ictus isquémico o ataque isquémico transitorio (AIT) en los 6 meses anteriores.
• Demencia.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO:
Intervención coronaria: Si está programada una intervención coronaria percutánea (ICP) de acuerdo con las guías actuales de tratamiento, no se debe administrar Tenecteplasa (ver sección Propiedades farmacodinámicas estudio ASSENT-4).
Los pacientes que no se pueden someter a una ICP primaria en 1 hora tal y como se recomienda en las guías y que reciben Tenecteplasa como tratamiento de recanalización coronaria primaria deben ser trasladados sin demora a un centro habilitado para intervención coronaria para angiografía e intervención coronaria adyuvante a tiempo en 6-24 horas o antes si el médico lo indica (ver sección Propiedades farmacodinámicas Estudio STREAM).
Hemorragia: Durante el tratamiento con Tenecteplasa, la complicación más común detectada es la hemorragia. Puede contribuir a esta hemorragia la administración concomitante de heparina como anticoagulante. Como durante el tratamiento con Tenecteplasa se produce lisis de fibrina, puede producirse hemorragia en el sitio de punción reciente. Por lo tanto, el tratamiento trombolítico requiere cuidadosa atención de todos los posibles puntos de hemorragia (incluyendo puntos de inserción de catéteres, puntos de punción arterial o venosa, zonas de corte y sitios de punción con aguja). Durante el tratamiento con Tenecteplasa debe evitarse el uso de catéteres rígidos, las inyecciones intramusculares y la manipulación innecesaria del paciente.
Las hemorragias observadas con mayor frecuencia se produjeron en el sitio de inyección, y ocasionalmente se observó hemorragia genitourinaria y gingival.
Si se produce una hemorragia grave, en particular hemorragia cerebral, debe suspenderse de inmediato la administración simultánea de heparina. Debe considerarse la administración de protamina si se ha administrado heparina durante las 4 horas precedentes al inicio de la hemorragia. En los pocos pacientes que no respondan a estas medidas conservadoras, puede estar indicada una administración cautelosa de perfusiones. Debe considerarse la perfusión de crioprecipitados, plasma fresco congelado y plaquetas, con una reevaluación clínica y de laboratorio después de cada administración. Con la perfusión de crioprecipitados es deseable obtener un nivel de fibrinógeno de 1g/L. Los fármacos antifibrinolíticos estarán disponibles como última alternativa. En las siguientes condiciones el riesgo del tratamiento con Tenecteplasa puede verse incrementado y debe ponderarse frente a los beneficios previstos:
• Presión arterial sistólica > 160 mmHg.
• Enfermedad cerebrovascular.
• Hemorragia gastrointestinal o genitourinaria reciente (durante los últimos 10 días).
• Elevada probabilidad de trombo cardíaco en el ventrículo izquierdo, por ej., estenosis mitral con fibrilación auricular.
• Cualquier inyección intramuscular reciente conocida (durante los últimos 2 días).
• Edad avanzada, por ej., mayor de 75 años.
• Bajo peso corporal < 60 kg.
• Pacientes que reciben anticoagulantes orales: el uso de METALYSE® se puede considerar cuando la dosis o el tiempo desde la última toma de tratamiento anticoagulante hace improbable que haya una eficacia residual y si las pruebas de actividad anticoagulante apropiadas para los correspondientes medicamentos no muestran actividad clínicamente relevante sobre el sistema de coagulación (p. ej., INR ≤ 1.3 para antagonistas de la vitamina K u otras pruebas pertinentes para otros anticoagulantes orales que estén dentro del correspondiente límite superior de la normalidad).
Arritmias: La trombólisis coronaria puede dar lugar a arritmias asociadas a la reperfusión. Se recomienda tener disponible un tratamiento antiarrítmico para la bradicardia y/o taquiarritmia ventricular (marcapasos, desfibrilador) cuando se administre Tenecteplasa.
Antagonistas GPIIb/IIIa: El uso concomitante de antagonistas GPIIb/IIIa aumenta el riesgo de hemorragia.
Hipersensibilidad/Re-administración: No se ha observado formación sostenida de anticuerpos a Tenecteplasa tras el tratamiento. Sin embargo, no se dispone de experiencia sistemática en la re-administración de Tenecteplasa. Tenecteplasa debe administrarse con precaución a individuos con hipersensibilidad conocida (distinta a reacciones anafilácticas) al principio activo, cualquier excipiente o a la gentamicina (sustancia residual del proceso de fabricación). Si se produce una reacción anafilactoide, debe interrumpirse inmediatamente la inyección y debe iniciarse un tratamiento adecuado. En cualquier caso, no debe re-administrarse la Tenecteplasa antes de la valoración de los factores hemostáticos tales como, el fibrinógeno, el plasminógeno y la α2-antiplasmina.
Población pediátrica: METALYSE® no está recomendado para uso en niños (menores de 18 años) debido a la ausencia de datos sobre seguridad y eficacia.
PRECAUCIONES ESPECIALES DE ELIMINACIÓN Y OTRAS MANIPULACIONES: METALYSE® debe reconstituirse añadiendo el volumen total de agua para inyectables de la jeringa precargada al vial que contiene el polvo para inyectable.
1. Asegurar que se ha elegido el tamaño del vial adecuado según el peso corporal del paciente:
|
Categoría de peso corporal del paciente (kg) |
Volumen de solución reconstituida (mL) |
Tenecteplasa (mg) |
Tenecteplasa (mg) |
|
< 60 |
6 |
6,000 |
30 |
|
≥ 60 a < 70 |
7 |
7,000 |
35 |
|
≥ 70 a < 80 |
8 |
8,000 |
40 |
|
≥ 80 a < 90 |
9 |
9,000 |
45 |
|
≥ 90 |
10 |
10,000 |
50 |
2. Verificar que el cierre del vial está todavía intacto.
3. Retirar el cierre “flip-off” del vial.
4. Retirar el cierre de la punta de la jeringa. Inmediatamente enroscar la jeringa precargada en el adaptador del vial e insertar el tapón del vial en el medio con la punta del adaptador.
5. Añadir el agua para inyectables al interior del vial empujando el émbolo de la jeringa hacia abajo lentamente para evitar la formación de espuma.
6. Reconstituir agitando suavemente.
7. La preparación reconstituida es una solución transparente, incolora o de color amarillo claro. Sólo debe ser administrada una solución transparente y sin partículas.
8. Inmediatamente antes de administrar la solución, invertir el vial con la jeringa todavía insertada, de forma que la jeringa se encuentre debajo del vial.
9. Transferir el volumen adecuado de solución reconstituida de METALYSE® a la jeringa, según el peso del paciente.
10. Desconectar la jeringa del adaptador del vial.
11. METALYSE® debe administrarse al paciente por vía intravenosa, en aproximadamente 10 segundos. No debe administrarse en un circuito que contenga dextrosa.
12. Debe desecharse la solución no utilizada.
Como alternativa, la reconstitución puede realizarse con la aguja que se incluye.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Fertilidad, embarazo y lactancia:
Embarazo: Hay datos limitados relativos al uso de METALYSE® en mujeres embarazadas. Los datos preclínicos obtenidos con Tenecteplasa mostraron hemorragias con mortalidad secundaria de los animales madre debido a la actividad farmacológica conocida del principio activo y en algunos casos se produjo aborto y reabsorción del feto (efectos sólo observados con una administración repetida de la dosis). Tenecteplasa no se considera teratogénica (ver sección Datos preclínicos sobre seguridad).
Se debe valorar el beneficio del tratamiento frente a los riesgos potenciales en caso de infarto de miocardio durante el embarazo.
Lactancia: Se desconoce si la Tenecteplasa se excreta en la leche materna. Se debe evitar la lactancia durante las primeras 24 horas después del tratamiento trombolítico.
Fertilidad: No se dispone de datos clínicos ni preclínicos en fertilidad con Tenecteplasa (METALYSE®).
REACCIONES ADVERSAS:
Resumen del perfil de seguridad: La hemorragia es una reacción adversa muy frecuente asociada al uso de la Tenecteplasa. El tipo de hemorragia es principalmente superficial en el sitio de inyección. Frecuentemente se han observado casos de equimosis que, normalmente, no requieren ninguna acción específica. Se han descrito muerte e incapacidad permanente en pacientes que han presentado ictus (incluyendo hemorragia intracraneal) y otros episodios graves de hemorragia.
Tabla de reacciones adversas:
Las reacciones adversas se clasifican según la frecuencias y según la clasificación por órganos y sistemas según las siguientes categorías: muy frecuente (≥ 1/10); frecuente (≥ 1/100 a < 1/10); poco frecuente (≥ 1/1,000 a < 1/100); raras (≥ 1/10,000 a < 1/1,000); muy raras (< 1/10,000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Tabla 1. Muestra de las frecuencias de las reacciones adversas
|
Clasificación por órganos y sistemas |
Reacción adversa |
|
Trastornos del sistema inmunológico |
|
|
Raras |
Reacción anafilactoide (incluyendo exantema, urticaria, broncoespasmo, edema laríngeo) |
|
Trastornos del sistema nervioso |
|
|
Poco frecuentes |
Hemorragia intracraneal (como hemorragia cerebral, hematoma cerebral, ictus hemorrágico, transformación hemorrágica del ictus, hematoma intracraneal, hemorragia subaracnoidea), incluyendo síntomas asociados como somnolencia, afasia, hemiparesia, convulsiones |
|
Trastornos oculares |
|
|
Poco frecuentes |
Hemorragia en el ojo |
|
Trastornos cardíacos |
|
|
Poco frecuentes |
La aparición de arritmias de reperfusión (como asistolia, arritmia idioventricular acelerada, arritmia, extrasístoles, fibrilación auricular, bloqueo aurículo-ventricular de primer grado a bloqueo aurículo-ventricular completo, bradicardia, taquicardia, arritmia ventricular, fibrilación ventricular, taquicardia ventricular) guarda una estrecha relación temporal con el tratamiento con Tenecteplasa. Las arritmias de reperfusión pueden conducir a un paro cardíaco, pueden ser una amenaza para la vida y pueden necesitar el uso de tratamiento antiarrítmico convencional |
|
Raras |
Hemorragia del pericardio |
|
Trastornos vasculares |
|
|
Muy frecuentes |
Hemorragia |
|
Raras |
Embolia (embolización trombótica) |
|
Trastornos respiratorios, torácicos y mediastínicos |
|
|
Frecuentes |
Epistaxis |
|
Raras |
Hemorragia pulmonar |
|
Trastornos gastrointestinales |
|
|
Frecuentes |
Hemorragia gastrointestinal (como hemorragia gástrica, úlcera gástrica sangrante, hemorragia rectal, hematemesis, melena, hemorragia bucal) |
|
Poco frecuentes |
Hemorragia retroperitoneal (como hematoma retroperitoneal) |
|
Frecuencia no conocida |
Náuseas, vómitos |
|
Trastornos de la piel y del tejido subcutáneo |
|
|
Frecuentes |
Equimosis |
|
Trastornos renales y urinarios |
|
|
Frecuentes |
Hemorragia urogenital (como hematuria, hemorragia en el tracto urinario) |
|
Trastornos generales y alteraciones en el lugar de administración |
|
|
Frecuentes |
Hemorragia en el lugar de inyección, hemorragia en el lugar de punción |
|
Exploraciones complementarias |
|
|
Raras |
Presión arterial disminuida |
|
Frecuencia no conocida |
Temperatura corporal aumentada |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
|
|
Frecuencia no conocida |
Embolia grasa, lo cual puede conducir a las correspondientes consecuencias en los órganos afectados |
Al igual que con otros agentes trombolíticos, se han descrito los siguientes acontecimientos como secuelas del infarto de miocardio y/o de la administración de trombolíticos:
• Muy frecuentes (> 1/10): hipotensión, trastornos del ritmo y frecuencia cardíacos, angina de pecho.
• Frecuentes (> 1/100, < 1/10): isquemia recurrente, insuficiencia cardíaca, infarto de miocardio, shock cardiogénico, pericarditis, edema pulmonar.
• Poco frecuentes (> 1/1,000, < 1/100): paro cardíaco, regurgitación de la válvula mitral, derrame pericárdico, trombosis venosa, taponamiento cardíaco, rotura del miocardio.
• Raras (> 1/10,000, > 1/1,000): embolia pulmonar.
Estas reacciones cardiovasculares pueden suponer un riesgo para la vida y conducir a la muerte.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: No procede.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacción con otros medicamentos y otras formas de interacción:
No se han realizado estudios formales de interacción con Tenecteplasa y los medicamentos administrados habitualmente en pacientes con IAM. No obstante, el análisis de datos de más de 12.000 pacientes tratados durante las Fases I, II y III no reveló interacciones clínicas importantes con medicamentos utilizados habitualmente en pacientes con IAM y administrados simultáneamente con Tenecteplasa.
Los medicamentos que afectan a la coagulación o aquellos que alteran la función plaquetaria (p.ej. ticlopidina, clopidogrel, heparinas de bajo peso molecular [LMWH]) pueden aumentar el riesgo de hemorragia antes, durante o después del tratamiento con Tenecteplasa.
El uso concomitante de antagonistas GPIIb/IIIa aumenta el riesgo de hemorragia.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Datos preclínicos sobre seguridad: La administración intravenosa de una dosis única en ratas, conejos y perros sólo produjo alteraciones dosis dependientes y reversibles de los parámetros de la coagulación, con hemorragia local en el sitio de inyección, que se consideró como una consecuencia del efecto farmacodinámico de la Tenecteplasa. Los estudios de toxicidad a dosis múltiples en ratas y perros, confirmaron las observaciones mencionadas anteriormente pero la duración del estudio se limitó a dos semanas por la formación de anticuerpos a la proteína humana Tenecteplasa, que produjeron anafilaxia.
Los datos farmacológicos de seguridad en monos cynomolgus revelaron una disminución de la presión arterial seguida de alteraciones del ECG, pero éstas se produjeron con exposiciones que eran considerablemente superiores a la exposición clínica.
En relación con la indicación y la administración de una dosis única en humanos, los estudios de toxicidad reproductiva se limitaron a estudios de embriotoxicidad en conejos, como especies sensibles. La Tenecteplasa indujo la muerte total de la descendencia durante el período embrionario medio. Cuando la tenecteplasa se administró durante el período embrionario medio o final, las hembras grávidas mostraron hemorragia vaginal en el día después de la primera dosis. La mortalidad secundaria se observó 1-2 días después. No se dispone de datos en el período fetal.
Para esta clase de proteínas recombinantes no son de esperar mutagenicidad ni carcinogenicidad y no fueron necesarios estudios de genotoxicidad ni carcinogenicidad.
No se observó irritación local del vaso sanguíneo después de la administración intravenosa, intraarterial o paravenosa de la formulación final de Tenecteplasa.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN:
Posología: METALYSE® debe ser prescrito por médicos expertos en la administración de tratamiento trombolítico y con medios para monitorizar esta administración.
El tratamiento con METALYSE® debe iniciarse lo antes posible, después de la aparición de los síntomas.
METALYSE® debe administrarse en función del peso corporal, con una dosis máxima de 10,000 unidades (50 mg de Tenecteplasa). El volumen requerido para administrar la dosis correcta puede calcularse a partir del siguiente esquema:
|
Categoría de peso corporal del paciente (kg) |
Tenecteplasa (U) |
Tenecteplasa (mg) |
Volumen correspondiente de solución reconstituida (mL) |
|
< 60 |
6,000 |
30 |
6 |
|
≥ 60 a < 70 |
7,000 |
35 |
7 |
|
≥ 70 a < 80 |
8,000 |
40 |
8 |
|
≥ 80 a < 90 |
9,000 |
45 |
9 |
|
≥ 90 |
10,000 |
50 |
10 |
|
Ver sección 6.6.: Precauciones especiales de eliminación y otras manipulaciones |
|||
Pacientes de edad avanzada (≥ 75 años): METALYSE® se debe administrar con precaución en pacientes de edad avanzada (≥ 75 años), ya que tienen un mayor riesgo de hemorragia (ver información sobre hemorragia en la sección Advertencias y precauciones especiales de empleo y en el estudio STREAM en la sección Propiedades farmacodinámicas).
Población pediátrica: No se ha establecido la seguridad y eficacia de METALYSE® en niños (menores de 18 años). No se dispone de datos.
Forma de administración: La dosis requerida debe administrarse como bolo intravenoso único en aproximadamente 10 segundos.
Puede utilizarse un circuito intravenoso pre-existente exclusivo para la administración de METALYSE® en solución de cloruro sódico 0.9%. METALYSE® es incompatible con soluciones de dextrosa.
No debe añadirse ningún otro medicamento a la solución inyectable.
Para consultar las instrucciones de reconstitución del medicamento antes de la administración, ver sección Precauciones especiales de eliminación y otras manipulaciones.
Tratamiento coadyuvante: De acuerdo con las guías actuales, debe administrarse tratamiento antitrombótico coadyuvante con inhibidores plaquetarios y de acuerdo con anticoagulantes para el tratamiento de pacientes con infarto de miocardio con elevación de ST.
Para intervención coronaria ver sección Advertencias y precauciones especiales de empleo.
Se ha utilizado heparina no fraccionada y enoxaparina como tratamiento antitrombótico coadyuvante en ensayos clínicos con METALYSE®.
Debe iniciarse el tratamiento con ácido acetilsalicílico lo antes posible tras la presentación de los síntomas y debe continuarse durante toda la vida a menos que esté contraindicado.
SOBREDOSIS:
En caso de sobredosis puede existir un riesgo aumentado de hemorragia. Si se produce una hemorragia prolongada grave, puede considerarse un tratamiento sustitutivo (plasma, plaquetas), ver también sección Advertencias y precauciones especiales de empleo.
DESCRIPCIÓN: METALYSE® 10,000 unidades. Polvo y disolvente para solución inyectable.
PRESENTACIÓN:
Naturaleza y contenido del envase: Caja por 1 vial de polvo liofilizado + 1 jeringuilla de solvente.
Hecho en Alemania por:
Boehringer Ingelheim Pharma GmbH & Co. KG,
Birkendorfer Str. 65 - 88397 Biberach a.d.R., Alemania
Para:
BOEHRINGER INGELHEIM PROMECO, S.A. de C.V.
Calle del Maíz No. 49, Col. Barrio Xaltocán,
C.P. 16090, Xochimilco, Ciudad de México, México.
Fecha de revisión de la monografía: 09 de diciembre de 2013.
® Marca registrada
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Período de validez:
Período de validez del envase para la venta: 2 años.
Solución reconstituida: Se ha demostrado una estabilidad química y física, en condiciones de uso de 24 horas a 2-8 °C y de 8 horas a 30 °C.
Desde un punto de vista microbiológico, el producto debe utilizarse inmediatamente después de su reconstitución. Si no se utiliza de inmediato, los tiempos de almacenamiento en condiciones de uso, y las condiciones previas a la utilización son responsabilidad del usuario y, normalmente, no deben ser superiores a 24 horas a 2-8 °C.
Precauciones especiales de conservación: Consérvese a no más de 30 ºC.


