
SPIRIVA RESPIMAT
BROMURO DE TIOTROPIO
Solución para inhalación
1 Caja, 1 Cartucho con dispositivo, 4.5 mL,
FORMA FARMACÉUTICA Y FORMULACIÓN:
Forma farmacéutica:
Solución para inhalación.
Solución para inhalación transparente, incolora.
INDICACIONES TERAPÉUTICAS:
EPOC: SPIRIVA® RESPIMAT® está indicado como tratamiento broncodilatador de mantenimiento para aliviar los síntomas en pacientes con enfermedad pulmonar obstructiva crónica (EPOC).
Asma: SPIRIVA® RESPIMAT® está indicado como tratamiento broncodilatador adicional de mantenimiento en pacientes a partir de 6 años con asma grave que hayan experimentado al menos una exacerbación grave de asma en el año anterior (ver secciones Posología y forma de administración y Propiedades farmacocinéticas).
PROPIEDADES FARMACOLÓGICAS:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Otros agentes contra padecimientos obstructivos de las vías respiratorias, inhalatorios, anticolinérgicos
Código ATC: R03B B04.
Mecanismo de acción: El bromuro de tiotropio es un antagonista específico de los receptores muscarínicos de acción prolongada. Tiene una afinidad similar por los diferentes subtipos M1 a M5. En las vías aéreas, el bromuro de tiotropio se une, de forma competitiva y reversible a los receptores M3 de la musculatura lisa bronquial, antagonizando el efecto colinérgico (broncodilatador) de la acetilcolina, provocando relajación de la musculatura lisa bronquial. El efecto fue dependiente de la dosis y duró más de 24 horas. Como anticolinérgico N-cuaternario, el bromuro de tiotropio es tópicamente (bronco-) selectivo cuando se administra por inhalación, demostrando un rango terapéutico aceptable antes de que aparezcan efectos anticolinérgicos sistémicos.
Efectos farmacodinámicos: La disociación del tiotropio especialmente de los receptores M3 es muy lenta, mostrando una semivida de disociación significativamente mayor que el ipratropio. La disociación de los receptores M2 es más rápida que la de los M3, hecho que en los estudios funcionales in vitro fue considerado como una selectividad (controlado cinéticamente) del subtipo de receptor, M3 sobre M2. La potencia elevada, la disociación muy lenta del receptor y la selectividad tópica por inhalación mostraron su correlación clínica en forma de una broncodilatación significativa y de larga duración en los pacientes con EPOC y asma.
Eficacia clínica y seguridad: El programa de desarrollo clínico en fase III incluyó dos estudios de 1 año, dos estudios de 12 semanas y dos estudios de 4 semanas, aleatorizados, doble ciego en 2901 pacientes con EPOC (1038 pacientes recibieron una dosis de 5 μg de tiotropio). El programa de 1 año consistió en dos ensayos controlados con placebo. Los dos ensayos de 12 semanas fueron ambos controlados con placebo y activo (ipratroprio). En los seis estudios se incluyeron determinaciones de la función pulmonar. Además, los estudios de 1 año incluyeron mediciones de los resultados de salud por lo que respecta a la disnea, calidad de vida relacionada con la salud y efecto sobre las exacerbaciones.
Estudios controlados con placebo:
Función pulmonar: La solución para inhalación de tiotropio, administrada una vez al día, produjo una mejoría significativa de la función pulmonar (volumen espiratorio forzado en un segundo y capacidad vital forzada) en los 30 minutos posteriores a la administración de la primera dosis comparado con placebo (mejoría media del FEV1 a los 30 minutos: 0,113 litros; intervalo de confianza (IC) 95%: 0,102 a 0,125 litros, p < 0,0001). La mejoría de la función pulmonar se mantuvo durante un período de 24 horas en estado estacionario comparado con placebo (mejoría media del FEV1 : 0,122 litros; IC 95%: 0,106 a 0,138 litros, p < 0,0001).
El estado de equilibrio farmacodinámico se alcanzó en una semana.
SPIRIVA® RESPIMAT® mejoró de forma significativa el PEFR (tasa de flujo espiratorio máximo) matinal y nocturno medido en los registros diarios de los pacientes comparado con placebo (mejoría media del PEFR: mejoría media por la mañana 22 L/min; IC 95%: 18 a 55 L/min, p < 0.0001; noche 26 L/min; IC 95%: 23 a 30 L/min, p < 0.0001). El uso de SPIRIVA® RESPIMAT® produjo una reducción del uso de broncodilatadores de rescate comparado con placebo (reducción media en la utilización en rescate 0.66 veces al día, IC 95%: 0.51 a 0.81 veces por día, p < 0,0001).
Los efectos broncodilatadores de SPIRIVA® RESPIMAT® se mantuvieron durante todo el período de administración de 1 año sin que se observaran signos de tolerancia.
Disnea, calidad de vida relacionada con la salud y exacerbaciones de la EPOC en estudios a largo plazo de 1 año:
Disnea: SPIRIVA® RESPIMAT® mejoró de forma significativa la disnea (tal como se evaluó utilizando el Índice de Transición de Disnea de Mahler) comparado con placebo (mejoría media 1.05 unidades; IC 95%: 0.73 a 1.38 unidades, p < 0.0001). La mejoría se mantuvo durante todo el período de tratamiento.
Calidad de vida relacionada con la salud: La mejoría media total de la evaluación de la calidad de vida de los pacientes (utilizando para su evaluación el cuestionario respiratorio de St. George) entre SPIRIVA® RESPIMAT® frente a placebo al final de los dos estudios de 1 año de duración fue de 3.5 unidades (IC 95%: 2.1 a 4.9, p < 0,0001). Una reducción de 4 unidades se considera clínicamente relevante.
Exacerbaciones de la EPOC: En tres ensayos clínicos de un año controlados con placebo, aleatorizados, doble ciego, el tratamiento con SPIRIVA® RESPIMAT® resultó en una reducción significativa del riesgo de exacerbación de la EPOC en comparación con el placebo. La exacerbación de la EPOC se define como un complejo de al menos dos acontecimientos/síntomas respiratorios con una duración de 3 días o más que requieran un cambio en el tratamiento (prescripción de antibióticos y/o corticosteroides sistémicos y/o un cambio significativo en la medicación respiratoria prescrita).
El tratamiento con SPIRIVA® RESPIMAT® resultó en una reducción del riesgo de hospitalización por exacerbación de la EPOC (que es significativa en el ensayo clínico suficientemente extenso sobre exacerbación).
En la Tabla 1 se muestra el análisis de datos recopilados en 2 ensayos clínicos fase III y un análisis separado del ensayo adicional de exacerbación. Se permitió el tratamiento simultáneo con otros medicamentos respiratorios excepto anticolinérgicos y beta-agonistas de larga acción, es decir, beta-agonistas de acción rápida, corticosteroides inhalados y xantinas. Los beta-agonistas de larga acción estaban permitidos adicionalmente en el ensayo de exacerbación.
Tabla 1: Análisis estadístico de exacerbaciones de la EPOC y hospitalizaciones por exacerbaciones de la EPOC en pacientes con EPOC moderada a muy severa.
|
Estudio (N SPIRIVA®, N placebo) |
Criterio de valoración |
SPIRIVA® RESPIMAT® |
Placebo |
% Reducción del riesgo (95% IC)a |
Valor p |
|
Análisis recopilado de los estudios fase III de 1 añod (670, 653) |
Días hasta la primera exacerbación de la EPOC |
160a |
86a |
29 (16 a 40)b |
< 0,0001b |
|
Tasa de incidencia promedio de exacerbaciones por paciente por año |
0.78c |
1,00c |
22 (8 a 33)c |
0,002c |
|
|
Tiempo hasta la primera hospitalización por exacerbación de la EPOC |
25 (-16 a 51)b |
0,20b |
|||
|
Tasa de incidencia promedio de hospitalización por exacerbación de EPOC |
0.09c |
0,11c |
20 (-4 a 38)c |
0,096c |
|
|
Estudio de 1 año fase IIIb de exacerbación (1939, 1953) |
Días hasta la primera exacerbación de la EPOC |
169a |
119a |
31 (23 a 37)b |
< 0,0001b |
|
Tasa de incidencia promedio de exacerbaciones por paciente por año |
0.69c |
0,87c |
21 (13 a 28)c |
< 0,0001c |
|
|
Tiempo hasta la primera hospitalización por exacerbación de la EPOC |
27 (10 a 41)b |
0,003b |
|||
|
Tasa de incidencia promedio de hospitalización por exacerbación de EPOC |
0.12c |
0,15c |
19 (7 a 30)c |
0,004c |
a Tiempo hasta el primer acontecimiento: días de tratamiento hasta que el 25% de los pacientes tuvo al menos una exacerbación de la EPOC/hospitalización por exacerbación de la EPOC. En el estudio A el 25% de los pacientes tratados con placebo tuvo una exacerbación el día 112, mientras que con SPIRIVA® RESPIMAT® el 25% tuvo una exacerbación el día 173, p=0,09; en el estudio B, el 25% de los pacientes tratados con placebo tuvo una exacerbación el día 74, mientras que con SPIRIVA® RESPIMAT® el 25% tuvo una exacerbación el día 149 (p < 0,0001).
b La razón de riesgo se estimó según el modelo de riesgo proporcional de Cox. El porcentaje de reducción de riesgo es 100(1–razón de riesgo).
c Regresión de Poisson: Reducción del riesgo es 100(1–razón de tasas).
d La recopilación se especificó cuando se diseñaron los ensayos. Los criterios de valoración de exacerbación mejoraban significativamente en el análisis individual de los dos estudios de 1 año.
Estudio a largo plazo de tiotropio con control activo: Se ha llevado a cabo un estudio aleatorizado, doble ciego, con control activo, a largo plazo y a gran escala, con un período de seguimiento de hasta 3 años para comparar la eficacia y seguridad de SPIRIVA® RESPIMAT® y SPIRIVA® HandiHaler (5,711 pacientes recibieron SPIRIVA® RESPIMAT®; 5,694 pacientes recibieron SPIRIVA® HandiHaler). Las variables principales fueron el tiempo hasta la primera exacerbación de la EPOC, el tiempo hasta mortalidad por cualquier causa y en un subestudio (906 pacientes) el FEV1 valle (predosis).
El tiempo hasta la primera exacerbación de la EPOC fue numéricamente similar durante el estudio de SPIRIVA® RESPIMAT® y SPIRIVA® HandiHaler (cociente de riesgos (SPIRIVA® RESPIMAT® /SPIRIVA® HandiHaler) 0.98 con un IC 95% de 0.93 a 1.03). El número medio de días hasta la primera exacerbación de EPOC fue de 756 días para SPIRIVA® RESPIMAT® y de 719 días para SPIRIVA® HandiHaler.
El efecto broncodilatador de SPIRIVA® RESPIMAT® se mantuvo durante las 120 semanas, y fue similar a SPIRIVA® HandiHaler. La diferencia promedio en el FEV1 valle de SPIRIVA® RESPIMAT® versus SPIRIVA® HandiHaler fue -0.010 L (IC 95%: -0.038-0.018 mL).
En el estudio poscomercialización TIOSPIR comparando SPIRIVA® RESPIMAT® y SPIRIVA® HandiHaler, la mortalidad por cualquier causa (incluyendo el seguimiento del estado vital) fue similar con un coeficiente de riesgos (SPIRIVA® RESPIMAT®/SPIRIVA® HandiHaler) = 0.96 con IC 95% de 0.84 a 1.09). La exposición al tratamiento fue de 13,135 y 13,050 paciente-año.
En los estudios previos controlados con placebo con seguimiento del estado vital hasta el final del período de intención de tratamiento, SPIRIVA® RESPIMAT® mostró un incremento numérico en todas las causas de mortalidad en comparación con placebo (razón de tasas (intervalo de confianza 95%) de 1.33 (0.93, 1.92) con una exposición al tratamiento de SPIRIVA® RESPIMAT® de 2.574 pacientes-año; el exceso de mortalidad se observó en pacientes con alteraciones conocidas del ritmo cardíaco. SPIRIVA®.
HandiHaler en estudios previos mostró una reducción del riesgo de muerte del 13% (coeficiente de riesgos incluyendo seguimiento del estado vital [tiotropio/placebo) = 0,87; IC 95%, 0,76 a 0,99]). La exposición al tratamiento de SPIRIVA® HandiHaler fue de 10,927 pacientes-año. No se observó un exceso del riesgo de mortalidad en el subgrupo de pacientes con alteraciones conocidas del ritmo cardíaco en el estudio controlado con placebo de SPIRIVA® HandiHaler así como en el comparativo de SPIRIVA® RESPIMAT® frente SPIRIVA® HandiHaler, TIOSPIR.
Eficacia clínica y seguridad en asma: El programa clínico de fase III para el asma persistente en adultos incluyó dos estudios de 1 año de duración, aleatorizados, doble ciego y controlados con placebo en un total de 907 pacientes con asma (453 recibieron SPIRIVA® RESPIMAT®) tratados con una combinación de ICS (≥ 800 mg budesonida/día o equivalente) y LABA. Los estudios incluyeron como principales criterios de valoración mediciones de la función pulmonar y de las exacerbaciones graves.
Estudios PrimoTinA-asma: En los dos estudios de 1 año en pacientes sintomáticos en tratamiento de mantenimiento con al menos ICS (≥ 800 mg budesonida/día o equivalente) más LABA, SPIRIVA® RESPIMAT® mostró una mejoría clínicamente relevante de la función pulmonar frente al placebo cuando se administró como terapia adicional al tratamiento de base.
En la semana 24, la mejoría media en FEV1 pico y valle fue de 0.110 litros (IC 95%: 0.063 a 0.158 litros, p < 0.0001) y 0.093 litros (IC 95%: 0.050 a 0,137 litros, p < 0.0001), respectivamente. La mejora de la función pulmonar en comparación con placebo se mantuvo durante 24 horas.
En los estudios PrimoTinA-asma, el tratamiento de los pacientes sintomáticos (N=453) con ICS más LABA más tiotropio redujo el riesgo de exacerbaciones graves de asma en un 21% con respecto al tratamiento de los pacientes sintomáticos (N=454) con ICS más LABA más placebo. La reducción del riesgo en el promedio de exacerbaciones graves de asma/paciente-año fue del 20%.
Esto fue respaldado por una reducción del 31% en el riesgo de empeoramiento del asma y una reducción del riesgo del 24% en el promedio de empeoramientos del asma/paciente-año (ver Tabla 2).
Tabla 2: Exacerbaciones en pacientes sintomáticos con ICS (≥ 800 mg budesonida/día o equivalente) más LABA (estudios de PrimoTinA- asma)
|
Estudio |
Criterios de valoración |
SPIRIVA® RESPIMAT®, como tratamiento adicional a ICSa/LABA (N=453) |
Placebo, como tratamiento adicional a ICSa/LABA (N=454) |
% Reducción del riesgo (95% CI) |
Valor-p |
|
Dos estudios de un año de fase III, análisis agrupados |
Días hasta la 1a exacerbación graves de asma |
282c |
226c |
21b (0,38) |
0.0343 |
|
Promedio de exacerbaciones graves de asma/paciente-año |
0.530 |
0.663 |
20d (0,36) |
0.0458 |
|
|
Días hasta el 1º empeoramiento del asma |
315c |
181c |
31b (18,42) |
< 0.0001 |
|
|
Promedio de empeoramientos del asma/paciente-año |
2.145 |
2.835 |
24d (9,37) |
0.0031 |
a ≥ 800 μg budesonida/día o equivalente.
b Razón de riesgo, intervalo de confianza y valor-p obtenido según el modelo de riesgo proporcional de Cox con sólo el tratamiento como efecto. El porcentaje de reducción de riesgo es 100 (1–razón de riesgo).
c Tiempo hasta el primer acontecimiento: días de tratamiento hasta que el 25/50% de los pacientes tuvo al menos una exacerbación grave de asma/empeoramiento del asma.
d La razón de tasas se obtuvo mediante la regresión de Poisson con una exposición log (en años) como compensación. El porcentaje de reducción de riesgo es 100 (1–razón de riesgo).
Población pediátrica:
EPOC: La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los estudios con SPIRIVA® RESPIMAT® en todos los subgrupos de población pediátrica en EPOC (ver sección 4.2 para consultar la información sobre el uso en población pediátrica).
Asma: Todos los estudios del programa clínico de Fase III para asma persistente en pacientes pediátricos (niños y adolescentes entre 1 y 17 años) fueron aleatorizados, doble ciego y controlados con placebo. Todos los pacientes estaban en tratamiento con ICS.
Asma grave:
Adolescentes entre 12 y 17 años: En el estudio PensieTinA-asma de doce semanas de duración se incluyeron un total de 392 pacientes (130 recibieron SPIRIVA® RESPIMAT®) sintomáticos a altas dosis de ICS y un controlador o a una dosis media de ICS y dos controladores.
En pacientes adolescentes entre 12 y 17 años, una dosis alta de ICS fue definida como una dosis de > 800-1600 μg budesonida/día o equivalente; y una dosis media de ICS fue definida como una dosis de 400-800 μg budesonida/día o equivalente. Además, pacientes adolescentes entre 12 y 14 años pueden recibir una dosis de ICS > 400 μg budesonida/día o equivalente con al menos un controlador, o una dosis ≥ 200 μg budesonida/día o equivalente con al menos dos controladores.
En este estudio, SPIRIVA® RESPIMAT® demostró mejoría en la función pulmonar frente a placebo utilizado como terapia de adición al tratamiento de base; sin embargo, la diferencia entre FEV1 pico y valle no fue estadísticamente significativa.
• En la semana 12, la mejoría media en FEV1 pico y valle fue de 0.090 litros (IC 95%: 0.019 a 0.198 litros, p < 0,1039) y 0,054 litros (IC 95%: 0,061 a 0,168 litros, p < 0,3065), respectivamente.
• En la semana 12, SPIRIVA® RESPIMAT® mejoró de manera significativa el PEFR matinal y nocturno (mañana 17.4 L/min; IC 95%: 5.1 a 29.6 L/min; noche 17.6 L/min; IC 95%: 5.9 a 29.6 L/min).
Niños entre 6 y 11 años: En el estudio VivaTinA-asma de doce semanas de duración se incluyeron un total de 400 pacientes (130 recibieron SPIRIVA® RESPIMAT®) sintomáticos a altas dosis de ICS y un controlador o a una dosis media de ICS y dos controladores. Una dosis alta de ICS fue definida como una dosis de > 400 μg budesonida/día o equivalente; y una dosis media de ICS fue definida como una dosis de 200-400 μg budesonida/día o equivalente.
En este estudio, SPIRIVA® RESPIMAT® demostró mejoría en la función pulmonar frente a placebo utilizado como terapia de adición al tratamiento de base.
• En la semana 12, la mejoría media en FEV1 pico y valle fue de 0.139 litros (IC 95%: 0.075 a 0.203 litros, p < 0,0001) y 0.087 litros (IC 95%: 0.019 a 0.154 litros, p < 0,0117), respectivamente.
Asma moderado:
Adolescentes entre 12 y 17 años: En el estudio RubaTinA-asma de un año de duración se incluyeron un total de 397 pacientes (134 recibieron SPIRIVA® RESPIMAT®) sintomáticos a dosis medias de ICS (definidas como una dosis de 200-800 μg budesonida/día o equivalente para pacientes adolescentes entre 12 y 14 años o de 400-800 μg budesonida/día o equivalente para pacientes adolescentes entre 15 y 17 años), SPIRIVA® RESPIMAT® demostró mejoría en la función pulmonar frente a placebo utilizado como terapia de adición al tratamiento de base.
Niños entre 6 y 11 años: En el estudio CanoTinA-asma de un año de duración se incluyeron un total de 401 pacientes (135 recibieron SPIRIVA® RESPIMAT®) sintomáticos a dosis medias de ICS (definidas como una dosis de 200-400 μg budesonida/día o equivalente), SPIRIVA® RESPIMAT® demostró mejoría en la función pulmonar frente a placebo utilizado como terapia de adición al tratamiento de base.
Niños entre 1 y 5 años: Se ha llevado a cabo un estudio en Fase II/III aleatorizado, doble ciego, controlado con placebo, de doce semanas de duración (NinoTinA-asma) en un total de 101 niños con asma (31 recibieron SPIRIVA® RESPIMAT®) y en tratamiento de base con ICS. Se utilizó un dispositivo espaciador (AeroChamber Plus Flow Vu®) con una máscara para administrar la medicación en 98 pacientes.
El objetivo principal del estudio fue la seguridad; la evaluación de la eficacia fue exploratoria.
En la Tabla 3 se muestra el número y el porcentaje de pacientes que han notificado acontecimientos adversos con independencia de su relación. El número de acontecimientos adversos de asma notificados con SPIRIVA® RESPIMAT® fue inferior al notificado con placebo. La evaluación de la eficacia exploratoria no mostró diferencias entre SPIRIVA® RESPIMAT® y placebo.
Tabla 3: Porcentaje de pacientes con acontecimientos adversos reportados al menos en 5 pacientes en el estudio NinoTinA-asma (niños entre 1 y 5 años)
|
Placebo N (%) |
SPIRIVA® RESPIMAT® N (%) |
||
|
Número de pacientes |
34 (100.0) |
31 (100.0) |
|
|
Pacientes con algún AE |
25 (73.5) |
18 (58.1) |
|
|
Nasofaringitis |
5 (14.7) |
2 (6.5) |
|
|
Infección respiratoria del tracto superior |
1 (2.9) |
5 (16.1) |
|
|
Asma* |
10 (29.4) |
2 (6.5) |
|
|
Pirexia |
6 (17.6) |
3 (9.7) |
|
* El nivel inferior de términos MedDRA de asma se refieren tanto a asma agravada o exacerbación de asma.
La Agencia Europea de Medicamentos ha eximido de la obligación presentar los resultados de los ensayos realizados con SPIRIVA® RESPIMAT® en uno o más grupos de la población pediátrica en el tratamiento del asma. (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
Eficacia clínica y seguridad en fibrosis quística (FQ): El programa de desarrollo clínico en FQ incluyó 3 estudios multicéntricos en 959 pacientes con edades de 5 meses o más. Los pacientes menores de 5 años utilizaron un dispositivo espaciador (AeroChamber Plus®) con una máscara y fueron incluidos solamente para la evaluación de seguridad. Los dos estudios principales (un estudio de fase II de determinación de dosis y un estudio confirmatorio de fase III) compararon los efectos de la función pulmonar (porcentaje predicho FEV1 AUC0-4h y FEV1 valle) de SPIRIVA® RESPIMAT® (5 μg de tiotroprio: 469 pacientes) frente a placebo (315 pacientes) en períodos de 12 semanas, aleatorizados y doble ciego; el estudio de fase III también incluyó una extensión abierta a largo plazo de hasta 12 meses. En estos estudios se permitían todas las medicaciones respiratorias, excepto anticolinérgicos, como tratamiento concomitante, p. ej., agonistas beta de acción prolongada, mucolíticos o antibióticos.
En la Tabla 4 se muestran los efectos sobre la función pulmonar. No se han observado mejorías significativas en los síntomas y en el estado de salud (exacerbaciones por el cuestionario de síntomas sistémicos y respiratorios y calidad de vida por el cuestionario de fibrosis quística).
Tabla 4: Diferencia media ajustada de placebo para cambios absolutos en el valor basal tras 12 semanas
|
Fase II |
Fase III |
|||||
|
Todos los pacientes (N SPIRIVA®=176, N placebo=168) |
Todos los pacientes(N SPIRIVA®=293, N placebo=147) |
≤11años (N SPIRIVA®=95, N placebo=47) |
≥12 años (N SPIRIVA®=198, N placebo=100) |
|||
|
Media (95% IC) |
Valor p |
Media (95% IC) |
Valor p |
Media (95% IC) |
Media (95% IC) |
|
|
FEV1 AUC0-4h (% predicho)a Cambios absolutos |
3.39 (1.67; 5.12) |
< 0.001 |
1.64 (-0.27; 3.55) |
0.092 |
-0.63 (-4.58; 3.32) |
2.58 (0.50; 4.65) |
|
FEV1 AUC0-4h (litros) Cambios absolutos |
0,09 (0.05; 0.14) |
< 0.001 |
0.07 (0.02; 0.12) |
0.010 |
0.01 (-0.07; 0.08) |
0.10 (0.03; 0.17) |
|
FEV1 valle (% predicho)a Cambios absolutos |
2,22 (0.38; 4.06) |
0.018 |
1.40 (-0.50; 3.30) |
0.150 |
-1.24 (-5.20; -2.71) |
2.56 (0.49; 4.62) |
|
FEV1 valle (litros) Cambios absolutos |
0,06 (0.01; 0.11) |
0.028 |
0.07 (0.02; 0.12) |
0.012 |
-0.01 (-0.08; 0.06) |
0.10 (0.03; 0.17) |
a Variables de eficacia co-primarias
Todas las reacciones adversas (RA) observadas en los ensayos en FQ son efectos secundarios conocidos de tiotropio (ver 4.8). Las reacciones adversas observadas más comúnmente durante las 12 semanas del período doble ciego fueron tos (4,1%) y sequedad de boca (2,8%).
En la Tabla 5 se muestra el número y el porcentaje de pacientes que han notificado acontecimientos adversos de especial interés en fibrosis quística con independencia de su relación. Los signos y síntomas considerados manifestaciones de fibrosis quística aumentaron numéricamente con tiotropio, aunque sin significación estadística, especialmente en pacientes con 11 años o menos.
Tabla 5. Porcentaje de pacientes con acontecimientos adversos de especial interés en fibrosis quística por grupo de edad tras 12 semanas de tratamiento con independencia de su relación (recopilación de fase II y fase III)
|
≤ 11 años |
≥ 12 años |
|||
|
N placebo=96 |
N SPIRIVA®=158 |
N placebo=215 |
N SPIRIVA®=307 |
|
|
Dolor abdominal |
7.3 |
7.0 |
5.1 |
6.2 |
|
Estreñimiento |
1.0 |
1.9 |
2.3 |
2.6 |
|
Síndrome de obstrucción intestinal |
0.0 |
0.0 |
1.4 |
1.3 |
|
Infecciones del tracto respiratorio |
34.4 |
36.7 |
28.4 |
28.3 |
|
Aumento del esputo |
1.0 |
5.1 |
5.6 |
6.2 |
|
Exacerbaciones |
10.4 |
14.6 |
18.6 |
17.9 |
El síndrome de obstrucción intestinal distal y el aumento de esputo son términos preferentes MedDRA. Las infecciones del tracto respiratorio pertenecen al nivel superior de términos MedDRA. El dolor abdominal, el estreñimiento y las exacerbaciones son colecciones de términos preferidos por MedDRA.
Treinta y cuatro (10.9%) de los pacientes aleatorizados a placebo y 56 (12.0%) de los pacientes aleatorizados a SPIRIVA® RESPIMAT® experimentaron acontecimientos adversos graves.
La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los ensayos con SPIRIVA® RESPIMAT® en los subgrupos de población pediátrica menores de 1 año.
Propiedades farmacocinéticas:
a) Introducción general:
El bromuro de tiotropio es un compuesto de amonio cuaternario no quiral y escasamente soluble en agua. El bromuro de tiotropio está disponible como solución para inhalación administrada mediante el inhalador RESPIMAT®. Aproximadamente el 40% de la dosis inhalada se deposita en los pulmones, órgano diana, el resto de la dosis se deposita en el tracto gastrointestinal. Algunos de los datos farmacocinéticos que se describen a continuación se obtuvieron con dosis superiores a las recomendadas para el tratamiento.
b) Características generales del principio activo después de la administración del medicamento:
Absorción: Después de la inhalación en voluntarios jóvenes sanos, los datos de excreción urinaria sugieren que aproximadamente el 33% de la dosis inhalada alcanza la circulación sistémica. Las soluciones orales de bromuro de tiotropio tienen una biodisponibilidad absoluta del 2-3%. No es de esperar una influencia del alimento sobre la absorción de este compuesto de amonio cuaternario.
Se observaron concentraciones plasmáticas máximas de tiotropio 5-7 minutos después de la inhalación.
En el estado de equilibrio estacionario, se alcanzaron niveles plasmáticos máximos de tiotropio en pacientes con EPOC de 10.5 pg/mL y disminuyeron rápidamente según un modelo multicompartimental. Las concentraciones plasmáticas valle en el estado de equilibrio estacionario fueron de 1.60 pg/mL. La concentración plasmática máxima de tiotropio de 5.15 pg/mL en el estado de equilibrio estacionario se alcanzó 5 minutos después de la administración de la misma dosis a los pacientes con asma.
La exposición sistémica a tiotropio tras la inhalación de tiotropio a través del dispositivo RESPIMAT® fue similar a la de tiotropio inhalado a través del dispositivo HandiHaler.
Distribución: El fármaco tiene una unión a proteínas plasmáticas del 72% y un volumen de distribución de 32 L/kg. Se desconocen las concentraciones locales en el pulmón pero la forma de administración sugiere concentraciones sustancialmente superiores en este órgano. Los estudios en ratas han mostrado que de tiotropio no atraviesa la barrera hematoencefálica en un grado significativo.
Biotransformación: El grado de biotransformación es bajo. Ello se hace evidente por una excreción urinaria del 74% de la sustancia inalterada, después de una dosis intravenosa en voluntarios jóvenes sanos. El éster del bromuro de tiotropio se fragmenta de manera no enzimática al alcohol (N-metilescopina) y al ácido (ácido ditienilglicólico), que son inactivos sobre los receptores muscarínicos. Los ensayos in vitro en microsomas hepáticos humanos y hepatocitos humanos sugieren que alguna proporción adicional del fármaco (< 20% de la dosis después de la administración intravenosa) se metaboliza mediante oxidación dependiente del citocromo P450 (CYP) y posterior conjugación con glutatión, a varios metabolitos de Fase II.
Los estudios in vitro en microsomas hepáticos revelan que la vía enzimática puede ser inhibida por los inhibidores del CYP 2D6 (y 3A4), quinidina, ketoconazol y gestodeno. Así pues, el CYP 2D6 y 3A4 están implicados en la vía metabólica responsable de la eliminación de una fracción inferior de la dosis.
Incluso a concentraciones supraterapéuticas, el bromuro de tiotropio no inhibe el CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 o 3A en microsomas hepáticos humanos.
Eliminación: La semivida efectiva de tiotropio oscila entre 27-45 h después de la inhalación en voluntarios sanos y en pacientes con EPOC. La semivida efectiva fue de 34 horas en pacientes con asma. El aclaramiento total fue de 880 mL/min después de la administración de una dosis intravenosa en voluntarios jóvenes sanos. El tiotropio administrado intravenosamente se excreta inalterado principalmente en la orina (74%).
Después de la inhalación de la solución por pacientes con EPOC hasta el estado estacionario, la excreción urinaria es de 18,6% (0,93 μg) de la dosis, permaneciendo el resto del fármaco sin absorber en el intestino, eliminándose por las heces.
En pacientes con asma, el 11,9% de la dosis (0.595 μg) se excreta inalterada en la orina durante 24 horas después de la dosificación en el estado estacionario. El aclaramiento renal de tiotropio es superior al aclaramiento de creatinina, indicando la existencia de una secreción a la orina.
Después de una inhalación una vez al día por pacientes con EPOC en tratamiento crónico, se alcanzó el estado de equilibrio farmacocinético al cabo del día 7, sin que se produjera una acumulación posterior.
Linealidad/no linealidad: El tiotropio muestra una farmacocinética lineal en el intervalo terapéutico independientemente de la formulación.
c) Características en pacientes:
Pacientes geriátricos: Tal como era de esperar para todos los fármacos excretados predominantemente por vía renal, una edad más avanzada se asoció a una reducción del aclaramiento renal de tiotropio (de 347 mL/min en pacientes con EPOC < 65 años hasta 275 mL/min en pacientes con EPOC > 65 años). Esto no dio lugar a un correspondiente incremento en la AUC0-6,SS o Cmáx,SS. La exposición a tiotropio en pacientes con asma no difirió con la edad.
Pacientes con insuficiencia renal: Después de la administración por inhalación una vez al día de tiotropio hasta el estado de equilibrio en pacientes con EPOC, la insuficiencia renal leve (CLCR 50-80 mL/min) resultó en un ligero aumento del AUC0-6,SS (entre 1.8-30% más alto) y valores similares de Cmáx,SS comparado con pacientes con función renal normal (CLCR > 80 mL/min).
En los pacientes con EPOC y con una insuficiencia renal de moderada a grave (CLCR < 50 mL/min), la administración intravenosa de una única dosis resultó en el doble de la exposición total (82% más alto de la AUC0-4h y 52% más alto de Cmáx) comparado con pacientes con EPOC con función renal normal, lo que fue confirmado por las concentraciones plasmáticas después de la inhalación del polvo seco. En pacientes asmáticos con insuficiencia renal leve (CLCR 50-80 mL/min) la inhalación de tiotropio no produjo aumentos relevantes en la exposición en comparación con pacientes con función renal normal.
Pacientes con insuficiencia hepática: No se espera que la insuficiencia hepática tenga ninguna influencia importante sobre la farmacocinética de tiotropio. El tiotropio se aclara predominantemente por eliminación renal (un 74% en los voluntarios jóvenes sanos) y por una fragmentación simple no enzimática del éster a productos farmacológicamente inactivos.
Pacientes japoneses con EPOC: En ensayos comparativos cruzados, las concentraciones plasmáticas pico medias 10 minutos tras la dosificación en el estadio estacionario fueron del 20 al 70% superiores en los pacientes con EPOC japoneses que en los caucásicos tras la inhalación de tiotropio, pero no hubo signos de mayor mortalidad o riesgo cardíaco en los pacientes japoneses en comparación con los pacientes caucásicos. Hay datos farmacocinéticos insuficientes de otras etnias o razas.
Pacientes pediátricos:
Asma: La exposición máxima y total (AUC y excreción urinaria) a tiotropio es comparable entre los pacientes con asma entre 6 y 11 años, entre 12 y 17 años y ≥ 18 años. Con base en la excreción urinaria, la exposición total a tiotropio en pacientes de 1 a 5 años de edad fue de 52 a 60% menor que en otros grupos de mayor edad. Los datos de exposición total ajustados para el área de superficie corporal fueron comparables en todos los grupos de edad. SPIRIVA® RESPIMAT® fue administrado con un dispositivo espaciador y una máscara en niños entre 1 y 5 años.
EPOC: No hubo pacientes pediátricos en el programa de EPOC (ver Posología y forma de administración).
Fibrosis quística: Tras la inhalación de 5 μg de bromuro de tiotropio, los niveles plasmáticos de tiotropio en pacientes con FQ de 5 años o más, fueron de 10.1 pg/mL a los 5 minutos de la dosificación en el estado estacionario y disminuyeron rápidamente. La fracción de dosis disponible en pacientes menores de 5 años con FQ que utilizaron un dispositivo espaciador y máscara fue aproximadamente de 3 a 4 veces más baja que la observada en pacientes con FQ de 5 años o más. La exposición a tiotropio se relacionó con el peso corporal en los pacientes con FQ menores de 5 años.
d) Relación(es) farmacocinética/farmacodinamia: No existe relación directa entre la farmacocinética y la farmacodinamia.
DATOS FARMACÉUTICOS:
Lista de excipientes: cloruro de benzalconio. Edetato disódico, agua purificada, ácido clorhídrico 3.6% (para ajuste de pH).
Precauciones especiales de conservación: Consérvese bien tapado a no más de 30 °C.
SPIRIVA® RESPIMAT® debe ser desechado máximo 3 meses después de la inserción del cartucho en el dispositivo inhalador aun cuando no se haya usado todo el medicamento.
No exponer al calor, no perforar o arrojar al fuego y evitar el contacto con los ojos.
No congelar.
CONTRAINDICACIONES:
Hipersensibilidad al bromuro de tiotropio, o alguno de los exipientes incluidos en la sección 6.1 o la atropina o a sus derivados, p.ej., ipratrtopio u oxitropio.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO:
Exipientes: El cloruro de benzalconio puede provocar sibilancias y dificultades respiratorias. Pacientes con asma tienen un mayor riesgo de sufrir estos efectos adversos.
El bromuro de tiotropio, como broncodilatador de mantenimiento de administración una vez al día, no debe ser utilizado para el tratamiento inicial de los episodios agudos de broncoespasmo, o para el alivio de los síntomas agudos. En caso de ataque agudo, debe utilizarse un agonista-beta-2 de acción rápida.
SPIRIVA® RESPIMAT® no debe ser utilizado como monoterapia para el asma. Los pacientes con asma deben continuar tomando el tratamiento antiinflamatorio, es decir corticosteroides inhalados, sin cambios después de la introducción de SPIRIVA® RESPIMAT®, aun cuando sus síntomas mejoren.Después de la administración de bromuro de tiotropio, solución para inhalación, pueden aparecer reacciones de hipersensibilidad inmediata.
De acuerdo con su actividad anticolinérgica, el bromuro de tiotropio debe utilizarse con precaución en pacientes con glaucoma de ángulo estrecho, hiperplasia prostática u obstrucción del cuello de la vejiga.
Los medicamentos inhalados pueden provocar broncoespasmo inducido por la inhalación.
El tiotropio se debe utilizar con precaución en pacientes con: infarto de miocardio reciente, hace menos de 6 meses; cualquier arritmia inestable o que ponga en riesgo la vida; o arritmia cardíaca que requiera intervención o un cambio en el tratamiento farmacológico en el año previo; hospitalización debido a fallo cardíaco (New York Heart Association [NYHA] Clase III o IV) en el año previo. Estos pacientes se excluyeron de los ensayos clínicos y pueden verse afectados por el mecanismo de acción anticolinérgico.
En pacientes con insuficiencia renal de moderada a grave (aclaramiento de creatinina ≤ 50 mL/min), el bromuro de tiotropio sólo debe utilizarse si el beneficio esperado supera el riesgo potencial, ya que la concentración plasmática aumenta cuando la función renal está disminuida. No existe experiencia a largo plazo en pacientes con insuficiencia renal grave (ver Propiedades farmacocinéticas).
Debe advertirse a los pacientes que eviten la introducción del aerosol en los ojos. Se les debe indicar que ello puede provocar o empeorar un glaucoma de ángulo estrecho, dolor o molestia ocular, visión borrosa transitoria, halos visuales o imágenes coloreadas, junto con enrojecimiento ocular por congestión de la conjuntiva y edema de la córnea. Si se desarrolla alguna combinación de estos síntomas oculares, los pacientes deben interrumpir el uso de bromuro de tiotropio y consultar inmediatamente a un especialista.
La sequedad de boca, observada con el tratamiento anticolinérgico, a largo plazo puede asociarse con caries dental.
El bromuro de tiotropio no debe utilizarse con una frecuencia superior a una vez al día (ver Sobredosis).
No se recomienda utilizar SPIRIVA® RESPIMAT® en fibrosis quística (FQ). Si se utiliza SPIRIVA® RESPIMAT® en pacientes con FQ los signos y síntomas de la FQ (por ejemplo reacciones adversas graves, exacerbaciones pulmonares, infecciones del tracto respiratorio) pueden aumentar.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Fertilidad, embarazo y lactancia:
Embarazo: Los datos sobre el uso de tiotropio en mujeres embarazadas son muy limitados. Los estudios en animales no muestran efectos nocivos directos o indirectos respecto a toxicidad reproductiva a dosis clínicamente relevantes (ver sección Datos preclínicos sobre seguridad). Como medida de precaución, es preferible evitar el uso de SPIRIVA® RESPIMAT® durante el embarazo.
Lactancia: Se desconoce si el bromuro de tiotropio se excreta en la leche materna. A pesar de que los estudios en roedores muestran que el bromuro de tiotropio se excreta sólo en pequeñas cantidades en la leche materna, el uso de SPIRIVA® RESPIMAT® no se recomienda durante la lactancia. El bromuro de tiotropio es un compuesto de acción prolongada. La decisión en cuanto a continuar/suspender la lactancia o continuar/suspender el tratamiento con SPIRIVA® RESPIMAT® debe tomarse considerando el beneficio de la lactancia para el niño y el beneficio del tratamiento con SPIRIVA® RESPIMAT® para la mujer.
Fertilidad: No se dispone de datos clínicos de tiotropio sobre la fertilidad. Un estudio preclínico con tiotropio no mostró indicios de ningún efecto adverso sobre la fertilidad (ver Datos preclínicos sobre seguridad)
REACCIONES ADVERSAS:
Resumen del perfil de seguridad: Muchas de las reacciones adversas listadas pueden atribuirse a las propiedades anticolinérgicas del bromuro de tiotropio.
Resumen tabulado de reacciones adversas: Las frecuencias asignadas a las reacciones adversas listadas a continuación se basan en porcentajes de incidencia bruta de reacciones adversas al fármaco (es decir, acontecimientos atribuidos a tiotropio) observadas en el grupo de tiotropio recopiladas de 7 ensayos clínicos controlados con placebo en EPOC (3,282 pacientes) y 12 ensayos clínicos controlados con placebo en pacientes adultos y pediátricos con asma (1,930 pacientes) con períodos de tratamiento en un rango desde 4 semanas a 1 año.
Las reacciones adversas han sido ordenadas según sus frecuencias utilizando la siguiente clasificación: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Clasificación por órganos y sistemas/término preferente MedDRA |
Frecuencia EPOC |
Frecuencia asma |
|
Trastornos del metabolismo y de la nutrición |
||
|
Deshidratación |
No conocida |
No conocida |
|
Trastornos del sistema nervioso |
||
|
Mareos |
Poco frecuente |
Poco frecuente |
|
Cefalea |
Poco frecuente |
Poco frecuente |
|
Insomnio |
Rara |
Poco frecuente |
|
Trastornos oculares |
||
|
Glaucoma |
Rara |
No conocida |
|
Aumento de la presión intraocular |
Rara |
No conocida |
|
Visión borrosa |
Rara |
No conocida |
|
Trastornos cardíacos |
||
|
Fibrilación auricular |
Rara |
No conocida |
|
Palpitaciones |
Rara |
Poco frecuente |
|
Taquicardia supraventricular |
Rara |
No conocida |
|
Taquicardia |
Rara |
No conocida |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Tos |
Poco frecuente |
Poco frecuente |
|
Faringitis |
Poco frecuente |
Poco frecuente |
|
Disfonía |
Poco frecuente |
Poco frecuente |
|
Epistaxis |
Rara |
Rara |
|
Broncoespasmo |
Rara |
Poco frecuente |
|
Laringitis |
Rara |
No conocida |
|
Sinusitis |
No conocida |
No conocida |
|
Trastornos gastrointestinales |
||
|
Sequedad de boca |
Frecuente |
Poco frecuente |
|
Estreñimiento |
Poco frecuente |
Rara |
|
Candidiasis orofaríngea |
Poco frecuente |
Poco frecuente |
|
Disfagia |
Rara |
No conocida |
|
Reflujo gastroesofágico |
Rara |
No conocida |
|
Caries dental |
Rara |
No conocida |
|
Gingivitis |
Rara |
Rara |
|
Glositis |
Rara |
No conocida |
|
Estomatitis |
No conocida |
Rara |
|
Obstrucción intestinal, incluyendo íleo paralítico |
No conocida |
No conocida |
|
Náuseas |
No conocida |
No conocida |
|
Trastornos de la piel y del tejido subcutáneo, trastornos del sistema inmunológico |
||
|
Erupción |
Poco frecuente |
Poco frecuente |
|
Prurito |
Poco frecuente |
Rara |
|
Edema angioneurótico |
Rara |
Rara |
|
Urticaria |
Rara |
Rara |
|
Infección de la piel/úlcera en la piel |
Rara |
No conocida |
|
Piel seca |
Rara |
No conocida |
|
Hipersensibilidad (incluyendo reacciones inmediatas) |
No conocida |
Rara |
|
Reacción anafiláctica |
No conocida |
No conocida |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Tumefacción en articulación |
No conocida |
No conocida |
|
Trastornos renales y urinarios |
||
|
Retención urinaria |
Poco frecuente |
No conocida |
|
Disuria |
Poco frecuente |
No conocida |
|
Infección del tracto urinario |
Rara |
Rara |
Descripción de reacciones adversas seleccionadas: Las reacciones adversas observadas frecuentemente en ensayos clínicos controlados en EPOC fueron las reacciones adversas de los anticolinérgicos, tales como la sequedad de boca que ocurre en aproximadamente el 2.9% de los pacientes. En asma la incidencia de sequedad de boca fue del 0.83%.
En 7 ensayos clínicos en EPOC, la sequedad de boca provocó el abandono de los estudios en 3 de los 3.282 pacientes tratados con tiotropio (0,1 %). En los 12 ensayos clínicos en asma (1.930 pacientes) no se notificaron abandonos debidos a la sequedad de boca.
Reacciones adversas graves relacionadas con efectos anticolinérgicos incluyen glaucoma, estreñimiento, obstrucción intestinal incluyendo íleo paralítico y retención urinaria.
Población pediátrica: La base de datos de seguridad incluye 560 pacientes (296 niños entre 1 y 11 años y 264 niños entre 12 y 17 años) en 5 ensayos clínicos controlados con placebo de duración comprendida entre 12 semanas y un año. La frecuencia, tipo y gravedad de las reacciones adversas en la población pediátrica son similares a las de los pacientes adultos.
Otra población especial: Puede darse un incremento de los efectos anticolinérgicos al aumentar la edad.
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas. La aparición de mareo o visión borrosa puede influir la capacidad para conducir y utilizar maquinaria.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN: Aunque no se han llevado a cabo estudios formales de interacción con otros fármacos, el bromuro de tiotropio ha sido utilizado conjuntamente con otros medicamentos habitualmente utilizados en el tratamiento de la EPOC y asma, incluyendo broncodilatadores simpaticomiméticos, metilxantinas, corticoides orales e inhalados, antihistamínicos, mucolíticos, modificadores de leucotrienos, cromonas, tratamiento con anti-IgE sin evidencia clínica de interacciones.
El uso de agonistas β de acción prolongada o corticosteroides inhalados (LABA o ICS) no se ha visto que alteren la exposición a tiotropio.
La administración simultánea de bromuro de tiotropio con otros medicamentos conteniendo anticolinérgicos no se ha estudiado y, por lo tanto, no se recomienda.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Datos preclínicos sobre seguridad: Muchos de los efectos observados en los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y toxicidad para la reproducción podrían explicarse por las propiedades anticolinérgicas del bromuro de tiotropio. En animales normalmente se observó reducción del consumo de alimentos, inhibición del aumento de peso corporal, sequedad de boca y nariz, reducción del lagrimeo y la salivación, midriasis y aumento de la frecuencia cardíaca. Otros efectos relevantes observados en los estudios de toxicidad de dosis repetidas fueron: irritación leve del tracto respiratorio en ratas y ratones, puesta de manifiesto por aparición de rinitis y cambios en el epitelio de la cavidad nasal y la laringe, y prostatitis junto con depósitos de proteínas y litiasis de la vejiga en ratas.
En ratas jóvenes expuestas desde los 7 días tras el nacimiento hasta la madurez sexual se observaron los mismos cambios farmacológicos directos e indirectos que en los estudios de toxicidad a dosis repetidas así como rinitis. No se observó toxicidad sistémica ni efectos toxicológicos relevantes en los parámetros principales del desarrollo, ni en el desarrollo traqueal o de órganos diana.
Los efectos nocivos sobre la gravidez, desarrollo embrional/fetal, parto o desarrollo post-natal únicamente se pudieron demostrar a niveles de dosis tóxicas para las madres. El bromuro de tiotropio no fue teratogénico en ratas ni conejos. En un estudio general de reproducción y fertilidad en ratas no se observaron indicios de ningún efecto adverso en la fertilidad o la conducta de apareamiento tanto en los progenitores tratados y su descendencia a ninguna dosis.
Los cambios respiratorios (irritación) y urogenitales (prostatitis) y la toxicidad sobre la reproducción, fueron observados con la exposición local o sistémica a dosis cinco veces superiores a la dosis terapéutica. Los estudios sobre genotoxicidad y potencial carcinogénico no mostraron un peligro especial para el ser humano.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN:
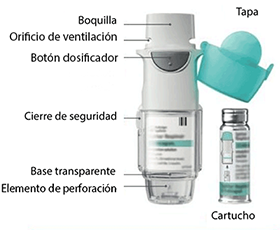
Posología: Este medicamento es únicamente para uso por vía inhalatoria. El cartucho sólo puede introducirse y utilizarse con el inhalador RESPIMAT®.
Una dosis son dos pulsaciones del inhalador RESPIMAT®.
La dosis recomendada para adultos es de 5 microgramos de tiotropio administrados en dos pulsaciones del inhalador RESPIMAT® una vez al día y a la misma hora.
No debe superarse la dosis recomendada.
En el tratamiento del asma el beneficio completo será evidente después de varias dosis del medicamento.
En pacientes adultos con asma grave, tiotropio se debe administrar con corticosteroides inhalados (≥ 800 μg budesonida/día o equivalente) y al menos un controlador.
Poblaciones especiales: Los pacientes geriátricos pueden utilizar el bromuro de tiotropio a la dosis recomendada.
Los pacientes con insuficiencia renal pueden utilizar el bromuro de tiotropio a la dosis recomendada. En pacientes con insuficiencia de moderada a grave (aclaramiento de creatinina ≤ 50 mL/min) ver Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas.
Los pacientes con insuficiencia hepática pueden utilizar el bromuro de tiotropio a la dosis recomendada (ver Propiedades farmacocinéticas).
Población pediátrica:
Asma: La dosis recomendada en pacientes entre 6 y 17 años es de 5 microgramos de tiotropio administrados en dos pulsaciones del inhalador RESPIMAT® una vez al día y a la misma hora.
En pacientes adolescentes entre 12 y 17 años con asma grave, tiotropio se debe administrar como adición a corticosteroides inhalados (> 800 μg-1600 budesonida/día o equivalente) y un controlador o como adición a corticosteroides inhalados (400-800 μg budesonida/día o equivalente) con dos controladores.
En niños entre 6 y 11 años con asma grave, tiotropio se debe administrar como adición a corticosteroides inhalados (>400 μg budesonida/día o equivalente) y un controlador o como adición a corticosteroides inhalados (200-400 μg budesonida/día o equivalente) con dos controladores.
No se ha establecido la seguridad y eficacia de SPIRIVA® RESPIMAT® en niños y adolescentes entre 6 y 17 años con asma moderado. No se ha establecido la seguridad y eficacia de SPIRIVA® RESPIMAT® en niños menores de 6 años. Los datos disponibles actualmente están descritos en las secciones 5.1 y 5.2 pero no se pueden establecer recomendaciones posológicas.
EPOC: No existe un uso relevante de SPIRIVA® RESPIMAT® en niños y adolescentes menores de 18 años.
Fibrosis quística: No se ha establecido la seguridad y eficacia de SPIRIVA® RESPIMAT® (ver secciones 4.4 y 5.1).
Forma de administración: Este medicamento es únicamente para uso por vía inhalatoria. El cartucho sólo puede introducirse y utilizarse con el inhalador RESPIMAT®. RESPIMAT® es un dispositivo inhalador que genera un aerosol para inhalación. Está diseñado para usarse en un único paciente y destinado a proporcionar varias dosis por cartucho.
Los pacientes deben leer las instrucciones de uso del inhalador RESPIMAT® antes de empezar a usar SPIRIVA® RESPIMAT®.
Para asegurar la correcta administración del medicamento, el paciente debe ser instruido por un médico u otros profesionales sanitarios en cómo usar el inhalador. Instrucciones para el manejo y uso del inhalador RESPIMAT®:
Los niños deben usar SPIRIVA® RESPIMAT® con la ayuda de un adulto.
Deberá utilizar este inhalador solo una vez al día. Cada vez que lo utilice tome dos pulsaciones.
• Si SPIRIVA® RESPIMAT® no se ha utilizado durante más de 7 días suelte una pulsación hacia el suelo.
• Si SPIRIVA® RESPIMAT® no se ha utilizado durante más de 21 días, repita los pasos del 4 al 6 de la sección Preparación para el primer uso, hasta que se vea una nube. Luego, repita los pasos del 4 al 6 tres veces más.
Cómo cuidar su SPIRIVA® RESPIMAT®:
Limpie la boquilla, incluida la parte interna metálica, solo con un paño humedo o un pañuelo de papel, al menos una vez a la semana.
Cualquier pequeña decoloración en la boquilla no afecta al desempeño de su inhalador SPIRIVA® RESPIMAT®.
Cuándo adquirir un nuevo SPIRIVA® RESPIMAT®
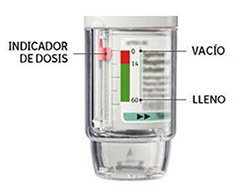
• Su inhalador SPIRIVA® RESPIMAT® contiene 60 pulsaciones (30 dosis) si se utiliza según las indicaciones (dos pulsaciones/una vez al día).
• El indicador de dosis muestra, aproximadamente, cuanto tiempo queda.
• Cuando el indicador de dosis entre en la zona roja de la escala derberá obtener una nueva receta; le queda aproximadamente medicamento para 7 días (14 pulsaciones).
• Una vez el indicador de dosis ha alcanzado el final de la zona roja, su SPIRIVA® RESPIMAT® se bloquea automáticamente: no se pueden liberar más dosis. En este punto, la base transparente ya no puede girarse más.
• Tres meses después del primer uso, el SPIRIVA® RESPIMAT® debe desecharse aunque no se haya utilizado.
Preparación para el primer uso:
1. Retirare la base transparente
• Mantenga la tapa cerrada.
• Presione el cierre de seguridad mientras tira firmemente la base transparente con su otra mano.

2. Inserte el cartucho
• Introduzca el extremo estrecho del cartucho en el inhalador.
• Coloque el inhalador sobre una superficie firme y empújelo hacia abajo con firmeza hasta que encaje en su sitio.
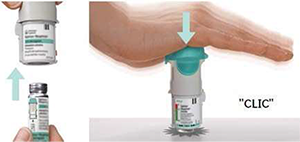
3. Vuelva a colocar la base transparente
• Vuelva a colocar la base transparente en su sitio hasta que haga clic.
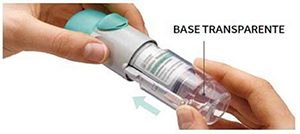
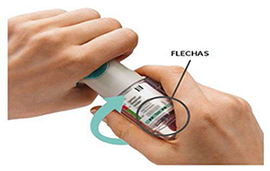
4. Girar
• Mantenga la tapa cerrada.
• Gire la base transparente en el sentido de las flechas de la etiqueta hasta que haga clic (media vuelta).
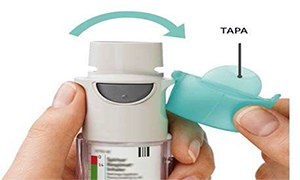
5. Abra
• Abra la tapa hasta que encaje por completo.
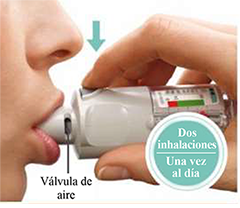
6. Presione
• Apunte el inhalador hacia el suelo.
• Presione el botón de liberación de dosis.
• Cierre la tapa.
• Repita los pasos 4 a 6 hastaque se vea una nube.
• Cuando se ve una nube, repita los pasos 4 a 6 tres veces más.

Uso diario
Girar
• Mantenga la tapa cerrada.
• Gire la base transparente en la dirección de las flechas de la etiqueta hasta que haga clic (media vuelta).
Abrir
• Abra la tapa completamente.

Presionar
• Exhale de forma lenta y profunda.
• Cierre los labios alrededor de la boquilla sin cubrir los orificios de ventilación.
• Mientras inhala lenta y profundamente por la boca, presione el botón de liberación de dosis y continúe inhalando.
• Contenga la respiración durante 10 segundos o el tiempo que le resulte cómodo.
• Repita los pasos girar, abrir y presionar hasta completar el total de 2 pulsaciones.
• Cierre la tapa hasta que vuelva a utilizar el inhalador.
SOBREDOSIS: Dosis elevadas de bromuro de tiotropio pueden provocar la aparición de signos y síntomas anticolinérgicos.
No obstante, después de la administración de una dosis única inhalada de hasta 340 microgramos de bromuro de tiotropio en voluntarios sanos, no se observaron efectos adversos anticolinérgicos sistémicos. Adicionalmente, no se han observado reacciones adversas relevantes, a parte de sequedad de boca/garganta y sequedad de la mucosa nasal, en voluntarios sanos que recibieron hasta 40 microgramos de solución de tiotropio para inhalación durante 14 días, con la excepción de una reducción pronunciada del flujo salivar a partir del séptimo día.
PRESENTACIONES:
Las presentaciones pueden variar dependiendo del país.
Caja con cartucho con 4.5 mL y dispositivo dosificador (RESPIMAT®).
Caja con cartucho con 4.5 mL y dispositivo dosificador (RESPIMAT®) muestra médica.
Hecho en Alemania por:
Boehringer Ingelheim Pharma GmbH & Co. KG.
Binger Str. 173
55216 Ingelheim am Rhein, Alemania
Para:
BOEHRINGER INGELHEIM PROMECO, S.A. DE C.V.
Calle del Maíz No. 49,
Col. Barrio Xaltocan, C.P. 16090,
Xochimilco, Ciudad de México, México
® Marca registrada
Fecha de la revisión del texto: Febrero 2023


