
TOPAMAX
TOPIRAMATO
Cápsulas
Frasco(s) , 28 Cápsulas , 15 Miligramos
Frasco(s) , 60 Cápsulas , 15 Miligramos
Frasco(s) , 28 Cápsulas , 25 Miligramos
Frasco(s) , 60 Cápsulas , 25 Miligramos
Frasco(s) , 28 Cápsulas , 50 Miligramos
Frasco(s) , 60 Cápsulas , 50 Miligramos
FORMAS FARMACÉUTICAS Y CONCENTRACIONES:
Comprimido recubierto con película.
Cápsula dispersable.
Comprimido de 25 mg: Comprimido grabado, redondo y de color blanco que contiene 25 mg de topiramato.
Comprimido de 50 mg: Comprimido grabado, redondo y de color amarillo claro que contiene 50 mg de topiramato.
Comprimido de 100 mg: Comprimido grabado, redondo y de color amarillo que contiene 100 mg de topiramato.
Comprimido de 200 mg: Comprimido grabado, redondo y de color salmón que contiene 200 mg de topiramato.
Cápsula de 15 mg: Esferas pequeñas de color blanco a blanquecino en una cápsula de gelatina que consta de un cuerpo blanco con una cubierta transparente. Cada CÁPSULA contiene 15 mg de topiramato.
Cápsula de 25 mg: Esferas pequeñas de color blanco a blanquecino en una cápsula de gelatina que consta de un cuerpo blanco con una cubierta transparente. Cada CÁPSULA contiene 25 mg de topiramato.
Cápsula de 50 mg: Esferas pequeñas de color blanco a blanquecino en una cápsula de gelatina que consta de un cuerpo blanco con una cubierta transparente. Cada CÁPSULA contiene 50 mg de topiramato.
Consulte los excipientes en la Lista de excipientes.
DATOS NO CLÍNICOS: La exposición aguda y a largo plazo al topiramato en ratones, ratas, perros y conejos fue bien tolerada. Se observó hiperplasia de las células epiteliales gástricas solamente en roedores, y en las ratas fue reversible luego de 9 semanas sin tratamiento.
Carcinogénesis y mutagénesis: Los tumores de músculo liso originados en la vejiga urinaria se observaron solamente en ratones (posologías orales de hasta 300 mg/kg durante 21 meses) y parecen afectar únicamente a esa especie. Dado que no existe un equivalente humano, no se consideraron como relevantes desde el punto de vista clínico. No hubo hallazgos de ese tipo en el estudio de carcinogénesis en ratas (posologías orales de hasta 120 mg/kg/día durante 24 meses). Otros efectos toxicológicos y patológicos del topiramato observados en estos estudios pueden estar relacionados con la inducción débil de las enzimas metabolizantes del medicamento o con la inhibición débil de la anhidrasa carbónica.
En una serie de ensayos in vitro e in vivo sobre mutagénesis, el topiramato no mostró potencial genotóxico.
Toxicología reproductiva y de desarrollo: En los estudios preclínicos, se ha demostrado que el topiramato tiene efectos teratogénicos en las especies estudiadas (ratones, ratas y conejos).En los ratones, los pesos fetales y la osificación del esqueleto se redujeron con dosis de 500 mg/kg/día en conjunción con la toxicidad materna. Los números globales de malformaciones fetales en ratones aumentaron para todos los grupos tratados con el medicamento (20, 100 y 500 mg/kg/día), pero no se observaron diferencias importantes o relaciones en respuesta a la posología para las malformaciones globales o específicas, lo que sugiere que otros factores tales como la toxicidad materna pueden estar implicados.
En las ratas, se observó una toxicidad maternal y del embrión/feto relacionada con la posología (pesos fetales y/u osificación esquelética reducidos) de tan solo 20 mg/kg/día y se observaron efectos teratogénicos (defectos en las extremidades y en los dedos) con la administración de 400 mg/kg/día o más. En los conejos, se observó una toxicidad materna relacionada con la posología con dosis de tan solo 10 mg/kg/día, una toxicidad del embrión/feto (letalidad aumentada) con la administración de 35 mg/kg/día, y efectos teratogénicos (malformaciones en las costillas y vertebrales) con 120 mg/kg/día.
Los efectos teratogénicos observados en ratas y conejos fueron similares a aquellos vistos con los inhibidores de la anhidrasa carbónica, que no se han asociado con las malformaciones en humanos. Los efectos en el crecimiento también se vieron reflejados por pesos más bajos en el nacimiento y durante la lactancia para las crías de las ratas hembras tratadas con 20 o 100 mg/kg/día durante la gestación y la lactancia. En las ratas, el topiramato atraviesa la barrera placentaria.
En las ratas jóvenes, la administración oral diaria de topiramato con dosis de hasta 300 mg/kg/día durante el período de desarrollo correspondiente a la infancia, la niñez y la adolescencia produjo toxicidades similares a las de los animales adultos (disminución del consumo de alimentos con disminución del aumento de peso, hipertrofia hepatocelular centrolobular e hiperplasia urotelial leve en la vejiga urinaria). No hubo efectos relevantes en el crecimiento de los huesos largos (tibia) o en la densidad mineral del hueso (fémur), en el desarrollo reproductivo y previo al destete, en el desarrollo neurológico (incluidas las evaluaciones sobre la memoria y el aprendizaje), ni en los parámetros de apareamiento y fertilidad o histerectomía.
Fertilidad: A pesar de una toxicidad materna y paterna con dosis de tan solo 8 mg/kg/día, no se observaron efectos sobre la fertilidad en las ratas machos o hembras con la administración de hasta 100 mg/kg/día.
INDICACIONES:
Epilepsia: TOPAMAX® está indicado como monoterapia en pacientes con diagnóstico reciente de epilepsia o para la conversión a monoterapia en pacientes con epilepsia.
TOPAMAX® está indicado como tratamiento adyuvante para adultos y niños con crisis de inicio parcial, crisis asociadas con el síndrome de Lennox-Gastaut y crisis generalizadas tónico-clónicas.
Migraña: TOPAMAX® está indicado en adultos para la profilaxis de la migraña. No se ha estudiado el beneficio de TOPAMAX® en el tratamiento agudo de la migraña.
PROPIEDADES FARMACOLÓGICAS:
El topiramato se designa químicamente como sulfamato de 2,3:4,5-bis-O-(1-metiletilideno)-ß-D-fructopiranosa. Su fórmula empírica es C12H21NO8S. y tiene un peso molecular de 339,36. Su fórmula estructural es la siguiente:
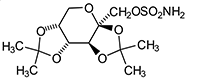
El topiramato es un polvo cristalino blanco de sabor amargo. El topiramato presenta una solubilidad máxima en las soluciones alcalinas que contienen hidróxido sódico o fosfato sódico que tienen un pH de 9 a 10. Es muy soluble en acetona, cloroformo, dimetilsulfóxido y etanol. Su solubilidad en agua es de 9,8 mg/mL. Su solución saturada presenta un pH de 6,3.
DATOS FARMACÉUTICOS:
Lista de excipientes:
— Comprimidos: Núcleo del comprimido: lactosa monohidrato, estearato de magnesio, celulosa microcristalina, almidón pregelatinizado, glicolato de almidón sódico.
• Película de recubrimiento: Cera carnauba; OPADRY® blanco, amarillo y rosa, que contiene hipromelosa, polietilenglicol, polisorbato, óxidos de hierro sintéticos (solamente el recubrimiento amarillo y el rosa) y dióxido de titanio.
— Cápsulas dispersables:
• Gránulos dispersables: Acetato de celulosa, povidona, esferas de azúcar.
• Cápsulas de gelatina: Gelatina, lauril sulfato de sodio, monolaurato de sorbitán, dióxido de titanio (para el cuerpo blanco y opaco).
Incompatibilidades: No aplicable.
Período de validez:
Comprimidos:
— Frascos y envases con blísteres: Observe la fecha de caducidad en el embalaje exterior.
Cápsulas dispersables:
— Frascos: Observe la fecha de caducidad en el embalaje exterior.
PROPIEDADES FARMACODINÁMICAS:
Grupo farmacoterapéutico: Otros antiepilépticos, código ATC: N03AX11.
El topiramato se clasifica como un monosacárido sustituido por el sulfamato. Algunos estudios electrofisiológicos y bioquímicos en neuronas cultivadas han identificado tres propiedades que pueden contribuir a la eficacia antiepiléptica del topiramato. El potencial de acción provocado en forma repetitiva por una despolarización sostenida de las neuronas se bloqueó con topiramato en un modo dependiente del tiempo; esto sugiere una acción bloqueadora del canal de sodio dependiente del estado. El topiramato aumentó la frecuencia en la que el ?-aminobutirato (GABA) activaba los receptores GABAA y mejoró la capacidad de GABA para inducir un flujo de iones de cloruro dentro de las neuronas; esto sugiere que el topiramato potencia la actividad de este neurotransmisor inhibidor. El flunamezil, un antagonista de las benzodiazepinas, no bloqueó este efecto; el topiramato tampoco aumenta la duración del tiempo de apertura del canal, lo que diferencia al topiramato de los barbitúricos que modulan a los receptores GABAA. Debido a que el perfil antiepiléptico del topiramato difiere considerablemente del de las benzodiazepinas, es posible que module un subtipo de receptor GABAA no sensible a las benzodiazepinas. El topiramato antagonizó la capacidad del kainato para activar el subtipo kainato/AMPA (a-amino-3-hidroxi-5-metilisoxazol-4-ácido propiónico) del receptor del aminoácido excitador (glutamato), pero no tuvo ningún efecto aparente en la actividad del N-metil-D-aspartato (NMDA) en el subtipo del receptor NMDA. Estos efectos del topiramato fueron dependientes de la concentración en un rango de 1 mcM a 200 mcM, con una actividad mínima observada entre 1 mcM y 10 mcM. Además, el topiramato inhibe algunas isoenzimas de la anhidrasa carbónica. Este efecto farmacológico es mucho más débil que el de la acetazolamida, un inhibidor conocido de la anhidrasa carbónica, y no se considera como un componente importante de la actividad antiepiléptica del topiramato. En los estudios con animales, el topiramato exhibe una actividad anticonvulsiva en las pruebas de crisis inducida por electrochoque máximo (MES, por sus siglas en inglés) en ratas y ratones y es efectivo en modelos de epilepsia de roedores, que incluyen crisis tónicas y casi ausentes en las ratas que son epilépticas espontáneamente (SER, por sus siglas en inglés) y crisis tónicas y clónicas inducidas en ratas mediante la inflamación de la amígdala o mediante isquemia global. El topiramato tiene una efectividad solamente débil en el bloqueo de crisis clónicas inducidas por el antagonista receptor GABAA, pentilentetrazol. Los estudios en ratones que recibían en forma concomitante la administración de topiramato y de carbamazepina o fenobarbital mostraron actividad anticonvulsiva sinérgica, mientras que la combinación con fenitoína mostró actividad anticonvulsiva aditiva. En ensayos adicionales bien controlados, no se ha demostrado que exista una correlación entre las concentraciones plasmáticas de valor mínimo del topiramato y su eficacia clínica. No se ha demostrado ninguna evidencia de tolerancia en el ser humano.
Ensayos clínicos de epilepsia: Los resultados de los ensayos clínicos controlados establecieron la eficacia de los comprimidos de TOPAMAX® y de las cápsulas dispersables de TOPAMAX® como monoterapia para adultos y niños (de 6 o más años de edad) con epilepsia, como tratamiento adyuvante en pacientes adultos y pediátricos de 2 a 16 años de edad con crisis de inicio parcial o crisis primarias tónico-clónicas generalizadas y en pacientes de 2 o más años de edad con crisis asociadas con el síndrome de Lennox-Gastaut.
Monoterapia: En 4 ensayos doble ciegos, aleatorizados y en grupos paralelos se estableció la eficacia del topiramato como monoterapia en adultos y niños de 6 o más años de edad con epilepsia de diagnóstico reciente. El estudio EPMN-106 se realizó en 487 pacientes (de 6 a 83 años de edad) con epilepsia de diagnóstico reciente (de inicio parcial o generalizada) o con un diagnóstico de epilepsia recurrente sin tratamiento con AED. Los pacientes fueron aleatorizados para recibir una dosis de topiramato de 50 mg/día o de 400 mg/día. Los pacientes permanecieron en la fase de doble ciego hasta la primera crisis de inicio parcial o tónico-clónica generalizada, hasta que finalizó la fase de doble ciego (6 meses después de la aleatorización del último sujeto) o hasta su retiro por motivos especificados en el protocolo. La evaluación de la eficacia primaria se llevó a cabo con base en una comparación entre los grupos de dosis de topiramato respecto del tiempo hasta la primera crisis de inicio parcial o tónico-clónica generalizada durante la fase de tratamiento doble ciego. La comparación de las curvas de sobrevida de Kaplan-Meier del tiempo hasta la primera crisis se decantó a favor de la dosis de topiramato de 400 mg/día, que se prefirió a la dosis de 50 mg/día (p=0,0002, prueba de rango logarítmico). La separación entre los grupos a favor del grupo de dosis más alta se produjo al principio de la fase de titulación y ya tenía importancia estadística tan solo 2 semanas después de la aleatorización (p = 0,046), cuando, al seguir el cronograma semanal de titulación, los pacientes del grupo de dosis más alta habían alcanzado una dosis máxima de topiramato de 100 mg/día. El grupo con la dosis más alta también aventajó al grupo de menor dosis en cuanto a la proporción de pacientes que no padecieron crisis, con base en las estimaciones de Kaplan-Meier, durante un mínimo de 6 meses de tratamiento (82,9% frente a 71,4%; p=0,005), y durante un mínimo de 1 año de tratamiento (75,7% frente a 58,8%; p=0,001).El cociente de riesgos instantáneos durante el tiempo transcurrido hasta la primera crisis fue de 0,516 (intervalo de confianza del 95%, 0,364 a 0,733). Los efectos del tratamiento con respecto al tiempo transcurrido hasta la primera crisis fueron coherentes entre los diferentes subgrupos de pacientes definidos por edad, sexo, región geográfica, peso corporal basal, tipo de crisis basal, tiempo transcurrido desde el diagnóstico y administración de AED basal.
En el estudio YI, un estudio unicéntrico, se cambió el tratamiento existente de pacientes de 15 a 63 años de edad con crisis de inicio parcial resistentes al tratamiento (N=48) a una monoterapia con TOPAMAX® de 100 o 1000 mg/día. Las variables de eficacia medidas en el grupo de dosis más alta fueron estadísticamente superiores a las del grupo de dosis más baja. Un 54% de los pacientes del grupo con la dosis más alta alcanzó la monoterapia, mientras que solamente un 17% de los pacientes del grupo de dosis más baja logró lo mismo, siendo la diferencia entre las dosis importante desde el punto de vista estadístico (p = 0,005). El tiempo medio hasta el alta fue considerablemente más prolongado en el grupo de dosis más alta (p = 0,002). Las evaluaciones de respuesta clínica globales del investigador y de los pacientes mostraron un valor estadísticamente significativo a favor del grupo de dosis más alta (?0,002).
En el estudio EPMN-104, los pacientes adultos y pediátricos (con edades de 6 a 85 años) con epilepsia de diagnóstico reciente (N=252) fueron distribuidos aleatoriamente al grupo de dosis baja (25 o 50 mg/día) o al de dosis alta (200 o 500 mg/día) con base en su peso corporal. En total, se informó que un 54% de los pacientes del grupo de dosis alta y un 39% de los pacientes del grupo de menor dosis no sufrieron crisis durante la fase de tratamiento doble ciego (p = 0,022). El grupo de dosis alta también presentaba un valor superior (p = 0,008) en la distribución de la frecuencia de crisis que el grupo de dosis baja, así como también en la diferencia en el tiempo transcurrido hasta la primera crisis en los tres estratos de concentraciones plasmáticas de topiramato (p = 0,015).
En el estudio EPMN-105, se aleatorizaron pacientes cuyas edades oscilaban entre 6 y 84 años que tenían epilepsia de diagnóstico reciente (n=613) para que recibieran 100 o 200 mg/día de TOPAMAX® o un tratamiento antiepiléptico estándar (carbamazepina o valproato).
TOPAMAX® demostró por lo menos la misma eficacia que la carbamazepina o el valproato para disminuir las crisis en estos pacientes; los intervalos de confianza del 95% correspondientes a la diferencia entre los dos grupos de tratamiento fueron estrechos e incluyeron el cero, lo cual indica la inexistencia de una diferencia estadísticamente significativa entre los grupos. Los dos grupos de tratamiento también fueron comparables en lo que respecta a todos los criterios de valoración de eficacia y utilidad clínicas, incluidos el tiempo hasta el alta, la proporción de pacientes sin crisis y el tiempo transcurrido hasta la primera crisis.
Los pacientes (N=207; 32 de ellos de ?16 años) que completaron la fase de doble ciego del estudio YI y EPMN-104 fueron inscritos en estudios complementarios a largo plazo en los cuales la mayoría los pacientes recibieron TOPAMAX® durante un período de 2 a 5 años. En dichos estudios, se demostró la eficacia sostenida de la administración a largo plazo de TOPAMAX® como monoterapia. No se realizó un cambio importante de dosis durante el período complementario ni se encontraron indicios de una disminución de la eficacia de la monoterapia con TOPAMAX® con la exposición prolongada.
Tratamiento adyuvante:
— Ensayos controlados en pacientes con crisis de inicio parcial:
• Adultos con crisis de inicio parcial: La eficacia del topiramato como tratamiento adyuvante para adultos con crisis de inicio parcial se demostró en seis ensayos doble ciegos, multicéntricos, aleatorizados y controlados con placebo, en dos de los cuales se compararon varias dosis de topiramato y placebo y en cuatro se comparó una dosis única con un placebo, en pacientes con antecedentes de crisis de inicio parcial, con o sin crisis generalizadas en forma secundaria.
Los pacientes de estos estudios podían recibir dos AED como máximo, además de los comprimidos de TOPAMAX® o el placebo. En cada uno de los estudios, los pacientes se estabilizaron con posologías óptimas de sus AED concomitantes durante la fase basal, que tuvo una duración de entre 4 y 12 semanas. Los pacientes que experimentaron un número mínimo previamente especificado de crisis de inicio parcial, con o sin generalización secundaria, durante la fase basal (12 crisis durante el período basal de 12 semanas, 8 durante el período basal de 8 semanas o 3 durante el período basal de 4 semanas) fueron asignados aleatoriamente al tratamiento con un placebo o una dosis especificada de comprimidos de TOPAMAX® además de sus otros AED.
Con posterioridad a la aleatorización,los pacientes iniciaron la fase del tratamiento doble ciego. En cinco de los seis estudios, los pacientes recibieron un medicamento activo, con una dosis inicial de 100 mg al día; posteriormente la dosis se aumentó en incrementos de 100 o 200 mg/día cada semana o cada dos semanas hasta alcanzar la dosis asignada, siempre y cuando no manifestaran una intolerancia que impidiera dichos incrementos. En el sexto estudio (119), las dosis iniciales de 25 o 50 mg/día de topiramato continuaron con incrementos semanales respectivos de 25 o 50 mg/día hasta alcanzar la dosis objetivo de 200 mg/día. Después de la titulación, los pacientes pasaron por un período de estabilización de 4, 8 o 12 semanas. El número de pacientes aleatorizados para cada dosis, y las dosis medias y medianas concretas del período de estabilización se muestran en las tablas 1 y 2.
• Pacientes pediátricos de 2 a 16 años de edad con crisis de inicio parcial: La eficacia de topiramato como tratamiento adyuvante para pacientes pediátricos de 2 a 16 años de edad con crisis de inicio parcial se demostró en un ensayo doble ciego, multicéntrico, aleatorizado y controlado con placebo, en el que se compararon el topiramato y el placebo en pacientes con antecedentes de crisis de inicio parcial, con o sin crisis generalizadas en forma secundaria.
Los pacientes de este estudio podían recibir dos AED como máximo, además de los comprimidos de TOPAMAX® o el placebo. En este estudio, los pacientes se estabilizaron con dosis óptimas de sus AED concomitantes durante una fase basal de 8 semanas. Los pacientes que experimentaron por lo menos seis crisis de inicio parcial, con o sin crisis generalizadas en forma secundaria, fueron asignados aleatoriamente durante la fase basal a un tratamiento con placebo o con comprimidos de TOPAMAX® además de sus otros AED.
Con posterioridad a la aleatorización, los pacientes iniciaron la fase del tratamiento doble ciego. Los pacientes recibieron un medicamento activo con una dosis inicial de 25 o 50 mg por día; posteriormente la dosis se aumentó en incrementos de 25 mg a 150 mg/día cada dos semanas hasta alcanzar la dosis asignada de 125, 175, 225 o 400 mg/día con base en el peso del paciente, hasta una posología aproximada de 6 mg/kg por día, siempre y cuando no manifestasen una intolerancia que impidiera dichos incrementos.Después de la titulación, los pacientes pasaron por un período de estabilización de 8 semanas.
• Ensayos controlados en pacientes con crisis primarias tónicoclónicas generalizadas: La eficacia de topiramato como tratamiento adyuvante para crisis primarias tónico-clónicas generalizadas en pacientes de 2 o más años de edad se demostró en un ensayo doble ciego, multicéntrico, aleatorizado y controlado con placebo, en el que se comparó una dosis única de topiramato con el placebo.
Los pacientes de este estudio podían recibir dos AED como máximo, además de TOPAMAX® o el placebo. Los pacientes se estabilizaron con posologías óptimas de sus AED concomitantes durante una fase basal de 8 semanas. Los pacientes que experimentaron por lo menos tres crisis primarias tónico-clónicas generalizadas durante la fase basal fueron asignados aleatoriamente a un tratamiento con un placebo o con TOPAMAX® además de sus otros AED.
Con posterioridad a la aleatorización, los pacientes iniciaron la fase del tratamiento doble ciego. Los pacientes recibieron un medicamento activo con una dosis inicial de 50 mg por día durante cuatro semanas; posteriormente la dosis se aumentó en incrementos de 50 a 150 mg/día cada dos semanas, hasta alcanzar la dosis asignada de 175, 225 o 400 mg/día con base en el peso del paciente, hasta una dosis aproximada de 6 mg/kg por día, siempre y cuando no manifestasen una intolerancia que impidiera dichos incrementos. Después de la titulación, los pacientes pasaron por un período de estabilización de 12 semanas.
• Ensayo controlado en pacientes con síndrome de Lennox-Gastaut: La eficacia del topiramato como tratamiento adyuvante para crisis asociadas con el síndrome de Lennox-Gastaut se demostró en un ensayo doble ciego, multicéntrico, aleatorizado y controlado con placebo, en el que se comparó una dosis única de topiramato con un placebo en pacientes de 2 o más años de edad.
Los pacientes de este estudio podían recibir dos AED como máximo, además de TOPAMAX® o el placebo. Los pacientes que experimentaron por lo menos 60 crisis por mes antes de participar en el estudio fueron estabilizados con dosis óptimas de sus AED concomitantes durante una fase basal de cuatro semanas. Después de la fase basal, los pacientes fueron asignados aleatoriamente a un tratamiento con placebo o con TOPAMAX® además de sus otros AED. Se tituló al medicamento activo con una dosis inicial de 1 mg/kg por día durante una semana. Después, la dosis se aumentó a 3 mg/kg por día durante una semana y luego, a 6 mg/kg por día. Después de la titulación, los pacientes pasaron por un período de estabilización de 8 semanas. Las medidas de eficacia principales fueron la disminución del porcentaje de crisis de caídas atónicas y una calificación global, realizada por los padres, de la gravedad de las crisis.
En todos los estudios adicionales, se midió la disminución de la tasa de crisis con respecto a la línea basal durante la totalidad de la fase de doble ciego. En la tabla 9 que se encuentra a continuación, se muestran las disminuciones porcentuales de la mediana de la tasa de crisis y el índice de pacientes que respondieron al tratamiento (fracción de los pacientes con una disminución del 50% como mínimo) por grupo de tratamiento para cada estudio. Como se ha explicado anteriormente, en el ensayo de Lennox-Gastaut también se evaluó una reducción global en la severidad de las crisis.
|
Tabla 9: Resultados de eficacia en estudios adicionales sobre epilepsia doble ciegos y controlados con placebo |
||||||||
|
Dosis objetivo de topiramato (mg/día) |
||||||||
|
Protocolo |
Resultados de eficacia |
Placebo |
200 |
400 |
600 |
800 |
1000 |
?6 mg/kg/día* |
|
Crisis de inicio parcial |
||||||||
|
Estudios en adultos |
||||||||
|
YD N |
45 |
45 |
45 |
46 |
-- |
-- |
-- |
|
|
% de reducción de la mediana |
11,6 |
27,2a |
47,5b |
44,7c |
-- |
-- |
-- |
|
|
% de pacientes que respondieron al tratamiento |
18 |
24 |
44d |
46d |
-- |
-- |
-- |
|
|
YE N |
47 |
-- |
-- |
48 |
48 |
47 |
-- |
|
|
% de reducción de la mediana |
1,7 |
-- |
-- |
40,8c |
41,0c |
36,0c |
-- |
|
|
% de pacientes que respondieron al tratamiento |
9 |
-- |
-- |
40c |
41c |
36d |
-- |
|
|
Y1 N |
24 |
-- |
23 |
-- |
-- |
-- |
-- |
|
|
% de reducción de la mediana |
1,1 |
-- |
40,7c |
-- |
-- |
-- |
-- |
|
|
% de pacientes que respondieron al tratamiento |
8 |
-- |
35d |
-- |
-- |
-- |
-- |
|
|
Y2 N |
30 |
-- |
-- |
30 |
-- |
-- |
-- |
|
|
% de reducción de la mediana |
-12,2 |
-- |
-- |
46,4f |
-- |
-- |
-- |
|
|
% de pacientes que respondieron al tratamiento |
10 |
-- |
-- |
47c |
-- |
-- |
-- |
|
|
Y3 N |
28 |
-- |
-- |
-- |
28 |
-- |
-- |
|
|
% de reducción de la mediana |
-20,6 |
-- |
-- |
-- |
24,3c |
-- |
-- |
|
|
% de pacientes que respondieron al tratamiento |
0 |
-- |
-- |
-- |
43c |
-- |
-- |
|
|
119 N |
91 |
168 |
-- |
-- |
-- |
-- |
-- |
|
|
% de reducción de la mediana |
20,0 |
44,2c |
-- |
-- |
-- |
-- |
-- |
|
|
% de pacientes que respondieron al tratamiento |
24 |
45c |
-- |
-- |
-- |
-- |
-- |
|
|
Estudios en pacientes pediátricos |
||||||||
|
YP N |
45 |
-- |
-- |
-- |
-- |
-- |
41 |
|
|
% de reducción de la mediana |
10,5 |
-- |
-- |
-- |
-- |
-- |
33,1d |
|
|
% de pacientes que respondieron al tratamiento |
20 |
-- |
-- |
-- |
-- |
-- |
39 |
|
|
Tónica-clónica primaria generalizadah |
||||||||
|
YTC N |
40 |
-- |
-- |
-- |
-- |
-- |
39 |
|
|
% de reducción de la mediana |
9,0 |
-- |
-- |
-- |
-- |
-- |
56,7d |
|
|
% de pacientes que respondieron al tratamiento |
20 |
-- |
-- |
-- |
-- |
-- |
56c |
|
|
Síndrome de Lennox-Gastauti |
||||||||
|
YL N |
49 |
-- |
-- |
-- |
-- |
-- |
46 |
|
|
% de reducción de la mediana |
-5,1 |
-- |
-- |
-- |
-- |
-- |
14,8d |
|
|
% de pacientes que respondieron al tratamiento |
14 |
-- |
-- |
-- |
-- |
-- |
28g |
|
|
Reducción en la gravedad de las crisisj |
28 |
-- |
-- |
-- |
-- |
-- |
52d |
|
|
Comparaciones con el placebo: a p = 0,080; b p<0,010; c p<0,001; d p<0,050; e p = 0,065; f p<0,005;g p = 0,071; h El % de reducción de la mediana y el % de pacientes que respondieron al tratamiento se informan para las crisis primarias tónico-clónicas generalizadas. i El % de reducción de la mediana y el % de pacientes que respondieron al tratamiento de crisis de caídas atónicas, es decir, crisis tónicas o atónicas. j Porcentaje de pacientes que presentaron una mejora mínima, importante o muy importante con respecto a los valores basales. * Para los protocolos YP e YTC, se asignaron dosis objetivo especificadas en el protocolo (<9,3 mg/kg/día) con base en el peso del paciente hasta una dosis aproximada de 6 mg/kg por día; dichas dosis correspondieron a dosis de 125, 175, 225 y 400 mg/día. |
||||||||
El análisis de subconjuntos de la eficacia antiepiléptica de los comprimidos de TOPAMAX® en estos estudios no mostró diferencias en función del sexo, raza, edad, tasa de crisis basal o AED concomitante.
Ensayos clínicos sobre la migraña: El programa de desarrollo clínico para evaluar la eficacia de TOPAMAX® en la prevención de la migraña incluyó dos ensayos doble ciegos, multicéntricos, aleatorizados y controlados con placebo, con grupos en paralelo y fundamentales que se realizaron en América del Norte (MIGR-001 y MIGR-002). El criterio de valoración de la eficacia principal fue la reducción en la frecuencia de la migraña, medida mediante el cambio en la tasa de migraña en 4 semanas desde la fase basal hasta la fase del tratamiento doble ciego en cada grupo de tratamiento con TOPAMAX® en comparación con el placebo en la población con intención de tratar (ITT, por sus siglas en inglés).
Los resultados combinados de los dos ensayos fundamentales que evaluaron las dosis de TOPAMAX® de 50 (N=233), 100 (N=244) y 200 mg/día (N=228) descubrieron una reducción porcentual mediana en la tasa del período mensual promedio de la migraña del 35%, 51% y 49%, respectivamente, en comparación con el 21% para el grupo con placebo (N=229). Los 100 y 200 mg/día de TOPAMAX® fueron mejores desde el punto de vista estadístico que el placebo. En particular, el 27% de los pacientes a los que se les administró TOPAMAX® en dosis de 100 mg/día alcanzaron como mínimo una reducción del 75% en la frecuencia de la migraña, mientras que el 52% alcanzó como mínimo una reducción del 50%.
Un estudio adicional ilustrativo, MIGR-003, demostró que TOPAMAX® en dosis de 100 mg/día era comparable en términos de eficacia con el propranolol en dosis de 160 mg/día. No hubo una diferencia de importancia estadística entre los dos grupos en el criterio de valoración de la eficacia principal.
PROPIEDADES FARMACOCINÉTICAS: Las formulaciones del comprimido y de la cápsula dispersable son bioequivalentes.
El perfil farmacocinético del topiramato comparado con otros AED muestra una vida media plasmática prolongada, una farmacocinética lineal, una eliminación predominantemente renal, la ausencia de unión proteica importante y la falta de metabolitos activos clínicamente relevantes.
El topiramato no es un inductor potente de las enzimas metabolizantes del medicamento, puede administrarse independientemente de las comidas y no es necesario un monitoreo de rutina de las concentraciones plasmáticas del topiramato. En los estudios clínicos, no hubo una relación coherente entre las concentraciones plasmáticas y la eficacia o los eventos adversos.
Absorción: El topiramato se absorbe bien y rápidamente. Luego de la administración oral de 100 mg de topiramato en sujetos sanos, se alcanzaron concentraciones plasmáticas máximas medias (Cmáx) de 1,5 mcg/mL en el plazo de 2 a 3 horas (Tmáx). Con base en la recuperación de la radiactividad de la orina, el alcance medio de absorción de una dosis oral de 100 mg de 14C-topiramato fue como mínimo del 81%. No hubo ningún efecto clínicamente importante de la comida en la biodisponibilidad del topiramato.
Distribución: En general, entre el 13 y el 17% del topiramato se une a la proteína plasmática. Se ha observado un lugar de unión de baja capacidad para el topiramato en/sobre los eritrocitos que es saturable por encima de las concentraciones plasmáticas de 4 mcg/mL. El volumen de distribución varió de una manera inversamente proporcional a la dosis. El volumen de distribución medio aparente fue de 0,80 a 0,55 L/kg para un rango de dosis única de 100 a 1200 mg. Se detectó un efecto del sexo sobre el volumen de distribución: valores para las mujeres fueron de aproximadamente el 50% de los valores para los hombres. Esto se atribuyó al porcentaje superior de grasa corporal en las pacientes de sexo femenino y no tiene importancia clínica.
Metabolismo: El topiramato no se metaboliza en forma extensiva(?20%) en voluntarios sanos Se metaboliza hasta el 50% en pacientes que reciben un tratamiento antiepiléptico concomitante con inductores conocidos de enzimas metabolizantes del medicamento. Se han aislado, caracterizado e identificado a partir del plasma, la orina y las heces de humanos, seis metabolitos formados mediante la hidroxilación, hidrólisis y glucuronidación. Cada metabolito representa menos del 3% de la radiactividad total excretada luego de la administración de14C-topiramato. Se evaluó a dos metabolitos, que retuvieron la mayoría de la estructura del topiramato, y se descubrió que presentan poca o ninguna actividad anticonvulsiva.
Eliminación: En los seres humanos, la vía de eliminación principal del topiramato no modificado y de sus metabolitos es a través de los riñones (al menos el 81% de la dosis). Se excretó aproximadamente el 66% de una dosis de 14C-topiramato no modificado en la orina en el plazo de cuatro días. Luego de una dosificación dos veces al día de 50 y 100 mg de topiramato, la eliminación media renal fue de aproximadamente 18 mL/min. y de 17 mL/min., respectivamente. Existe evidencia de reabsorción tubular renal del topiramato. Esto se encuentra respaldado por estudios en ratas, en los cuales se administró topiramato con probenecida, y se observó un aumento importante en la eliminación renal del topiramato. En general, la eliminación plasmática es de aproximadamente 20 a 30 mL/min. en los seres humanos luego de la administración oral.
El topiramato exhibe una variabilidad reducida entre pacientes en las concentraciones plasmáticas y, por lo tanto, tiene una farmacocinética predecible. La farmacocinética del topiramato es lineal, su eliminación plasmática permanece constante y el área bajo la curva de las concentraciones plasmáticas aumenta de una forma proporcional a la dosis en un rango de dosis oral única de 100 a 400 mg en sujetos sanos. En los pacientes con función renal normal, el alcance de concentraciones plasmáticas en estado de equilibrio puede llevar de 4 a 8 días. La Cmáx media luego de dosis orales múltiples de 100 mg dos veces al día en sujetos sanos fue de 6,76 mcg/mL. Luego de la administración de dosis múltiples de 50 y 100 mg de topiramato dos veces al día, la vida media de eliminación plasmática fue de aproximadamente 21 horas.
Consumo con otros AED: La administración concomitante de dosis múltiples de topiramato en dosis de 100 a 400 mg dos veces al día, con fenitoína o carbamazepina, muestra aumentos proporcionales a la dosis en las concentraciones plasmáticas del topiramato.
Poblaciones especiales:
— Población pediátrica (hasta 12 años de edad): La farmacocinética del topiramato en niños, así como también en adultos que reciben un tratamiento adicional, es lineal, y tiene una eliminación independiente de la dosis y concentraciones plasmáticas en estado de equilibrio que aumentan en proporción a la dosis. Sin embargo, los niños tienen una eliminación superior y una vida media de eliminación más corta.En consecuencia, las concentraciones plasmáticas del topiramato para la misma dosis mg/kg puede ser inferior en los niños en comparación con los adultos. Al igual que en los adultos, la enzima hepática que induce los AED disminuye las concentraciones plasmáticas en estado de equilibrio.
— Ancianos: La eliminación plasmática del topiramato no presenta modificaciones en los pacientes ancianos en ausencia de enfermedad renal subyacente.
— Insuficiencia renal: La eliminación plasmática y renal del topiramato disminuyó en los pacientes con insuficiencia moderada y severa de la función renal (CLCR<70 mL/min.). Como resultado, se esperan concentraciones plasmáticas en estado de equilibrio del topiramato superiores para una dosis dada en pacientes con insuficiencia renal, en comparación con aquellos con función renal normal. Además, los pacientes con insuficiencia renal necesitan un tiempo más prolongado para alcanzar un estado de equilibrio con cada dosis. En pacientes con insuficiencia renal moderada y severa, se recomienda la mitad de la dosis inicial habitual y de mantenimiento (consulte Posología y forma de administración: Poblaciones especiales, Insuficiencia renal).
El topiramato se elimina del plasma en forma efectiva mediante hemodiálisis. Un período prolongado de hemodiálisis puede causar que la concentración de topiramato descienda por debajo de los niveles que se necesitan para mantener un efecto anticonvulsivo. Para evitar descensos rápidos en la concentración plasmática del topiramato durante la hemodiálisis, se puede requerir una dosis complementaria de topiramato. El ajuste real debe tener en cuenta 1) la duración del período de diálisis, 2) la tasa de eliminación del sistema de diálisis que se está utilizando, y 3) la eliminación renal efectiva del topiramato en el paciente que está siendo dializado.
— Insuficiencia hepática: La eliminación plasmática del topiramato disminuyó en una media del 26% en pacientes con insuficiencia hepática de moderada a severa. Por lo tanto, el topiramato debe utilizarse con precaución en pacientes con insuficiencia hepática.
CONTRAINDICACIONES: Hipersensibilidad a cualquier componente de este producto.
ADVERTENCIAS Y PRECAUCIONES:
Interrupción de TOPAMAX®: En pacientes con o sin antecedentes de crisis o epilepsia, los AED, incluido el TOPAMAX®, deben retirarse gradualmente para minimizar las posibilidades de crisis o el aumento en la frecuencia de las crisis. En los ensayos clínicos, se disminuyeron las posologías diarias en intervalos semanales de 50 a 100 mg en adultos con epilepsia y de 25 a 50 mg en adultos que recibían TOPAMAX® en dosis de hasta 100 mg/día para la profilaxis de la migraña. En los ensayos clínicos en niños, se retiró el TOPAMAX® gradualmente en un período de 2 a 8 semanas. En las situaciones donde el TOPAMAX® sea necesario desde el punto de vista médico, se recomienda un monitoreo apropiado.
Insuficiencia renal: La vía de eliminación principal del topiramato no modificado y de sus metabolitos se realiza a través de los riñones. La eliminación renal depende de la función renal y es independiente de la edad. Los pacientes con insuficiencia renal moderada o severa pueden demorar de 10 a 15 días en alcanzar concentraciones plasmáticas en estado de equilibrio en comparación con los 4 a 8 días en pacientes con función renal normal.
Al igual que con todos los pacientes, el cronograma de titulación debe guiarse según el resultado clínico (es decir, control de crisis, prevención de efectos secundarios) con el conocimiento de que en los pacientes con insuficiencia renal conocida puede ser necesario un tiempo más prolongado para alcanzar un estado de equilibrio en cada dosis (consulte Posología y forma de administración: Poblaciones especiales, Insuficiencia renal y Propiedades farmacocinéticas: Poblaciones especiales, Deterioro renal).
Hidratación: Se han informado oligohidrosis (sudoración disminuida) y anhidrosis en relación con el uso de topiramato. La sudoración disminuida y la hipertermia (aumento de la temperatura corporal) pueden ocurrir especialmente en niños pequeños expuestos a temperaturas ambientales elevadas (consulte Reacciones adversas).
Es muy importante una hidratación adecuada mientras se consume topiramato. La hidratación puede reducir el riesgo de nefrolitiasis (consulte Advertencias y precauciones: Nefrolitiasis). Una hidratación apropiada antes y durante las actividades tales como el ejercicio o la exposición a temperaturas cálidas puede reducir el riesgo de eventos adversos relacionados con el calor (consulte Reacciones adversas).
Trastornos en el estado de ánimo/Depresión: Se ha observado un aumento en la incidencia de trastornos en el estado de ánimo y de depresión durante el tratamiento con topiramato.
Suicido/Ideación suicida: Los AED, incluido el TOPAMAX®,aumentan el riesgo de pensamientos o comportamientos suicidas en los pacientes que consumen estos medicamentos debido a cualquier indicación. Un metanálisis de ensayos aleatorizados controlados con placebo de AED ha demostrado un aumento en el riesgo de ideación y comportamiento suicida (0,43% con los AED frente a 0,24% con placebo). Se desconoce el mecanismo de este riesgo.
En los ensayos clínicos doble ciego, los eventos relacionados con el suicido (ideación suicida, intentos suicidas y suicido) ocurrieron con una frecuencia del 0,5% en los pacientes tratados con topiramato (46 de 8652 pacientes tratados) en comparación con el 0,2% en los tratados con placebo (8 de 4045 pacientes tratados). Se informó un suicido cometido en un ensayo doble ciego de trastorno bipolar en un paciente que consumía topiramato.
Por lo tanto, los pacientes deben ser monitoreados en busca de signos de ideación y comportamiento suicida y se debe considerar un tratamiento apropiado. Se debe aconsejar a los pacientes (y, cuando sea necesario, a quienes están a cargo del cuidado de los pacientes) de que busquen asistencia médica inmediata si surgen signos de ideación o comportamiento suicida.
Nefrolitiasis: Algunos pacientes, en especial aquellos con una predisposición a la nefrolitiasis, pueden presentar mayor riesgo de formación de cálculos renales y de signos y síntomas asociados, tales como cólicos renales, dolor renal o dolor lateral.
Los factores de riesgo para la nefrolitiasis incluyen formación anterior de cálculos,antecedentes familiares de nefrolitiasis e hipercalcinuria. Ninguno de estos factores de riesgo pueden predecir con seguridad la formación de cálculos durante el tratamiento con topiramato. Además, los pacientes que consumen otros medicamentos asociados con la nefrolitiasis pueden presentar un mayor riesgo (consulte Interacciones: Otras formas de interacción, Agentes que predisponen a la nefrolitiasis).
Insuficiencia hepática: En pacientes con insuficiencia hepática, el topiramato debe administrarse con precaución debido a que puede disminuir la eliminación de topiramato (consulte Posología y forma de administración: Poblaciones especiales, Insuficiencia hepática y Propiedades farmacocinéticas: Poblaciones especiales, Insuficiencia hepática).
Miopía aguda y glaucoma secundario de ángulo cerrado: Se ha informado un síndrome que consiste en miopía aguda asociada con glaucoma secundario de ángulo cerrado en los pacientes que reciben TOPAMAX®. Los síntomas incluyen la aparición aguda de disminución de la agudeza visual y/o dolor ocular. Los hallazgos oftalmológicos pueden incluir miopía, aplanamiento de la cámara anterior, hiperemia ocular (enrojecimiento) y presión intraocular aumentada. Puede aparecer o no midriasis. Este síndrome puede estar asociado con efusión supraciliar que tiene como consecuencia el desplazamiento anterior de las lentes y del iris, con glaucoma secundario de ángulo cerrado. Los síntomas habitualmente ocurren en el plazo de un mes desde el inicio del tratamiento con TOPAMAX®. A diferencia del glaucoma primario de ángulo estrecho, que es raro en las personas de menos de 40 años de edad, se ha informado glaucoma secundario de ángulo cerrado asociado con el topiramato tanto en pacientes pediátricos como en adultos. El tratamiento incluye la interrupción de TOPAMAX®, lo más rápido posible a criterio del médico tratante, y las medidas apropiadas para reducir la presión intraocular. En general, estas medidas producen una disminución de la presión intraocular.
La presión intraocular elevada de cualquier etiología, si no se trata, puede llevar a secuelas serias, incluida la pérdida permanente de visión.
Defectos en el campo visual: Se han informado defectos en el campo visual que no dependen de la presión intraocular elevada en pacientes que reciben topiramato. En los ensayos clínicos, la mayoría de estos eventos fueron reversibles luego de la interrupción del tratamiento con topiramato. Si ocurren problemas visuales en cualquier momento durante el tratamiento con topiramato, se debe considerar la interrupción de la administración del medicamento.
Acidosis metabólica: La acidosis metabólica hiperclorémica sin diferencia aniónica (es decir, la disminución del bicarbonato sérico por debajo del rango normal de referencia en ausencia de alcalosis respiratoria) se asocia con el tratamiento con topiramato. Esta disminución del bicarbonato sérico se debe al efecto inhibitorio del topiramato en la anhidrasa carbónica renal. En general, la disminución del bicarbonato ocurre al comienzo del tratamiento aunque puede producirse en cualquier momento durante el tratamiento. Estas disminuciones son habitualmente de leves a moderadas (disminución promedio de 4 mmol/L en dosis de 100 mg/día o superiores en adultos y de aproximadamente 6 mg/kg/día en pacientes pediátricos). Los pacientes han presentado disminuciones a valores por debajo de los 10 mmol/L con muy poca frecuencia. Las afecciones o tratamientos que predisponen a la acidosis (tales como enfermedad renal, trastornos respiratorios severos, estado epiléptico, diarrea, cirugía, dieta cetogénica o ciertos medicamentos) pueden incrementar los niveles de bicarbonato, de modo que disminuyen los efectos del topiramato.
La acidosis metabólica crónica en pacientes pediátricos puede reducir las tasas de crecimiento. No se ha investigado en forma sistemática el efecto del topiramato en el crecimiento ni las secuelas relacionadas con los huesos en poblaciones pediátricas o adultas.
Dependiendo de las afecciones subyacentes, se recomienda una evaluación apropiada que incluya los niveles de bicarbonato sérico con el tratamiento con topiramato. Si la acidosis metabólica se desarrolla y persiste, se debe considerar una reducción de la dosis o la interrupción del tratamiento con topiramato (mediante la reducción gradual de la dosis).
Complementos nutricionales: Se puede considerar la administración de un suplemento dietario o un aumento de la ingesta alimenticia si el paciente pierde peso mientras toma este medicamento.
EMBARAZO Y LACTANCIA:
Embarazo: Los estudios en animales han demostrado toxicidad reproductiva (consulte Datos no clínicos: Toxicología reproductiva y del desarrollo). En las ratas, el topiramato atraviesa la barrera placentaria.
No hay estudios adecuados y bien controlados de la administración de TOPAMAX® en mujeres embarazadas.
TOPAMAX® puede causar daño fetal cuando se administra a mujeres embarazadas. Los datos de los registros de embarazos indican que los lactantes expuestos al topiramato in utero tienen mayor riesgo de padecer malformaciones congénitas (p. ej., anomalías craneofaciales, tales como paladar/labio leporino, hipospadias y anomalías que implican varias partes del sistema corporal).
Esto se ha informado con la monoterapia de topiramato y con topiramato como parte de un régimen de politerapia.
Además, los datos de otros estudios indican que, en comparación con la monoterapia, hay un incremento en el riesgo de efectos teratogénicos asociados con el consumo de AED en tratamientos combinados.
En comparación con un grupo de referencia al que no se le administraron AED, los datos de los registros de la monoterapia con TOPAMAX® arrojaron una prevalencia más elevada de peso bajo al nacer (<2500 gramos). Un registro de embarazo informó un incremento en la frecuencia de neonatos que fueron pequeños para la edad gestacional (SGA, definido por el peso al nacer por debajo del percentil 10 corregido por su edad gestacional, estratificado por sexo) entre aquellos expuestos a la monoterapia de topiramato in utero. No se pudieron determinar las consecuencias a largo plazo de los hallazgos de SGA. No se estableció una relación causal para el bajo peso al nacer y el SGA.
TOPAMAX® debe administrarse durante el embarazo, solamente si el beneficio potencial justifica el riesgo potencial para el feto. Al tratar y aconsejar mujeres en edad fértil, el médico que prescribe debe ponderar los beneficios del tratamiento contra los riesgos y considerar las opciones alternativas de tratamiento. Si se administra este medicamento durante el embarazo o si la paciente queda embarazada durante el tratamiento con este, se le debe informar sobre el daño potencial al feto.
Lactancia: El topiramato se excreta en la leche de ratas lactantes. La excreción de topiramato en la leche humana no se ha evaluado en los estudios controlados. Las observaciones limitadas en pacientes sugieren una excreción amplia de topiramato en la leche materna. Dado que muchos medicamentos se excretan a través de la leche humana, se debe tomar una decisión de interrumpir la lactancia o interrumpir el medicamento, teniendo en cuenta la importancia del medicamento para la madre.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: TOPAMAX® actúa sobre el sistema nervioso central y puede producir somnolencia, mareos u otros síntomas relacionados. También puede causar alteraciones visuales y/o visión borrosa. Estos eventos adversos podrían ser potencialmente peligrosos en pacientes que conducen un vehículo u operan máquinas, en particular hasta el momento en que se establece la experiencia individual del paciente con el medicamento.
REACCIONES ADVERSAS: En esta sección, se presentan las reacciones adversas. Las reacciones adversas son eventos adversos que se consideran razonablemente asociados con el consumo de topiramato con base en la evaluación completa de la información disponible sobre eventos adversos. No se puede establecer con seguridad una relación causal con el topiramato en casos individuales. Además, dado que los ensayos clínicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un medicamento no pueden compararse directamente con las tasas de los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica clínica.
Datos de ensayos clínicos: Se evaluó la seguridad de TOPAMAX® a partir de una base de datos de ensayos clínicos que consta de 4111 pacientes (3182 tratados con TOPAMAX® y 929 con placebo) que participaron en 20 ensayos doble ciegos y 2847 pacientes que participaron en 34 ensayos de etiqueta abierta, respectivamente, para el tratamiento de crisis generalizadas tónico-clónicas primarias, crisis de inicio parcial, crisis asociadas con el síndrome de Lennox-Gastaut, con diagnóstico reciente de epilepsia o migraña. La información proporcionada en esta sección se ha obtenido de los datos combinados.
La mayoría de las reacciones adversas fueron de intensidad leve a moderada.
Ensayos sobre epilepsia doble ciegos, de datos controlados con placebo y adyuvantes – Pacientes adultos: En la Tabla 1, se muestran las reacciones adversas informadas en ?1% de los pacientes adultos tratados con TOPAMAX® en ensayos sobre epilepsia doble ciegos, controlados con placebo y adyuvantes. Las reacciones adversas que tuvieron una incidencia >5% en el rango de dosis recomendada (200 a 400 mg/día) en adultos en estudios sobre epilepsia doble ciegos, controlados con placebo y adyuvantes incluyeron somnolencia, mareos, fatiga, irritabilidad, pérdida de peso, bradifrenia, parestesia, diplopía, coordinación anormal, náuseas, nistgamo, letargo, anorexia, disartria, visión borrosa, reducción del apetito, deterioro de la memoria y diarrea.
|
Tabla 1: Reacciones adversas informadas por ≥1% de los pacientes adultos tratados con TOPAMAX® en ensayos sobre epilepsia doble ciegos, controlados con placebo y adyuvantes |
|||
|
TOPAMAX® 200-400 mg/día |
TOPAMAX® 600-1000 mg/día |
PLACEBO |
|
|
Clase de órgano o sistema Reacción adversa |
(N=354) % |
(N=437) % |
(N=382) % |
|
Trastornos metabólicos y nutricionales |
|||
|
Anorexia |
5,4 |
6,2 |
1,8 |
|
Reducción del apetito |
5,1 |
8,7 |
3,7 |
|
Trastornos psiquiátricos |
|||
|
Bradifrenia |
8,2 |
19,5 |
3,1 |
|
Trastorno de la producción del habla |
4,5 |
9,4 |
1,6 |
|
Estado de confusión |
3,1 |
5,0 |
0,8 |
|
Depresión |
3,1 |
11,7 |
3,4 |
|
Insomnio |
3,1 |
6,4 |
4,5 |
|
Agresión |
2,8 |
3,2 |
1,8 |
|
Agitación |
1,7 |
2,3 |
1,3 |
|
Ira |
1,7 |
2,1 |
0,5 |
|
Ansiedad |
1,7 |
6,6 |
2,9 |
|
Desorientación |
1,7 |
3,2 |
1,0 |
|
Alteraciones del estado de ánimo |
1,7 |
4,6 |
1,0 |
|
Trastornos del sistema nervioso |
|||
|
Somnolencia |
17,8 |
17,4 |
8,4 |
|
Mareos |
16,4 |
34,1 |
13,6 |
|
Parestesia |
8,2 |
17,2 |
3,7 |
|
Coordinación anormal |
7,1 |
11,4 |
4,2 |
|
Nistagmo |
6,2 |
11,7 |
6,8 |
|
Letargo |
5,6 |
8,0 |
2,1 |
|
Disartria |
5,4 |
6,2 |
1,0 |
|
Deterioro de la memoria |
5,1 |
10,8 |
1,8 |
|
Trastornos de la atención |
4,5 |
11,9 |
1,8 |
|
Temblores |
4,0 |
9,4 |
5,0 |
|
Amnesia |
3,4 |
5,3 |
1,0 |
|
Trastornos del equilibrio |
3,4 |
3,9 |
2,4 |
|
Hipoestesia |
3,1 |
5,9 |
1,0 |
|
Temblor intencional |
3,1 |
4,8 |
2,9 |
|
Disgeusia |
1,4 |
4,3 |
0,8 |
|
Deterioro mental |
1,4 |
5,0 |
1,3 |
|
Alteraciones del habla |
1,1 |
2,7 |
0,5 |
|
Trastornos oculares |
|||
|
Diplopía |
7,3 |
12,1 |
5,0 |
|
Visión borrosa |
5,4 |
8,9 |
2,4 |
|
Trastorno visual |
2,0 |
1,4 |
0,3 |
|
Trastornos gastrointestinales |
|||
|
Náuseas |
6,8 |
15,1 |
8,4 |
|
Diarrea |
5,1 |
14,0 |
5,2 |
|
Dolor abdominal superior |
3,7 |
3,9 |
2,1 |
|
Estreñimiento |
3,7 |
3,2 |
1,8 |
|
Molestias gástricas |
3,1 |
3,2 |
1,3 |
|
Dispepsia |
2,3 |
3,0 |
2,1 |
|
Boca seca |
1,7 |
3,7 |
0,3 |
|
Dolor abdominal |
1,1 |
2,7 |
0,8 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|||
|
Mialgia |
2,0 |
2,5 |
1,3 |
|
Espasmos musculares |
1,7 |
2,1 |
0,8 |
|
Dolor musculoesquelético en el pecho |
1,1 |
1,8 |
0,3 |
|
Trastornos generales y alteraciones en el lugar de administración |
|||
|
Fatiga |
13,0 |
30,7 |
11,8 |
|
Irritabilidad |
9,3 |
14,6 |
3,7 |
|
Astenia |
3,4 |
3,0 |
1,8 |
|
Trastornos de la marcha |
1,4 |
2,5 |
1,3 |
|
Estudios complementarios |
|||
|
Pérdida de peso |
9,0 |
11,9 |
4,2 |
La dosis recomendada para el tratamiento adyuvante de epilepsia en adultos es de 200 a 400 mg/día.
Ensayos sobre epilepsia doble ciegos, de datos controlados con placebo y adyuvantes – Pacientes pediátricos: En la Tabla 2, se muestran las reacciones adversas informadas en ≥2% de los pacientes pediátricos tratados con TOPAMAX® (2 a 16 años de edad) en ensayos sobre epilepsia doble ciegos, controlados con placebo y adyuvantes. Las reacciones adversas que tuvieron una incidencia >5% en el rango de dosis recomendada (5 a 9 mg/kg/día) en orden descendiente de frecuencia incluyeron reducción del apetito, fatiga, somnolencia, letargo, irritabilidad, trastornos de la atención, pérdida de peso, agresión, erupción, comportamiento anormal, anorexia, trastornos del equilibrio y estreñimiento.
|
Tabla 2: Reacciones adversas informadas por ≥2% de los pacientes pediátricos tratados con TOPAMAX® en ensayos sobre epilepsia doble ciegos, controlados con placebo y adyuvantes |
||
|
TOPAMAX® |
Placebo |
|
|
Clase de órgano o sistema Reacción adversa |
(N=104) % |
(N=102) % |
|
Trastornos metabólicos y nutricionales |
||
|
Reducción del apetito |
19,2 |
12,7 |
|
Anorexia |
5,8 |
1,0 |
|
Trastornos psiquiátricos |
||
|
Agresión |
8,7 |
6,9 |
|
Comportamiento anormal |
5,8 |
3,9 |
|
Estado de confusión |
2,9 |
2,0 |
|
Alteraciones del estado de ánimo |
2,9 |
2,0 |
|
Trastornos del sistema nervioso |
||
|
Somnolencia |
15,4 |
6,9 |
|
Letargo |
13,5 |
8,8 |
|
Trastornos de la atención |
10,6 |
2,0 |
|
Trastornos del equilibrio |
5,8 |
2,0 |
|
Mareos |
4,8 |
2,9 |
|
Deterioro de la memoria |
3,8 |
1,0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Epistaxis |
4,8 |
1,0 |
|
Trastornos gastrointestinales |
||
|
Estreñimiento |
5,8 |
4,9 |
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Erupción |
6,7 |
5,9 |
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Fatiga |
16,3 |
4,9 |
|
Irritabilidad |
11,5 |
8,8 |
|
Trastornos de la marcha |
4,8 |
2,0 |
|
Estudios complementarios |
||
|
Pérdida de peso |
9,6 |
1,0 |
La dosis recomendada para el tratamiento adyuvante de epilepsia en niños (2 a 16 niños) es de 5 a 9 mg/kg/día.
Ensayos sobre epilepsia doble ciegos, de datos controlados y de monoterapia – Pacientes adultos: En la Tabla 3, se muestran las reacciones adversas informadas en ?1% de los pacientes adultos tratados con TOPAMAX® en ensayos sobre epilepsia doble ciegos, controlados y de monoterapia Las reacciones adversas que tuvieron una incidencia >5% en el rango de dosis recomendada (400 mg/día) en orden descendiente de frecuencia incluyeron parestesia, pérdida de peso, fatiga, anorexia, depresión, deterioro de la memoria, ansiedad, diarrea, astenia, disgeusia e hipoestesia.
|
Tabla 3: Reacciones adversas informadas por ≥1% de los pacientes adultos tratados con TOPAMAX® en ensayos sobre epilepsia doble ciegos, controlados y de monoterapia |
||
|
Clase de órgano o sistema Reacción adversa |
TOPAMAX® 50 mg/día (N=257) % |
TOPAMAX® 400 mg/día (N=153) % |
|
Trastornos de la sangre y del sistema linfático |
||
|
Anemia |
0,8 |
2,0 |
|
Trastornos metabólicos y nutricionales |
||
|
Anorexia |
3,5 |
12,4 |
|
Reducción del apetito |
2,3 |
2,6 |
|
Trastornos psiquiátricos |
||
|
Depresión |
4,3 |
8,5 |
|
Ansiedad |
3,9 |
6,5 |
|
Bradifrenia |
2,3 |
4,6 |
|
Trastorno de la producción del habla |
3,5 |
4,6 |
|
Ánimo decaído |
0,8 |
2,6 |
|
Alteraciones del estado de ánimo |
0,4 |
2,0 |
|
Cambios de humor |
1,6 |
2,0 |
|
Trastornos del sistema nervioso |
||
|
Parestesia |
18,7 |
40,5 |
|
Deterioro de la memoria |
1,2 |
7,2 |
|
Disgeusia |
2,3 |
5,9 |
|
Hipoestesia |
4,3 |
5,2 |
|
Trastornos del equilibrio |
1,6 |
3,3 |
|
Disartria |
1,6 |
2,6 |
|
Trastornos cognitivos |
0,4 |
2,0 |
|
Letargo |
1,2 |
2,0 |
|
Deterioro mental |
0,8 |
2,0 |
|
Deterioro de las capacidad psicomotoras |
0 |
2,0 |
|
Sedación |
0 |
1,3 |
|
Defectos en el campo visual |
0,4 |
1,3 |
|
Trastornos oculares |
||
|
Ojos secos |
0 |
1,3 |
|
Trastornos del oído y del laberinto |
||
|
Dolor de oídos |
0 |
1,3 |
|
Tinnitus |
1,6 |
1,3 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Disnea |
1,2 |
2,0 |
|
Rinorrea |
0 |
1,3 |
|
Trastornos gastrointestinales |
||
|
Diarrea |
5,4 |
6,5 |
|
Parestesia oral |
1,2 |
3,3 |
|
Boca seca |
0,4 |
2,6 |
|
Gastritis |
0,8 |
2,6 |
|
Dolor abdominal |
1,2 |
2,0 |
|
Reflujo gastroesofágico |
0,4 |
2,0 |
|
Sangrado gingival |
0 |
1,3 |
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Erupción |
0,4 |
3,9 |
|
Alopecia |
1,6 |
3,3 |
|
Prurito |
0,4 |
3,3 |
|
Hipoestesia facial |
0,4 |
2,0 |
|
Prurito generalizado |
0 |
1,3 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Espasmos musculares |
2,7 |
3,3 |
|
Artralgia |
1,9 |
2,0 |
|
Contracciones musculares |
0,4 |
1,3 |
|
Trastornos renales y urinarios |
||
|
Nefrolitiasis |
0 |
2,6 |
|
Disuria |
0,8 |
2,0 |
|
Polaquiuria |
0,8 |
2,0 |
|
Trastornos del sistema reproductivo y de las mamas |
||
|
Disfunción eréctil |
0,8 |
1,3 |
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Fatiga |
15,2 |
14,4 |
|
Astenia |
3,5 |
5,9 |
|
Irritabilidad |
3,1 |
3,3 |
|
Estudios complementarios |
||
|
Pérdida de peso |
7,0 |
17,0 |
La dosis recomendada para el tratamiento con monoterapia en adultos es de 400 mg/día.
Ensayos sobre epilepsia doble ciegos, de datos controlados y de monoterapia – Pacientes pediátricos: En la Tabla 4, se muestran las reacciones adversas informadas en ?2% de los pacientes pediátricos tratados con TOPAMAX® (10 a 16 años de edad) en ensayos sobre epilepsia doble ciegos, controlados y de monoterapia. Las reacciones adversas que tuvieron una incidencia >5% con la dosis recomendada (400 mg/día) en orden descendiente de frecuencia incluyeron pérdida de peso, parestesia, diarrea, trastornos de la atención, pirexia y alopecia.
|
Tabla 4: Reacciones adversas informadas por ≥2% de los pacientes pediátricos tratados con TOPAMAX® en ensayos sobre epilepsia doble ciegos, controlados y de monoterapia |
||
|
Clase de órgano o sistema Reacción adversa |
TOPAMAX® 50 mg/día (N=77) % |
TOPAMAX® 400 mg/día (N= 63) % |
|
Trastornos metabólicos y nutricionales |
||
|
Reducción del apetito |
1,3 |
4,8 |
|
Trastornos psiquiátricos |
||
|
Bradifrenia |
0 |
4,8 |
|
Alteraciones del estado de ánimo |
1,3 |
4,8 |
|
Depresión |
0 |
3,2 |
|
Trastornos del sistema nervioso |
||
|
Parestesia |
3,9 |
15,9 |
|
Trastornos de la atención |
3,9 |
7,9 |
|
Trastornos del oído y del laberinto |
||
|
Vértigo |
0 |
3,2 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Epistaxis |
0 |
3,2 |
|
Trastornos gastrointestinales |
||
|
Diarrea |
3,9 |
9,5 |
|
Vómitos |
3,9 |
4,8 |
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Alopecia |
0 |
6,3 |
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Pirexia |
0 |
6,3 |
|
Astenia |
0 |
4,8 |
|
Estudios complementarios |
||
|
Pérdida de peso |
7,8 |
20,6 |
|
Circunstancias sociales |
||
|
Dificultad de aprendizaje |
0 |
3,2 |
La dosis recomendada para el tratamiento con monoterapia en niños de 10 o más años de edad es de 400 mg/día.
Ensayos sobre prevención de la migraña doble ciegos y de datos controlados con placebo – Pacientes adultos: En la Tabla 5, se muestran las reacciones adversas informadas en ?1% de los pacientes adultos tratados con TOPAMAX® en ensayos sobre prevención de la migraña doble ciegos y controlados con placebo. Las reacciones adversas que tuvieron una incidencia >5% con la dosis recomendada (100 mg/día) en orden descendiente de frecuencia incluyeron parestesia, fatiga, náuseas, diarrea, pérdida de peso, disgeusia, anorexia, reducción del apetito, insomnio, hipoestesia, trastornos de la atención, ansiedad, somnolencia y trastorno de la producción del habla.
|
Tabla 5: Reacciones adversas informadas por ≥1% de los pacientes adultos tratados con TOPAMAX® en ensayos sobre prevención de la migraña doble ciegos y controlados con placebo |
||||
|
Clase de órgano o sistema Reacción adversa |
TOPAMAX® 50 mg/día (N=227) % |
TOPAMAX® 100 mg/día (N=374) % |
TOPAMAX® 200 mg/día (N=501) % |
PLACEBO (N=436) % |
|
Trastornos metabólicos y nutricionales |
||||
|
Anorexia |
3,5 |
7,5 |
7,2 |
3,0 |
|
Reducción del apetito |
5,7 |
7,0 |
6,8 |
3,0 |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
4,8 |
7,0 |
5,6 |
3,9 |
|
Ansiedad |
4,0 |
5,3 |
5,0 |
1,8 |
|
Trastorno de la producción del habla |
6,6 |
5,1 |
5,2 |
1,4 |
|
Depresión |
3,5 |
4,8 |
7,4 |
4,1 |
|
Ánimo decaído |
0,4 |
2,9 |
2,0 |
0,9 |
|
Estado de confusión |
0,4 |
1,6 |
2,0 |
1,1 |
|
Cambios de humor |
1,8 |
1,3 |
1,0 |
0,2 |
|
Labilidad afectiva |
0,4 |
1,1 |
0,2 |
0,2 |
|
Bradifrenia |
1,8 |
1,1 |
3,4 |
1,4 |
|
Trastornos del sistema nervioso |
||||
|
Parestesia |
35,7 |
50,0 |
48,5 |
5,0 |
|
Disgeusia |
15,4 |
8,0 |
12,6 |
0,9 |
|
Hipoestesia |
5,3 |
6,7 |
7,4 |
1,4 |
|
Trastornos de la atención |
2,6 |
6,4 |
9,2 |
2,3 |
|
Somnolencia |
6,2 |
5,1 |
6,8 |
3,0 |
|
Deterioro de la memoria |
4,0 |
4,5 |
6,2 |
1,6 |
|
Amnesia |
3,5 |
2,9 |
5,2 |
0,5 |
|
Temblores |
1,3 |
1,9 |
2,4 |
1,4 |
|
Trastornos del equilibrio |
0,4 |
1,3 |
0,4 |
0 |
|
Deterioro mental |
0,4 |
1,1 |
1,8 |
0,9 |
|
Trastornos oculares |
||||
|
Visión borrosa |
4,0 |
2,4 |
4,4 |
2,5 |
|
Trastornos del oído y del laberinto |
||||
|
Tinnitus |
0,4 |
1,3 |
1,6 |
0,7 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Disnea |
1,3 |
2,7 |
1,6 |
1,4 |
|
Epistaxis |
0,4 |
1,1 |
0,6 |
0,5 |
|
Trastornos gastrointestinales |
||||
|
Náuseas |
9,3 |
13,6 |
14,6 |
8,3 |
|
Diarrea |
9,3 |
11,2 |
10,0 |
4,4 |
|
Boca seca |
1,8 |
3,2 |
5,0 |
2,5 |
|
Parestesia oral |
1,3 |
2,9 |
1,6 |
0,5 |
|
Estreñimiento |
1,8 |
2,1 |
1,8 |
1,4 |
|
Distensión abdominal |
0 |
1,3 |
0,2 |
0,2 |
|
Molestias gástricas |
2,2 |
1,3 |
1,0 |
0,2 |
|
Reflujo gastroesofágico |
0,4 |
1,1 |
1,2 |
0,5 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Contracciones musculares |
1,8 |
1,3 |
1,8 |
0,7 |
|
Trastornos generales y alteraciones en el lugar de administración |
||||
|
Fatiga |
15,0 |
15,2 |
19,2 |
11,2 |
|
Astenia |
0,9 |
2,1 |
2,6 |
0,5 |
|
Irritabilidad |
3,1 |
1,9 |
2,4 |
0,9 |
|
Sed |
1,3 |
1,6 |
1,0 |
0,5 |
|
Estudios complementarios |
||||
|
Pérdida de peso |
5,3 |
9,1 |
10,8 |
1,4 |
La dosis recomendada para la profilaxis de la migraña es de 100 mg/día.
Otros datos de ensayos clínicos – Pacientes adultos: En la Tabla 6, se muestran las reacciones adversas informadas en los ensayos clínicos doble ciegos y controlados en <1% de los pacientes adultos tratados con TOPAMAX® o de cualquier tasa en ensayos clínicos de etiqueta abierta con pacientes adultos tratados con TOPAMAX®.
|
Tabla 6. Reacciones adversas informadas en los ensayos clínicos doble ciegos y controlados en <1% de los pacientes adultos tratados con TOPAMAX® o de cualquier tasa en ensayos clínicos de etiqueta abierta con pacientes adultos tratados con TOPAMAX® |
|
Trastornos de la sangre y del sistema linfático Leucopenia, linfadenopatía, trombocitopenia |
|
Trastornos del sistema inmunitario Hipersensibilidad |
|
Trastornos metabólicos y nutricionales Acidosis hiperclorémica, hipocalemia, aumento del apetito, acidosis metabólica, polidipsia. |
|
Trastornos psiquiátricos Comportamiento anormal, anorgasmia, apatía, llanto, dificultades de concentración, trastornos en el deseo sexual, disfemias, despertar temprano, humor elevado, humor eufórico, afecto aplanado, alucinaciones, alucinación auditiva, alucinación visual, hipomanía, insomnio inicial, falta de habla espontánea, disminución de la libido, indiferencia, pérdida de la libido, manía, insomnio medio, disminución de sensación orgásmica, ataque de pánico, trastorno de pánico, reacciones de pánico, paranoia, perseveración, trastorno de la lectura, inquietud, trastornos del sueño, ideación suicida, intento de suicidio, lacrimosidad, pensamientos anormales |
|
Trastornos del sistema nervioso Ageusia, acinesia, anosmia, afasia, apraxia, aura, sensación de ardor, síndrome cerebeloso, trastornos del ritmo circadiano del sueño, torpeza, crisis parcial compleja, crisis, disminución del nivel de conciencia, mareo postural, babeo, disestesia, disgrafía, diskinesia, disfasia,distonía, temblor esencial, hormigueo, crisis convulsivas de tipo gran mal, hiperestesia, hipersomnia, hipogeusia, hipocinesia, hiposmia, neuropatía periférica, parosmia, mala calidad de sueño, presíncope, tartamudeo, alteración sensorial, pérdida sensorial, estupor, síncope, falta de respuesta a los estímulos |
|
Trastornos oculares Trastorno de acomodación, percepción visual de la profundidad alterada, ambliopía, blefaroespasmo, ceguera transitoria, ceguera unilateral, glaucoma, aumento del lagrimeo, midriasis, ceguera nocturna, fotopsia, presbicia, escotoma centelleante, escotoma, agudeza visual reducida |
|
Trastornos del oído y del laberinto Sordera, sordera neurosensorial, sordera unilateral, malestar auditivo, trastornos auditivos |
|
Trastornos cardíacos Bradicardia, bradicardia sinusal, palpitaciones |
|
Trastornos vasculares Bochornos, sofocación, hipotensión ortostática, fenómeno de Raynaud |
|
Trastornos respiratorios, torácicos y mediastínicos Disfonía, disnea de esfuerzo, congestión nasal, hipersecreción sinusal de los senos paranasales |
|
Trastornos gastrointestinales Malestar abdominal, dolor abdominal bajo, sensibilidad abdominal, halitosis, malestar epigástrico, flatulencia, glosodinia, hipoestesia oral, dolor oral, pancreatitis, hipersecreción salival |
|
Trastornos de la piel y del tejido subcutáneo Anhidrosis, dermatitis alérgica, eritema, erupción macular, decoloración de la piel, olor anormal en la piel, hinchazón del rostro, urticaria, urticaria localizada |
|
Trastornos musculoesqueléticos y del tejido conjuntivo Dolor lateral, fatiga muscular, debilidad muscular, rigidez musculoesquelética |
|
Trastornos renales y urinarios Cálculo ureteral, cálculo urinario, hematuria, incontinencia, urgencia miccional, cólico renal, dolor renal, incontinencia urinaria |
|
Trastornos del sistema reproductivo y de las mamas Disfunción sexual |
|
Trastornos generales Calcinosis, edema facial, sensación anormal, sensación de embriaguez, sensación de inquietud, malestar, enfriamiento periférico, aletargamiento |
|
Estudios complementarios Disminución de bicarbonato en sangre, presencia de cristales en la orina, prueba de marcha en tándem anormal, disminución en el recuento de leucocitos |
Otros datos de ensayos clínicos – Pacientes pediátricos: En la Tabla 7, se muestran las reacciones adversas informadas en los ensayos clínicos doble ciegos y controlados en <2% de los pacientes pediátricos tratados con TOPAMAX® o de cualquier tasa en ensayos clínicos de etiqueta abierta con pacientes pediátricos tratados con TOPAMAX®.
|
Tabla 7. Reacciones adversas informadas en los ensayos clínicos doble ciegos y controlados en <2% de los pacientes pediátricos tratados con TOPAMAX® o de cualquier tasa en ensayos clínicos de etiqueta abierta con pacientes pediátricos tratados con TOPAMAX® |
|
Trastornos de la sangre y del sistema linfático Eosinofilia, leucopenia, linfadenopatía, trombocitopenia |
|
Trastornos del sistema inmunitario Hipersensibilidad |
|
Trastornos metabólicos y nutricionales Acidosis hiperclorémica, hipocalemia, aumento del apetito |
|
Trastornos psiquiátricos Ira, apatía, llanto, dificultades de concentración, trastorno de la producción del habla, insomnio inicial, insomnio, insomnio medio, cambios de humor, perseveración, trastornos del sueño, ideación suicida, intento de suicidio |
|
Trastornos del sistema nervioso Trastornos del ritmo circadiano del sueño, crisis, disartria, disgeusia, crisis convulsivas generalizadas, hipoestesia, deterioro mental, nistagmo, parosmia, mala calidad de sueño, hiperactividad psicomotriz, disminución de la capacidad psicomotriz, síncope, temblor |
|
Trastornos oculares Diplopía, aumento del lagrimeo, visión borrosa |
|
Trastornos del oído y del laberinto Dolor de oídos |
|
Trastornos cardíacos Palpitaciones, bradicardia sinusal |
|
Trastornos vasculares Hipotensión ortostática |
|
Trastornos respiratorios, torácicos y mediastínicos Congestión nasal, hipersecreción sinusal de los senos paranasales, rinorrea |
|
Trastornos gastrointestinales Malestar abdominal, dolor abdominal, sequedad bucal, flatulencia, gastritis, reflujo gastroesofágico, hemorragia gingival, glosodinia, pancreatitis, parestesia oral, malestar estomacal |
|
Trastornos musculoesqueléticos y del tejido conjuntivo Artralgia, rigidez musculoesquelética, mialgia |
|
Trastornos renales y urinarios Incontinencia, urgencia miccional, polaquiuria |
|
Trastornos generales Sensación anormal, hipertermia, malestar, aletargamiento |
Datos posteriores a la comercialización: Los eventos adversos que se identificaron en primera instancia como reacciones adversas durante el período de postcomercialización con TOPAMAX® se incluyen en la Tabla 8. Las frecuencias se presentan por categoría de frecuencia con base en las tasas de informes espontáneos, de acuerdo con la siguiente convención:
— Muy común ≥1/10
— Frecuentes ≥1/100 a <1/10
— Poco frecuentes ≥1/1000 a <1/100
— Raros ≥1/10000 a <1/1000
— Muy raros <1/10000, incluye los informes aislados
|
Tabla 8: Reacciones adversas identificadas durante la experiencia de postcomercialización con TOPAMAX® por categoría de frecuencia estimada a partir de las tasas de informes espontáneos |
|
|
Infecciones e infestaciones |
|
|
Muy raras |
Nasofaringitis |
|
Trastornos de la sangre y del sistema linfático |
|
|
Muy raros |
Neutropenia |
|
Trastornos del sistema inmunitario |
|
|
Muy raros |
Edema alérgico |
|
Trastornos psiquiátricos |
|
|
Muy raros |
Sentimientos de desesperación |
|
Trastornos oculares |
|
|
Muy raros |
Sensación anormal en el ojo |
|
Muy raros |
Glaucoma de ángulo cerrado |
|
Muy raros |
Edema conjuntival |
|
Muy raros |
Trastornos de los movimientos oculares |
|
Muy raros |
Edema de párpados |
|
Muy raros |
Maculopatía |
|
Muy raros |
Miopía |
|
Trastornos respiratorios, torácicos y mediastínicos |
|
|
Muy raros |
Tos |
|
Trastornos de la piel y del tejido subcutáneo |
|
|
Muy raros |
Eritema multiforme |
|
Muy raros |
Edema periorbitario |
|
Muy raros |
Síndrome de StevensJohnson |
|
Muy raros |
Necrólisis epidérmica tóxica |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|
|
Muy raros |
Inflamación de las articulaciones |
|
Muy raros |
Malestar en las extremidades |
|
Trastornos renales y urinarios |
|
|
Muy raros |
Acidosis tubular renal |
|
Trastornos generales y reacciones en el lugar de la administración |
|
|
Muy raros |
Edema generalizado |
|
Muy raros |
Enfermedad tipo gripe |
|
Estudios complementarios |
|
|
Muy raros |
Aumento de peso |
INTERACCIONES: Para el propósito de esta sección, se define a la dosis sin efecto como un cambiodel ≤15%.
Efectos de otros AED en TOPAMAX®: La fenitoína y la carbamazepina disminuyen la concentración plasmática de TOPAMAX®.El agregado o la interrupción de la administración de fenitoína o de la carbamazepina al tratamiento con TOPAMAX® puede requerir de un ajuste en la posología de este último. Para realizar este ajuste, debe utilizarse la titulación hasta alcanzar un efecto clínico. El agregado o la interrupción de la administración de ácido valproico no produce cambios significativos a nivel clínico en las concentraciones plasmáticas de TOPAMAX® y, por lo tanto, no justifican el ajuste de la posología de TOPAMAX®.
Los resultados de estas interacciones se resumen a continuación:
|
AED coadministrados |
Concentración de AED |
Concentración de TOPAMAX® |
|
Fenitoína |
?** |
?(48%) |
|
Carbamazepina (CBZ) |
? |
?(40%) |
|
Ácido valproico |
? |
? |
|
Lamotrigina |
? |
? |
|
Fenobarbital |
? |
NE |
|
Primidona |
? |
NE |
|
? =Sin efecto en la concentración plasmática (cambio?15%) ** = Las concentraciones plasmáticas aumentan en los pacientes ? = Las concentraciones plasmáticas disminuyen NE = No estudiado AED = Medicamento antiepiléptico |
||
Efectos del TOPAMAX® en otros AED: El agregado de TOPAMAX® al tratamiento con otros AED (fenitoína, carbamazepina, ácido, fenobarbital, primidona) no tiene efecto en sus concentraciones plasmáticas en estado de equilibrio, excepto en muy pocos pacientes, para quienes el agregado de TOPAMAX® al tratamiento con fenitoína puede provocar un aumento de concentraciones plasmáticas de fenitoína. Esto es posible debido a la inhibición de una isoforma específica polimorfa de una enzima (CYP2C19). En consecuencia, deben monitorearse los niveles de fenitoína en cualquier paciente que consuma fenitoína y que muestre signos clínicos o síntomas de toxicidad.
Un estudio de interacción farmacocinética en pacientes con epilepsia indicó que el agregado de topiramato no tuvo efectos en la concentración plasmática en estado de equilibrio de la lamotrigina con dosis de topiramato de 100 a 400 mg/día. Además, no hubo cambios en la concentración plasmática en estado de equilibrio del topiramato durante ni luego de la interrupción del tratamiento con lamotrigina (dosis media de 327 mg/día).
Otras interacciones medicamentosas:
— Digoxina: En un estudio de dosis única, el área de digoxina sérica bajo la curva de concentración plasmática (AUC) disminuyó en un 12% debido a la administración concomitante de TOPAMAX®. No se ha establecido la relevancia clínica de esta observación. Cuando se agrega o se suspende la administración de TOPAMAX® en los pacientes bajo tratamiento con digoxina, se debe prestar mucha atención al monitoreo de rutina de la digoxina sérica.
— Anticonceptivos orales:En un estudio de interacción farmacocinética en voluntarios sanos con una combinación administrada en forma concomitante de un producto anticonceptivo oral con un contenido de 1 mg de noretindrona (NET) más 35 mcg de etinilestradiol (EE), no se asoció al TOPAMAX® administrado en ausencia de otros medicamentos en dosis de 50 a 200 mg/día con cambios de importancia estadística en la exposición media (AUC) a ninguno de los componentes del anticonceptivo oral.En otro estudio, la exposición al EE disminuyó en forma significativa desde el punto de vista estadístico en dosis de 200, 400 y 800 mg/día (18%, 21% y 30%, respectivamente) cuando se administró como tratamiento adyuvante en pacientes que consumían ácido valproico. En ambos estudios, el TOPAMAX® (50 mg/día a 800 mg/día) no afectó en forma significativa la exposición a la NET. Aunque hubo una disminución dependiente de la dosis en la exposición al EE para las dosis de 200 a 800 mg/día, no hubo un cambio importante dependiente de la dosis en la exposición al EE para las dosis de 50 a 200 mg/día. Se desconoce la importancia clínica de los cambios observados. La posibilidad de una disminución en la eficacia del anticonceptivo y de hemorragia intermenstrual debe tenerse en cuenta en pacientes que consumen una combinación de productos anticonceptivos orales con TOPAMAX®. Debe pedirse a los pacientes que consumen anticonceptivos que contienen estrógeno que informen cualquier cambio en sus patrones de hemorragia. La eficacia anticonceptiva puede disminuir incluso en ausencia de la hemorragia intermenstrual.
— Litio: En voluntarios sanos, se observó una reducción (18% para AUC) en la exposición sistémica al litio durante la administración concomitante con topiramato de 200 mg/día. En pacientes con trastorno bipolar, la farmacocinética del litio no resultó afectada durante el tratamiento con topiramato en dosis de 200 mg/día; sin embargo,se observó un aumento en la exposición sistémica (26% para AUC) luego de las dosis de topiramato de hasta 600 mg/día. Los niveles de litio deben monitorearse cuando se coadministra con topiramato.
— Risperidona: Los estudios de interacciones medicamentosas llevados a cabo bajo condiciones de dosis única y múltiple en voluntarios sanos y en pacientes con trastorno bipolar arrojaron resultados similares. Cuando se administró en forma concomitante con topiramato en dosis escalonadas de 100, 250 y 400 mg/día, hubo una reducción en la exposición sistémica (16% y 33% para AUC en las dosis de 250 y 400 mg/día, respectivamente) de la risperidona (administrada en dosis que oscilaban de 1 a 6 mg/día). Se observaron alteraciones mínimas en la farmacocinética de la fracción activa total (risperidona más 9-hidroxi-risperidona) y no se observaron alteraciones para la 9-hidroxi-risperidona. No hubo cambios importantes desde el punto de vista clínico en la exposición sistémica del componente activo total de la risperidona ni del topiramato; por lo tanto, es probable que esta interacción no tenga importancia clínica.
— Hidroclorotiazida (HCTZ): Un estudio de interacciones medicamentosas realizado en voluntarios sanos evaluó la farmacocinética en estado de equilibrio de la HCTZ (25 mg cada 24 h) y del topiramato (96 mg cada 12 h) cuando se administraron solos y en forma concomitante. Los resultados de este estudio indican que la Cmáx del topiramato aumentó un 27% y que la AUC aumentó un 29% cuando se agregó HCTZ al topiramato. Se desconoce la relevancia clínica de este cambio. El agregado de HCTZ al tratamiento con topiramato puede requerir de un ajuste de la dosis de topiramato. La farmacocinética en estado de equilibrio de la HCTZ no se vio influenciada en forma significativa por la administración concomitante de topiramato. Los resultados clínicos de laboratorio indicaron disminuciones en el potasio sérico luego de la administración de topiramato o de HCTZ, que fueron superiores cuando la HCTZ y el topiramato se administraron en combinación.
— Metformina: Un estudio de interacciones medicamentosas realizado en voluntarios sanos evaluó la farmacocinética en estado de equilibrio de la metformina y el topiramato en plasma cuando se administró solamente la metformina y cuando la metformina y el topiramato se administraron simultáneamente. Los resultados de este estudio indicaron que la Cmáx media y la AUC0-12h media de la metformina aumentaron un 18% y 25%, respectivamente, mientras que el CL/F medio disminuyó un 20% cuando se coadministró metformina con topiramato. El topiramato no afectó el tmáx de la metformina. No es clara la importancia clínica del efecto del topiramato en la farmacocinética de la metformina. Aparentemente, la eliminación oral plasmática del topiramato disminuye cuando se administra con metformina. Se desconoce el alcance del cambio en la eliminación. No es clara la importancia clínica del efecto de la metformina en la farmacocinética del topiramato. Cuando se agrega o se suspende la administración de TOPAMAX® en los pacientes bajo tratamiento con metformina, se debe prestar mucha atención al monitoreo de rutina para un control adecuado del estado de su diabetes.
— Pioglitazona: Un estudio de interacciones medicamentosas realizado en voluntarios sanos evaluó la farmacocinética en estado de equilibrio del topiramato y de la pioglitazona cuando se administraron solos y en forma concomitante. Se observó una disminución del 15% en la AUC?,ss de la pioglitazona sin alteración en la Cmáx,ss. Este hallazgo no tuvo importancia estadística. Además, se observó una disminución del 13 y del 16% en la Cmáx,ss y en la AUC?,ss, respectivamente, del hidroxi metabolito activo, así como también una disminución del 60% en la Cmáx,ss y en la AUC?,ss del keto metabolito activo. Se desconoce la importancia clínica de estos hallazgos. Cuando se agrega TOPAMAX® al tratamiento con pioglitazona o cuando se agrega pioglitazona al tratamiento con TOPAMAX®,se debe prestar mucha atención al monitoreo de rutina de los pacientes para un control adecuado del estado de su diabetes.
— Gliburida: Un estudio de interacciones medicamentosas realizado en pacientes con diabetes de tipo 2 evaluó la farmacocinética en estado de equilibrio de la gliburida (5 mg/día) sola y administrada en forma concomitante con topiramato (150 mg/día). Hubo una reducción del 25% en la AUC24 de la gliburida durante la administración de topiramato. La exposición sistémica de los metabolitos activos, 4-trans-hidroxi-gliburida (M1) and 3-cis-hidroxigliburida (M2), también se redujo un 13 y 15%, respectivamente. La farmacocinética en estado de equilibrio del topiramato no se vio afectada por la administración concomitante de gliburida. Cuando se agrega topiramato al tratamiento con gliburida o cuando se agrega gliburida al tratamiento con topiramato, se debe prestar mucha atención al monitoreo de rutina de los pacientes para un control adecuado del estado de su diabetes.
Otras formas de interacciones:
— Agentes que predisponen a la nefrolitiasis: TOPAMAX®, cuando se administra en forma concomitante con otros agentes que predisponen a la nefrolitiasis, puede aumentar el riesgo de nefrolitiasis. Mientras se administra TOPAMAX®, se deben evitar los agentes de este tipo dado que pueden crear un ambiente fisiológico que aumente el riesgo de formación de cálculos renales.
— Ácido valproico: Se ha asociado a la administración concomitante de topiramato y de ácido valproico con la hiperamonemia con o sin encefalopatía en pacientes que han tolerado ambos medicamentos solos. En la mayoría de los casos, los síntomas y los signos disminuyeron con la interrupción de cualquiera de los dos medicamentos. Este evento adverso no se debe a una interacción farmacocinética. No se ha establecido asociación de la hiperamonemia con la monoterapia de topiramato ni con el tratamiento concomitante con otros AED.
Se ha informado hipotermia, definida como una disminución no intencional de la temperatura corporal central a <35 °C, en relación con el consumo concomitante de topiramato y de ácido valproico (VPA) tanto en conjunción con hiperamonemia como en ausencia de hiperamonemia. Estos eventos adversos en pacientes que consumen topiramato y valproato en forma concomitante pueden ocurrir luego de comenzar el tratamiento con topiramato o después de aumentar la dosis diaria de topiramato.
— Estudios adicionales de las interacciones medicamentosas farmacocinéticas: Se han realizado estudios clínicos para evaluar las potenciales interacciones medicamentosas farmacocinéticas entre el topiramato y otros agentes: A continuación se resumen los cambios en la Cmáx o en la AUC como resultado de las interacciones. La segunda columna (concentración del medicamento concomitante) describe lo que sucede con la concentración del medicamento concomitante detallado en la primera columna cuando se agrega topiramato. La tercera columna (concentración de topiramato) describe cómo la coadministración de un medicamento detallado en la primera columna modifica la concentración del topiramato.
Resumen de resultados de estudios clínicos adicionales de interacciones medicamentosas farmacocinéticas:
|
Medicamento concomitante |
Medicamento concomitante Concentracióna |
Concentración de topiramatoa |
|
Amitriptilina |
? Aumento del 20% en la Cmáx y en la AUC del metabolito nortriptilina |
NE |
|
Dihidroergotamina (Oral y subcutáneo) |
? |
? |
|
Haloperidol |
? Aumento del 31% en la AUC del metabolito reducido |
NE |
|
Propranolol |
? Aumento del 17% en Cmáx para 4-OH propranolol (TPM 50 mg q12h) |
Aumento del 9 y del 16% en la Cmáx, aumento del 9 y del 17% en la AUC (propranolol 40 mg y 80 mg q12h, respectivamente) |
|
Sumatriptán (oral y subcutáneo) |
? |
NE |
|
Pizotifeno |
? |
? |
|
Diltiazem |
Disminución del 25% en la AUC de diltiazem y disminución del 18% en la DEA, y ? para DEM* |
Aumento del 20% en la AUC |
|
Venlafaxina |
? |
? |
|
Flunarizina |
Aumento del 16% en la AUC (TPM50 mg q12h)b |
? |
|
a = Los valores expresados como % son los cambios en la Cmáx o la AUC medias del tratamiento con respecto a la monoterapia. ? = Sin efecto en la Cmáx ni en la AUC (cambio ?15%) del compuesto parental. NE = No estudiado *DEA = Desacetil diltiazem, DEM = Ndemetil diltiazem b = La AUC de la flunarizina aumentó un 14% en sujetos que consumían solamente flunarizina.El aumento en la exposición se puede atribuir a la acumulación durante el alcance del estado de equilibrio. |
||
INSTRUCCIONES DE USO Y MANIPULACIÓN: No aplicable.
Instrucciones para la eliminación: Cualquier producto no utilizado o material de desecho debe eliminarse de acuerdo con las normas locales.
Fabricado por: Janssen Ortho LLC, Puerto Rico
Fecha de revisión del texto: 15 octubre 2014.
Para información de producto, reporte de eventos adversos y quejas de calidad contáctenos a infojanssen@janpa.jnj.com y/o Costa Rica: 8000521607, El Salvador: 8006987, Guatemala: 18008350399, Honduras: 80001226155, Nicaragua: 18002260464, Panamá: 8000521419, República Dominicana: 18887600103, Jamaica: 18003712474, Venezuela: infojanssen@janve.jnj.com y/o 08001005742
JANSSEN PHARMACEUTICAL COMPANIES
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN:
Posología: Se recomienda que el tratamiento se inicie con una dosis baja, seguida de titulación hasta una dosis efectiva.
— Epilepsia – Tratamiento adyuvante:
• Adultos: El tratamiento debe iniciarse con 25 a 50 mg todas las noches durante una semana. Se ha informado el uso de dosis iniciales más bajas, pero no se ha estudiado de forma sistemática. Posteriormente, en intervalos de una o de dos semanas, se debe aumentar la dosis entre 25 y 50 mg/día y administrarse en dos dosis divididas. La titulación de la dosis debe guiarse según el resultado clínico. Algunos pacientes pueden alcanzar la eficacia con una dosificación de una vez al día.
En los ensayos clínicos como tratamiento adyuvante, 200 mg fueron efectivos y fueron la posología más baja que se estudió. Por lo tanto, se considera que esta es la dosis efectiva mínima. La dosis diaria habitual es de 200 a 400 mg en dos dosis divididas. Algunos pacientes han recibido dosis de hasta 1600 mg/día.
Estas recomendaciones de dosificación se aplican a todos los adultos, incluso los ancianos, en ausencia de enfermedad renal subyacente (consulte Advertencias y precauciones: Insuficiencia renal).
• Niños de 2 o más años de edad: La dosis diaria total recomendada de TOPAMAX® como tratamiento adyuvante es de aproximadamente 5 a 9 mg/kg/día en dos dosis divididas. La titulación debe comenzar con 25 mg (o menos, con base en un rango de 1 a 3 mg/kg/día) todas las noches durante la primera semana. Luego, la posología debe aumentarse a intervalos de una o de dos semanas en incrementos de 1 a 3 mg/kg/día (administrados en dos dosis divididas) para alcanzar una respuesta clínica óptima. La titulación de la dosis debe guiarse según el resultado clínico.
Se han estudiado dosis diarias de hasta 30 mg/kg/día y en general fueron bien toleradas.
— Epilepsia – Monoterapia: Cuando se suspenden los medicamentos antiepilépticos concomitantes (AED, por sus siglas en inglés) para lograr una monoterapia con topiramato, se deben tener en cuenta los efectos que esto puede tener en el control de las crisis. A menos que por cuestiones de seguridad sea necesaria una interrupción abrupta de los AED, se recomienda una interrupción gradual con una tasa de aproximadamente un tercio de la dosis del AED concomitante cada dos semanas (consulte Advertencias y precauciones: Interrupción de TOPAMAX®).
Cuando se suspenden los medicamentos de inducción enzimática, aumentan los niveles de topiramato. Es posible que sea necesaria una disminución en la posología de TOPAMAX® si se indica clínicamente.
• Adultos: La titulación debe comenzar con 25 mg todas las noches durante una semana. Luego, la posología debe aumentarse a intervalos de una o de dos semanas en incrementos de 25 a 50 mg/kg/día administrados en dos dosis divididas. Si el paciente no puede tolerar el régimen de titulación, se pueden utilizar incrementos más pequeños o intervalos más prolongados entre los incrementos. La dosis y la tasa de titulación deben guiarse según el resultado clínico.
La dosis objetivo inicial recomendada para la monoterapia de topiramato en adultos es de 100 mg/día y la dosis diaria recomendada máxima es de 500 mg. Algunos pacientes con tipos de epilepsia resistentes al tratamiento han tolerado la monoterapia de topiramato con dosis de 1000 mg/día. Estas recomendaciones de dosificación se aplican a todos los adultos, incluso los ancianos, en ausencia de enfermedad renal subyacente.
• Niños de 2 o más años de edad: El tratamiento de niños de 2 o más años de edad debe iniciarse con 0,5 a 1 mg/kg todas las noches durante la primera semana. Luego, la posología debe aumentarse a intervalos de una o de dos semanas en incrementos de 0,5 a 1 mg/kg/día administrados en dos dosis divididas. Si el niño no puede tolerar el régimen de titulación, se pueden utilizar incrementos más pequeños o intervalos más prolongados entre los incrementos de las dosis. La dosis y la titulación de la dosis deben guiarse según el resultado clínico.
El rango objetivo inicial recomendado para la monoterapia de topiramato en niños de dos o más años de edad es de 100 a 400 mg/día. Los niños con diagnóstico reciente de crisis de inicio parcial han recibido dosis de hasta 500 mg/día.
— Migraña:
• Adultos: La dosis diaria total recomendada de topiramato para la profilaxis de la migraña es de 100 mg/día administrados en dos dosis divididas. La titulación debe comenzar con 25 mg todas las noches durante una semana. Luego, la posología debe aumentarse en incrementos de 25 mg/día administrados en intervalos de una semana. Si el paciente no puede tolerar el régimen de titulación, se pueden utilizar intervalos más prolongados entre los ajustes de las dosis.
Algunos pacientes pueden notar beneficios con dosis totales de 50 mg/día. Los pacientes han recibido dosis diarias totales de hasta 200 mg/día. La dosis y la tasa de titulación deben guiarse según el resultado clínico (consulte Propiedades farmacodinámicas: Ensayos clínicos sobre migrañas).
— Poblaciones especiales:
• Insuficiencia renal: Es posible que los pacientes con insuficiencia renal moderada y severa (CLCR<70 mL/min.) necesiten una reducción de la dosis. Se recomienda la mitad de la dosis inicial y de mantenimiento habitual (consulte Propiedades farmacocinéticas: Poblaciones especiales, Insuficiencia renal).
Dado que TOPAMAX® se elimina del plasma con la hemodiálisis, se debe administrar una dosis complementaria de TOPAMAX® equivalente a aproximadamente la mitad de la dosis diaria en los días de hemodiálisis. La dosis complementaria debe administrarse en dosis divididas al comienzo y la finalización del procedimiento de hemodiálisis. La dosis complementaria puede diferir con base en las características del equipo de diálisis que se utilice (consulte Propiedades farmacocinéticas: Poblaciones especiales, Insuficiencia renal).
• Insuficiencia hepática: El topiramato debe administrarse con precaución en pacientes con insuficiencia hepática (consulte Propiedades farmacocinéticas: Poblaciones especiales, Insuficiencia hepática).
Forma de administración: TOPAMAX® está disponible en comprimidos y en una formulación en cápsulas dispersables para administración oral. Se recomienda no romper los comprimidos TOPAMAX®. La formulación dispersable se proporciona a aquellos pacientes que no pueden tragar comprimidos, por ejemplo, los pacientes pediátricos y los ancianos.
Las cápsulas dispersables TOPAMAX® pueden tragarse enteras o puede abrirse con cuidado la cápsula y dispersar todo el contenido en una pequeña cantidad de comida blanda (del tamaño de una cucharita de té) para su administración. Esta mezcla de medicamento/comida debe tragarse de inmediato y no se debe masticar. No debe almacenarse para su consumo futuro.
TOPAMAX® puede consumirse independientemente de las comidas.
SOBREDOSIS:
Signos y síntomas: Se han informado sobredosis de topiramato. Los signos y síntomas incluyeron crisis, somnolencia, trastornos en el habla, visión borrosa, diplopía, deterioro de la actividad mental, letargo, coordinación anormal, estupor, hipotensión, dolor abdominal, agitación, mareos y depresión. En la mayoría de los casos, las consecuencias clínicas no fueron severas, pero se han informado casos de muerte después de sobredosis con múltiples medicamentos entre los cuales estaba incluido el topiramato.
La sobredosis con topiramato puede provocar acidosis metabólica severa (consulte Advertencias y precauciones – Acidosis metabólica).
Se calculó que la sobredosis de topiramato más alta estuvo entre los 96 y 110 g y que provocó un coma que duró de 20 a 24 horas, seguido de una recuperación total luego de 3 a 4 días.
Tratamiento: Si la ingesta es reciente en una sobredosis aguda de topiramato, se debe vaciar de inmediato el estómago mediante lavado o por inducción de emesis. Se ha demostrado que el carbón activado absorbe el topiramato in vitro. El tratamiento debe ser de apoyo. Se ha demostrado que la hemodiálisis es un medio efectivo para eliminar al topiramato del organismo. El paciente debe estar bien hidratado.
CONDICIONES DE ALMACENAMIENTO: Manténgase fuera del alcance de los niños.
Comprimidos: Almacene a una temperatura de 30 °C o inferior y proteja de la humedad. Conserve en el empaque original.
Cápsulas dispersables: Almacene a una temperatura de 25 °C o inferior y proteja de la humedad.
No almacene el medicamento mezclado con la comida.
NATURALEZA Y CONTENIDO DEL ENVASE:
Frascos que contienen 20, 28, 30, 56, 60 o 100 comprimidos con desecante.
Frascos que contienen 28 o 60 cápsulas dispersables.
Envases con blísteres de 10, 20, 28, 56 o 60 comprimidos.
Dispensado en las farmacias con receta médica.


