

TRAYENTA
LINAGLIPTINA
Tabletas recubiertas
1 Caja, 30 Tabletas recubiertas, 5 mg
1 Caja, 10 Tabletas recubiertas, 5 mg
1 Caja, 3 Tabletas recubiertas, 5 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Tabletas recubiertas
Cada TABLETA RECUBIERTA contiene:
5 mg de linagliptina
Código ATC: A10BH05
Leyenda exclusiva para Cuba:
Contiene manitol, puede provocar un ligero efecto laxante
Formas farmacéuticas y concentraciones:
Tabletas de 5 mg de color rojo claro, redondas, biconvexas, biseladas y recubiertas, grabadas en un lado con “D5” y en el otro lado con el logo de Boehringer Ingelheim
INDICACIONES TERAPÉUTICAS:
Indicaciones y uso: TRAYENTA® está indicado como un complemento de la dieta y el ejercicio para mejorar el control glucémico en adultos con diabetes mellitus tipo 2.
Limitaciones para su uso: No se recomienda TRAYENTA® en pacientes con diabetes mellitus tipo 1, ya que no sería efectivo.
TRAYENTA® no se ha estudiado en pacientes con antecedentes de pancreatitis. Se desconoce si los pacientes con antecedentes de pancreatitis tienen un mayor riesgo de padecer pancreatitis al usar TRAYENTA® [ver Advertencias y precauciones (Pancreatitis)].
FARMACOCINÉTICA Y FARMACODINAMIA:
Mecanismo de acción: La linagliptina es un inhibidor de la DPP-4, una enzima que degrada las hormonas incretinas péptido similar al glucagón tipo 1 (GLP-1) y polipéptido insulinotrópico dependiente de glucosa (GIP). Por consiguiente, la linagliptina incrementa la concentración de las hormonas incretinas activas, lo que estimula la liberación de insulina de manera dependiente de glucosa y disminuye los niveles de glucagón en la circulación. Ambas hormonas incretinas participan en la regulación fisiológica de la homeostasis de la glucosa. El organismo segrega hormonas incretinas a un nivel basal bajo a lo largo del día, y estos niveles aumentan inmediatamente después de la ingestión de alimentos. El GLP-1 y el GIP incrementan la biosíntesis de la insulina y la secreción de esta desde las células beta pancreáticas en presencia de niveles normales y elevados de glucosa en sangre. Además, el GLP-1 también reduce la secreción de glucagón desde las células alfa pancreáticas, lo que causa una reducción de la producción hepática de glucosa.
Farmacodinámica: La linagliptina se une a la DPP-4 en una forma reversible, y de esta manera aumenta las concentraciones de hormonas incretinas. La linagliptina aumenta la secreción de insulina y disminuye la secreción de glucagón en una forma que es dependiente de la glucosa, y, por lo tanto, produce una mejor regulación de la homeostasis de la glucosa. La linagliptina se une de manera selectiva a la DPP-4 e inhibe de manera selectiva la actividad de la DPP-4, pero no la actividad in vitro de DPP-8 o DPP-9 a concentraciones que se aproximan a las exposiciones terapéuticas.
Electrofisiología cardíaca: En un estudio aleatorizado, con diseño cruzado de 4 vías, controlado con un placebo y comparado con un fármaco activo, a 36 sujetos sanos se les administró una sola dosis oral de linagliptina 5 mg, linagliptina 100 mg (20 veces la dosis recomendada), moxifloxacino y un placebo. No se observó ningún aumento en el QTc ni con la dosis recomendada de 5 mg ni con la dosis de 100 mg. Con la dosis de 100 mg, las concentraciones plasmáticas máximas de linagliptina fueron aproximadamente 38 veces más altas que las concentraciones máximas luego de la dosis de 5 mg.
Farmacocinética: La farmacocinética de la linagliptina ha sido caracterizada en sujetos sanos y en pacientes con diabetes mellitus tipo 2. Después de la administración por vía oral de una dosis única de 5 mg a sujetos sanos, las concentraciones plasmáticas máximas de linagliptina se registraron aproximadamente una hora y media después de la administración de la dosis (Tmáx); la media del área bajo la curva (ABC) de la concentración plasmática fue de 139 nmol*h/L y la concentración máxima (Cmáx) fue de 8.9 nmol/L.
Las concentraciones plasmáticas de linagliptina disminuyen en por lo menos una manera bifásica con una vida media terminal larga (mayor de 100 horas), que se relaciona con la unión saturable de la linagliptina a la DPP-4. La prolongada fase de eliminación no contribuye a la acumulación del fármaco. La vida media efectiva para la acumulación de linagliptina, determinada mediante la administración oral de dosis múltiples de linagliptina 5 mg, es de aproximadamente 12 horas. Luego de la dosificación una vez al día, las concentraciones plasmáticas en el estado estacionario se alcanzan alrededor de la tercera dosis de linagliptina 5 mg, y la Cmáx y el ABC aumentaron en un factor de 1.3 en el estado estacionario, en comparación con la primera dosis. Los coeficientes de variación intra-sujetos e inter-sujetos para el ABC de linagliptina fueron pequeños (12.6% y 28.5%, respectivamente). El ABC de la concentración plasmática de linagliptina aumentó de una manera menor que proporcional a la dosis, en el intervalo de dosis de 1 a 10 mg. La farmacocinética de la linagliptina es similar en los sujetos sanos y en los pacientes con diabetes mellitus tipo 2.
Absorción: La biodisponibilidad absoluta de la linagliptina es de aproximadamente 30%. Las comidas con alto contenido de grasas redujeron la Cmáx en 15% y aumentaron el ABC en 4%; este efecto no es clínicamente relevante. TRAYENTA ® se puede tomar con o sin alimentos.
Distribución: La media del volumen aparente de distribución en el estado estacionario luego de una dosis intravenosa única de linagliptina 5 mg a sujetos sanos es de aproximadamente 1,110 L, lo que indica que la linagliptina se distribuye ampliamente a los tejidos. La unión de la linagliptina a las proteínas plasmáticas depende de la concentración; disminuye de aproximadamente 99% a 1 nmol/L hasta 75%-89% a ≥30 nmol/L, lo que refleja una saturación de la unión a la DPP-4 conforme aumenta la concentración de linagliptina. A concentraciones altas, donde la DPP-4 está totalmente saturada, el 70% al 80% de la linagliptina permanece unida a las proteínas plasmáticas, y el 20% al 30% está libre en el plasma. La unión a las proteínas plasmáticas no se ve afectada en los pacientes con insuficiencia renal o hepática.
Eliminación: La linagliptina tiene una vida media terminal de aproximadamente 200 horas en estado estacionario, aunque la vida media de acumulación es de aproximadamente 11 horas. La depuración renal en el estado estacionario fue de aproximadamente 70 mL/min.
Metabolismo: Luego de la administración oral, la mayor parte (alrededor del 90%) de la linagliptina se excreta sin cambio, lo que indica que el metabolismo representa una vía menor de eliminación. Una pequeña fracción de la linagliptina absorbida es metabolizada y produce un metabolito farmacológicamente inactivo, que muestra una exposición en el estado estacionario del 13.3% en relación con la linagliptina.
Excreción: Después de la administración por vía oral de una dosis de [14C]-linagliptina a sujetos sanos, aproximadamente el 85% de la radioactividad administrada se eliminó a través del sistema enterohepático (80%) o a través de la orina (5%) dentro de 4 días a partir de la administración de la dosis.
Poblaciones específicas:
Pacientes con insuficiencia renal: Un estudio farmacocinético abierto evaluó la farmacocinética de linagliptina 5 mg en pacientes de ambos sexos con diferentes grados de insuficiencia renal crónica. El estudio incluyó 6 sujetos sanos con un funcionamiento renal normal (depuración de creatinina [CrCl] ≥80 mL/min), 6 pacientes con insuficiencia renal leve (CrCl 50 a <80 mL/min), 6 pacientes con insuficiencia renal moderada (CrCl 30 a <50 mL/min), 10 pacientes con diabetes tipo 2 e insuficiencia renal grave (CrCl <30 mL/min) y 11 pacientes con diabetes mellitus tipo 2 y un funcionamiento renal normal. La depuración de creatinina se determinó mediante la cuantificación de la depuración de creatinina en orina de 24 horas o se calculó a partir de la creatinina sérica usando la fórmula de Cockcroft-Gault.
En el estado estacionario, la exposición a la linagliptina en los pacientes con insuficiencia renal leve fue similar a la de los sujetos sanos.
En los pacientes con insuficiencia renal moderada en condiciones del estado estacionario, se vio un aumento del valor promedio de la exposición a la linagliptina (ABCτ,ss del 71% y Cmáx del 46%) en comparación con los sujetos sanos. Este aumento no estuvo asociado con una vida media de acumulación o una vida media terminal prolongadas, o con un factor de acumulación aumentado. La excreción renal de linagliptina fue de menos del 5% de la dosis administrada, y no se vio afectada por un funcionamiento renal disminuido.
Los pacientes con diabetes mellitus tipo 2 e insuficiencia renal grave mostraron una exposición en el estado estacionario aproximadamente 40% mayor que la de los pacientes con diabetes mellitus tipo 2 y funcionamiento renal normal (aumento en el ABCτ,ss del 42% y en la Cmáx del 35%). Para ambos grupos con diabetes mellitus tipo 2, la eliminación renal fue de menos del 7% de la dosis administrada.
Estos resultados fueron respaldados adicionalmente por los resultados de los análisis de farmacocinética poblacional.
Pacientes con insuficiencia hepática: En los pacientes con insuficiencia hepática leve (Child-Pugh clase A), la exposición en el estado estacionario (ABCτ,ss) de la linagliptina fue aproximadamente 25% menor, y la Cmáx,ss fue aproximadamente 36% menor que en los sujetos sanos. En los pacientes con insuficiencia hepática moderada (Child-Pugh clase B), el ABCss de la linagliptina fue aproximadamente 14% menor y la Cmáx,ss fue aproximadamente 8% menor que en los sujetos sanos. Los pacientes con insuficiencia hepática grave (Child-Pugh clase C) tuvieron una exposición similar a la linagliptina, en términos del ABC0-24, y una Cmáx aproximadamente 23% menor en comparación con los sujetos sanos. Las reducciones en los parámetros farmacocinéticos vistas en los pacientes con insuficiencia hepática no causaron reducciones en la inhibición de la DPP-4.
Efectos de la edad, el índice de masa corporal (IMC), el sexo y la raza: Con base en el análisis farmacocinético poblacional, la edad, el IMC, el sexo y la raza no tienen un efecto clínicamente significativo sobre la farmacocinética de la linagliptina [ver Uso en poblaciones específicas (Uso geriátrico)].
Estudios de interacciones medicamentosas:
Evaluación de las interacciones medicamentosas in vitro: La linagliptina es un inhibidor débil a moderado de CYP3A4, una isoenzima CYP, pero no inhibe otras isoenzimas CYP y no es un inductor de las isoenzimas CYP, entre ellas CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 y 4A11.
La linagliptina es un sustrato de la glicoproteína P (P-gp), y a concentraciones altas inhibe el transporte de digoxina mediado por la P-gp. Con base en estos resultados y en estudios in vivo de interacciones medicamentosas, se considera muy poco probable que la linagliptina produzca interacciones con otros sustratos de la P-gp a concentraciones terapéuticas.
Evaluación de las interacciones medicamentosas in vivo: Los inductores potentes de CYP3A4 o P-gp (p. ej., rifampicina) disminuyen la exposición a la linagliptina a concentraciones por debajo de las terapéuticas y probablemente inefectivas [ver Interacciones medicamentosas]. Estudios in vivo mostraron indicios de una predisposición baja a la aparición de interacciones medicamentosas con sustratos de CYP3A4, CYP2C9, CYP2C8, P-gp y transportadores de cationes orgánicos (OCT).
En la tabla 3 se describe el efecto de los fármacos administrados simultáneamente sobre la exposición sistémica de la linagliptina.
Tabla 3. Efecto de los fármacos administrados simultáneamente sobre la exposición sistémica de la linagliptina
|
Fármaco administrado simultáneamente |
Dosis del fármaco administrado simultáneamente* |
Dosis de linagliptina* |
Cociente de la media geométrica (cociente con/sin el fármaco administrado simultáneamente) Sin efecto=1.0 |
|
|
ABC† |
Cmáx |
|||
|
Metformina |
850 mg tres veces al día |
10 mg una vez al día |
1.20 |
1.03 |
|
Gliburida |
1.75 mg# |
5 mg una vez al día |
1.02 |
1.01 |
|
Pioglitazona |
45 mg una vez al día |
10 mg una vez al día |
1.13 |
1.07 |
|
Ritonavir |
200 mg dos veces al día |
5 mg# |
2.01 |
2.96 |
|
Rifampicina** |
600 mg una vez al día |
5 mg una vez al día |
0.60 |
0.56 |
* Dosis múltiples (estado estacionario) a menos que se indique otra cosa.
** Para información con respecto a recomendaciones clínicas [ver Interacciones medicamentosas (Inductores de la glucoproteína P o de las enzimas CYP3A4)].
# Dosis única.
† ABC = ABC(0 a 24 horas) para los tratamientos con dosis única y ABC=ABC(TAU) para los tratamientos con dosis múltiples.
En la tabla 4 se describe el efecto de la linagliptina sobre la exposición sistémica de los fármacos administrados simultáneamente.
Tabla 4. Efecto de la linagliptina en la exposición sistémica de los fármacos administrados simultáneamente
|
Fármaco administrado simultáneamente |
Dosis del fármaco administrado simultáneamente* |
Dosis de linagliptina* |
Cociente de la media geométrica (cociente con/sin el fármaco administrado simultáneamente) Sin efecto=1.0 |
||
|
ABC† |
Cmáx |
||||
|
Metformina |
850 mg tres veces al día |
10 mg una vez al día |
Metformina |
1.01 |
0.89 |
|
Gliburida |
1.75 mg# |
5 mg una vez al día |
Gliburida |
0.86 |
0.86 |
|
Pioglitazona |
45 mg una vez al día |
10 mg una vez al día |
Pioglitazona Metabolito M-III Metabolito M-IV |
0.94 0.98 1.04 |
0.86 0.96 1.05 |
|
Digoxina |
0.25 mg una vez al día |
5 mg una vez al día |
Digoxina |
1.02 |
0.94 |
|
Simvastatina |
40 mg una vez al día |
10 mg una vez al día |
Simvastatina Simvastatina ácida |
1.34 1.33 |
1.10 1.21 |
|
Warfarina |
10 mg# |
5 mg una vez al día |
R-warfarina S-warfarina IIN TP |
0.99 1.03 0.93** 1.03** |
1.00 1.01 1.04** 1.15** |
|
Etinilestradiol y levonorgestrel |
Etinilestradiol 0.03 mg y levonorgestrel 0.150 mg una vez al día |
5 mg una vez al día |
Etinilestradiol Levonorgestrel |
1.01 1.09 |
1.08 1.13 |
* Dosis múltiples (estado estacionario) a menos que se indique otra cosa.
# Dosis única
† ABC=ABC(INF) para los tratamientos con dosis única y ABC=ABC(TAU) para los tratamientos con dosis múltiples.
** ABC=ABC(0-168) y Cmáx=Emáx para los criterios de valoración farmacodinámicos.
IIN=Índice internacional normalizado.
TP=Tiempo de protrombina.
CONTRAINDICACIONES:
TRAYENTA® está contraindicado en pacientes con reacciones de hipersensibilidad a la linagliptina o a cualquiera de los excipientes de TRAYENTA®, tales como anafilaxia, angioedema, exfoliación cutánea, urticaria o hiperreactividad bronquial [ver Advertencias y precauciones (Reacciones de hipersensibilidad) y Reacciones adversas].
PRECAUCIONES Y ADVERTENCIAS:
Advertencias y precauciones:
Pancreatitis: Se ha informado pancreatitis aguda, incluida pancreatitis mortal, en pacientes tratados con TRAYENTA®. En el ensayo CARMELINA [ver Estudios clínicos (Ensayos de seguridad cardiovascular en pacientes con diabetes mellitus tipo 2)], se notificó pancreatitis aguda en 9 (0.3%) pacientes tratados con TRAYENTA® y en 5 (0.1%) pacientes tratados con placebo. Dos pacientes tratados con TRAYENTA® en el ensayo CARMELINA tuvieron pancreatitis aguda con un desenlace mortal. Ha habido informes posteriores a la comercialización de pancreatitis aguda, incluida pancreatitis mortal, en pacientes tratados con TRAYENTA®.
Se deben tener en cuenta los posibles signos y síntomas de pancreatitis. Si se sospecha pancreatitis, suspender inmediatamente la administración de TRAYENTA® e iniciar el tratamiento adecuado. Se desconoce si los pacientes con antecedentes de pancreatitis tienen un mayor riesgo de desarrollar pancreatitis mientras toman TRAYENTA®.
Hipoglucemia con uso concomitante con insulina y secretagogos de insulina: Se sabe que los secretagogos de insulina y la insulina causan hipoglucemia. El riesgo de hipoglucemia aumenta cuando TRAYENTA® se usa en combinación con un secretagogo de insulina (p. ej., sulfonilurea) o con insulina [ver Reacciones adversas (Experiencia en ensayos clínicos)]. El uso de TRAYENTA® en combinación con insulina en sujetos con insuficiencia renal grave se asoció con una mayor tasa de hipoglucemia [ver Reacciones adversas (Experiencia en ensayos clínicos)]. Por consiguiente, es posible que sea necesario una dosificación más baja del secretagogo de insulina o de insulina para reducir el riesgo de hipoglucemia cuando se administran en combinación con TRAYENTA®.
Reacciones de hipersensibilidad: Posterior a la comercialización se han informado reacciones graves de hipersensibilidad en pacientes tratados con TRAYENTA®. Entre estas reacciones se encuentran anafilaxia, angioedema y exfoliación cutánea. La aparición de estas reacciones ocurrió predominantemente dentro de los tres primeros meses después de iniciar el tratamiento con TRAYENTA®, y algunos informes se presentaron después de la primera dosis. Si se sospecha una reacción de hipersensibilidad grave, suspender la administración de TRAYENTA®, evaluar otras posibles causas del acontecimiento y establecer un tratamiento alternativo para la diabetes mellitus.
También se han informado casos de angioedema con otros inhibidores de la dipeptidil peptidasa-4 (DPP-4). Se debe tener precaución en pacientes con antecedentes de angioedema con otro inhibidor de la DPP-4, ya que se desconoce si estos pacientes tendrán predisposición a sufrir angioedema con TRAYENTA®.
Artralgia grave e incapacitante: Posterior a la comercialización se han informado casos de artralgia grave e incapacitante en pacientes que toman TRAYENTA® [ver Reacciones adversas]. El tiempo hasta la aparición de los síntomas luego del inicio del tratamiento con el medicamento varía de un día hasta años. Los pacientes experimentaron el alivio de los síntomas tras la interrupción de la medicación. Un subconjunto de pacientes experimentó una recurrencia de los síntomas al reiniciar el mismo medicamento o un inhibidor de la DPP-4 diferente. Se debe considerar al fármaco como una posible causa del dolor articular intenso y se debe interrumpir el medicamento, en caso necesario.
Penfigoide ampolloso: El penfigoide ampolloso se informó en 7 (0.2%) pacientes tratados con TRAYENTA®, en comparación con ninguno en los pacientes tratados con placebo en el ensayo CARMELINA [ver Estudios clínicos (Ensayos de seguridad cardiovascular en pacientes con diabetes mellitus tipo 2)], y 3 de estos pacientes fueron hospitalizados debido a penfigoide ampolloso. Luego de la comercialización, se han informado casos de penfigoide ampolloso que requirieron hospitalización en pacientes tratados con un inhibidor de la DPP-4. En los casos informados, los pacientes generalmente se recuperaron con tratamiento inmunosupresor tópico o sistémico y la suspensión del inhibidor de la DPP-4. Se debe indicar a los pacientes que informen sobre el desarrollo de ampollas o erosiones mientras reciben TRAYENTA®. Si se sospecha penfigoide ampolloso, se debe suspender la administración de TRAYENTA® y se debe considerar la derivación a un dermatólogo para el diagnóstico y tratamiento adecuado.
Insuficiencia cardíaca: En ensayos de resultados cardiovasculares para otros dos miembros de la clase de inhibidores de la DPP-4 se ha observado una asociación entre el tratamiento con inhibidores de la DPP-4 y la insuficiencia cardíaca. Estos ensayos evaluaron pacientes con diabetes mellitus tipo 2 y enfermedad cardiovascular aterosclerótica.
Se deben considerar los riesgos y beneficios de TRAYENTA® antes de iniciar el tratamiento en pacientes con riesgo de insuficiencia cardíaca, como los que tienen antecedentes de insuficiencia cardíaca y antecedentes de insuficiencia renal, y durante el tratamiento se debe observar a estos pacientes en busca de signos y síntomas de insuficiencia cardíaca. Se debe informar a los pacientes sobre los síntomas característicos de la insuficiencia cardíaca y decirles que inmediatamente reporten dichos síntomas. Si se presenta insuficiencia cardíaca, evaluar y tratar de acuerdo con los estándares actuales de atención y considerar la interrupción de TRAYENTA®.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
Toxicología preclínica:
Carcinogénesis, mutagénesis y alteración de la fertilidad: La linagliptina no incrementó la incidencia de tumores en ratas macho y hembra en un estudio de 2 años, a dosis de 6, 18 y 60 mg/kg. La dosis más alta de 60 mg/kg es aproximadamente 418 veces la dosis clínica de 5 mg/día, con base en la exposición expresada como el ABC. La linagliptina no incrementó la incidencia de tumores en ratones en un estudio de 2 años a dosis de hasta 80 mg/kg (machos) y 25 mg/kg (hembras), o aproximadamente 35 y 270 veces la dosis clínica, con base en la exposición expresada como el ABC. Dosis más altas de linagliptina en ratones hembra (80 mg/kg) incrementaron la incidencia de linfoma a dosis aproximadamente 215 veces la dosis clínica, con base en la exposición expresada como el ABC.
La linagliptina no resultó ser mutagénica ni clastogénica con o sin activación metabólica en la prueba de Ames de mutagenicidad bacteriana, en una prueba de aberraciones cromosómicas en linfocitos humanos, y en un estudio de micronúcleos in vivo.
En estudios de fertilidad realizados en ratas, la linagliptina no tuvo efectos adversos en el desarrollo embrionario temprano, en el apareamiento, la fertilidad o la gestación de crías vivas hasta la dosis más alta de 240 mg/kg (aproximadamente 943 veces la dosis clínica con base en la exposición expresada como el ABC).
REACCIONES ADVERSAS:
Las siguientes reacciones adversas graves se describen a continuación o en otra parte de la información para prescribir:
• Pancreatitis [ver Advertencias y precauciones (Pancreatitis)]
• Hipoglucemia con uso concomitante con insulina y secretagogos de insulina [ver Advertencias y precauciones (Hipoglucemia con uso concomitante con insulina y secretagogos de insulina [ver Advertencias y precauciones)]
• Reacciones de hipersensibilidad [ver Advertencias y precauciones (Reacciones de hipersensibilidad)]
• Artralgia grave e incapacitante [ver Advertencias y precauciones (Artralgia grave e incapacitante)]
• Penfigoide ampolloso [ver Advertencias y precauciones (Penfigoide ampolloso)]
• Insuficiencia cardíaca [ver Advertencias y precauciones (Insuficiencia cardíaca)]
Experiencia en ensayos clínicos: Debido a que los ensayos clínicos se realizan en condiciones sumamente variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse de manera directa con las tasas obtenidas en los ensayos clínicos de otro fármaco, y es posible que no reflejen las tasas observadas en la práctica.
La evaluación de la seguridad de TRAYENTA® 5 mg una vez al día en pacientes con diabetes mellitus tipo 2 se basa en 14 ensayos controlados con un placebo, 1 ensayo controlado con un fármaco activo y un ensayo en pacientes con insuficiencia renal grave. En los 14 ensayos controlados con un placebo, un total de 3625 pacientes fueron asignados aleatoriamente y tratados con TRAYENTA® 5 mg al día, y 2176 tratados con un placebo. La exposición promedio en los pacientes tratados con TRAYENTA® en los distintos estudios fue de 29.6 semanas. El período máximo de seguimiento fue de 78 semanas.
TRAYENTA® 5 mg una vez al día se estudió como monoterapia en tres estudios controlados con un placebo, de 18 y 24 semanas de duración, y en cinco estudios adicionales controlados con un placebo, con una duración ≤ 18 semanas. El uso de TRAYENTA® en combinación con otros antihiperglucemiantes se estudió en seis ensayos controlados con un placebo: dos con metformina (duración del tratamiento de 12 y 24 semanas); uno con una sulfonilurea (duración del tratamiento de 18 semanas); uno con metformina y sulfonilurea (duración del tratamiento de 24 semanas); uno con pioglitazona (duración del tratamiento de 24 semanas); y uno con insulina (criterio principal de valoración a las 24 semanas).
En la Tabla 1 se muestran las reacciones adversas registradas en ≥ 2% de los pacientes que recibieron TRAYENTA (n = 3,625) y con mayor frecuencia que en los pacientes que recibieron el placebo (n = 2,176), provenientes de un conjunto de datos combinados de 14 ensayos clínicos controlados con un placebo.
Tabla 1. Reacciones adversas informadas en ≥2% de los pacientes tratados con TRAYENTA® y más frecuentes que con el placebo en los estudios clínicos controlados con un placebo, de TRAYENTA® administrado como monoterapia o en combinación
|
Reacciones adversas |
TRAYENTA® 5 mg (%) n = 3,625 |
Placebo (%) n = 2,176 |
|
Nasofaringitis |
7.0 |
6.1 |
|
Diarrea |
3.3 |
3.0 |
|
Tos |
2.1 |
1.4 |
Las tasas de otras reacciones adversas con TRAYENTA® 5 mg frente al placebo cuando TRAYENTA® se administró en combinación con antidiabéticos específicos fueron: infección de las vías urinarias (3.1% frente a 0%) e hipertrigliceridemia (2.4% frente a 0%) cuando TRAYENTA® se administró como terapia complementaria a una sulfonilurea; hiperlipidemia (2.7% frente a 0.8%) y aumento de peso (2.3% frente a 0.8%) cuando TRAYENTA® se administró como terapia complementaria a la pioglitazona; y estreñimiento (2.1% frente a 1%) cuando TRAYENTA® se administró como terapia complementaria a la insulina basal. Otras reacciones adversas notificadas en estudios clínicos con tratamiento con TRAYENTA® fueron: hipersensibilidad (p. ej., urticaria, angioedema, exfoliación cutánea localizada o hiperreactividad bronquial) y mialgia.
Después de 104 semanas de tratamiento en un ensayo controlado que comparó TRAYENTA® con glimepirida, en el que todos los pacientes también recibieron metformina, las reacciones adversas registradas en ≥5% de los pacientes tratados con TRAYENTA® (n = 776) y con mayor frecuencia que en los pacientes tratados con una sulfonilurea (n=775) fueron dolor de espalda (9.1% frente a 8.4%), artralgia (8.1% frente a 6.1%), infección de las vías respiratorias altas (8.0% frente a 7.6%), cefalea (6.4% frente a 5.2%), tos (6.1% frente a 4.9%) y dolor en las extremidades (5.3% frente a 3.9%).
En el programa de ensayos clínicos se reportó pancreatitis en 15.2 casos por 10,000 años paciente de exposición durante el tratamiento con TRAYENTA®, en comparación con 3.7 casos por 10,000 años paciente de exposición durante el tratamiento con el comparador (placebo y comparador activo sulfonilurea). Después de la última dosis administrada de linagliptina se registraron tres casos adicionales de pancreatitis.
Otras reacciones adversas:
Hipoglucemia:
En la Tabla 2 se resume la incidencia de hipoglucemia en estudios de TRAYENTA® controlados con placebo. La incidencia de hipoglucemia aumentó cuando TRAYENTA® se administró con sulfonilurea o insulina.
Tabla 2: Incidencia (%) de hipoglucemia en estudios clínicos de TRAYENTA® controlados con placebo en pacientes con diabetes mellitus tipo 2
|
Adicionada a sulfonilurea (18 semanas) |
Placebo (N = 84) |
TRAYENTA® (N = 161) |
|
Hipoglucemia con glucosa plasmática <54 mg/dL (%) |
1.2 |
1.9 |
|
Hipoglucemia grave* (%) |
0 |
0 |
|
Adicionada a metformina y sulfonilurea (24 semanas) |
Placebo (N = 263) |
TRAYENTA® (N = 792) |
|
Hipoglucemia con glucosa plasmática <54 mg/dL (%) |
5.3 |
8.1 |
|
Hipoglucemia grave* (%) |
0.8 |
0.6 |
|
Adicionada a insulina basal (52 semanas) |
Placebo (N = 630) |
TRAYENTA® (N = 631) |
|
Hipoglucemia con glucosa plasmática <54 mg/dL (%) |
21.6 |
19.8 |
|
Hipoglucemia grave* (%) |
1.1 |
1.7 |
* Hipoglucemia que requiere la asistencia de otra persona para administrar activamente carbohidratos, glucagón u otras acciones de reanimación.
En un ensayo de seguridad cardiovascular (CAROLINA) con TRAYENTA®, controlado con un activo (glimepirida), con una duración mediana de 5.9 años de tratamiento, la incidencia de hipoglucemia grave fue del 0.3% en el grupo de TRAYENTA® (N = 3,014) y del 2.2% en el grupo de glimepirida (N = 3,000).
Uso en pacientes con insuficiencia renal: TRAYENTA® se comparó con un placebo, como terapia complementaria a un tratamiento antidiabético preexistente durante 52 semanas en 133 pacientes con insuficiencia renal grave (valor estimado de TFG <30 mL/min). Durante las 12 primeras semanas del ensayo, el tratamiento antidiabético de base se mantuvo estable, e incluyó insulina, sulfonilurea, glinidas y pioglitazona. Para el resto del ensayo se permitieron ajustes de la dosificación en el tratamiento antidiabético de base.
En general, la incidencia de eventos adversos, entre ellos, hipoglucemia grave, fue similar a la reportada en otros ensayos con TRAYENTA®. La incidencia observada de hipoglucemia fue mayor (TRAYENTA®, 63% en comparación con el placebo, 49%) debido a un incremento de los eventos hipoglucémicos asintomáticos, en especial durante las 12 primeras semanas cuando los tratamientos de base para el control de la glucosa plasmática se mantuvieron estables. Diez pacientes tratados con TRAYENTA® (15%) y 11 pacientes que recibieron el placebo (17%) informaron haber experimentado al menos un episodio hipoglucémico sintomático confirmado (acompañado de una prueba de glucosa en sangre por pinchazo en el dedo con un resultado ≤54 mg/dL). Durante el mismo período se informaron eventos hipoglucémicos graves, definidos como todo evento que requiera de la ayuda de otra persona para administrar activamente carbohidratos, glucagón u otras medidas de reanimación en 3 (4.4%) pacientes tratados con TRAYENTA® y en 3 (4.6%) pacientes que recibieron el placebo. Los eventos que fueron considerados potencialmente mortales o que requirieron hospitalización se reportaron en 2 (2.9%) pacientes tratados con TRAYENTA® y en 1 (1.5%) paciente que recibió el placebo.
El funcionamiento renal, medido a través del valor de la TFGe promedio y de la depuración de creatinina no varió durante las 52 semanas de tratamiento en comparación con el placebo.
Anomalías en pruebas de laboratorio en los ensayos clínicos: Los cambios en los datos de laboratorio fueron similares en los pacientes tratados con TRAYENTA® 5 mg en comparación con los pacientes que recibieron el placebo.
Aumento del ácido úrico: Los cambios en los valores de laboratorio que ocurrieron con mayor frecuencia en el grupo de TRAYENTA® y ≥1% más que en el grupo del placebo fueron aumentos en el ácido úrico (1.3% en el grupo del placebo, 2.7% en el grupo de TRAYENTA®).
Aumento de la lipasa: En un ensayo clínico de TRAYENTA® controlado con un placebo en pacientes con diabetes mellitus tipo 2 y micro o macroalbuminuria, se observó un incremento promedio del 30% en las concentraciones de lipasa desde el inicio hasta las 24 semanas en el grupo de TRAYENTA®, en comparación con una disminución promedio del 2% en el grupo del placebo. Se observaron niveles de lipasa superiores a 3 veces el límite superior del rango normal en 8.2% de los pacientes del grupo de TRAYENTA®, en comparación con 1.7% de los pacientes del grupo del placebo, respectivamente.
Aumento de la amilasa: En un ensayo de seguridad cardiovascular que comparó TRAYENTA® contra glimepirida en pacientes con diabetes mellitus tipo 2, se observaron niveles de amilasa por encima de 3 veces el límite superior normal en el 1.0%, en comparación con el 0.5% de los pacientes en los grupos de TRAYENTA® y glimepirida respectivamente.
Se desconoce la significancia clínica de las elevaciones de la lipasa y la amilasa con TRAYENTA® en ausencia de posibles signos y síntomas de pancreatitis [ver Advertencias y precauciones (Pancreatitis)].
Signos vitales: No se observaron cambios clínicamente significativos en los signos vitales en los pacientes tratados con TRAYENTA®.
Experiencia posterior a la comercialización: Se han identificado otras reacciones adversas durante el uso de TRAYENTA® después de su aprobación. Como estas reacciones se informan de forma voluntaria por parte de una población cuyo tamaño se desconoce, por lo general no resulta posible estimar de manera fiable su frecuencia ni establecer una relación causal con la exposición al fármaco.
• Trastornos gastrointestinales: Pancreatitis aguda, incluso pancreatitis mortal [ver Indicaciones y uso (Indicaciones y uso)], úlceras bucales, estomatitis.
• Trastornos del sistema inmunitario: Reacciones de hipersensibilidad que incluyen anafilaxia, angioedema y exfoliación cutánea.
• Trastornos musculoesqueléticos y del tejido conjuntivo: Rabdomiólisis, artralgia grave e incapacitante.
• Trastornos de la piel y tejido subcutáneo: Penfigoide ampolloso, erupción cutánea.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones medicamentosas:
Inductores de la glucoproteína P o de las enzimas CYP3A4: La rifampicina disminuyó la exposición a la linagliptina, lo que indica que la eficacia de TRAYENTA® puede verse reducida cuando se administra en combinación con un inductor potente de la P-gp o de las enzimas CYP3A4. Por consiguiente, se recomienda enfáticamente el uso de tratamientos alternativos cuando linagliptina se administre con un inductor potente de la P-gp o de las enzimas CYP3A4 [ver Farmacología clínica (Farmacocinética)].
Secretagogos de insulina o insulina: Se sabe que la insulina y los secretagogos de insulina causan hipoglucemia. El riesgo de hipoglucemia aumenta cuando la linagliptina se usa en combinación con un secretagogo de insulina (p. ej., una sulfonilurea) o insulina. La coadministración de TRAYENTA® con un secretagogo de insulina (p. ej., una sulfonilurea) o con insulina puede requerir dosificaciones menores del secretagogo de insulina o de insulina para reducir el riesgo de hipoglucemia [ver Advertencias y precauciones (Hipoglucemia con uso concomitante con insulina y secretagogos de insulina)].
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Uso en poblaciones específicas:
Emba razo:
Resumen de los riesgos: Los datos limitados sobre el uso de TRAYENTA® en mujeres embarazadas no son suficientes para informar un riesgo de defectos congénitos importantes y de aborto espontáneo asociados al fármaco. Existen riesgos para la madre y el feto asociados a la diabetes mal controlada durante el embarazo [ver Consideraciones clínicas].
En estudios sobre la reproducción en animales, no se observaron efectos adversos en el desarrollo cuando se administró linagliptina a ratas preñadas durante el período de organogénesis en dosis similares a la dosis clínica máxima recomendada, con base en la exposición [ver Datos].
El riesgo subyacente estimado de defectos congénitos importantes es del 6 al 10% en mujeres con diabetes pregestacional con HbA1c >7, y se ha informado que es del 20 al 25% en mujeres con HbA1c >10. Se desconoce el riesgo de fondo estimado de aborto espontáneo para la población indicada. En la población general estadounidense, el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en los embarazos clínicamente reconocidos es del 2 al 4% y del 15 al 20%, respectivamente.
Consideraciones clínicas:
Riesgo maternal y/o embriofetal asociado a la enfermedad: La diabetes mal controlada durante el embarazo aumenta el riesgo maternal de cetoacidosis diabética, preeclampsia, abortos espontáneos, partos prematuros y complicaciones en el parto. La diabetes mal controlada aumenta el riesgo fetal de defectos congénitos importantes, mortinatos y morbilidad relacionada con macrosomía.
Datos:
Datos en animales: No se observaron resultados adversos en el desarrollo cuando se administró linagliptina a ratas Wistar Han y conejas himalayas preñadas durante el período de organogénesis, en dosis de hasta 240 mg/kg/día y 150 mg/kg/día, respectivamente. Estas dosis representan alrededor de 943 veces la dosis clínica de 5 mg en ratas y 1,943 veces la dosis clínica de 5 mg en conejas, con base en la exposición. No se observaron resultados adversos funcionales, conductuales ni en la reproducción en las crías tras la administración de linagliptina a ratas Wistar Han desde el día 6 de gestación hasta el día 21 de lactancia con dosis 49 veces la dosis máxima recomendada para humanos, con base en la exposición.
La linagliptina atraviesa la placenta hacia el feto después de la administración oral en ratas y conejas preñadas.
Lactancia:
Resumen de los riesgos: No hay información acerca de la presencia de linagliptina en la leche materna, los efectos que tiene en los lactantes o los efectos en la producción de leche. No obstante, la linagliptina está presente en la leche de rata. Por lo tanto, deben tenerse en cuenta los beneficios de la lactancia materna para el desarrollo y la salud ,junto con la necesidad clínica de la madre de recibir TRAYENTA®, así como los posibles efectos adversos de TRAYENTA® o de la enfermedad materna subyacente en el niño lactante.
Uso pediátrico: No se ha establecido la seguridad y eficacia de TRAYENTA® en pacientes pediátricos.
La eficacia de TRAYENTA® no se demostró en un ensayo aleatorizado de 26 semanas, doble ciego, controlado con placebo en 157 pacientes pediátricos con edad de 10 a 17 años con diabetes mellitus tipo 2 no controlada adecuadamente.
Uso geriátrico: En los estudios de linagliptina, 1085 pacientes tratados con linagliptina tenían 65 años o más y 131 pacientes tenían 75 años o más. En estos estudios de linagliptina, no se observaron diferencias generales en la seguridad o eficacia de la linagliptina entre los pacientes geriátricos y los pacientes adultos más jóvenes.
Insuficiencia renal: No se recomienda ningún ajuste de la dosificación en los pacientes con insuficiencia renal [ver Farmacología clínica (Farmacocinética)].
En el grupo de tratamiento con TRAYENTA® del ensayo CARMELINA [ver Estudios clínicos (Estudios de seguridad cardiovascular de linagliptina)], 2200 (63%) pacientes tenían insuficiencia renal (TFGe <60 mL/min/1.73 m2). Aproximadamente 20% de la población tenía una TFGe ≥45 a <60 mL/min/1.73 m2, 28% de la población tenía una TFGe ≥30 a <45 mL/min/1.73 m2 y 15% tenía una TFGe <30 mL/min/1.73 m2. La incidencia global de reacciones adversas generalmente fue similar entre los grupos de tratamiento con TRAYENTA® y con el placebo.
Insuficiencia hepática: No se recomienda ningún ajuste de la dosis en los pacientes con insuficiencia hepática [ver Farmacología clínica (Farmacocinética)].
ESTUDIOS CLÍNICOS:
Ensayos del control glucémico en adultos con diabetes mellitus tipo 2: TRAYENTA® se ha estudiado como monoterapia y en combinación con metformina, sulfonilurea, pioglitazona e insulina. TRAYENTA® también se ha estudiado en pacientes con diabetes mellitus tipo 2 e insuficiencia renal crónica grave.
En pacientes con diabetes mellitus tipo 2, el tratamiento con TRAYENTA® produjo mejorías clínicamente significativas en los valores de hemoglobina A1c (A1C), glucosa plasmática en ayunas (GPA) y glucosa postprandial (GPP) a las 2 horas, en comparación con el placebo.
Monoterapia: Un total de 730 pacientes con diabetes mellitus tipo 2 participaron en dos estudios doble ciego, controlados con un placebo, uno de 18 semanas y el otro de 24 semanas de duración, para evaluar la eficacia y seguridad de la monoterapia con TRAYENTA®. En ambos estudios con monoterapia, los pacientes que estaban tomando un antihiperglucemiante lo suspendieron y fueron sometidos a un período de dieta y ejercicio, y a un período de lavado farmacológico de aproximadamente 6 semanas que incluyó un período abierto de preinclusión con placebo durante las 2 últimas semanas. Los pacientes que tenían un control deficiente de la glucosa plasmática (A1C 7% a 10%) después del período de lavado fueron asignados aleatoriamente; los pacientes que no estaban tomando ningún antihiperglucemiante en ese momento (sin tratamiento durante por lo menos 8 semanas) y que tenían un control deficiente de la glucosa plasmática (A1C 7 a 10%) fueron asignados aleatoriamente después de completar el período abierto de preinclusión con placebo de 2 semanas. En el ensayo de 18 semanas solo se incluyeron pacientes que no eran aptos para tomar metformina. En el ensayo de 18 semanas, 76 pacientes fueron asignados aleatoriamente para recibir el placebo y 151 para recibir TRAYENTA® 5 mg; en el ensayo de 24 semanas, 167 pacientes fueron asignados aleatoriamente para recibir el placebo, y 336 para recibir TRAYENTA® 5 mg. Los pacientes que no alcanzaron los objetivos específicos de glucosa plasmática durante el ensayo de 18 semanas recibieron tratamiento de rescate con pioglitazona y/o insulina; en el ensayo de 24 semanas se utilizó metformina para el tratamiento de rescate.
El tratamiento con TRAYENTA® 5 mg al día produjo mejoras estadísticamente significativas en la A1C, la glucosa plasmática en ayunas y la glucosa plasmática postprandial a las 2 horas, en comparación con el placebo (Tabla 5). En el ensayo de 18 semanas, 12% de los pacientes tratados con TRAYENTA® 5 mg y 18% de los que recibieron el placebo necesitaron el tratamiento de rescate. En el ensayo de 24 semanas, 10.2% de los pacientes tratados con TRAYENTA® 5 mg y 20.9% de los que recibieron el placebo necesitaron el tratamiento de rescate. La mejoría en la A1C comparada con el placebo no se vio afectada por el sexo, la edad, la raza, el tratamiento previo con antihiperglucemiantes, el IMC basal o un índice estándar de resistencia a la insulina (HOMA-IR). Como suele ocurrir en los ensayos de fármacos para tratar la diabetes mellitus tipo 2, la reducción promedio de la A1C con TRAYENTA® parece estar relacionada con el grado de elevación de la A1C al inicio del estudio. En estos estudios de 18 y 24 semanas, los cambios de la A1C con respecto a los valores al inicio del estudio fueron -0.4 y -0.4%, respectivamente, para los pacientes que recibieron TRAYENTA®, y 0.1 y 0.3%, respectivamente, para los que recibieron el placebo. El cambio en el peso corporal, respecto a los valores basales, no difirió de manera significativa entre los grupos.
Tabla 5. Parámetros glucémicos en los ensayos de monoterapia con TRAYENTA® controlados con un placebo*
|
Ensayos de 18 semanas |
Ensayos de 24 semanas |
|||
|---|---|---|---|---|
|
TRAYENTA® 5 mg |
Placebo |
TRAYENTA® 5 mg |
Placebo |
|
|
A1C (%) |
||||
|
Número de pacientes |
n = 147 |
n = 73 |
n = 333 |
n = 163 |
|
Valores iniciales (promedio) |
8.1 |
8.1 |
8.0 |
8.0 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-0.4 |
0.1 |
-0.4 |
0.3 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
-0.6 (-0.9, -0.3) |
- |
-0.7 (-0.9, -0.5) |
- |
|
Pacientes [n (%)] que alcanzaron una A1C <7%** |
32 (23.5) |
8 (11.8) |
77 (25) |
17 (12) |
|
Glucosa plasmática en ayunas (mg/dL) |
||||
|
Número de pacientes |
n = 138 |
n = 66 |
n = 318 |
n = 149 |
|
Valores iniciales (promedio) |
178 |
176 |
164 |
166 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-13 |
7 |
-9 |
15 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
-21 (-31, -10) |
- |
-23 (-30, -16) |
- |
|
Glucosa plasmática postprandial a las 2 horas (mg/dL) |
||||
|
Número de pacientes |
Datos no disponibles |
Datos no disponibles |
n = 67 |
n = 24 |
|
Valores iniciales (promedio) |
- |
- |
258 |
244 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
- |
- |
-34 |
25 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
- |
- |
-58 (-82, -34) |
- |
* Población completa para el análisis utilizando la última observación en el ensayo.
** Ensayo de 18 semanas: Placebo, n = 68; TRAYENTA®, n = 136 Estudio de 24 semanas: Placebo, n = 147; TRAYENTA®, n = 306.
*** Ensayo de 18 semanas. HbA1c: el modelo ANCOVA incluyó el tratamiento, el motivo de la intolerancia a la metformina y el número de medicamentos antidiabéticos orales previos como efectos de clase, y el valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas: el modelo ANCOVA incluyó el tratamiento, el motivo de la intolerancia a la metformina y número de medicamentos antidiabéticos orales previos como efectos de clase, y el valor de la HbA1c inicial y la glucosa plasmática en ayunas inicial como covariables continuas.
Ensayo de 24 semanas. HbA1C: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, y el valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, así como el valor de la HbA1c inicial y el valor de la glucosa plasmática en ayunas inicial como covariables continuas. Glucosa plasmática postprandial: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, así como el valor de la HbA1c inicial y el valor inicial de la glucosa plasmática postprandial a las dos horas como covariable.
Terapia de combinación complementaria con metformina: Un total de 701 pacientes con diabetes mellitus tipo 2 participaron en un ensayo de 24 semanas aleatorizado, doble ciego y controlado con un placebo, diseñado para evaluar la eficacia de TRAYENTA® en combinación con metformina. Los pacientes que ya recibían metformina (n = 491) a una dosificación de por lo menos 1500 mg por día fueron asignados aleatoriamente después de completar un período abierto de preinclusión con placebo de 2 semanas. Los pacientes que recibían metformina y otro antihiperglucemiante (n = 207) fueron asignados aleatoriamente después de un período de preinclusión de aproximadamente 6 semanas con administración de metformina (a una dosificación de por lo menos 1500 mg por día) en monoterapia. Los pacientes fueron asignados aleatoriamente para añadir la administración de TRAYENTA® 5 mg o de un placebo, administrados una vez al día. Los pacientes que no alcanzaron los objetivos específicos para la glucosa plasmática durante estos estudios recibieron tratamiento de rescate con glimepirida.
En combinación con metformina, TRAYENTA® produjo mejoras estadísticamente significativas en la A1C, en la glucosa plasmática en ayunas y en la glucosa plasmática postprandial a las 2 horas, en comparación con el placebo (Tabla 6). El tratamiento de rescate para el control de la glucosa plasmática se utilizó en 7.8% de los pacientes tratados con TRAYENTA® 5 mg y en 18.9% de los pacientes tratados con el placebo. En ambos grupos de tratamiento se observó una disminución similar del peso corporal.
Tabla 6. Parámetros glucémicos para TRAYENTA® en combinación con metformina en un ensayo controlado con un placebo*
|
TRAYENTA® 5 mg + Metformina |
Placebo + Metformina |
|
|---|---|---|
|
A1C (%) |
||
|
Número de pacientes |
n = 513 |
n = 175 |
|
Valores iniciales (promedio) |
8.1 |
8.0 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-0.5 |
0.15 |
|
Diferencia respecto al placebo + metformina (promedio ajustado) (IC 95%) |
-0.6 (-0.8, -0.5) |
- |
|
Pacientes [n (%)] que alcanzaron A1C <7%** |
127 (26.2) |
15 (9.2) |
|
Glucosa plasmática en ayunas (mg/dL) |
||
|
Número de pacientes |
n = 495 |
n = 159 |
|
Valores iniciales (promedio) |
169 |
164 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-11 |
11 |
|
Diferencia respecto al placebo + metformina (promedio ajustado) (IC 95%) |
-21 (-27, -15) |
- |
|
Glucosa plasmática postprandial a las 2 horas (mg/dL) |
||
|
Número de pacientes |
n = 78 |
n = 21 |
|
Valores iniciales (promedio) |
270 |
274 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-49 |
18 |
|
Diferencia respecto al placebo + metformina (promedio ajustado) (IC 95%) |
-67 (-95, -40) |
- |
* Población completa para el análisis utilizando la última observación en el ensayo.
** TRAYENTA® 5 mg + metformina, n=485; placebo + metformina, n=163
*** HbA1c: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial y el valor de la glucosa plasmática en ayunas inicial como covariables continuas. Glucosa plasmática postprandial: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial y de la glucosa plasmática postprandial a las dos horas como covariables.
Terapia inicial de combinación con metformina: Un total de 791 pacientes con diabetes mellitus tipo 2 y con un control deficiente de la glucosa plasmática con dieta y ejercicio participaron en la porción de 24 semanas aleatorizada y doble ciega de este ensayo factorial y controlado con un placebo, diseñado para evaluar la eficacia de TRAYENTA® como tratamiento inicial con metformina. Los pacientes que recibían un antihiperglucemiante (52%) fueron sometidos a un período de lavado farmacológico durante 4 semanas. Después del período de lavado, y tras completar un período de preinclusión con placebo de 2 semanas, ciego simple, los pacientes que presentaban un control deficiente de la glucosa plasmática (A1C ≥7.0% a ≤10.5%) fueron asignados aleatoriamente. Los pacientes que presentaban un control deficiente de la glucosa plasmática (A1C ≥7.5% a <11.0%) y que no recibían antihiperglucemiantes al ingresar al ensayo (48%) entraron de inmediato al período de preinclusión con placebo de 2 semanas, ciego simple, y luego fueron asignados aleatoriamente. La aleatorización fue estratificada según el valor inicial de la A1C (<8.5% frente a ≥8.5%) y el uso previo de antidiabéticos orales (ninguno frente a monoterapia). Los pacientes fueron asignados aleatoriamente en una proporción de 1:2:2:2:2:2 al grupo del placebo o a uno de los 5 grupos de tratamiento activo. Aproximadamente el mismo número de pacientes fueron asignados aleatoriamente para recibir tratamiento inicial con 5 mg de TRAYENTA® una vez al día, 500 mg o 1000 mg de metformina dos veces al día, o 2.5 mg de linagliptina dos veces al día en combinación con 500 mg o 1000 mg de metformina dos veces al día. Los pacientes que no alcanzaron los objetivos específicos para la glucosa plasmática durante el ensayo recibieron tratamiento de rescate con una sulfonilurea, tiazolidindiona o insulina.
El tratamiento inicial con la combinación de linagliptina y metformina produjo mejoras significativas en la A1C y en la glucosa plasmática en ayunas GPA), en comparación con el placebo, con la metformina sola y con la linagliptina sola (Tabla 7).
El promedio ajustado de la diferencia entre los tratamientos en el valor de A1C desde el inicio hasta la semana 24 (LOCF) fue -0.5% (IC 95% -0.7, -0.3; p <0.0001) para el grupo de linagliptina 2.5 mg/metformina 1000 mg dos veces al día, en comparación con metformina 1000 mg dos veces al día; -1.1% (IC 95% -1.4, -0.9; p <0.0001) para el grupo de linagliptina 2.5 mg/metformina 1000 mg dos veces al día, en comparación con TRAYENTA® 5 mg una vez al día; -0.6% (IC 95% -0.8, -0.4; p <0.0001) para el grupo de linagliptina 2.5 mg/metformina 500 mg dos veces al día, en comparación con metformina 500 mg dos veces al día; y -0.8% (IC 95% -1.0, -0.6; p <0.0001) para el grupo de linagliptina 2.5 mg/metformina 500 mg dos veces al día, en comparación con TRAYENTA® 5 mg una vez al día.
Los efectos sobre los lípidos generalmente fueron neutros. No se observó ningún cambio importante en el peso corporal en ninguno de los 6 grupos de tratamiento.
Tabla 7. Parámetros glucémicos en la visita final (ensayo de 24 semanas) para linagliptina y metformina, solos y en combinación, en pacientes asignados aleatoriamente que padecían diabetes mellitus tipo 2 no controlada adecuadamente con la dieta y ejercicio**
|
Placebo |
TRAYENTA® 5 mg una vez al día* |
Metformina 500 mg dos veces al día |
Linagliptina 2.5 mg dos veces al día* + Metformina 500 mg dos veces al día |
Metformina 1000 mg dos veces al día |
Linagliptina 2.5 mg dos veces al día* + Metformina 1000 mg dos veces al día |
|
|
A1C (%) |
||||||
|
Número de pacientes |
n = 65 |
n = 135 |
n = 141 |
n = 137 |
n = 138 |
n = 140 |
|
Valores iniciales (promedio) |
8.7 |
8.7 |
8.7 |
8.7 |
8.5 |
8.7 |
|
Cambio respecto a los valores iniciales (promedio ajustado****) |
0.1 |
-0.5 |
-0.6 |
-1.2 |
-1.1 |
-1.6 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
- |
-0.6 (-0.9, -0.3) |
-0.8 (-1.0, -0.5) |
-1.3 (-1.6, -1.1) |
-1.2 (-1.5, -0.9) |
-1.7 (-2.0, -1.4) |
|
Pacientes [n (%)] que alcanzaron A1C <7%*** |
7 (10.8) |
14 (10.4) |
26 (18.6) |
41 (30.1) |
42 (30.7) |
74 (53.6) |
|
Pacientes (%) que recibieron medicamentos de rescate |
29.2 |
11.1 |
13.5 |
7.3 |
8.0 |
4.3 |
|
Glucosa plasmática en ayunas (mg/dL) |
||||||
|
Número de pacientes |
n = 61 |
n = 134 |
n = 136 |
n = 135 |
n = 132 |
n = 136 |
|
Valores iniciales (promedio) |
203 |
195 |
191 |
199 |
191 |
196 |
|
Cambio respecto a los valores iniciales (promedio ajustado****) |
10 |
-9 |
-16 |
-33 |
-32 |
-49 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
- |
-19 (-31, -6) |
-26 (-38, -14) |
-43 (-56, -31) |
-42 (-55, -30) |
-60 (-72, -47) |
* La dosificación diaria total de TRAYENTA® es igual a 5 mg
** Población completa para el análisis utilizando la última observación en el ensayo.
***Metformina 500 mg dos veces al día, n=140; linagliptina 2.5 mg dos veces al día + metformina 500 mg dos veces al día, n=136; metformina 1000 mg dos veces al día, n=137; linagliptina 2.5 mg dos veces al día + metformina 1000 mg dos veces al día, n=138
****HbA1c: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial y el valor de la glucosa plasmática en ayunas inicial como covariables continuas.
Ensayo controlado con un fármaco activo frente a glimepirida en combinación con metformina: La eficacia de TRAYENTA® se evaluó en un ensayo de no inferioridad de 104 semanas, doble ciego, controlado con glimepirida, en pacientes con diabetes tipo 2 y con un control insuficiente de la glucosa plasmática a pesar de recibir tratamiento con metformina. Los pacientes que recibían tratamiento solo con metformina ingresaron a un período de preinclusión de 2 semanas de duración, mientras que los pacientes con tratamiento previo con metformina y otro antihiperglucemiante ingresaron a un período de pretratamiento de 6 semanas de duración con monoterapia con metformina (dosificación ≥1,500 mg/día) y lavado del otro fármaco. Después de un período adicional de preinclusión de 2 semanas con placebo, los pacientes que presentaban un control deficiente de la glucosa plasmática (A1C 6.5 a 10%) fueron asignados aleatoriamente en una proporción de 1:1 para la adición de TRAYENTA® 5 mg una vez al día o glimepirida. La aleatorización fue estratificada según el valor de la HbA1c inicial (<8.5% frente a ≥8.5%) y el uso previo de antidiabéticos (metformina sola frente a metformina más otro antidiabético oral). A los pacientes que recibían glimepirida se les dio una dosificación inicial de 1 mg/día y luego se les ajustó la dosis de manera opcional durante las siguientes 12 semanas hasta una dosificación máxima de 4 mg/día, según fuera necesario para optimizar el control de la glucosa plasmática. A partir de ahí, la dosificación de glimepirida debía mantenerse constante, excepto en los momentos en que debía disminuirse la dosis para prevenir una hipoglucemia.
Después de 52 y 104 semanas, tanto TRAYENTA® como glimepirida produjeron disminuciones del valor de A1C inicial (52 semanas: -0.4% para TRAYENTA®, -0.6% para glimepirida; 104 semanas: -0.2% para TRAYENTA®, -0.4% para glimepirida) desde un valor promedio inicial de 7.7% (Tabla 8). La diferencia promedio entre los grupos en el cambio del valor de A1C desde el inicio fue de 0.2% con un intervalo de confianza bilateral al 97.5% (0.1%, 0.3%) para el análisis de la población por intención de tratar, utilizando la última observación considerada. Estos resultados fueron congruentes con el análisis de los pacientes que completaron el estudio.
Tabla 8. Parámetros glucémicos a las 52 y a las 104 semanas en un ensayo que comparó TRAYENTA® con glimepirida como tratamiento complementario en pacientes no controlados adecuadamente con metformina**
|
Semana 52 |
Semana 104 |
|||
|
TRAYENTA® 5 mg + Metformina |
Glimepirida + Metformina (dosificación media de glimepirida 3 mg) |
TRAYENTA® 5 mg + Metformina |
Glimepirida + Metformina (dosificación media de glimepirida 3 mg) |
|
|
A1C (%) |
||||
|
Número de pacientes |
n = 764 |
n = 755 |
n = 764 |
n = 755 |
|
Valores iniciales (promedio) |
7.7 |
7.7 |
7.7 |
7.7 |
|
Cambio respecto a los valores iniciales (promedio ajustado****) |
-0.4 |
-0.6 |
-0.2 |
-0.4 |
|
Diferencia respecto a la glimepirida (promedio ajustado) (IC 97.5%) |
0.2 (0.1, 0.3) |
– |
0.2 (0.1, 0.3) |
– |
|
Glucosa plasmática en ayunas (mg/dL) |
||||
|
Número de pacientes |
n = 733 |
n = 725 |
n = 733 |
n = 725 |
|
Valores iniciales (promedio) |
164 |
166 |
164 |
166 |
|
Cambio respecto a los valores iniciales (promedio ajustado****) |
-8* |
-15 |
-2† |
-9 |
|
Incidencia de hipoglucemia (%)*** |
||||
|
Número de pacientes |
n = 776 |
n = 775 |
n = 776 |
n = 775 |
|
Incidencia**** |
5.3* |
31.1 |
7.5* |
36.1 |
* p <0.0001 frente a glimepirida; †p=0.0012 frente a glimepirida
** Población completa para el análisis utilizando la última observación en el ensayo.
*** La incidencia de hipoglucemia incluyó eventos asintomáticos (no acompañados por los síntomas característicos y una concentración plasmática de glucosa ≤70 mg/dL) y eventos sintomáticos con los síntomas característicos de hipoglucemia y una concentración plasmática de glucosa ≤70 mg/dL.
**** HbA1c: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial y el valor de la glucosa plasmática en ayunas inicial como covariables continuas. Incidencia de hipoglucemia (%): se realizó la prueba de Cochran-Mantel-Haenszel en la población de pacientes que formaban parte del conjunto de pacientes tratados, para comparar la proporción de pacientes con eventos de hipoglucemia entre los pacientes tratados con linagliptina y los pacientes tratados con glimepirida.
Los pacientes tratados con linagliptina tuvieron un peso corporal inicial promedio de 86 kg, y se observó que tuvieron una disminución del promedio ajustado del peso corporal de 1.1 kg a las 52 semanas y de 1.4 kg a las 104 semanas. Los pacientes que recibieron glimepirida tuvieron un peso corporal inicial promedio de 87 kg, y se observó que tuvieron un incremento del promedio ajustado del peso corporal desde el valor inicial de 1.4 kg a las 52 semanas y de 1.3 kg a las 104 semanas (diferencia entre los tratamientos p <0.0001 para ambos tiempos de medición).
Terapia de combinación complementaria con pioglitazona: Un total de 389 pacientes con diabetes mellitus tipo 2 participaron en un ensayo de 24 semanas aleatorizado, doble ciego y controlado con un placebo, diseñado para evaluar la eficacia de TRAYENTA® en combinación con pioglitazona. El tratamiento se suspendió en los pacientes que estaban recibiendo tratamiento con antihiperglucemiantes orales durante un período de 6 semanas (4 semanas seguidas por un período abierto de preinclusión con placebo de 2 semanas). Los pacientes que no habían recibido tratamiento previo ingresaron directamente al período de preinclusión con placebo de 2 semanas. Después del período de preinclusión, los pacientes fueron asignados aleatoriamente para recibir TRAYENTA® 5 mg o el placebo, ambos sumados a la administración de pioglitazona 30 mg diarios. Los pacientes que no alcanzaron los objetivos específicos de glucosa plasmática durante los estudios recibieron tratamiento de rescate con metformina. Los criterios de valoración glucémicos determinados fueron el valor de A1C y la glucosa plasmática en ayunas.
En la combinación inicial con pioglitazona 30 mg, TRAYENTA® 5 mg produjo mejoras estadísticamente significativas en la A1C y en la glucosa plasmática en ayunas, en comparación con el placebo con pioglitazona (Tabla 9). El tratamiento de rescate se utilizó en 7.9% de los pacientes tratados con TRAYENTA® 5 mg/pioglitazona 30 mg y en 14.1% de los pacientes tratados con placebo/pioglitazona 30 mg. El peso de los pacientes aumentó en ambos grupos durante el ensayo, con un cambio promedio ajustado respecto a los valores iniciales de 2.3 kg y 1.2 kg en los grupos tratados con TRAYENTA 5 mg/pioglitazona 30 mg y placebo/pioglitazona 30 mg, respectivamente (p = 0.0141).
Tabla 9. Parámetros glucémicos para TRAYENTA® en combinación con pioglitazona en un ensayo controlado con un placebo*
|
TRAYENTA® 5 mg + Pioglitazona |
Placebo + Pioglitazona |
|
|
A1C (%) |
||
|
Número de pacientes |
n = 252 |
n = 128 |
|
Valores iniciales (promedio) |
8.6 |
8.6 |
|
Cambio respecto a los valores iniciales (promedio ajustado**) |
-1.1 |
-0.6 |
|
Diferencia respecto al placebo + pioglitazona (promedio ajustado) (IC 95%) |
-0.5 (-0.7, -0.3) |
- |
|
Pacientes [n (%)] que alcanzaron A1C <7% |
108 (42.9) |
39 (30.5) |
|
Glucosa plasmática en ayunas (mg/dL) |
||
|
Número de pacientes |
n = 243 |
n = 122 |
|
Valores iniciales (promedio) |
188 |
186 |
|
Cambio respecto a los valores iniciales (promedio ajustado**) |
-33 |
-18 |
|
Diferencia respecto al placebo + pioglitazona (promedio ajustado) (IC 95%) |
-14 (-21, -7) |
- |
* Población completa para el análisis utilizando la última observación en el ensayo
** HbA1c: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial y el valor de la glucosa plasmática en ayunas inicial como covariables continuas.
Terapia de combinación complementaria con sulfonilureas: Un total de 245 pacientes con diabetes mellitus tipo 2 participaron en un ensayo de 18 semanas aleatorizado, doble ciego y controlado con un placebo, diseñado para evaluar la eficacia de TRAYENTA® en combinación con una sulfonilurea (SU). Los pacientes que recibían monoterapia con una sulfonilurea (n = 142) fueron asignados aleatoriamente después de completar un período de preinclusión con placebo, ciego simple, de 2 semanas. Los pacientes que recibían una sulfonilurea más otro antihiperglucemiante oral (n = 103) fueron asignados aleatoriamente después de un período de lavado farmacológico de 4 semanas y un período de preinclusión con placebo, ciego simple, de 2 semanas. Los pacientes fueron asignados aleatoriamente para añadir la administración de TRAYENTA® 5 mg o de un placebo, cada uno administrado una vez al día. Los pacientes que no alcanzaron los objetivos específicos de glucosa plasmática durante los estudios recibieron tratamiento de rescate con metformina. Los criterios de valoración glucémicos determinados incluyeron la A1C y la glucosa plasmática en ayunas.
En combinación con una sulfonilurea, TRAYENTA® produjo mejoras estadísticamente significativas en la A1C, en comparación con el placebo después de 18 semanas de tratamiento; las mejorías que se observaron en la glucosa plasmática en ayunas con TRAYENTA® no fueron estadísticamente significativas en comparación con el placebo (Tabla 10). Se utilizó tratamiento de rescate en el 7.6% de los pacientes tratados con TRAYENTA® 5 mg y en el 15.9% de los pacientes tratados con el placebo. No se observó una diferencia significativa entre TRAYENTA® y el placebo en cuanto al peso corporal.
Tabla 10. Parámetros glucémicos para TRAYENTA® en combinación con una sulfonilurea en un ensayo controlado con un placebo*
|
TRAYENTA® 5 mg + SU |
Placebo + SU |
|
|---|---|---|
|
A1C (%) |
||
|
Número de pacientes |
n = 158 |
n = 82 |
|
Valores iniciales (promedio) |
8.6 |
8.6 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-0.5 |
-0.1 |
|
Diferencia respecto al placebo + SU (promedio ajustado) (IC 95%) |
-0.5 (-0.7, -0.2) |
– |
|
Pacientes [n (%)] que alcanzaron A1C <7%** |
23 (14.7) |
3 (3.7) |
|
Glucosa plasmática en ayunas (mg/dL) |
||
|
Número de pacientes |
n = 155 |
n = 78 |
|
Valores iniciales (promedio) |
180 |
171 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-8 |
-2 |
|
Diferencia respecto al placebo + SU (promedio ajustado) (IC 95%) |
-6 (-17, 4) |
– |
SU = sulfonilurea
* Población completa para el análisis utilizando la última observación en el ensayo.
** TRAYENTA® 5 mg + SU, n = 156; placebo + SU, n = 82
*** HbA1c: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas: el modelo ANCOVA incluyó el tratamiento y el número de medicamentos antidiabéticos orales previos como efectos de clase, además del valor de la HbA1c inicial y el valor de la glucosa plasmática en ayunas inicial como covariables continuas.
Terapia de combinación complementaria con metformina y una sulfonilurea: Un total de 1058 pacientes con diabetes mellitus tipo 2 participaron en un ensayo de 24 semanas aleatorizado, doble ciego, controlado con un placebo, diseñado para evaluar la eficacia de TRAYENTA® en combinación con una sulfonilurea y metformina. Las sulfonilureas utilizadas con mayor frecuencia por los pacientes del ensayo fueron: glimepirida (31%), glibenclamida (26%) y gliclazida (26%, no está disponible en los Estados Unidos). Los pacientes que recibían una sulfonilurea y metformina fueron asignados aleatoriamente para recibir TRAYENTA® 5 mg o un placebo, cada uno administrado una vez al día. Los pacientes que no alcanzaron los objetivos específicos de glucosa plasmática durante el ensayo recibieron tratamiento de rescate con pioglitazona. Los criterios de valoración glucémicos determinados incluyeron la A1C y la glucosa plasmática en ayunas.
En combinación con una sulfonilurea y metformina, TRAYENTA® produjo mejoras estadísticamente significativas en la A1C y en la glucosa plasmática en ayunas, en comparación con el placebo (Tabla 11). En la población total del ensayo (pacientes que recibieron TRAYENTA® en combinación con una sulfonilurea y metformina), se observó una disminución promedio de los valores iniciales de A1C de -0.6% y de la glucosa plasmática en ayunas de -13 mg/dL, en comparación con el placebo. Se utilizó tratamiento de rescate en 5.4% de los pacientes tratados con TRAYENTA® 5 mg y en 13% de los pacientes tratados con el placebo. El cambio respecto a los valores iniciales del peso corporal no difirió de manera significativa entre los grupos.
Tabla 11. Parámetros glucémicos para TRAYENTA® en combinación con metformina y una sulfonilurea en un ensayo controlado con un placebo*
|
TRAYENTA® 5 mg + Metformina + SU |
Placebo + Metformina + SU |
|
|
A1C (%) |
||
|
Número de pacientes |
n = 778 |
n = 262 |
|
Valores iniciales (promedio) |
8.2 |
8.1 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-0.7 |
-0.1 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
-0.6 (-0.7, -0.5) |
– |
|
Pacientes [n (%)] que alcanzaron A1C <7%** |
217 (29.2) |
20 (8.1) |
|
Glucosa plasmática en ayunas (mg/dL) |
||
|
Número de pacientes |
n = 739 |
n = 248 |
|
Valores iniciales (promedio) |
159 |
163 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-5 |
8 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
-13 (-18, -7) |
– |
SU=sulfonilurea
* Población completa para el análisis utilizando la última observación en el ensayo
** TRAYENTA® 5 mg + metformina + SU, n=742; placebo + metformina + SU, n=247
*** HbA1c: el modelo ANCOVA incluyó el tratamiento como efectos de clase y el valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas; el modelo ANCOVA incluyó el tratamiento como efectos de clase, además del valor de la HbA1c inicial y el valor de la glucosa plasmática en ayunas inicial como covariables continuas.
Terapia de combinación complementaria con insulina:
Un total de 1261 pacientes con diabetes mellitus tipo 2 no controlados adecuadamente con insulina basal sola o insulina basal en combinación con fármacos orales, participaron en un ensayo aleatorizado, doble ciego, controlado con un placebo, diseñado para evaluar la eficacia de TRAYENTA® como tratamiento complementario a la insulina basal durante 24 semanas. La aleatorización fue estratificada según el valor de la HbA1c inicial (<8.5% frente a ≥8.5%), la determinación del estado de insuficiencia renal (según el valor de la TFGe inicial) y el uso concomitante de antidiabéticos orales (ninguno, solo metformina, solo pioglitazona, metformina + pioglitazona). Los pacientes con un valor de A1C inicial ≥7% y ≤10% fueron incluidos en el ensayo, entre ellos 709 pacientes con insuficiencia renal (TFGe <90 mL/min), clasificada en la mayoría de los casos (n = 575) como insuficiencia renal leve (TFGe 60 a <90 mL/min). Los pacientes ingresaron a un período de preinclusión con placebo de 2 semanas bajo tratamiento con insulina basal (p. ej., insulina glargina, insulina detemir o NPH insulina) con o sin tratamiento de base con metformina y/o pioglitazona. Después del período de preinclusión, los pacientes con un control deficiente de la glucosa plasmática fueron asignados aleatoriamente para añadir la administración de 5 mg de TRAYENTA® o un placebo, administrados una vez al día. Se mantuvo a los pacientes con una dosificación estable de insulina antes de la inscripción en el estudio, durante el período de preinclusión y durante las primeras 24 semanas de tratamiento. Los pacientes que no alcanzaron los objetivos específicos de glucosa plasmática durante el período de tratamiento doble ciego recibieron tratamiento de rescate mediante el incremento de la dosificación de insulina de base.
TRAYENTA® utilizado en combinación con insulina (con o sin metformina y/o pioglitazona) produjo mejoras estadísticamente significativas en la A1C y en la glucosa plasmática en ayunas, en comparación con el placebo (Tabla 12) después de 24 semanas de tratamiento. La dosificación diaria total promedio de insulina al inicio fue de 42 unidades para los pacientes tratados con TRAYENTA®, y de 40 unidades para los pacientes a los que se les administró el placebo. El tratamiento de base inicial para la diabetes mellitus incluyó la administración de: insulina sola (16.1%), insulina combinada con metformina solamente (75.5%), insulina combinada con metformina y pioglitazona (7.4%) e insulina combinada con pioglitazona solamente (1%). El cambio promedio desde el inicio del estudio hasta la semana 24 en la dosificación diaria de insulina fue de + 1.3 UI en el grupo del placebo y de + 0.6 UI en el grupo de TRAYENTA®. El cambio promedio del peso corporal desde el inicio del estudio hasta la semana 24 fue similar en los dos grupos de tratamiento.
Tabla 12. Parámetros glucémicos para TRAYENTA® en combinación con insulina en un ensayo controlado con un placebo*
|
TRAYENTA® 5 mg + Insulina |
Placebo + Insulina |
|
|
A1C (%) |
||
|
Número de pacientes |
n = 618 |
n = 617 |
|
Valores iniciales (promedio) |
8.3 |
8.3 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-0.6 |
0.1 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
-0.7 (-0.7, -0.6) |
– |
|
Pacientes [n (%)] que alcanzaron A1C <7%** |
116 (19.5) |
48 (8.1) |
|
Glucosa plasmática en ayunas (mg/dL) |
||
|
Número de pacientes |
n = 613 |
n = 608 |
|
Valores iniciales (promedio) |
147 |
151 |
|
Cambio respecto a los valores iniciales (promedio ajustado***) |
-8 |
3 |
|
Diferencia respecto al placebo (promedio ajustado) (IC 95%) |
-11 (-16, -6) |
– |
* Población completa para el análisis utilizando el método de la última observación considerada (LOCF) en el ensayo.
** TRAYENTA® + insulina, n=595; placebo + insulina, n=593
*** HbA1c: el modelo ANCOVA incluyó el tratamiento, la determinación de categoría de estado de insuficiencia renal y el uso concomitante de antidiabéticos orales como efectos de clase, además del valor de la HbA1c inicial como covariables continuas. Glucosa plasmática en ayunas: el modelo ANCOVA incluyó el tratamiento, la determinación de categoría del estado de insuficiencia renal y el uso concomitante de antidiabéticos orales como efectos de clase, además del valor de la HbA1c inicial y el valor de la glucosa plasmática en ayunas inicial como covariables continuas.
La diferencia entre el tratamiento con linagliptina y con el placebo en términos del cambio promedio ajustado de cambio en la HbA1c inicial después de 24 semanas fue similar para los pacientes sin insuficiencia renal (TFGe ≥90 mL/min, n = 539), con insuficiencia renal leve (TFGe 60 a <90 mL/min, n = 565) o con insuficiencia renal moderada (TFGe 30 a <60 mL/min, n = 124).
Insuficiencia renal: Un total de 133 pacientes con diabetes mellitus tipo 2 participaron en un ensayo de 52 semanas aleatorizado, doble ciego, controlado con un placebo, diseñado para evaluar la eficacia y la seguridad de TRAYENTA® en pacientes con diabetes mellitus tipo 2 e insuficiencia renal crónica grave. Los participantes con un valor estimado (con base en la ecuación de cuatro variables del estudio de la Modificación de la Dieta en la Enfermedad Renal [MDRD]) de TFG <30 mL/min fueron elegibles para participar en el ensayo. La aleatorización fue estratificada por valor inicial de la HbA1c (≤8% y >8%) y tratamiento antidiabético de base (insulina o cualquier combinación con insulina, SU o glinidas como monoterapia y pioglitazona o cualquier otro antidiabético, excepto cualquier otro inhibidor de DPP-4). En las 12 primeras semanas del ensayo, se mantuvo estable el tratamiento antidiabético de base, el cual incluyó insulina, sulfonilureas, glinidas y pioglitazona. Durante el resto del ensayo se permitieron ajustes de la dosificación en el tratamiento antidiabético de base. Al inicio del ensayo, el 62.5% de los pacientes recibían solo insulina como tratamiento para la diabetes mellitus de base, y el 12.5% recibían sólo una sulfonilurea.
Después de 12 semanas de tratamiento, TRAYENTA® 5 mg proporcionó una mejora estadísticamente significativa en los niveles de A1C en comparación con el placebo, con un cambio promedio ajustado de cambio de -0.6% en comparación con el placebo (intervalo de confianza al 95%: -0.9, -0.3) con base en el análisis que utiliza la última observación considerada (LOCF). Con ajustes al tratamiento antidiabético de base después de las primeras 12 semanas, la eficacia se mantuvo durante 52 semanas, con un promedio ajustado de cambio en A1C de -0.7% desde el inicio del estudio en comparación con el placebo (intervalo de confianza al 95%: - 1.0, -0.4) con base en el análisis que utiliza la LOCF.
Ensayos de seguridad cardiovascular en pacientes condiabetes mellitus tipo 2:
CARMELINA: El riesgo cardiovascular de TRAYENTA® se evaluó en CARMELINA, un ensayo multinacional, multicéntrico, controlado con un placebo, doble ciego y con grupos paralelos, en el que se comparó TRAYENTA® (N = 3494) con un placebo (N = 3485) en pacientes adultos con diabetes mellitus tipo 2 y antecedentes de enfermedad renal y/o macrovascular establecida. El ensayo comparó el riesgo de eventos adversos cardiovasculares mayores (MACE, major adverse cardiovascular events) entre TRAYENTA® y el placebo cuando se agregaron a los tratamientos estándar de atención para la diabetes mellitus y otros factores de riesgo cardiovascular. El ensayo fue un estudio de duración determinada por eventos, la mediana de la duración del seguimiento fue de 2.2 años, y se obtuvo el estado vital del 99.7% de los pacientes.
Los pacientes eran elegibles para ingresar en el ensayo si eran adultos con diabetes mellitus tipo 2, con HbA1c de 6.5% a 10%, y tenían albuminuria o enfermedad macrovascular previa (39% de la población inscrita), o indicios de deterioro de la función renal según los criterios de la TFGe y del cociente de albúmina/creatinina en orina (UACR, Urinary Albumin Creatinine Ratio) (42% de la población inscrita), o ambos (18% de la población inscrita).
Al inicio del estudio la edad promedio era de 66 años, y el 63% de la población era de sexo masculino, 80% caucásica, 9% asiática y 6% de raza negra o afroamericana y 36% hispana o latina. El valor promedio de la HbA1c era del 8.0% y la duración promedio de la diabetes mellitus tipo 2 era de 15 años. La población del ensayo incluyó 17% de los pacientes ≥ 75 años de edad y 62% de los pacientes con insuficiencia renal definida como TFGe <60 mL/min/1.73 m2. La TFGe promedio era de 55 mL/min/1.73 m2, y 27% de los pacientes tenían insuficiencia renal leve (TFGe de 60 a 90 mL/min/1.73 m2), 47% de los pacientes tenían insuficiencia renal moderada (TFGe de 30 a <60 mL/min/1.73 m2) y 15% de los pacientes tenían insuficiencia renal grave (TFGe <30 mL/min/1.73 m2). Los pacientes estaban tomando al menos un fármaco antidiabético (97%), y los utilizados con mayor frecuencia fueron insulina y análogos (57%), metformina (54%) y sulfonilurea (32%). Los pacientes también estaban tomando antihipertensivos (96%), antilipemiantes (76%) con estatinas en el 72% de los casos, y aspirina (62%).
El criterio principal de valoración, MACE, fue el tiempo hasta la primera aparición de uno de los tres resultados compuestos que incluyeron muerte cardiovascular, infarto de miocardio no mortal o accidente cerebrovascular no mortal. El ensayo fue diseñado como un estudio de no inferioridad con un margen de riesgo preespecificado de 1.3 para el cociente de riesgo de MACE.
Los resultados de CARMELINA, incluida la contribución de cada componente al criterio principal de valoración compuesto, se presentan en la Tabla 13. El cociente de riesgos estimado para MACE asociados a TRAYENTA® en comparación con el placebo fue de 1.02 con un intervalo de confianza al 95% de (0.89, 1.17). El límite superior del intervalo de confianza, 1.17, excluyó el margen de riesgo de 1.3. La curva de Kaplan-Meier que representa el tiempo hasta la primera aparición de MACE se muestra en la Figura 1.
Tabla 13. Eventos cardiovasculares adversos mayores (MACE) por grupo de tratamiento en el ensayo CARMELINA
|
TRAYENTA® 5 mg n = 3494 |
Placebo n = 3485 |
Cociente de riesgos |
|||
|
Número de sujetos (%) |
Tasa de incidencia por 1000 AP* |
Número de sujetos (%) |
Tasa de incidencia por 1000 AP* |
(IC 95%) |
|
|
Compuesto de primer evento de muerte CV, infarto de miocardio (IM) no mortal, o accidente cerebrovascular no mortal (MACE) |
434 (12.4) |
57.7 |
420 (12.1) |
56.3 |
1.02 (0.89, 1.17) |
|
Muerte CV** |
255 (7.3) |
32.6 |
264 (7.6) |
34.0 |
0.96 (0.81, 1.14) |
|
IM no mortal** |
156 (4.5) |
20.6 |
135 (3.9) |
18.0 |
1.15 (0.91, 1.45) |
|
Accidente cerebrovascular no mortal** |
65 (1.9) |
8.5 |
73 (2.1) |
9.6 |
0.88 (0.63, 1.23) |
* AP=años-paciente
** Un paciente puede haber experimentado más de un componente, por lo que la suma de los componentes es mayor que el número de pacientes que experimentaron el resultado compuesto.
Figura 1. Kaplan-Meier: Tiempo hasta el primer MACE en el ensayo CARMELINA

CAROLINA: El riesgo cardiovascular de la TRAYENTA® se evaluó en CAROLINA, un ensayo multicéntrico, multinacional, aleatorizado, doble ciego, de grupos paralelos que comparó TRAYENTA® (N = 3023) con glimepirida (N = 3010) en pacientes adultos con diabetes mellitus tipo 2 y antecedentes de enfermedad cardiovascular establecida y/o múltiples factores de riesgo cardiovascular. El ensayo comparó el riesgo de eventos cardiovasculares adversos mayores (MACE, major adverse cardiovascular events) entre TRAYENTA® y glimepirida cuando estos fármacos se agregaron a los tratamientos estándar para la diabetes mellitus y otros factores de riesgo cardiovascular. El ensayo se basó en eventos, la duración mediana del seguimiento fue de 6.23 años y el estado vital se obtuvo para el 99.3% de los pacientes.
Los pacientes eran elegibles para participar en el ensayo si eran adultos con diabetes mellitus tipo 2 con control glucémico insuficiente (definido como HbA1c de 6.5% a 8.5% o de 6.5% a 7.5%, dependiendo de si no habían recibido tratamiento previo, si estaban siendo tratados con monoterapia o con terapia combinada), y se definieron como de alto riesgo cardiovascular con enfermedad vascular previa, evidencia de daño vascular relacionado con el órgano afectado, edad ≥70 años y/o dos factores de riesgo cardiovascular (duración de la diabetes mellitus >10 años, presión arterial sistólica >140 mmHg, fumador actual, colesterol LDL ≥135 mg dL).
Al inicio del estudio, la edad promedio fue de 64 años y 60% de la población era masculina, 73% caucásica, 18% asiática y 5% de raza negra o afroamericana y 17% hispana o latina. La HbA1c promedio fue de 7.15% y la duración promedio de la diabetes mellitus tipo 2 fue de 7.6 años. La población del ensayo incluyó 34% de pacientes ≥ 70 años y 19% de pacientes con insuficiencia renal definida como TFGe < 60 mL/min/1.73 m2. La TFGe promedio fue de 77 mL/ min/1.73 m2. Los pacientes tomaban al menos un fármaco antidiabético (91%), y los más frecuentes eran metformina (83%) y sulfonilurea (28%). Los pacientes también tomaban antihipertensivos (89 %), hipolipemiantes (70 %) con 65 % que tomaban estatina y aspirina (47%).
El criterio principal de valoración, MACE, fue el tiempo transcurrido hasta la primera aparición de uno de los tres resultados compuestos que incluyeron muerte cardiovascular, infarto de miocardio no mortal o accidente cerebrovascular no mortal. El ensayo se diseñó como un ensayo de no inferioridad con un margen de riesgo preespecificado de 1.3 para el límite superior del IC al 95% para el cociente de riesgos (HR) de MACE.
Los resultados de CAROLINA, incluida la contribución de cada componente al criterio principal de valoración compuesto, se muestran en la Tabla 14. La curva de Kaplan-Meier que representa el tiempo hasta la primera aparición de MACE se muestra en la Figura 2.
Tabla 14. Eventos cardiovasculares adversos mayores (MACE) por grupo de tratamiento en el ensayo CAROLINA
|
Linagliptina 5 mg n = 3023 |
Glimepirida (1 mg a 4 mg) n = 3010 |
Cociente de riesgos (HR) |
|||
|
Número de sujetos (%) |
Tasa de incidencia por 1000 AP* |
Número de sujetos (%) |
Tasa de incidencia por 1000 AP* |
(IC 95%) |
|
|
Compuesto del primer evento de muerte CV, infarto de miocardio (IM) no mortal o accidente cerebrovascular no mortal (MACE) |
356 (11.8) |
20.7 |
362 (12.0) |
21.2 |
0.98 (0.84, 1.14) |
|
Muerte CV** |
169 (5.6) |
9.2 |
168 (5.6) |
9.2 |
1.00 (0.81, 1.24) |
|
IM no mortal** |
145 (4.8) |
8.3 |
142 (4.7) |
8.2 |
1.01 (0.80, 1.28) |
|
Accidente cerebrovascular no mortal** |
91 (3.0) |
5.2 |
104 (3.5) |
6.0 |
0.87 (0.66, 1.15) |
* AP=años paciente
** Un paciente puede haber experimentado más de un componente; por lo que, la suma de los componentes es mayor que el número de pacientes que experimentaron el resultado compuesto.
Figura 2. Tiempo hasta la primera aparición de 3P-MACE en el ensayo CAROLINA

DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología y administración:
Dosificación y administración recomendada: La dosificación recomendada de TRAYENTA® es de 5 mg tomada de forma oral una vez al día con o sin alimentos.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis: En caso de una sobredosis con TRAYENTA®, comunicarse con la autoridad local. La eliminación de linagliptina por hemodiálisis o diálisis peritoneal resulta poco probable.
DESCRIPCIÓN:
TRAYENTA® tabletas para uso oral contiene linagliptina un inhibidor de la enzima (DPP-4).
El nombre químico de la linagliptina es 1H-purina-2,6-diona, 8-[(3R)-3-amino-1-piperidinil]-7-(2-butin-1-il)-3,7-dihidro-3-metil-1-[(4-metil-2- quinazolinil)metil].
La fórmula empírica es C25H28N8O2 y el peso molecular es 472.54 g/mol. La fórmula estructural es:
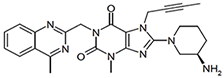
La linagliptina es una sustancia sólida blanca a amarillenta, no higroscópica o solo ligeramente higroscópica. Es muy poco soluble en agua (0.9 mg/mL). La linagliptina es soluble en metanol (aprox. 60 mg/mL), moderadamente soluble en etanol (aprox. 10 mg/mL), muy poco soluble en isopropanol (<1 mg/mL) y muy poco soluble en acetona (aprox. 1 mg/mL).
Cada tableta recubierta de TRAYENTA® contiene 5 mg de linagliptina base libre y los siguientes excipientes: manitol, almidón pregelatinizado, almidón de maíz, copovidona y estearato de magnesio. Además, la cubierta pelicular contiene los siguientes excipientes: hipromelosa, dióxido de titanio, talco, polietilenglicol y óxido de hierro rojo.
PRESENTACIÓN:
La disponibilidad de las presentaciones y depende de cada país.
Caja con blíster de 30 tabletas recubiertas de 5 mg e instructivo anexo.
Caja con blíster de 10 tabletas recubiertas de 5 mg e instructivo anexo.
Caja con blíster de 3 tabletas recubiertas de 5 mg e instructivo anexo (muestra médica).
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 30 °C.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje al alcance de los niños. No se use en el embarazo, en la lactancia ni en niños menores de 18 años.
Hecho en México por:
BOEHRINGER INGELHEIM PROMECO S.A. de C.V.
Calle del Maíz No. 49
Col. Barrio Xaltocan, C.P. 16090
Xochimilco, Ciudad de México, México
o
Hecho en Estados Unidos de América por:
West-Ward Columbus Inc.
Wilson Road 1809, 43228
Columbus, Ohio, EUA
Para:
Boehringer Ingelheim Promeco S.A. de C.V.
Calle del Maíz No. 49
Col. Barrio Xaltocan, C.P. 16090
Xochimilco, Ciudad de México, México
®Marca registrada
Fecha de revisión de la monografía: 20 de Junio de 2023


